
Severe COVID-19 Shares a Common Neutrophil Activation Signature with Other Acute Inflammatory States

 , , ,
, , ,  ,
,  , ,
, ,  , , , , ,
, , , , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Curation
2.2. Differential Expression Analysis and Visualization of Transcriptional Overlap
2.3. Single Cell RNAseq Analysis
2.4. Interactome Analysis
2.5. Enrichment Analysis and Data Visualization
2.6. Correlation Analysis
2.7. Proteome Data Analysis
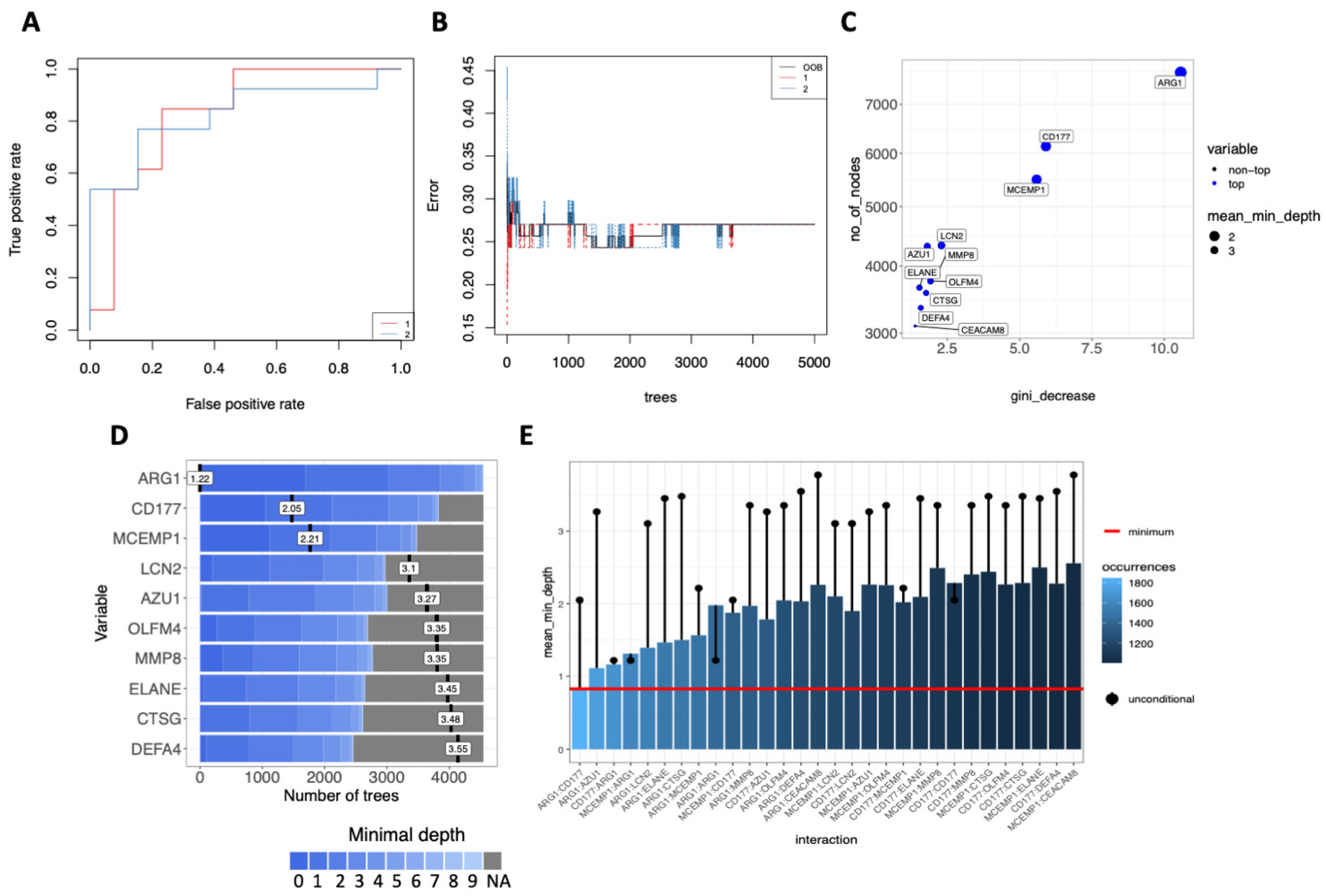
2.8. Decision-Tree Classification and Machine Learning Model Predictors
3. Results
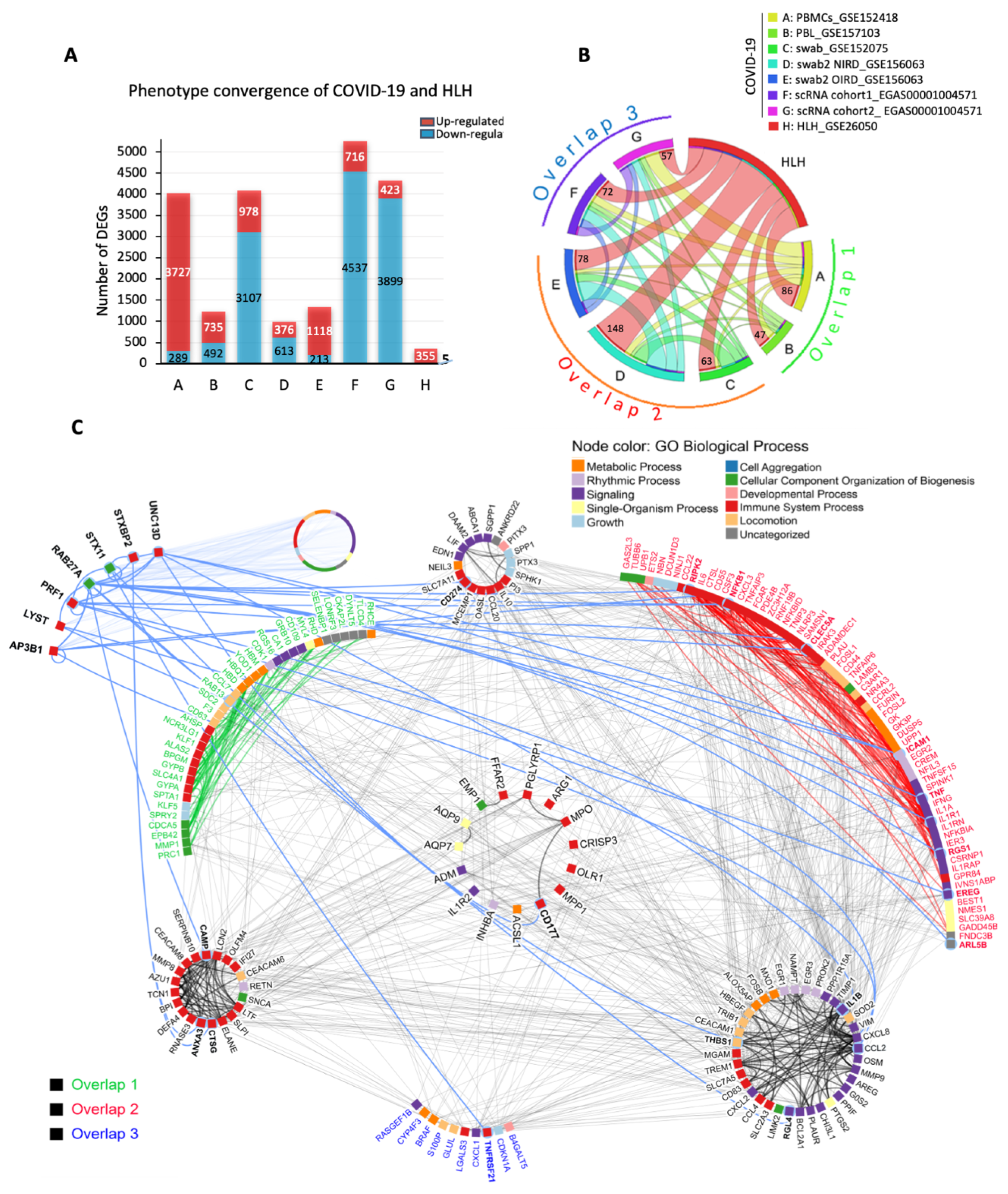
3.1. The Transcriptional Overlap between COVID-19 and HLH
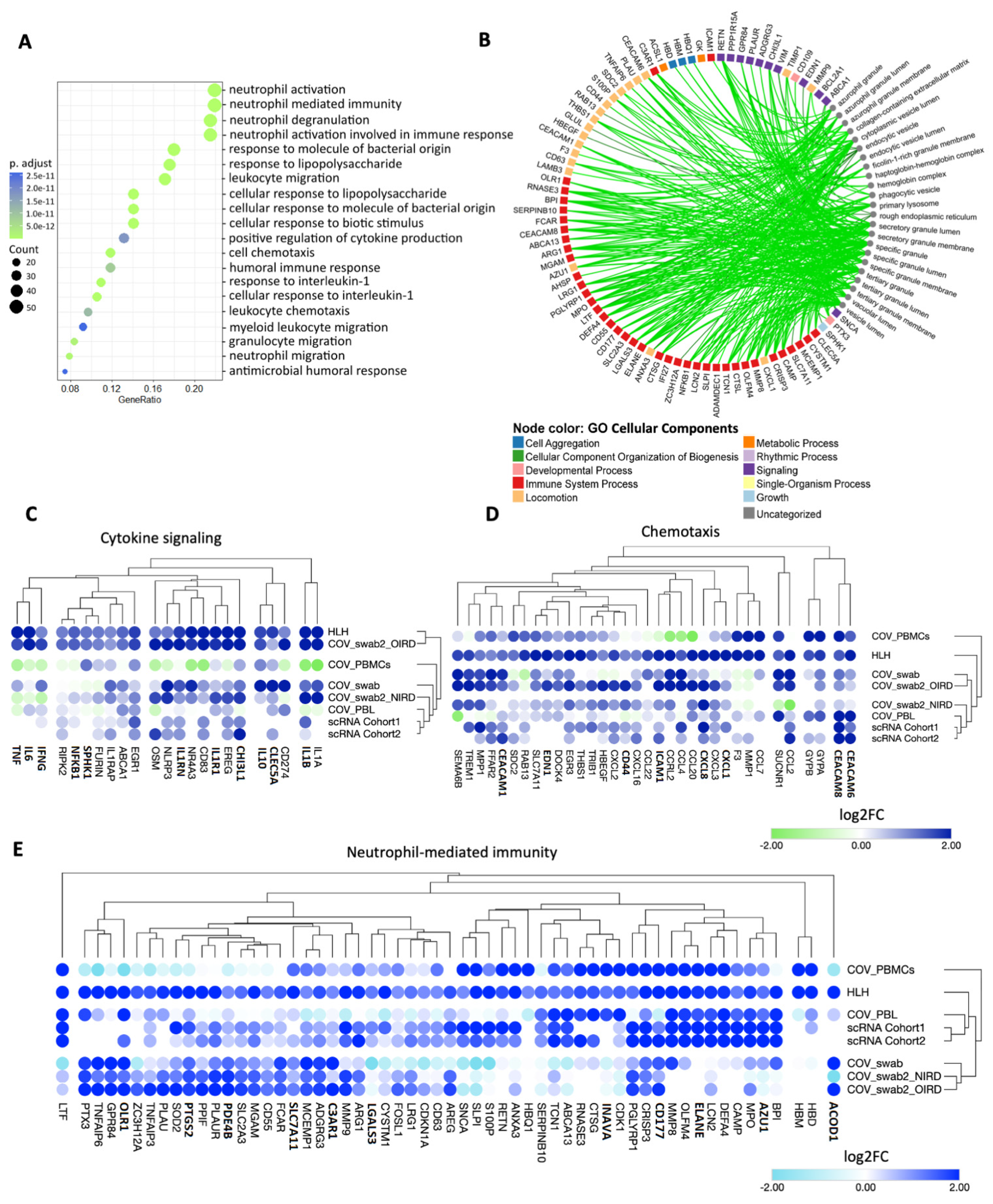
3.2. Cytokine/Chemotaxis and Neutrophil Signatures Predominate in COVID-19 and HLH
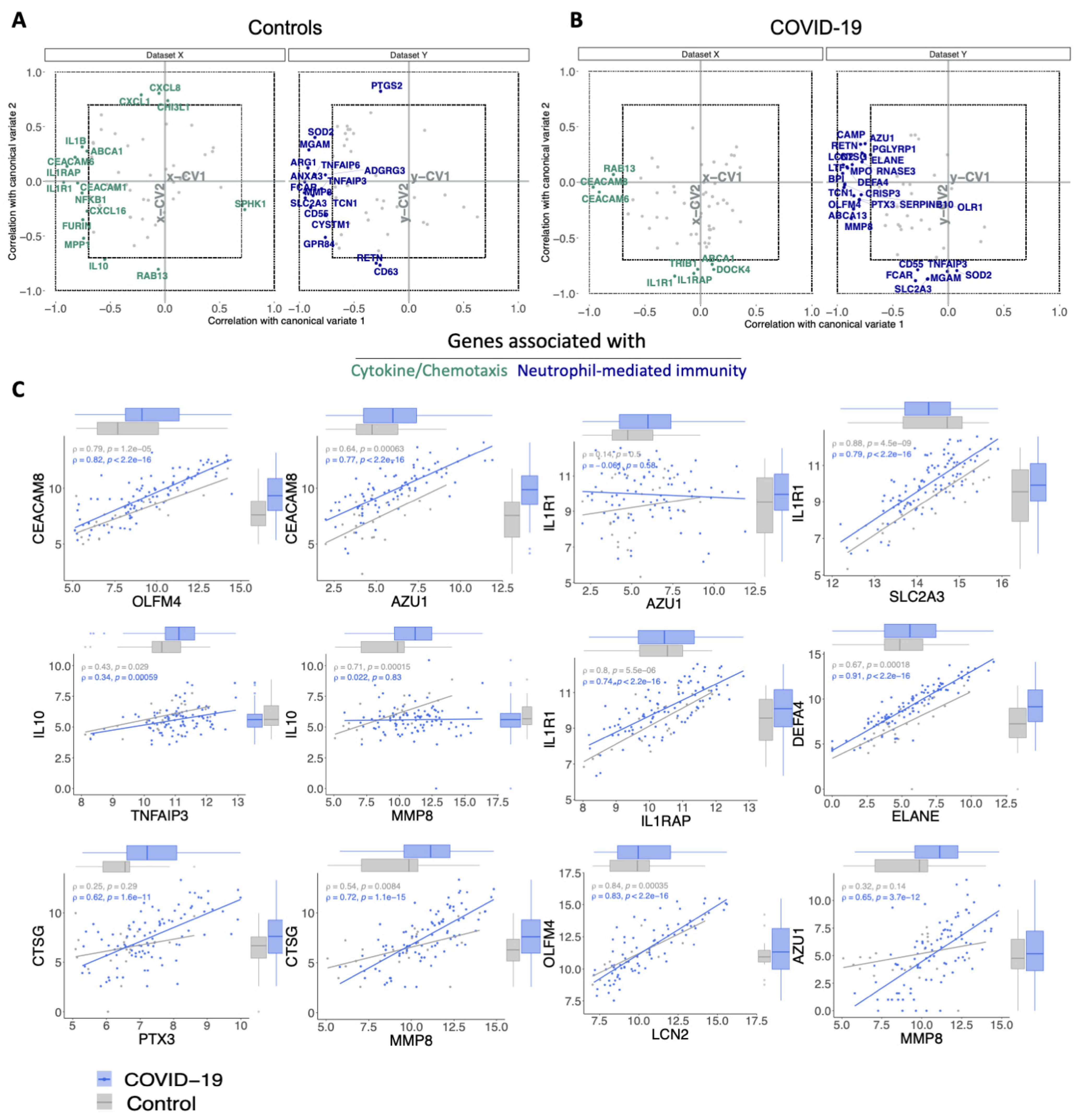
3.3. The Relationship between Cytokine/Chemotaxis and Neutrophil-Mediated Immunity Gene Signatures
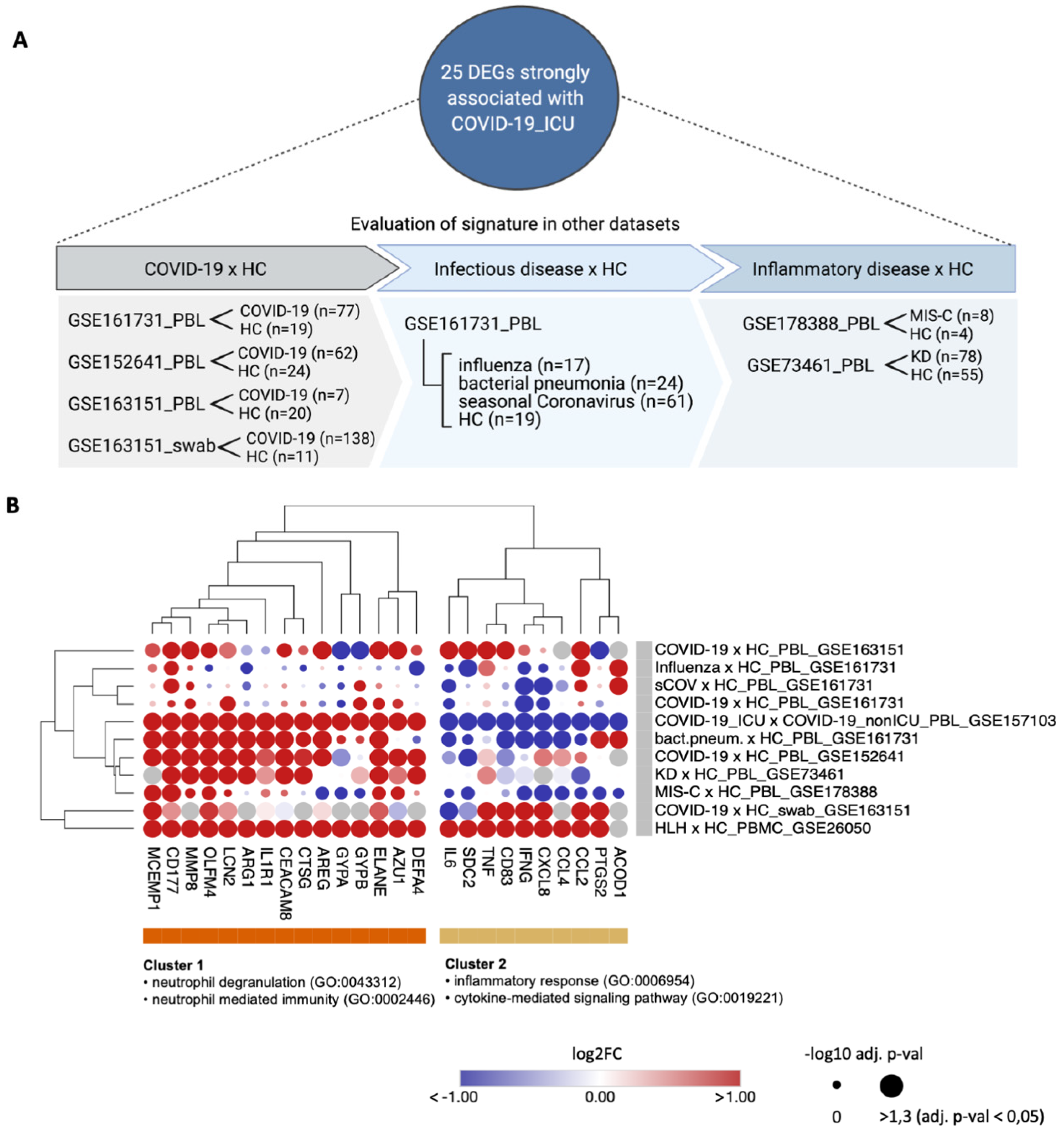
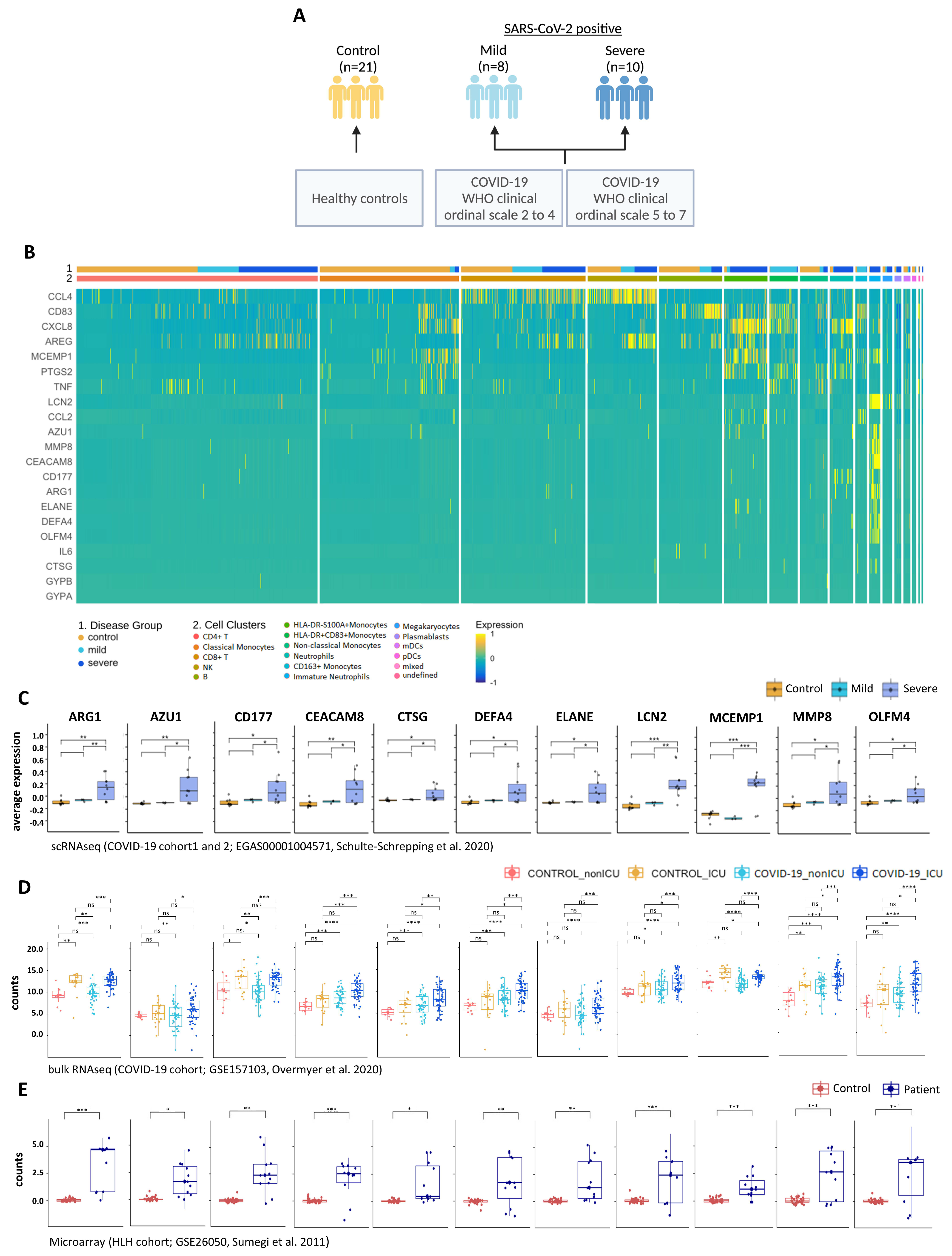
3.4. Transcripts Stratifying Severe COVID-19 from Other Respiratory Diseases Are Highly Dysregulated in HLH and Other Acute Inflammatory States
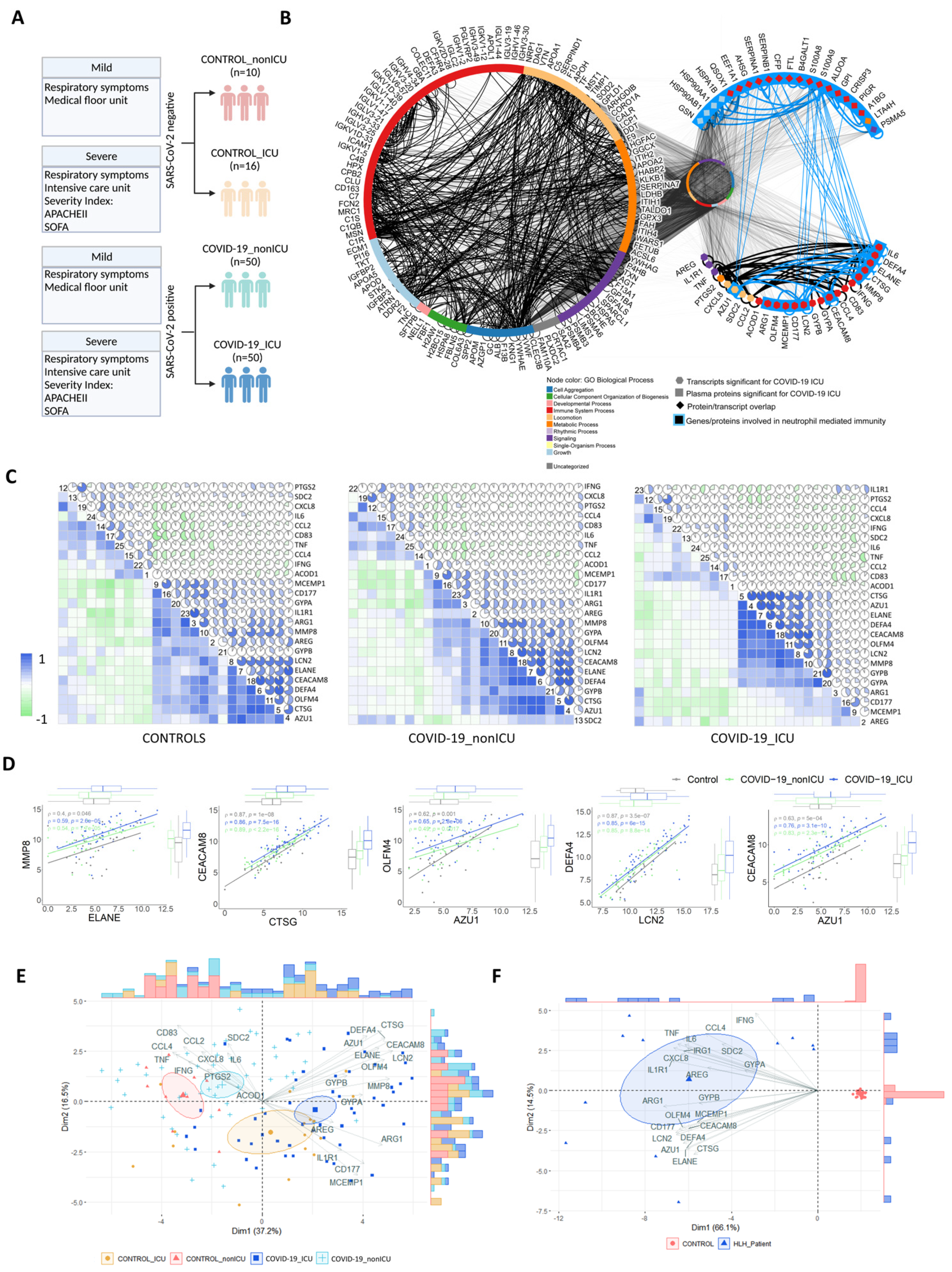
3.5. Multi-Layered Transcriptomic Analysis Associates Neutrophil Activation Signature with COVID-19 Severity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Chen, L.Y.C.; Quach, T.T.T. COVID-19 cytokine storm syndrome: A threshold concept. Lancet Microbe 2021, 2, e49. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Santoso, A.; Pranata, R.; Wibowo, A.; Al-Farabi, M.J.; Biomed, M.; Huang, I.; Antariksa, B. Cardiac injury is associated with mortality and critically ill pneumonia in COVID-19: A meta-analysis. Am. J. Emerg. Med. 2021, 44, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Ørskov, S.; Frost Nielsen, B.; Føns, S.; Sneppen, K.; Simonsen, L. The COVID-19 pandemic: Key considerations for the epidemic and its control. APMIS 2021, 129, 408–420. [Google Scholar] [CrossRef]
- Pandit, B.; Bhattacharjee, S.; Bhattacharjee, B. Association of clade-G SARS-CoV-2 viruses and age with increased mortality rates across 57 countries and India. Infect. Genet. Evol. 2021, 90, 104734. [Google Scholar] [CrossRef]
- Garcia-Beltran, W.F.; Lam, E.C.; St. Denis, K.; Nitido, A.D.; Garcia, Z.H.; Hauser, B.M.; Feldman, J.; Pavlovic, M.N.; Gregory, D.J.; Poznansky, M.C.; et al. Multiple SARS-CoV-2 variants escape neutralization by vaccine-induced humoral immunity. Cell 2021, 184, 2372–2383.e9. [Google Scholar] [CrossRef]
- Kalfaoglu, B.; Almeida-Santos, J.; Tye, C.A.; Satou, Y.; Ono, M. T-Cell Hyperactivation and Paralysis in Severe COVID-19 Infection Revealed by Single-Cell Analysis. Front. Immunol. 2020, 11, 2605. [Google Scholar] [CrossRef]
- Wang, E.Y.; Mao, T.; Klein, J.; Dai, Y.; Huck, J.D.; Jaycox, J.R.; Liu, F.; Zhou, T.; Israelow, B.; Wong, P.; et al. Diverse functional autoantibodies in patients with COVID-19. Nature 2021, 595, 283–288. [Google Scholar] [CrossRef]
- Cabral-Marques, O.; Halpert, G.; Schimke, L.F.; Ostrinski, Y.; Zyskind, I.; Lattin, M.T.; Tran, F.; Schreiber, S.; Marques, A.H.C.; Filgueiras, I.S.; et al. The relationship between autoantibodies targeting GPCRs and the renin-angiotensin system associates with COVID-19 severity. medRxiv 2021. [Google Scholar] [CrossRef]
- Mathew, D.; Giles, J.R.; Baxter, A.E.; Oldridge, D.A.; Greenplate, A.R.; Wu, J.E.; Alanio, C.; Kuri-Cervantes, L.; Pampena, M.B.; D’Andrea, K.; et al. Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science 2020, 369, eaba8511. [Google Scholar] [CrossRef] [PubMed]
- Laing, A.G.; Lorenc, A.; del Molino del Barrio, I.; Das, A.; Fish, M.; Monin, L.; Muñoz-Ruiz, M.; McKenzie, D.R.; Hayday, T.S.; Francos-Quijorna, I.; et al. A dynamic COVID-19 immune signature includes associations with poor prognosis. Nat. Med. 2020, 26, 1623–1635. [Google Scholar] [CrossRef]
- Azkur, A.K.; Akdis, M.; Azkur, D.; Sokolowska, M.; van de Veen, W.; Brüggen, M.C.; O’Mahony, L.; Gao, Y.; Nadeau, K.; Akdis, C.A. Immune response to SARS-CoV-2 and mechanisms of immunopathological changes in COVID-19. Allergy Eur. J. Allergy Clin. Immunol. 2020, 75, 1564–1581. [Google Scholar] [CrossRef] [PubMed]
- Koutsakos, M.; Rowntree, L.C.; Hensen, L.; Chua, B.Y.; van de Sandt, C.E.; Habel, J.R.; Zhang, W.; Jia, X.; Kedzierski, L.; Ashhurst, T.M.; et al. Integrated immune dynamics define correlates of COVID-19 severity and antibody responses. Cell Reports Med. 2021, 2, 100208. [Google Scholar] [CrossRef]
- Webb, B.J.; Peltan, I.D.; Jensen, P.; Hoda, D.; Hunter, B.; Silver, A.; Starr, N.; Buckel, W.; Grisel, N.; Hummel, E.; et al. Clinical criteria for COVID-19-associated hyperinflammatory syndrome: A cohort study. Lancet Rheumatol. 2020, 2, e754–e763. [Google Scholar] [CrossRef]
- Fajgenbaum, D.C.; June, C.H. Cytokine Storm. N. Engl. J. Med. 2020, 383, 2255–2273. [Google Scholar] [CrossRef] [PubMed]
- Yongzhi, X. COVID-19-associated cytokine storm syndrome and diagnostic principles: An old and new Issue. Emerg. Microbes Infect. 2021, 10, 266–276. [Google Scholar] [CrossRef] [PubMed]
- George, M. Hemophagocytic lymphohistiocytosis: Review of etiologies and management. J. Blood Med. 2014, 69. [Google Scholar] [CrossRef]
- Soy, M.; Atagündüz, P.; Atagündüz, I.; Sucak, G.T. Hemophagocytic lymphohistiocytosis: A review inspired by the COVID-19 pandemic. Rheumatol. Int. 2021, 41, 7–18. [Google Scholar] [CrossRef]
- Lorenz, G.; Moog, P.; Bachmann, Q.; La Rosée, P.; Schneider, H.; Schlegl, M.; Spinner, C.; Heemann, U.; Schmid, R.M.; Algül, H.; et al. Title: Cytokine release syndrome is not usually caused by secondary hemophagocytic lymphohistiocytosis in a cohort of 19 critically ill COVID-19 patients. Sci. Rep. 2020, 10, 18277. [Google Scholar] [CrossRef]
- Freire, P.P.; Marques, A.H.C.; Baiocchi, G.C.; Schimke, L.F.; Fonseca, D.L.M.; Salgado, R.C.; Filgueiras, I.S.; Napoleao, S.M.S.; Plaça, D.R.; Akashi, K.T.; et al. The relationship between cytokine and neutrophil gene network distinguishes SARS-CoV-2–infected patients by sex and age. JCI Insight 2021, 6, e147535. [Google Scholar] [CrossRef] [PubMed]
- Alqutami, F.; Senok, A.; Hachim, M. COVID-19 Transcriptomic Atlas: A Comprehensive Analysis of COVID-19 Related Transcriptomics Datasets. Front. Genet. 2021, 12, 2500. [Google Scholar] [CrossRef] [PubMed]
- Delorey, T.M.; Ziegler, C.G.K.; Heimberg, G.; Normand, R.; Yang, Y.; Segerstolpe, Å.; Abbondanza, D.; Fleming, S.J.; Subramanian, A.; Montoro, D.T.; et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature 2021, 595, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Daamen, A.R.; Bachali, P.; Owen, K.A.; Kingsmore, K.M.; Hubbard, E.L.; Labonte, A.C.; Robl, R.; Shrotri, S.; Grammer, A.C.; Lipsky, P.E. Comprehensive transcriptomic analysis of COVID-19 blood, lung, and airway. Sci. Rep. 2021, 11, 1–19. [Google Scholar] [CrossRef]
- Arunachalam, P.S.; Wimmers, F.; Mok, C.K.P.; Perera, R.A.P.M.; Scott, M.; Hagan, T.; Sigal, N.; Feng, Y.; Bristow, L.; Tsang, O.T.Y.; et al. Systems biological assessment of immunity to mild versus severe COVID-19 infection in humans. Science 2020, 369, 1210–1220. [Google Scholar] [CrossRef]
- Overmyer, K.A.; Shishkova, E.; Miller, I.J.; Balnis, J.; Bernstein, M.N.; Peters-Clarke, T.M.; Meyer, J.G.; Quan, Q.; Muehlbauer, L.K.; Trujillo, E.A.; et al. Large-Scale Multi-omic Analysis of COVID-19 Severity. Cell Syst. 2021, 12, 23–40. [Google Scholar] [CrossRef]
- Lieberman, N.A.P.; Peddu, V.; Xie, H.; Shrestha, L.; Huang, M.-L.; Mears, M.C.; Cajimat, M.N.; Bente, D.A.; Shi, P.-Y.; Bovier, F.; et al. In vivo antiviral host transcriptional response to SARS-CoV-2 by viral load, sex, and age. PLoS Biol. 2020, 18, e3000849. [Google Scholar] [CrossRef]
- Mick, E.; Kamm, J.; Pisco, A.O.; Ratnasiri, K.; Babik, J.M.; Calfee, C.S.; Castaneda, G.; DeRisi, J.L.; Detweiler, A.M.; Hao, S.; et al. Upper airway gene expression differentiates COVID-19 from other acute respiratory illnesses and reveals suppression of innate immune responses by SARS-CoV-2. medRxiv 2020. [Google Scholar] [CrossRef]
- Schulte-Schrepping, J.; Reusch, N.; Paclik, D.; Baßler, K.; Schlickeiser, S.; Zhang, B.; Krämer, B.; Krammer, T.; Brumhard, S.; Bonaguro, L.; et al. Severe COVID-19 Is Marked by a Dysregulated Myeloid Cell Compartment. Cell 2020, 182, 1419–1440. [Google Scholar] [CrossRef]
- Sumegi, J.; Barnes, M.G.; Nestheide, S.V.; Molleran-Lee, S.; Villanueva, J.; Zhang, K.; Risma, K.A.; Grom, A.A.; Filipovich, A.H. Gene expression profiling of peripheral blood mononuclear cells from children with active hemophagocytic lymphohistiocytosis. Blood 2011, 117, e151–e160. [Google Scholar] [CrossRef]
- Ng, D.; Granados, A.; Santos, Y.; Servellita, V.; Goldgof, G.; Meydan, C.; Sotomayor-Gonzalez, A.; Levine, A.; Balcerek, J.; Han, L.; et al. A diagnostic host response biosignature for COVID-19 from RNA profiling of nasal swabs and blood. Sci. Adv. 2021, 7, eabe5984. [Google Scholar] [CrossRef] [PubMed]
- Thair, S.A.; He, Y.D.; Hasin-Brumshtein, Y.; Sakaram, S.; Pandya, R.; Toh, J.; Rawling, D.; Remmel, M.; Coyle, S.; Dalekos, G.N.; et al. Transcriptomic similarities and differences in host response between SARS-CoV-2 and other viral infections. iScience 2021, 24, 101947. [Google Scholar] [CrossRef] [PubMed]
- McClain, M.T.; Constantine, F.J.; Henao, R.; Liu, Y.; Tsalik, E.L.; Burke, T.W.; Steinbrink, J.M.; Petzold, E.; Nicholson, B.P.; Rolfe, R.; et al. Dysregulated transcriptional responses to SARS-CoV-2 in the periphery. Nat. Commun. 2021, 12, 1079. [Google Scholar] [CrossRef] [PubMed]
- Beckmann, N.D.; Comella, P.H.; Cheng, E.; Lepow, L.; Beckmann, A.G.; Tyler, S.R.; Mouskas, K.; Simons, N.W.; Hoffman, G.E.; Francoeur, N.J.; et al. Downregulation of exhausted cytotoxic T cells in gene expression networks of multisystem inflammatory syndrome in children. Nat. Commun. 2021, 12, 4854. [Google Scholar] [CrossRef]
- Wright, V.J.; Herberg, J.A.; Kaforou, M.; Shimizu, C.; Eleftherohorinou, H.; Shailes, H.; Barendregt, A.M.; Menikou, S.; Gormley, S.; Berk, M.; et al. Diagnosis of Kawasaki Disease Using a Minimal Whole-Blood Gene Expression Signature. JAMA Pediatr. 2018, 172, e182293. [Google Scholar] [CrossRef]
- Clough, E.; Barrett, T. The Gene Expression Omnibus database. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2016; Volume 1418, pp. 93–110. [Google Scholar]
- Athar, A.; Füllgrabe, A.; George, N.; Iqbal, H.; Huerta, L.; Ali, A.; Snow, C.; Fonseca, N.A.; Petryszak, R.; Papatheodorou, I.; et al. ArrayExpress update—From bulk to single-cell expression data. Nucleic Acids Res. 2019, 47, D711–D715. [Google Scholar] [CrossRef]
- Sanchis, P.; Lavignolle, R.; Abbate, M.; Lage-Vickers, S.; Vazquez, E.; Cotignola, J.; Bizzotto, J.; Gueron, G. Analysis workflow of publicly available RNA-sequencing datasets. STAR Protoc. 2021, 2, 100478. [Google Scholar] [CrossRef]
- Humaidan, P.; Polyzos, N.P. (Meta)analyze this: Systematic reviews might lose credibility. Nat. Med. 2012, 18, 1321. [Google Scholar] [CrossRef]
- Zhou, G.; Soufan, O.; Ewald, J.; Hancock, R.E.W.; Basu, N.; Xia, J. NetworkAnalyst 3.0: A visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019, 47, W234–W241. [Google Scholar] [CrossRef]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. Voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef]
- Bardou, P.; Mariette, J.; Escudié, F.; Djemiel, C.; Klopp, C. Jvenn: An interactive Venn diagram viewer. BMC Bioinform. 2014, 15, 293. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M.; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zagar, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587. [Google Scholar] [PubMed]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M.; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef]
- Brown, K.R.; Otasek, D.; Ali, M.; McGuffin, M.J.; Xie, W.; Devani, B.; van Toch, I.L.; Jurisica, I. NAViGaTOR: Network Analysis, Visualization and Graphing Toronto. Bioinformatics 2009, 25, 3327–3329. [Google Scholar] [CrossRef]
- Kotlyar, M.; Pastrello, C.; Ahmed, Z.; Chee, J.; Varyova, Z.; Jurisica, I. IID 2021: Towards context-specific protein interaction analyses by increased coverage, enhanced annotation and enrichment analysis. Nucleic Acids Res. 2022, 50, D640–D647. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. ClusterProfiler: An R package for comparing biological themes among gene clusters. Omi. A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef]
- Starruß, J.; de Back, W.; Brusch, L.; Deutsch, A. Morpheus: A user-friendly modeling environment for multiscale and multicellular systems biology. Bioinformatics 2014, 30, 1331–1332. [Google Scholar] [CrossRef]
- Kassambara, A.; Mundt, F. Multivariate Analysis II, Practical Guide to Principal Component Methods in R. Available online: http://http://www.sthda.com/english/ (accessed on 9 February 2022).
- Jendoubi, T.; Strimmer, K. A whitening approach to probabilistic canonical correlation analysis for omics data integration. BMC Bioinform. 2019, 20, 15. [Google Scholar] [CrossRef] [PubMed]
- Konietschke, F.; Placzek, M.; Schaarschmidt, F.; Hothorn, L.A. nparcomp: An R Software Package for Nonparametric Multiple Comparisons and Simultaneous Confidence Intervals. J. Stat. Softw. 2015, 64, 1–17. [Google Scholar] [CrossRef]
- Burchett, W.W.; Ellis, A.R.; Harrar, S.W.; Bathke, A.C. Nonparametric Inference for Multivariate Data: The R Package npmv. J. Stat. Softw. 2017, 76, 1–18. [Google Scholar] [CrossRef]
- Liaw, A.; Wiener, M. Classification and Regression by randomForest. R News 2002, 2, 18–22. [Google Scholar]
- Usmani, G.N.; Woda, B.A.; Newburger, P.E. Advances in understanding the pathogenesis of HLH. Br. J. Haematol. 2013, 161, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1.30.1–1.30.33. [Google Scholar] [CrossRef]
- Lebre, M.C.; Burwell, T.; Vieira, P.L.; Lora, J.; Coyle, A.J.; Kapsenberg, M.L.; Clausen, B.E.; De Jong, E.C. Differential expression of inflammatory chemokines by Th1- and Th2-cell promoting dendritic cells: A role for different mature dendritic cell populations in attracting appropriate effector cells to peripheral sites of inflammation. Immunol. Cell Biol. 2005, 83, 525–535. [Google Scholar] [CrossRef]
- Charo, I.F.; Ransohoff, R.M. The Many Roles of Chemokines and Chemokine Receptors in Inflammation. N. Engl. J. Med. 2006, 354, 610–621. [Google Scholar] [CrossRef]
- Blandino-Rosano, M.; Perez-Arana, G.; Mellado-Gil, J.M.; Segundo, C.; Aguilar-Diosdado, M. Anti-proliferative effect of pro-inflammatory cytokines in cultured β cells is associated with extracellular signal-regulated kinase 1/2 pathway inhibition: Protective role of glucagon-like peptide -1. J. Mol. Endocrinol. 2008, 41, 35–44. [Google Scholar] [CrossRef]
- Zhang, J.M.; An, J. Cytokines, inflammation, and pain. Int. Anesthesiol. Clin. 2007, 45, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Aratani, Y. Myeloperoxidase: Its role for host defense, inflammation, and neutrophil function. Arch. Biochem. Biophys. 2018, 640, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Pohl, J.; Pereira, H.A.; Martin, N.M.; Spitznagel, J.K. Amino acid sequence of CAP37, a human neutrophil granule-derived antibacterial and monocyte-specific chemotactic glycoprotein structurally similar to neutrophil elastase. FEBS Lett. 1990, 272, 200–204. [Google Scholar] [CrossRef]
- Korkmaz, B.; Horwitz, M.S.; Jenne, D.E.; Gauthier, F. Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol. Rev. 2010, 62, 726–759. [Google Scholar] [CrossRef]
- Tralau, T.; Meyer-Hoffert, U.; Schröder, J.M.; Wiedow, O. Human leukocyte elastase and cathepsin G are specific inhibitors of C5a-dependent neutrophil enzyme release and chemotaxis. Exp. Dermatol. 2004, 13, 316–325. [Google Scholar] [CrossRef]
- Lehrer, R.I.; Lu, W. α-Defensins in human innate immunity. Immunol. Rev. 2012, 245, 84–112. [Google Scholar] [CrossRef]
- Bradley, L.M.; Douglass, M.F.; Chatterjee, D.; Akira, S.; Baaten, B.J.G. Matrix metalloprotease 9 mediates neutrophil migration into the airways in response to influenza virus-induced toll-like receptor signaling. PLoS Pathog. 2012, 8, e1002641. [Google Scholar] [CrossRef]
- Lin, W.C.; Fessler, M.B. Regulatory mechanisms of neutrophil migration from the circulation to the airspace. Cell. Mol. Life Sci. 2021, 1, 3. [Google Scholar] [CrossRef]
- Cabral-Marques, O.; Schimke, L.F.; de Oliveira, E.B.; El Khawanky, N.; Ramos, R.N.; Al-Ramadi, B.K.; Segundo, G.R.S.; Ochs, H.D.; Condino-Neto, A. Flow Cytometry Contributions for the Diagnosis and Immunopathological Characterization of Primary Immunodeficiency Diseases With Immune Dysregulation. Front. Immunol. 2019, 10, 2742. [Google Scholar] [CrossRef]
- Marsh, R.A.; Satake, N.; Biroschak, J.; Jacobs, T.; Johnson, J.; Jordan, M.B.; Bleesing, J.J.; Filipovich, A.H.; Zhang, K. STX11 mutations and clinical phenotypes of familial hemophagocytic lymphohistiocytosis in North America. Pediatr. Blood Cancer 2010, 55, 134–140. [Google Scholar] [CrossRef]
- Wada, T.; Sakakibara, Y.; Nishimura, R.; Toma, T.; Ueno, Y.; Horita, S.; Tanaka, T.; Nishi, M.; Kato, K.; Yasumi, T.; et al. Down-regulation of CD5 expression on activated CD8+ T cells in familial hemophagocytic lymphohistiocytosis with perforin gene mutations. Hum. Immunol. 2013, 74, 1579–1585. [Google Scholar] [CrossRef] [PubMed]
- Janka, G.E. Familial and acquired hemophagocytic lymphohistiocytosis. Eur. J. Pediatr. 2007, 166, 95–109. [Google Scholar] [CrossRef]
- Dell’Acqua, F.; Saettini, F.; Castelli, I.; Badolato, R.; Notarangelo, L.D.; Rizzari, C. Hermansky-Pudlak syndrome type II and lethal hemophagocytic lymphohistiocytosis: Case description and review of the literature. J. Allergy Clin. Immunol. Pract. 2019, 7, 2476–2478.e5. [Google Scholar] [CrossRef]
- Zhao, X.W.; Gazendam, R.P.; Drewniak, A.; Van Houdt, M.; Tool, A.T.J.; Van Hamme, J.L.; Kustiawan, I.; Meijer, A.B.; Janssen, H.; Russell, D.G.; et al. Defects in neutrophil granule mobilization and bactericidal activity in familial hemophagocytic lymphohistiocytosis type 5 (FHL-5) syndrome caused by STXBP2/Munc18-2 mutations. Blood 2013, 122, 109–111. [Google Scholar] [CrossRef]
- D’Orlando, O.; Zhao, F.; Kasper, B.; Orinska, Z.; Müller, J.; Hermans-Borgmeyer, I.; Griffiths, G.M.; Zur Stadt, U.; Bulfone-Paus, S. Syntaxin 11 is required for NK and CD8+ T-cell cytotoxicity and neutrophil degranulation. Eur. J. Immunol. 2013, 43, 194–208. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tang, W.; Zhang, H.; Niu, X.; Xu, Y.; Zhang, J.; Gao, K.; Pan, W.; Boggon, T.J.; Toomre, D.; et al. A Network of interactions enables CCM3 and STK24 to coordinate UNC13D-driven vesicle exocytosis in neutrophils. Dev. Cell 2013, 27, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Catz, S.D. The role of Rab27a in the regulation of neutrophil function. Cell. Microbiol. 2014, 16, 1301–1310. [Google Scholar] [CrossRef] [PubMed]
- Rickman, J.M.; Wang, Y.; Rollett, A.D.; Harmer, M.P.; Compson, C. Data analytics using canonical correlation analysis and Monte Carlo simulation. NPJ Comput. Mater. 2017, 3, 26. [Google Scholar] [CrossRef]
- Ferreira, F.; Bota, D.; Bross, A.; Mélot, C.; Vincent, J. Serial evaluation of the SOFA score to predict outcome in critically ill patients. JAMA 2001, 286, 1754–1758. [Google Scholar] [CrossRef]
- Lever, J.; Krzywinski, M.; Altman, N. Points of Significance: Principal component analysis. Nat. Methods 2017, 14, 641–642. [Google Scholar] [CrossRef]
- Ringnér, M. What is principal component analysis? Nat. Biotechnol. 2008, 26, 303–304. [Google Scholar] [CrossRef] [PubMed]
- Moser, B. T-cell memory: The importance of chemokine-mediated cell attraction. Curr. Biol. 2006, 16, R504–R507. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Castellino, F.; Huang, A.Y.; Altan-Bonnet, G.; Stoll, S.; Scheinecker, C.; Germain, R.N. Chemokines enhance immunity by guiding naive CD8+ T cells to sites of CD4+ T cell-dendritic cell interaction. Nature 2006, 440, 890–895. [Google Scholar] [CrossRef] [PubMed]
- Pinho, M.P.; Migliori, I.K.; Flatow, E.A.; Barbuto, J.A.M. Dendritic cell membrane CD83 enhances immune responses by boosting intracellular calcium release in T lymphocytes. J. Leukoc. Biol. 2014, 95, 755–762. [Google Scholar] [CrossRef]
- Krzyzak, L.; Seitz, C.; Urbat, A.; Hutzler, S.; Ostalecki, C.; Gläsner, J.; Hiergeist, A.; Gessner, A.; Winkler, T.H.; Steinkasserer, A.; et al. CD83 Modulates B Cell Activation and Germinal Center Responses. J. Immunol. 2016, 196, 3581–3594. [Google Scholar] [CrossRef]
- Silvestre-Roig, C.; Fridlender, Z.G.; Glogauer, M.; Scapini, P. Neutrophil Diversity in Health and Disease. Trends Immunol. 2019, 40, 565–583. [Google Scholar] [CrossRef]
- Ostendorf, L.; Mothes, R.; van Koppen, S.; Lindquist, R.L.; Bellmann-Strobl, J.; Asseyer, S.; Ruprecht, K.; Alexander, T.; Niesner, R.A.; Hauser, A.E.; et al. Low-Density Granulocytes Are a Novel Immunopathological Feature in Both Multiple Sclerosis and Neuromyelitis Optica Spectrum Disorder. Front. Immunol. 2019, 10, 2725. [Google Scholar] [CrossRef]
- Carmona-Rivera, C.; Kaplan, M.J. Low-density granulocytes: A distinct class of neutrophils in systemic autoimmunity. Semin. Immunopathol. 2013, 35, 455–463. [Google Scholar] [CrossRef]
- Singh, A.K.; Jena, A.; Kumar-M, P.; Sharma, V.; Sebastian, S. Risk and outcomes of coronavirus disease in patients with inflammatory bowel disease: A systematic review and meta-analysis. United Eur. Gastroenterol. J. 2021, 9, 159–176. [Google Scholar] [CrossRef]
- Gerayeli, F.V.; Milne, S.; Cheung, C.; Li, X.; Yang, C.W.T.; Tam, A.; Choi, L.H.; Bae, A.; Sin, D.D. COPD and the risk of poor outcomes in COVID-19: A systematic review and meta-analysis. EClinicalMedicine 2021, 33, 100789. [Google Scholar] [CrossRef]
- Almansa, R.; Socias, L.; Sanchez-Garcia, M.; Martín-Loeches, I.; Del Olmo, M.; Andaluz-Ojeda, D.; Bobillo, F.; Rico, L.; Herrero, A.; Roig, V.; et al. Critical COPD respiratory illness is linked to increased transcriptomic activity of neutrophil proteases genes. BMC Res. Notes 2012, 5, 401. [Google Scholar] [CrossRef] [PubMed]
- Sng, J.H.J.; Prazakova, S.; Thomas, P.S.; Herbert, C. MMP-8, MMP-9 and Neutrophil Elastase in Peripheral Blood and Exhaled Breath Condensate in COPD. COPD J. Chronic Obstr. Pulm. Dis. 2017, 14, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Zhu, H.; Zuo, X.; Luo, H. Cathepsin g and its role in inflammation and autoimmune diseases. Arch. Rheumatol. 2018, 33, 748–749. [Google Scholar] [CrossRef]
- Asad, S.; Wegler, C.; Ahl, D.; Bergström, C.A.S.; Phillipson, M.; Artursson, P.; Teleki, A. Proteomics-Informed Identification of Luminal Targets For In Situ Diagnosis of Inflammatory Bowel Disease. J. Pharm. Sci. 2021, 110, 239–250. [Google Scholar] [CrossRef]
- Reusch, N.; De Domenico, E.; Bonaguro, L.; Schulte-Schrepping, J.; Baßler, K.; Schultze, J.L.; Aschenbrenner, A.C. Neutrophils in COVID-19. Front. Immunol. 2021, 12, 952. [Google Scholar] [CrossRef] [PubMed]
- Metzemaekers, M.; Cambier, S.; Blanter, M.; Vandooren, J.; Carvalho, A.C.; Malengier-Devlies, B.; Vanderbeke, L.; Jacobs, C.; Coenen, S.; Martens, E.; et al. Kinetics of peripheral blood neutrophils in severe coronavirus disease 2019. Clin. Transl. Immunol. 2021, 10, e1271. [Google Scholar] [CrossRef] [PubMed]
- Meizlish, M.L.; Pine, A.B.; Bishai, J.D.; Goshua, G.; Nadelmann, E.R.; Simonov, M.; Chang, C.H.; Zhang, H.; Shallow, M.; Bahel, P.; et al. A neutrophil activation signature predicts critical illness and mortality in COVID-19. Blood Adv. 2021, 5, 1164–1177. [Google Scholar] [CrossRef]
- Ackermann, M.; Anders, H.-J.; Bilyy, R.; Bowlin, G.L.; Daniel, C.; De Lorenzo, R.; Egeblad, M.; Henneck, T.; Hidalgo, A.; Hoffmann, M.; et al. Patients with COVID-19: In the dark-NETs of neutrophils. Cell Death Differ. 2021, 11, 12. [Google Scholar] [CrossRef]
- Albeituni, S.; Verbist, K.C.; Tedrick, P.E.; Tillman, H.; Picarsic, J.; Bassett, R.; Nichols, K.E. Mechanisms of action of ruxolitinib in murine models of hemophagocytic lymphohistiocytosis. Blood 2019, 134, 147–159. [Google Scholar] [CrossRef]
- Ding, J.; Hostallero, D.E.; El Khili, M.R.; Fonseca, G.J.; Milette, S.; Noorah, N.; Guay-Belzile, M.; Spicer, J.; Daneshtalab, N.; Sirois, M.; et al. A network-informed analysis of SARS-CoV-2 and hemophagocytic lymphohistiocytosis genes’ interactions points to Neutrophil extracellular traps as mediators of thrombosis in COVID-19. PLoS Comput. Biol. 2021, 17, e1008810. [Google Scholar] [CrossRef]
- Takeshita, S.; Kawamura, Y.; Kanai, T.; Yoshida, Y.; Tsujita, Y.; Nonoyama, S. The Role of Neutrophil Activation in the Pathogenesis of Kawasaki Disease. Pediatr. Infect. Dis. Open Access 2017, 3, 1. [Google Scholar] [CrossRef]
- Biezeveld, M.H.; Van Mierlo, G.; Lutter, R.; Kuipers, I.M.; Dekker, T.; Hack, C.E.; Newburger, J.W.; Kuijpers, T.W. Sustained activation of neutrophils in the course of Kawasaki disease: An association with matrix metalloproteinases. Clin. Exp. Immunol. 2005, 141, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Qian, W.; Yu, Z.; Xu, T.; Ju, L.; Hua, Q.; Wang, Y.; Ling, J.J.; Lv, H. Increased Neutrophil Respiratory Burst Predicts the Risk of Coronary Artery Lesion in Kawasaki Disease. Front. Pediatr. 2020, 8, 391. [Google Scholar] [CrossRef] [PubMed]
- Balamayooran, G.; Batra, S.; Fessler, M.B.; Happel, K.I.; Jeyaseelan, S. Mechanisms of Neutrophil Accumulation in the Lungs Against Bacteria. Am. J. Respir. Cell Mol. Biol. 2010, 43, 5. [Google Scholar] [CrossRef] [PubMed]
- Margraf, A.; Ley, K.; Zarbock, A. Neutrophil Recruitment: From Model Systems to Tissue-Specific Patterns. Trends Immunol. 2019, 40, 613–634. [Google Scholar] [CrossRef] [PubMed]
- Mortaz, E.; Alipoor, S.D.; Adcock, I.M.; Mumby, S.; Koenderman, L. Update on neutrophil function in severe inflammation. Front. Immunol. 2018, 9, 2171. [Google Scholar] [CrossRef]
- Lucas, C.; Wong, P.; Klein, J.; Castro, T.B.R.; Silva, J.; Sundaram, M.; Ellingson, M.K.; Mao, T.; Oh, J.E.; Israelow, B.; et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 2020, 584, 463–469. [Google Scholar] [CrossRef]
- Li, Y.; Duche, A.; Sayer, M.R.; Roosan, D.; Khalafalla, F.G.; Ostrom, R.S.; Totonchy, J.; Roosan, M.R. SARS-CoV-2 early infection signature identified potential key infection mechanisms and drug targets. BMC Genom. 2021, 22, 125. [Google Scholar] [CrossRef]
- Gardinassi, L.G.; Souza, C.O.S.; Sales-Campos, H.; Fonseca, S.G. Immune and Metabolic Signatures of COVID-19 Revealed by Transcriptomics Data Reuse. Front. Immunol. 2020, 11, 1636. [Google Scholar] [CrossRef]
- Andonegui-Elguera, S.; Taniguchi-Ponciano, K.; Gonzalez-Bonilla, C.R.; Torres, J.; Mayani, H.; Herrera, L.A.; Peña-Martínez, E.; Silva-Román, G.; Vela-Patiño, S.; Ferreira-Hermosillo, A.; et al. Molecular Alterations Prompted by SARS-CoV-2 Infection: Induction of Hyaluronan, Glycosaminoglycan and Mucopolysaccharide Metabolism. Arch. Med. Res. 2020, 51, 645–653. [Google Scholar] [CrossRef]
- Protasio Veras, F.; Pontelli, M.; Silva, C.; Toller-Kawahisa, J.; de Lima, M.; Nascimento, D.; Schneider, A.; Caetité, D.; Tavares, L.; Paiva, I.; et al. SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J. Exp. Med. 2020, 217, e20201129. [Google Scholar] [CrossRef] [PubMed]
- Derakhshani, A.; Hemmat, N.; Asadzadeh, Z.; Ghaseminia, M.; Shadbad, M.A.; Jadideslam, G.; Silvestris, N.; Racanelli, V.; Baradaran, B. Arginase 1 (Arg1) as an Up-Regulated Gene in COVID-19 Patients: A Promising Marker in COVID-19 Immunopathy. J. Clin. Med. 2021, 10, 1051. [Google Scholar] [CrossRef] [PubMed]
- Akgun, E.; Tuzuner, M.B.; Sahin, B.; Kilercik, M.; Kulah, C.; Cakiroglu, H.N.; Serteser, M.; Unsal, I.; Baykal, A.T. Proteins associated with neutrophil degranulation are upregulated in nasopharyngeal swabs from SARS-CoV-2 patients. PLoS ONE 2020, 15, e0240012. [Google Scholar] [CrossRef]
- Agresti, N.; Lalezari, J.P.; Amodeo, P.P.; Mody, K.; Mosher, S.F.; Seethamraju, H.; Kelly, S.A.; Pourhassan, N.Z.; Sudduth, C.D.; Bovinet, C.; et al. Disruption of CCR5 signaling to treat COVID-19-associated cytokine storm: Case series of four critically ill patients treated with leronlimab. J. Transl. Autoimmun. 2021, 4, 100083. [Google Scholar] [CrossRef] [PubMed]
- Guaraldi, G.; Meschiari, M.; Cozzi-Lepri, A.; Milic, J.; Tonelli, R.; Menozzi, M.; Franceschini, E.; Cuomo, G.; Orlando, G.; Borghi, V.; et al. Tocilizumab in patients with severe COVID-19: A retrospective cohort study. Lancet Rheumatol. 2020, 2, e474–e484. [Google Scholar] [CrossRef]
- Robinson, P.C.; Liew, D.F.L.; Liew, J.W.; Monaco, C.; Richards, D.; Shivakumar, S.; Tanner, H.L.; Feldmann, M. The Potential for Repurposing Anti-TNF as a Therapy for the Treatment of COVID-19. Med 2020, 1, 90–102. [Google Scholar] [CrossRef]
- Huet, T.; Beaussier, H.; Voisin, O.; Jouveshomme, S.; Dauriat, G.; Lazareth, I.; Sacco, E.; Naccache, J.M.; Bézie, Y.; Laplanche, S.; et al. Anakinra for severe forms of COVID-19: A cohort study. Lancet Rheumatol. 2020, 2, e393–e400. [Google Scholar] [CrossRef]
- Gozzetti, A.; Capochiani, E.; Bocchia, M. The Janus kinase 1/2 inhibitor ruxolitinib in COVID-19. Leukemia 2020, 34, 2815–2816. [Google Scholar] [CrossRef]
- Rizk, J.G.; Kalantar-Zadeh, K.; Mehra, M.R.; Lavie, C.J.; Rizk, Y.; Forthal, D.N. Pharmaco-Immunomodulatory Therapy in COVID-19. Drugs 2020, 80, 1267–1292. [Google Scholar] [CrossRef]
- Chiang, C.C.; Korinek, M.; Cheng, W.J.; Hwang, T.L. Targeting Neutrophils to Treat Acute Respiratory Distress Syndrome in Coronavirus Disease. Front. Pharmacol. 2020, 11, 1576. [Google Scholar] [CrossRef]
- Seery, V.; Raiden, S.C.; Algieri, S.C.; Grisolía, N.A.; Filippo, D.; De Carli, N.; Di Lalla, S.; Cairoli, H.; Chiolo, M.J.; Meregalli, C.N.; et al. Blood neutrophils from children with COVID-19 exhibit both inflammatory and anti-inflammatory markers. EBioMedicine 2021, 67, 103357. [Google Scholar] [CrossRef] [PubMed]
- Dennison, D.; Al Khabori, M.; Al Mamari, S.; Aurelio, A.; Al Hinai, H.; Al Maamari, K.; Alshekaili, J.; Al Khadouri, G. Circulating activated neutrophils in COVID-19: An independent predictor for mechanical ventilation and death. Int. J. Infect. Dis. 2021, 106, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Gu, R.; Mao, T.; Lu, Q.; Tianjiao Su, T.; Wang, J. Myeloid dysregulation and therapeutic intervention in COVID-19. Semin. Immunol. 2021, 55, 101524. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data-Base | Dataset ID | Seq. Method | Sample Type | Disease Type of Patients (Sample Size) | Type of Controls (Sample Size) | Original Study |
|---|---|---|---|---|---|---|
| GEO | GSE152418 | bulk-RNA seq | PBMC | COVID-19 (n = 17) | healthy controls (n = 17) | Arunachalam et al., 2020 [25] |
| GEO | GSE157103 | bulk-RNA seq | PBL | COVID-19_ICU (n = 50) COVID-19_nonICU (n = 50) | SARS-CoV-2 negative ICU (n = 16), SARS-CoV-2 negative nonICU (n = 10) | Overmyer et al., 2020 [26] |
| GEO | GSE152075 | bulk-RNA seq | nph swab | COVID-19 (n = 430) | SARS-CoV-2 negative (n = 54) | Liebermann et al., 2020 [27] |
| GEO | GSE156063 | bulk-RNA seq | nph swab | COVID-19 (n = 93) | NIRD (n = 100) OIRD (n = 41) | Mick et al., 2020 [28] |
| EGA | EGAS 00001004571 | scRNA seq | PBL/ PBMC | Cohort1: COVID-19 mild (n = 8), COVID-19 severe (n = 10) Cohort2: COVID-19 (n = 17) | healthy controls (n = 21) healthy controls (n = 13) | Schulte-Schrepping et al., 2020 [29] |
| GEO | GSE26050 | microarray | PBMC | HLH (n = 11) | healthy controls (n = 33) | Sumegi et al., 2011 [30] |
| GEO | GSE163151 | bulk-RNA seq | nph swab PBL | COVID-19 (n = 138) COVID-19 (n = 7) | healthy controls (n = 11) healthy controls (n = 20) | Ng et al., 2021 [31] |
| GEO | GSE152641 | bulk-RNA seq | PBL | COVID-19 (n = 62) | healthy controls (n = 24) | Thair et al., 2021 [32] |
| GEO | GSE161731 | bulk-RNA seq | PBL | COVID-19 (n = 77) influenza (n = 17) bact. pneum. (n = 24) seasonal CoV (n = 61) | healthy controls (n = 19) | McClain et al., 2021 [33] |
| GEO | GSE178388 | bulk-RNA seq | PBL | MIS-C (n = 8) | healthy controls (n = 4) | Beckmann et al., 2021 [34] |
| GEO | GSE73461 | microarray | PBL | KD (n = 78) | healthy controls (n = 55) | Wright et al., 2018 [35] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schimke, L.F.; Marques, A.H.C.; Baiocchi, G.C.; de Souza Prado, C.A.; Fonseca, D.L.M.; Freire, P.P.; Rodrigues Plaça, D.; Salerno Filgueiras, I.; Coelho Salgado, R.; Jansen-Marques, G.; et al. Severe COVID-19 Shares a Common Neutrophil Activation Signature with Other Acute Inflammatory States. Cells 2022, 11, 847. https://doi.org/10.3390/cells11050847
Schimke LF, Marques AHC, Baiocchi GC, de Souza Prado CA, Fonseca DLM, Freire PP, Rodrigues Plaça D, Salerno Filgueiras I, Coelho Salgado R, Jansen-Marques G, et al. Severe COVID-19 Shares a Common Neutrophil Activation Signature with Other Acute Inflammatory States. Cells. 2022; 11(5):847. https://doi.org/10.3390/cells11050847
Chicago/Turabian StyleSchimke, Lena F., Alexandre H. C. Marques, Gabriela Crispim Baiocchi, Caroline Aliane de Souza Prado, Dennyson Leandro M. Fonseca, Paula Paccielli Freire, Desirée Rodrigues Plaça, Igor Salerno Filgueiras, Ranieri Coelho Salgado, Gabriel Jansen-Marques, and et al. 2022. "Severe COVID-19 Shares a Common Neutrophil Activation Signature with Other Acute Inflammatory States" Cells 11, no. 5: 847. https://doi.org/10.3390/cells11050847
APA StyleSchimke, L. F., Marques, A. H. C., Baiocchi, G. C., de Souza Prado, C. A., Fonseca, D. L. M., Freire, P. P., Rodrigues Plaça, D., Salerno Filgueiras, I., Coelho Salgado, R., Jansen-Marques, G., Rocha Oliveira, A. E., Peron, J. P. S., Cabral-Miranda, G., Barbuto, J. A. M., Camara, N. O. S., Calich, V. L. G., Ochs, H. D., Condino-Neto, A., Overmyer, K. A., ... Cabral-Marques, O. (2022). Severe COVID-19 Shares a Common Neutrophil Activation Signature with Other Acute Inflammatory States. Cells, 11(5), 847. https://doi.org/10.3390/cells11050847