
MG132 Induces Progerin Clearance and Improves Disease Phenotypes in HGPS-like Patients’ Cells

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Samples
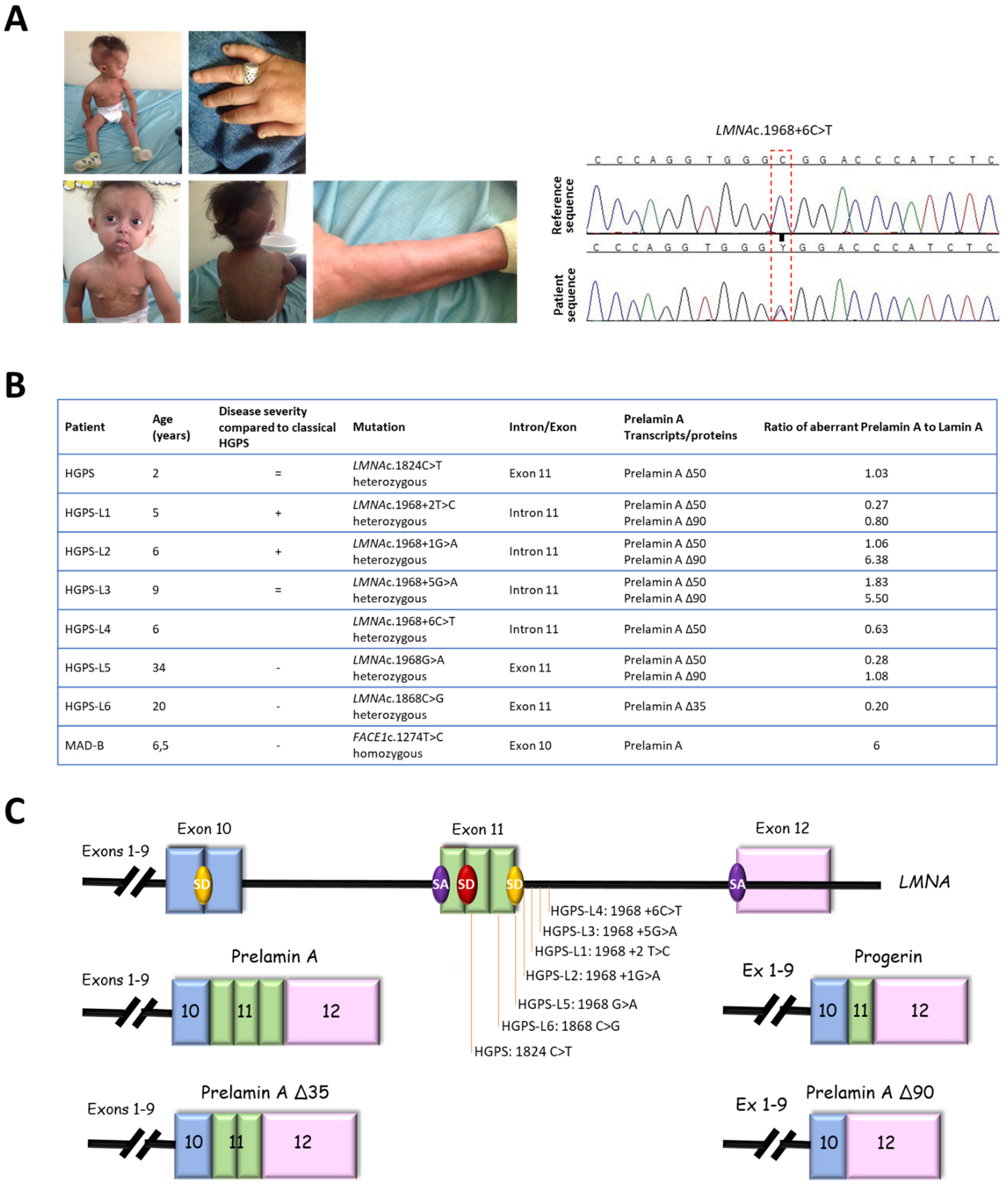
2.2. Genomic Characterization of LMNA Variants
2.3. Cell Culture
2.4. RNA Sequencing (ArrayExpress Accession Number: E-MTAB-5807)
2.4.1. Quality Control
2.4.2. Unsupervised Analysis
2.4.3. Differential Expression Analysis
2.5. RNA Isolation, Reverse Transcription, and Real-Time PCR
2.6. Western Blot
2.7. Fluorescence Microscopy
2.8. Antibodies
2.9. Measurement of Senescence
2.10. Proliferation Assay
2.11. Wound Healing Assay
2.12. Multi-Analyte ELISA Array
2.13. Statistics
3. Results
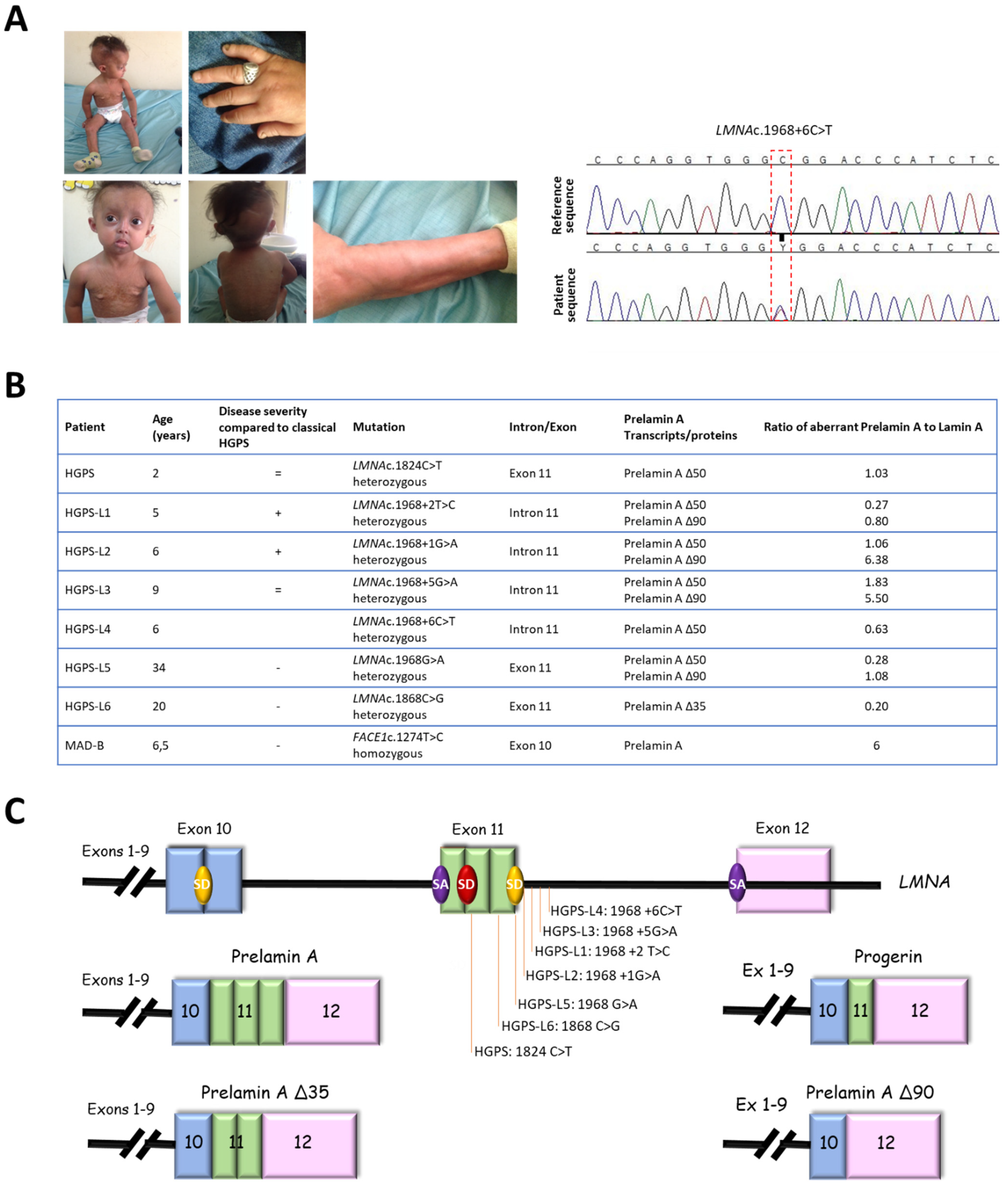
3.1. Patients’ Molecular and Clinical Features
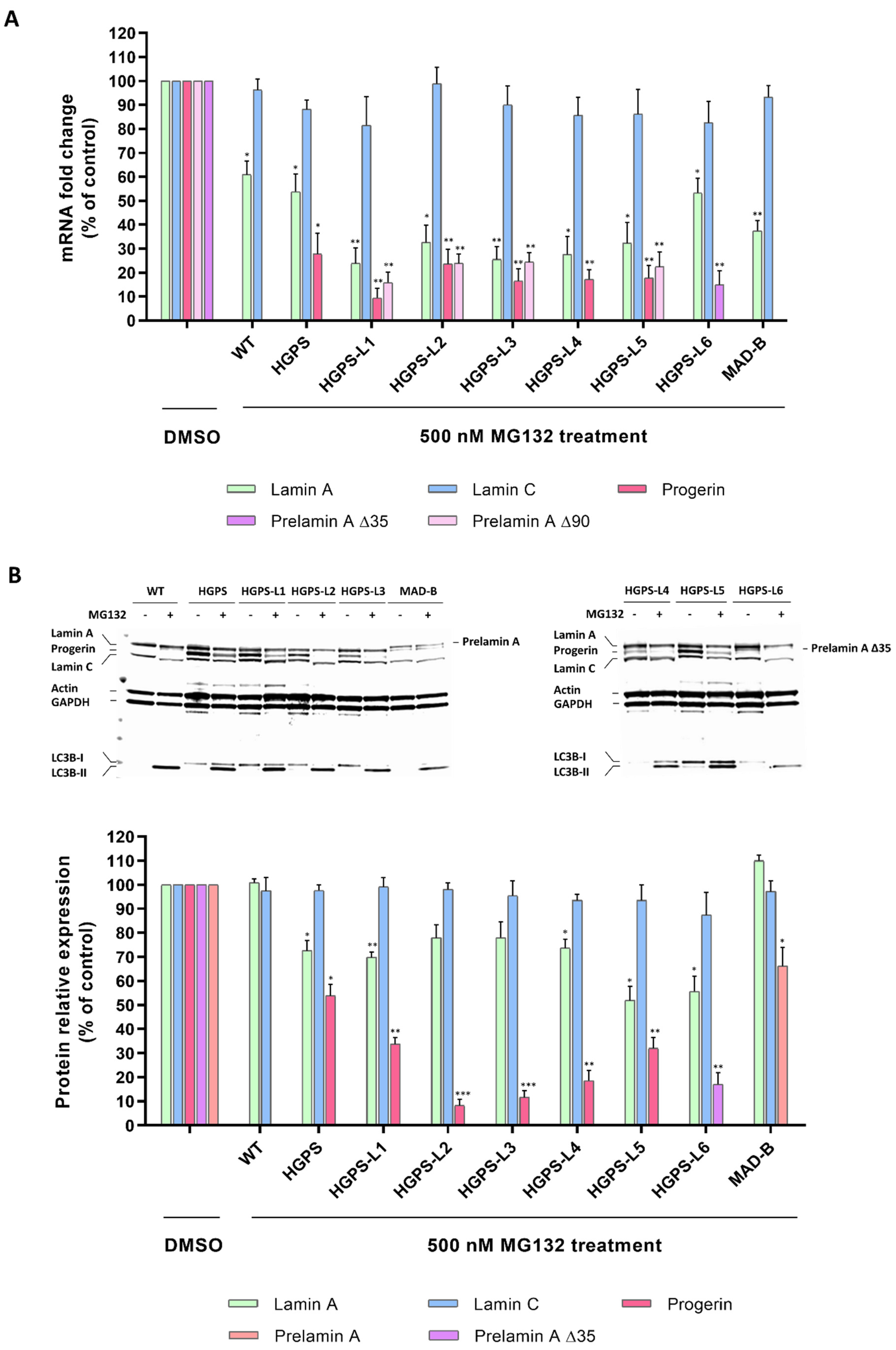
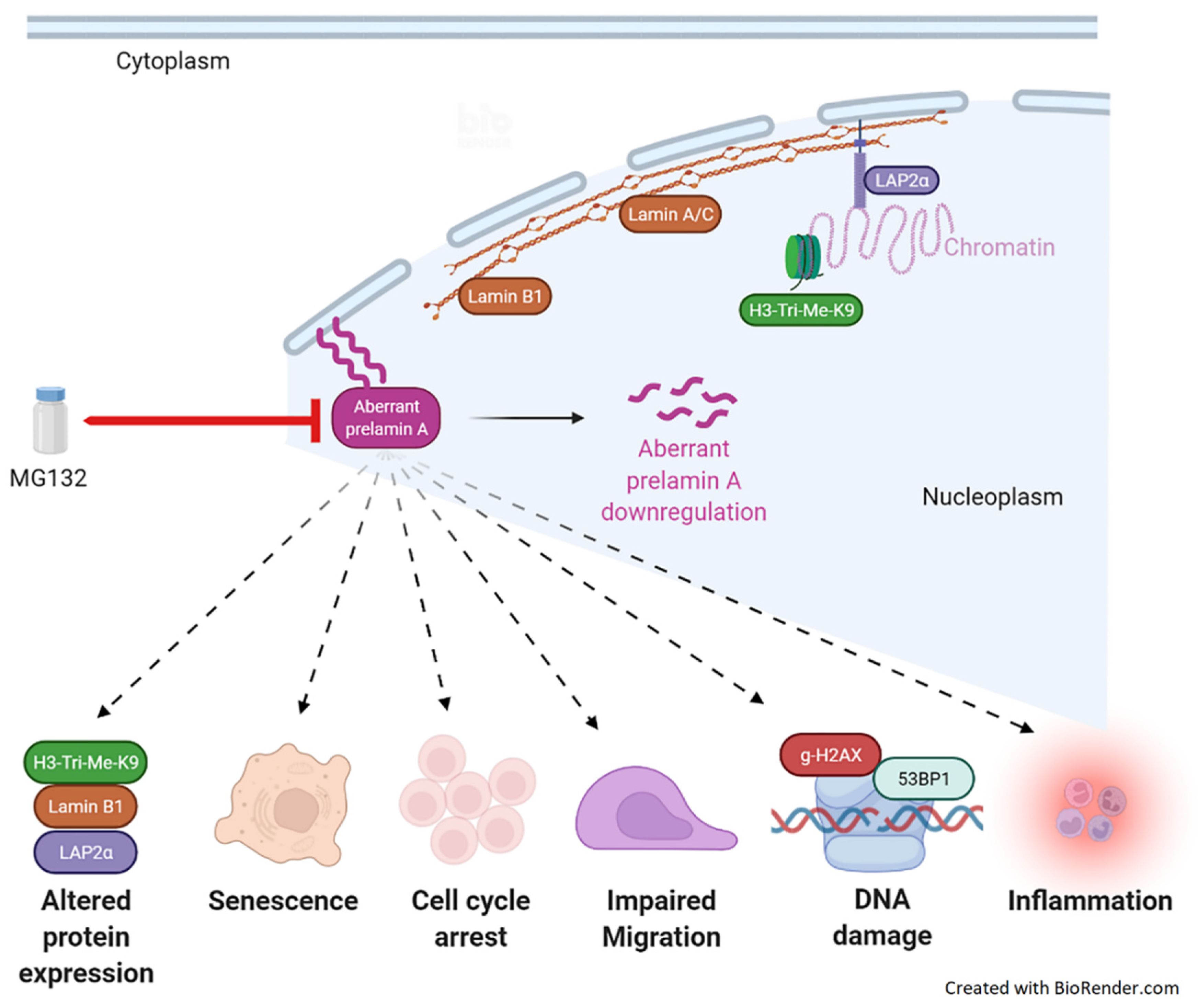
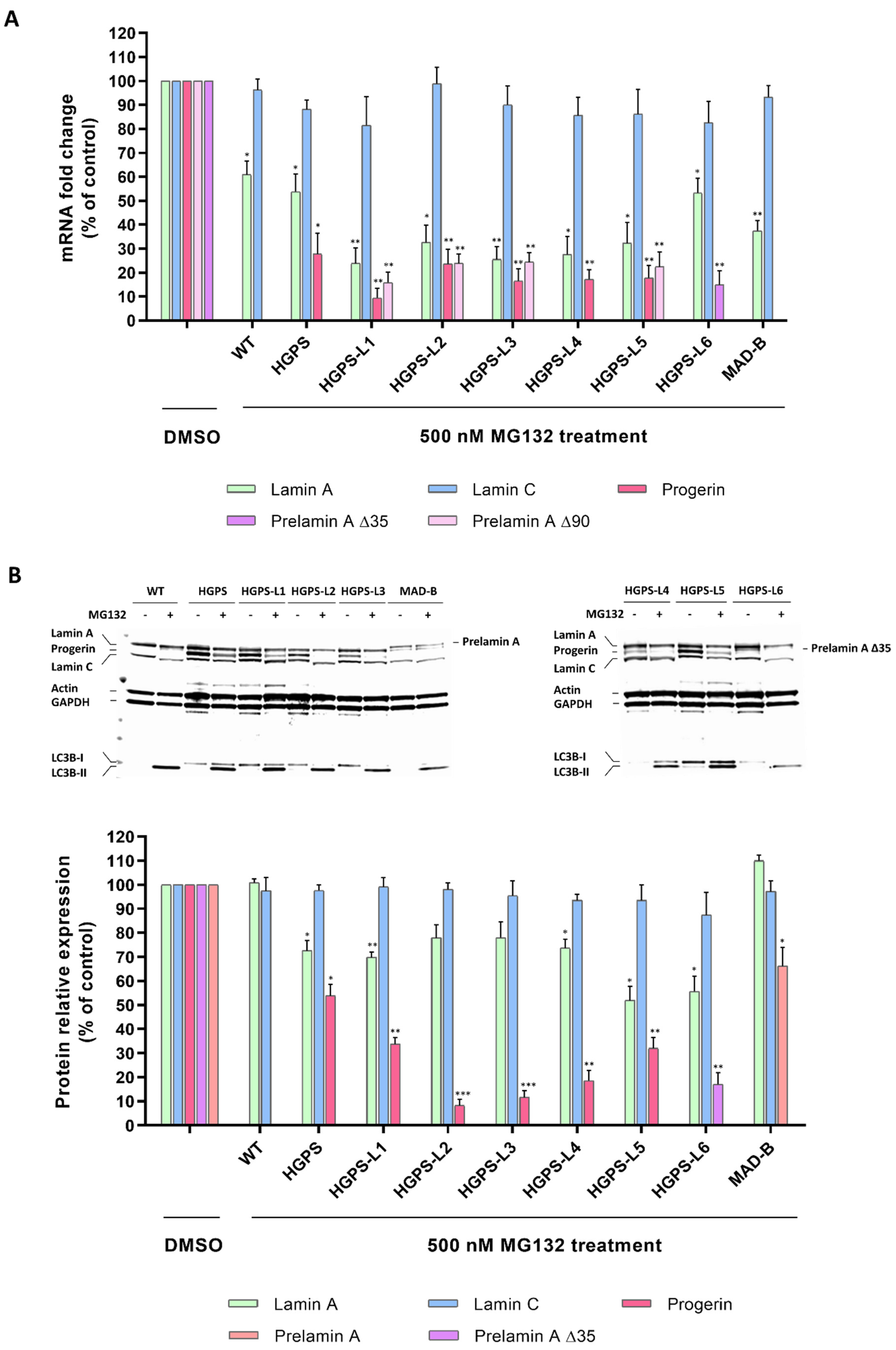
3.2. MG132 Reduces Aberrant Prelamin A Levels in HGPS-like and MAD-B Fibroblasts
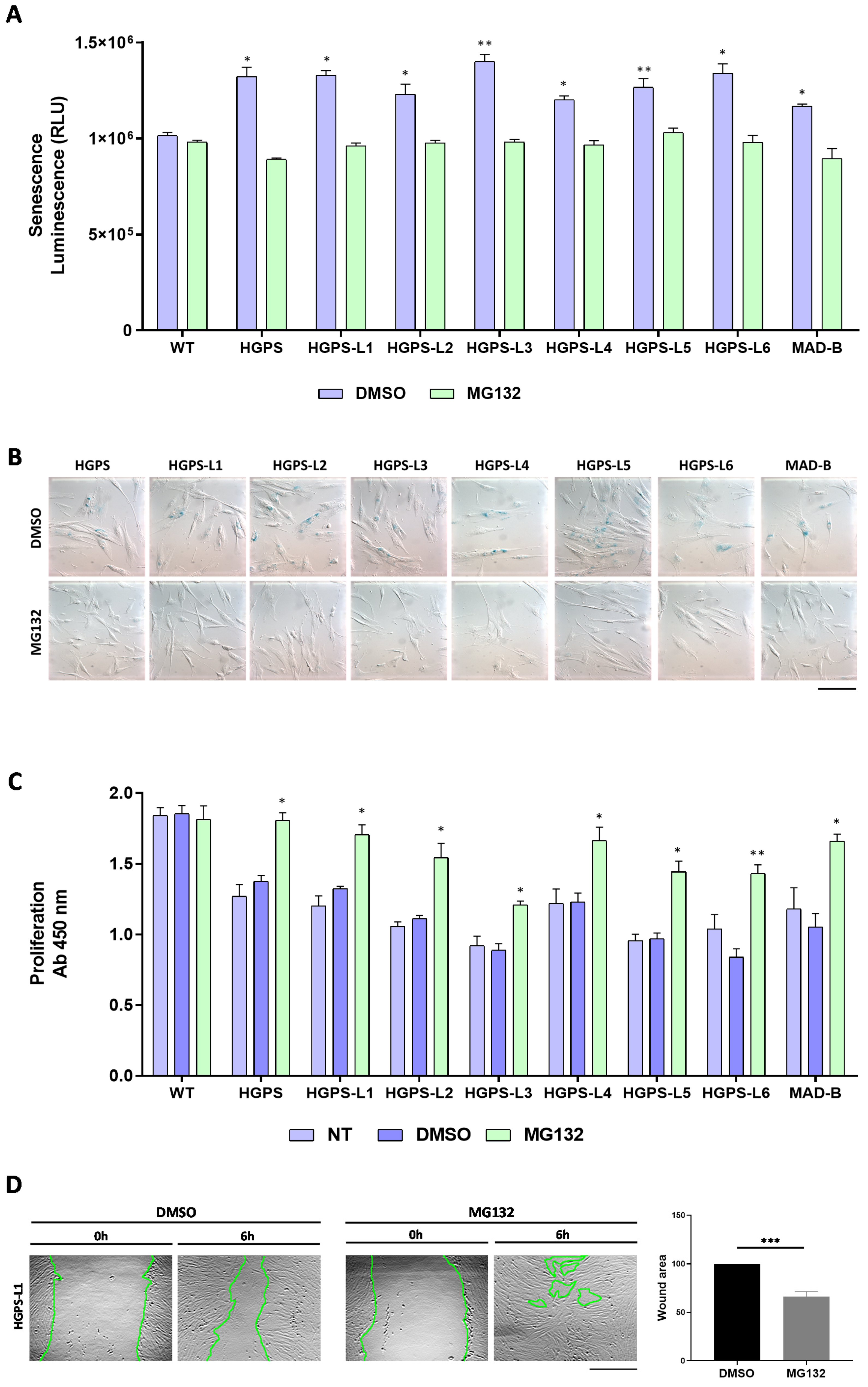
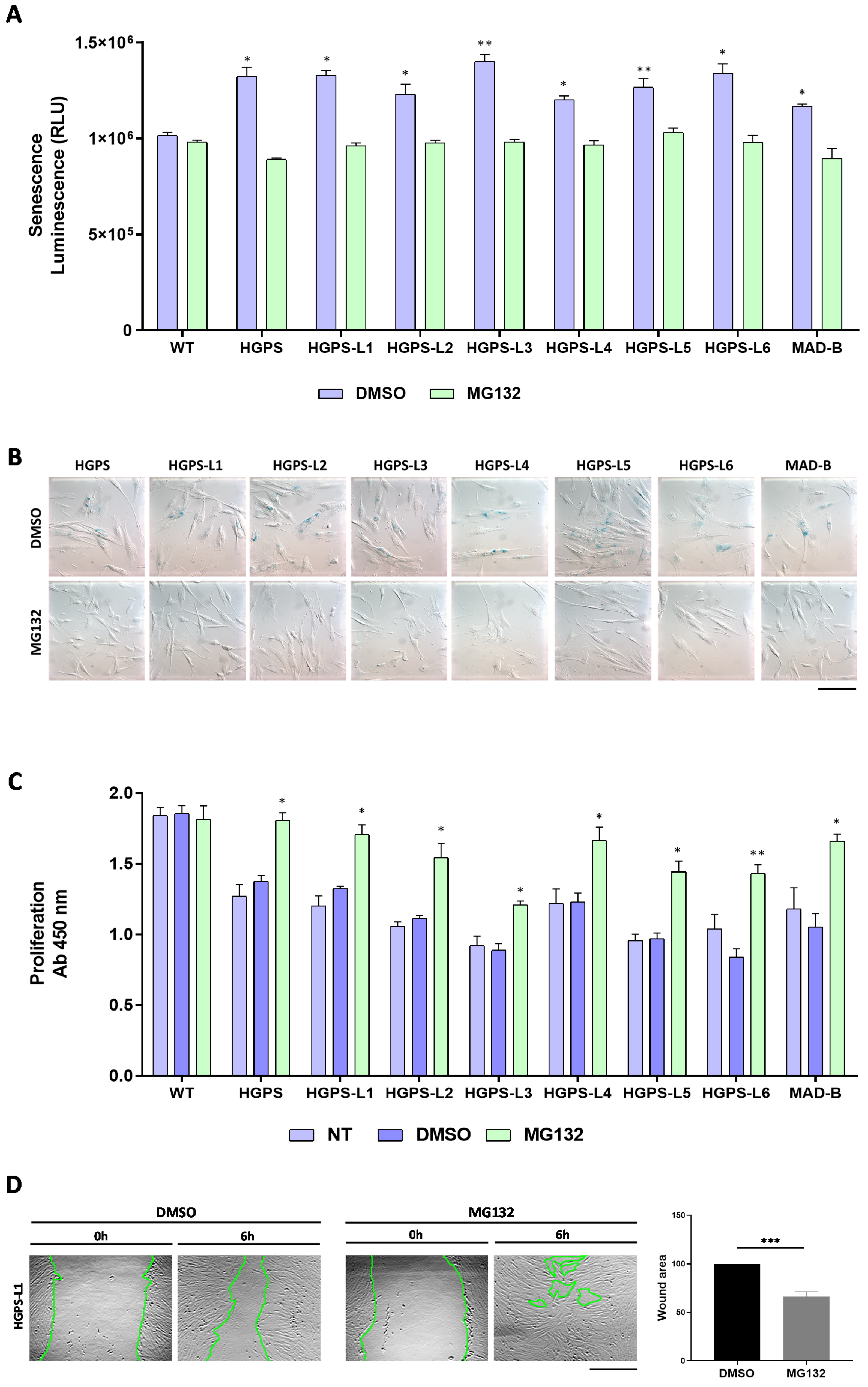
3.3. MG132 Reduces Senescence, Enhances Proliferation and Migration in HGPS-like and MAD-B Patient Cells
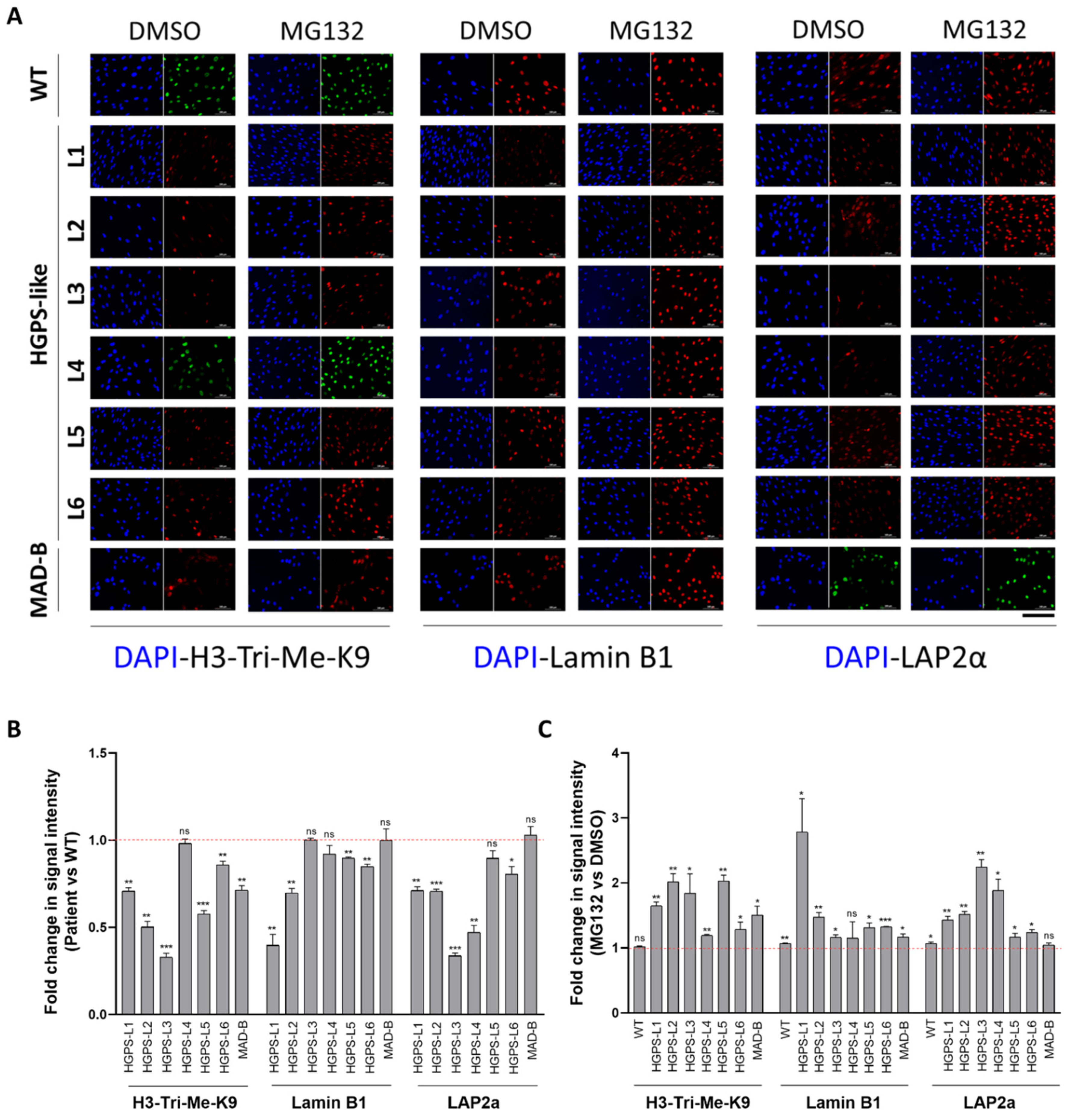
3.4. MG132 Treatment Rescues the Level of Proteins Whose Expression Is Altered in HGPS-like and MAD-B Cells
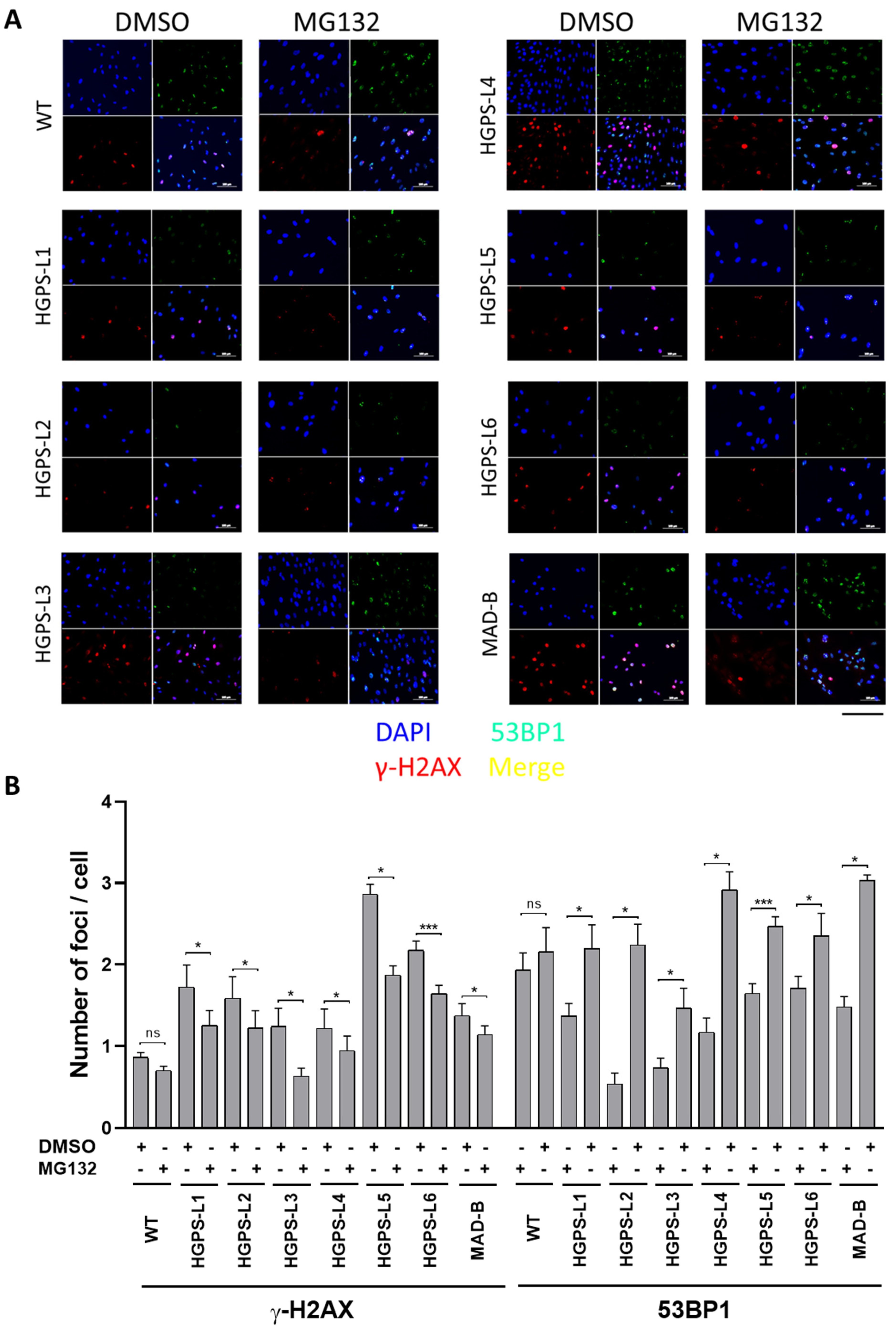
3.5. Treatment of HGPS-like and MAD-B Cells with MG132 Reduces the Levels of DNA Damage
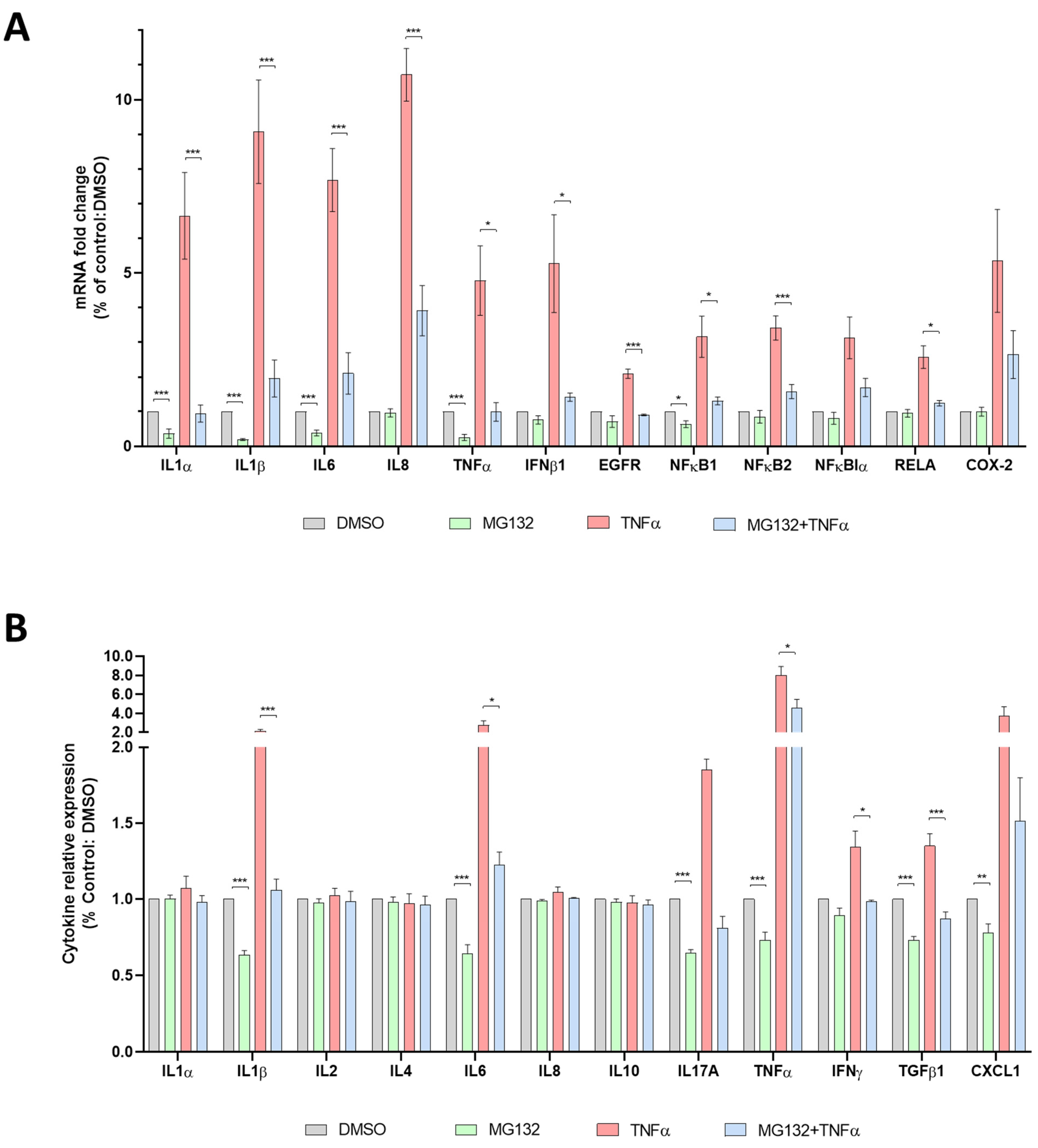
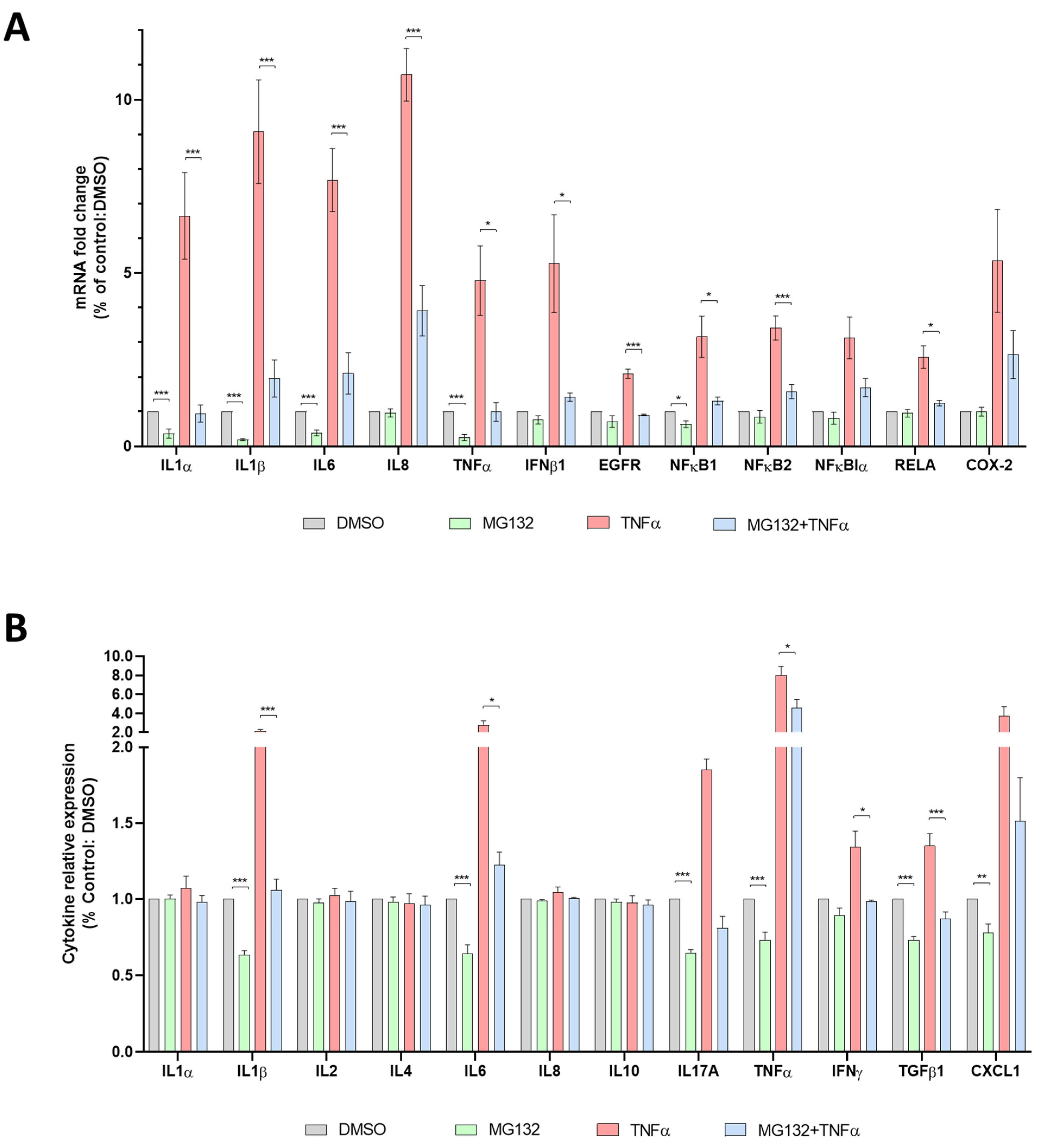
3.6. Anti-Inflammatory Effects of MG132 in HGPS-like and MAD-B Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hennekam, R.C. Hutchinson-Gilford progeria syndrome: Review of the phenotype. Am. J. Med. Genet. A 2006, 140, 2603–2624. [Google Scholar] [CrossRef] [Green Version]
- Gordon, L.B.; Massaro, J.; D’Agostino, R.B., Sr.; Campbell, S.E.; Brazier, J.; Brown, W.T.; Kleinman, M.E.; Kieran, M.W. Impact of farnesylation inhibitors on survival in Hutchinson-Gilford progeria syndrome. Circulation 2014, 130, 27–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Sandre-Giovannoli, A.; Bernard, R.; Cau, P.; Navarro, C.; Amiel, J.; Boccaccio, I.; Lyonnet, S.; Stewart, C.L.; Munnich, A.; Le Merrer, M.; et al. Lamin a truncation in Hutchinson-Gilford progeria. Science 2003, 300, 2055. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, M.; Brown, W.T.; Gordon, L.B.; Glynn, M.W.; Singer, J.; Scott, L.; Erdos, M.R.; Robbins, C.M.; Moses, T.Y.; Berglund, P.; et al. Recurrent de novo point mutations in lamin A cause Hutchinson–Gilford progeria syndrome. Nature 2003, 423, 293–298. [Google Scholar] [CrossRef] [Green Version]
- Karoutas, A.; Akhtar, A. Functional mechanisms and abnormalities of the nuclear lamina. Nat. Cell Biol. 2021, 23, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Mejia, I.C.; Vautrot, V.; De Toledo, M.; Behm-Ansmant, I.; Bourgeois, C.F.; Navarro, C.L.; Osorio, F.G.; Freije, J.M.P.; Stévenin, J.; De Sandre-Giovannoli, A.; et al. A conserved splicing mechanism of the LMNA gene controls premature aging. Hum. Mol. Genet. 2011, 20, 4540–4555. [Google Scholar] [CrossRef] [Green Version]
- Gonzalo, S.; Kreienkamp, R.; Askjaer, H.-G. Progeria Syndrome: A premature aging disease caused by LMNA gene mutations. Ageing Res. Rev. 2017, 33, 18–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Cau, P.; Navarro, C.; Harhouri, K.; Roll, P.; Sigaudy, S.; Kaspi, E.; Perrin, S.; De Sandre-Giovannoli, A.; Lévy, N. Nuclear matrix, nuclear envelope and premature aging syndromes in a translational research perspective. Semin. Cell Dev. Biol. 2014, 29, 125–147. [Google Scholar] [CrossRef]
- Harhouri, K.; Frankel, D.; Bartoli, C.; Roll, P.; De Sandre-Giovannoli, A.; Lévy, N. An overview of treatment strategies for Hutchinson-Gilford Progeria syndrome. Nucleus 2018, 9, 246–257. [Google Scholar] [CrossRef] [Green Version]
- Zhu, K.; Dunner, K., Jr.; McConkey, D.J. Proteasome Inhibitors Activate Autophagy as a Cytoprotective Response in Human Prostate Cancer Cells. Oncogene 2010, 29, 451–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zang, Y.; Thomas, S.M.; Chan, E.T.; Kirk, C.J.; Freilino, M.L.; DeLancey, H.M.; Grandis, J.R.; Li, C.Y.; Johnson, D.E. The Next Generation Proteasome Inhibitors Carfilzomib and Oprozomib Activate Prosurvival Autophagy Via Induction of the Unfolded Protein Response and Atf4. Autophagy 2010, 8, 1873–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.J.; Yu, J.; Wong, S.H.; Cheng, A.S.; Chan, F.K.; Ng, S.S.; Cho, C.H.; Sung, J.J.; Wu, W.K. A Novel Crosstalk between Two Major Protein Degradation Systems Regulation of Proteasomal Activity by Autophagy. Autophagy 2013, 9, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Harhouri, K.; Navarro, C.; Depetris, D.; Mattei, M.; Nissan, X.; Cau, P.; De Sandre-Giovannoli, A.; Levy, N. Mg132-Induced Progerin Clearance Is Mediated by Autophagy Activation and Splicing Regulation. EMBO Mol Med 2017, 9, 1294–1313. [Google Scholar] [CrossRef] [PubMed]
- Barthelemy, F.; Navarro, C.; Fayek, R.; Da Silva, N.; Roll, P.; Sigaudy, S.; Oshima, J.; Bonne, G.; Legbelou, K.P.; Evangeliou, E.A.; et al. Truncated prelamin A expression in HGPS-like patients: A transcriptional study. Eur. J. Hum. Genet. 2015, 23, 1051–1061. [Google Scholar] [CrossRef]
- Harhouri, K.; Navarro, C.; Baquerre, C.; Da Silva, N.; Bartoli, C.; Casey, F.; Mawuse, G.K.; Doubaj, Y.; Lévy, N.; De Sandre-Giovannoli, A. Antisense-Based Progerin Downregulation in HGPS-Like Patients’ Cells. Cells 2016, 5, 31. [Google Scholar] [CrossRef] [Green Version]
- Merideth, M.A.; Gordon, L.B.; Clauss, S.; Sachdev, V.; Smith, A.C.; Perry, M.B.; Brewer, C.C.; Zalewski, C.; Kim, H.J.; Solomon, B.; et al. Phenotype and Course of Hutchinson–Gilford Progeria Syndrome. N. Engl. J. Med. 2008, 358, 592–604. [Google Scholar] [CrossRef] [Green Version]
- Ben Yaou, R.; Navarro, C.; Quijano-Roy, S.; Bertrand, A.T.; Massart, C.; De Sandre-Giovannoli, A.; Cadiñanos, J.; Mamchaoui, K.; Butler-Browne, G.; Estournet, B.; et al. Type B mandibuloacral dysplasia with congenital myopathy due to homozygous ZMPSTE24 missense mutation. Eur. J. Hum. Genet. 2011, 19, 647–654. [Google Scholar] [CrossRef] [Green Version]
- Goldman, R.D.; Shumaker, D.K.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Gordon, L.B.; Gruenbaum, Y.; Khuon, S.; Mendez, M.; Varga, R.; et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson–Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2004, 101, 8963–8968. [Google Scholar] [CrossRef] [Green Version]
- Scaffidi, P.; Misteli, T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat. Med. 2005, 11, 440–445. [Google Scholar] [CrossRef]
- Vidak, S.; Foisner, R. Molecular insights into the premature aging disease progeria. Histochem. Cell Biol. 2016, 145, 401–417. [Google Scholar] [CrossRef] [Green Version]
- Chang, W.; Wang, Y.; Luxton, G.W.G.; Östlund, C.; Worman, H.J.; Gundersen, G.G. Imbalanced nucleocytoskeletal connections create common polarity defects in progeria and physiological aging. Proc. Natl. Acad. Sci. USA 2019, 116, 3578–3583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pienta, K.; Coffey, D.S. Characterization of the subtypes of cell motility in ageing human skin fibroblasts. Mech. Ageing Dev. 1990, 56, 99–105. [Google Scholar] [CrossRef]
- Gonzalo, S.; Kreienkamp, R. DNA repair defects and genome instability in Hutchinson–Gilford Progeria Syndrome. Curr. Opin. Cell Biol. 2015, 34, 75–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scaffidi, P.; Misteli, T. Lamin A-Dependent Nuclear Defects in Human Aging. Science 2006, 312, 1059–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Wang, J.; Chan, K.M.; Tjia, W.M.; Deng, W.; Guan, X.; Huang, J.-D.; Li, K.M.; Chau, P.Y.; Chen, D.J.; et al. Genomic instability in laminopathy-based premature aging. Nat. Med. 2005, 11, 780–785. [Google Scholar] [CrossRef]
- Zhang, H.; Sun, L.; Wang, K.; Wu, D.; Trappio, M.; Witting, C.; Cao, K. Loss of H3K9me3 Correlates with ATM Activation and Histone H2AX Phosphorylation Deficiencies in Hutchinson-Gilford Progeria Syndrome. PLoS ONE 2016, 11, e0167454. [Google Scholar] [CrossRef]
- Osorio, F.G.; Bárcena, C.; Soria-Valles, C.; Ramsay, A.J.; de Carlos, F.; Cobo, J.; Fueyo, A.; Freije, J.M.; López-Otín, C. Nuclear lamina defects cause ATM-dependent NF-kappaB activation and link accelerated aging to a systemic inflammatory response. Genes Dev. 2012, 26, 2311–2324. [Google Scholar] [CrossRef] [Green Version]
- Ortiz-Lazareno, P.C.; Hernandez-Flores, G.; Dominguez-Rodriguez, J.R.; Lerma-Diaz, J.M.; Jave-Suarez, L.F.; Aguilar-Lemarroy, A.; Gomez-Contreras, P.C.; Scott-Algara, D.; Bravo-Cuellar, A. MG132 proteasome inhibitor modulates proinflammatory cytokines production and expression of their receptors in U937 cells: Involvement of nuclear factor-kappa B and activator protein-1. Immunology 2008, 124, 534–541. [Google Scholar] [CrossRef]
- Hozhabri, N.S.T.; Kim, H.; Varanasi, V. NF-kappa B inhibitor MG132 enhances differentiation and collagen expression of dental pulp stem cells. FASEB J. 2014, 28. [Google Scholar]
- Mathes, E.; O’dea, E.L.; Hoffmann, A.; Ghosh, G. NF-kappa B dictates the degradation pathway of I kappa B alpha (vol 27, pg 1357, 2008). EMBO J. 2008, 27, 1421. [Google Scholar] [CrossRef] [Green Version]
- Mahmoudi, S.; Mancini, E.; Xu, L.; Moore, A.; Jahanbani, F.; Hebestreit, K.; Srinivasan, R.; Li, X.; Devarajan, K.; Prélot, L.; et al. Heterogeneity in old fibroblasts is linked to variability in reprogramming and wound healing. Nature 2019, 574, 553–558. [Google Scholar] [CrossRef]
- Moulson, C.L.; Fong, L.G.; Gardner, J.M.; Farber, E.A.; Go, G.; Passariello, A.; Grange, D.K.; Young, S.G.; Miner, J.H. Increased progerin expression associated with unusual LMNA mutations causes severe progeroid syndromes. Hum. Mutat. 2007, 28, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Martins, F.; Sousa, J.; Pereira, C.D.I.; Silva, O.A.B.D.C.E.; Rebelo, S. Nuclear envelope dysfunction and its contribution to the aging process. Aging Cell 2020, 19, e13143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Röhrl, J.; Arnold, R.; Djabali, K. Nuclear Pore Complexes Cluster in Dysmorphic Nuclei of Normal and Progeria Cells during Replicative Senescence. Cells 2021, 10, 153. [Google Scholar] [CrossRef]
- Serebryannyy, L.; Misteli, T. Protein sequestration at the nuclear periphery as a potential regulatory mechanism in premature aging. J. Cell Biol. 2017, 217, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Osorio, F.G.; Soria-Valles, C.; Santiago-Fernández, O.; Freije, J.M.P.; López-Otín, C. NF-kappa B Signaling as a Driver of Ageing. Int. Rev. Cell Mol. Biol. 2016, 326, 133–174. [Google Scholar] [PubMed]
- Ashapkin, V.V.; Kutueva, L.I.; Kurchashova, S.Y.; Kireev, I.I. Are There Common Mechanisms Between the Hutchinson-Gilford Progeria Syndrome and Natural Aging? Front. Genet. 2019, 10, 405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreesen, O. Towards delineating the chain of events that cause premature senescence in the accelerated aging syndrome Hutchinson–Gilford progeria (HGPS). Biochem. Soc. Trans. 2020, 48, 981–991. [Google Scholar] [CrossRef]
- Zhang, L.; Tang, H.; Kou, Y.; Li, R.; Zheng, Y.; Wang, Q.; Zhou, X.; Jin, L. MG132-mediated inhibition of the ubiquitin–proteasome pathway ameliorates cancer cachexia. J. Cancer Res. Clin. Oncol. 2013, 139, 1105–1115. [Google Scholar] [CrossRef]
- Nakajima, S.; Kato, H.; Takahashi, S.; Johno, H.; Kitamura, M. Inhibition of NF-kappa B by MG132 through ER stress-mediated induction of LAP and LIFebs. Letters 2011, 585, 2249–2254. [Google Scholar]
- Neves, J.; Sousa-Victor, P. Regulation of inflammation as an anti-aging intervention. FEBS J. 2019, 287, 43–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olive, M.; Harten, I.; Mitchell, R.; Beers, J.K.; Djabali, K.; Cao, K.; Erdos, M.R.; Blair, C.; Funke, B.; Smoot, L.; et al. Cardiovascular Pathology in Hutchinson-Gilford Progeria: Correlation With the Vascular Pathology of Aging. Arter. Thromb. Vasc. Biol. 2010, 30, 2301–2309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamczyk, M.R.; Villa-Bellosta, R.; Gonzalo, P.; Andrés-Manzano, M.J.; Nogales, P.; Bentzon, J.F.; López-Otín, C.; Andrés, V. Vascular Smooth Muscle–Specific Progerin Expression Accelerates Atherosclerosis and Death in a Mouse Model of Hutchinson-Gilford Progeria Syndrome. Circulation 2018, 138, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Kubben, N.; Zhang, W.; Wang, L.; Voss, T.C.; Yang, J.; Qu, J.; Liu, G.-H.; Misteli, T. Repression of the Antioxidant NRF2 Pathway in Premature Aging. Cell 2016, 165, 1361–1374. [Google Scholar] [CrossRef] [Green Version]
- Cui, W.; Bai, Y.; Luo, P.; Miao, L.; Cai, L. Preventive and Therapeutic Effects of MG132 by Activating Nrf2-ARE Signaling Pathway on Oxidative Stress-Induced Cardiovascular and Renal Injury. Oxidative Med. Cell. Longev. 2013, 2013, 306073. [Google Scholar] [CrossRef]
- Dreger, H.; Westphal, K.; Wilck, N.; Baumann, G.; Stangl, V.; Stangl, K.; Meiners, S. Protection of vascular cells from oxidative stress by proteasome inhibition depends on Nrf2. Cardiovasc. Res. 2009, 85, 395–403. [Google Scholar] [CrossRef] [Green Version]
- Miao, X.; Cui, W.; Sun, W.; Xin, Y.; Wang, B.; Tan, Y.; Cai, L.; Miao, L.; Fu, Y.; Su, G.; et al. Therapeutic Effect of MG132 on the Aortic Oxidative Damage and Inflammatory Response in OVE26 Type 1 Diabetic Mice. Oxidative Med. Cell. Longev. 2013, 2013, 879516. [Google Scholar] [CrossRef] [Green Version]
- Feng, B.; Zhang, Y.; Mu, J.; Ye, Z.; Zeng, W.; Qi, W.; Luo, Z.; Guo, Y.; Yang, X.; Yuan, F. Preventive Effect of a Proteasome Inhibitor on the Formation of Accelerated Atherosclerosis in Rabbits With Uremia. J. Cardiovasc. Pharmacol. 2010, 55, 129–138. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, W.; Du, B.; Miao, X.; Bai, Y.; Xin, Y.; Tan, Y.; Cui, W.; Liu, B.; Cui, T.; et al. Therapeutic effect of MG-132 on diabetic cardiomyopathy is associated with its suppression of proteasomal activities: Roles of Nrf2 and NF-kappa B. Am. J. Physiol.-Heart Circ. Physiol. 2013, 304, H567–H578. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, A.S.; Li, J.; Ahmed, M.; Hua, L.; Yakovleva, T.; Ossipov, M.H.; Bakalkin, G.; Stark, A. Attenuation of pain and inflammation in adjuvant-induced arthritis by the proteasome inhibitor MG132. Arthritis Care Res. 2010, 62, 2160–2169. [Google Scholar] [CrossRef]
- Harten, I.A.; Zahr, R.S.; Lemire, J.M.; Machan, J.T.; Moses, M.A.; Doiron, R.J.; Curatolo, A.S.; Rothman, F.G.; Wight, T.N.; Toole, B.P.; et al. Age-Dependent Loss of MMP-3 in Hutchinson-Gilford Progeria Syndrome. J. Gerontol. Ser. Biol. Sci. Med. Sci. 2011, 66, 1201–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, N.; Hu, M.; Khalil, R.A. Biochemical and Biological Attributes of Matrix Metalloproteinases. Matrix Metalloproteinses and Tissue Remodeling. Health Dis. Cardiovasc. Remodel. 2017, 147, 1–73. [Google Scholar]
- Yang, H.; Liang, J.; Zhou, J.; Mi, J.; Ma, K.; Fan, Y.; Ning, J.; Wang, C.; Wei, X.; Li, E. Knockdown of RHOC by shRNA suppresses invasion and migration of cholangiocellular carcinoma cells via inhibition of MMP2, MMP3, MMP9 and epithelial-mesenchymal transition. Mol. Med. Rep. 2016, 13, 5255–5261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goffin, L.; Seguin-Estévez, Q.; Alvarez, M.; Reith, W.; Chizzolini, C. Transcriptional regulation of matrix metalloproteinase-1 and collagen 1A2 explains the anti-fibrotic effect exerted by proteasome inhibition in human dermal fibroblasts. Arthritis Res. Ther. 2010, 12, R73. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | ID |
|---|---|
| 18s rRNA | Hs99999901_s1 |
| IL-1α | Hs00174092_m1 |
| IL-1β | Hs01555410_m1 |
| IL-6 | Hs00174131_m1 |
| IL-8 | Hs00174103_m1 |
| TNFα | Hs00174128_m1 |
| IFN-β1 | Hs01077958_s1 |
| EGFR | Hs01076090_m1 |
| NFκB1 | Hs00765730_m1 |
| NFκB2 | Hs01028890_g1 |
| NFκBIα | Hs00355671_g1 |
| RELA | Hs01042014_m1 |
| COX-2 | Hs00153133_m1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harhouri, K.; Cau, P.; Casey, F.; Guedenon, K.M.; Doubaj, Y.; Van Maldergem, L.; Mejia-Baltodano, G.; Bartoli, C.; De Sandre-Giovannoli, A.; Lévy, N. MG132 Induces Progerin Clearance and Improves Disease Phenotypes in HGPS-like Patients’ Cells. Cells 2022, 11, 610. https://doi.org/10.3390/cells11040610
Harhouri K, Cau P, Casey F, Guedenon KM, Doubaj Y, Van Maldergem L, Mejia-Baltodano G, Bartoli C, De Sandre-Giovannoli A, Lévy N. MG132 Induces Progerin Clearance and Improves Disease Phenotypes in HGPS-like Patients’ Cells. Cells. 2022; 11(4):610. https://doi.org/10.3390/cells11040610
Chicago/Turabian StyleHarhouri, Karim, Pierre Cau, Frank Casey, Koffi Mawuse Guedenon, Yassamine Doubaj, Lionel Van Maldergem, Gerardo Mejia-Baltodano, Catherine Bartoli, Annachiara De Sandre-Giovannoli, and Nicolas Lévy. 2022. "MG132 Induces Progerin Clearance and Improves Disease Phenotypes in HGPS-like Patients’ Cells" Cells 11, no. 4: 610. https://doi.org/10.3390/cells11040610
APA StyleHarhouri, K., Cau, P., Casey, F., Guedenon, K. M., Doubaj, Y., Van Maldergem, L., Mejia-Baltodano, G., Bartoli, C., De Sandre-Giovannoli, A., & Lévy, N. (2022). MG132 Induces Progerin Clearance and Improves Disease Phenotypes in HGPS-like Patients’ Cells. Cells, 11(4), 610. https://doi.org/10.3390/cells11040610