
Neuronal Rubicon Represses Extracellular APP/Amyloid β Deposition in Alzheimer’s Disease

 ,
,  , , , , , , and add
Show full author list
, , , , , , and add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human-Induced Pluripotent Stem Cells (hiPSCs)
2.2. Human Tissues
2.3. Transgenic Mice
2.4. Mouse Tissue Preparation
2.5. Histology Analysis
2.6. Cell Culture Experiments
2.7. Images Analysis
2.8. RNA Extraction and Quantitative Real-Time PCR
2.9. Western Blot and Dot Plot Analysis
2.10. Statistical Analysis
3. Results
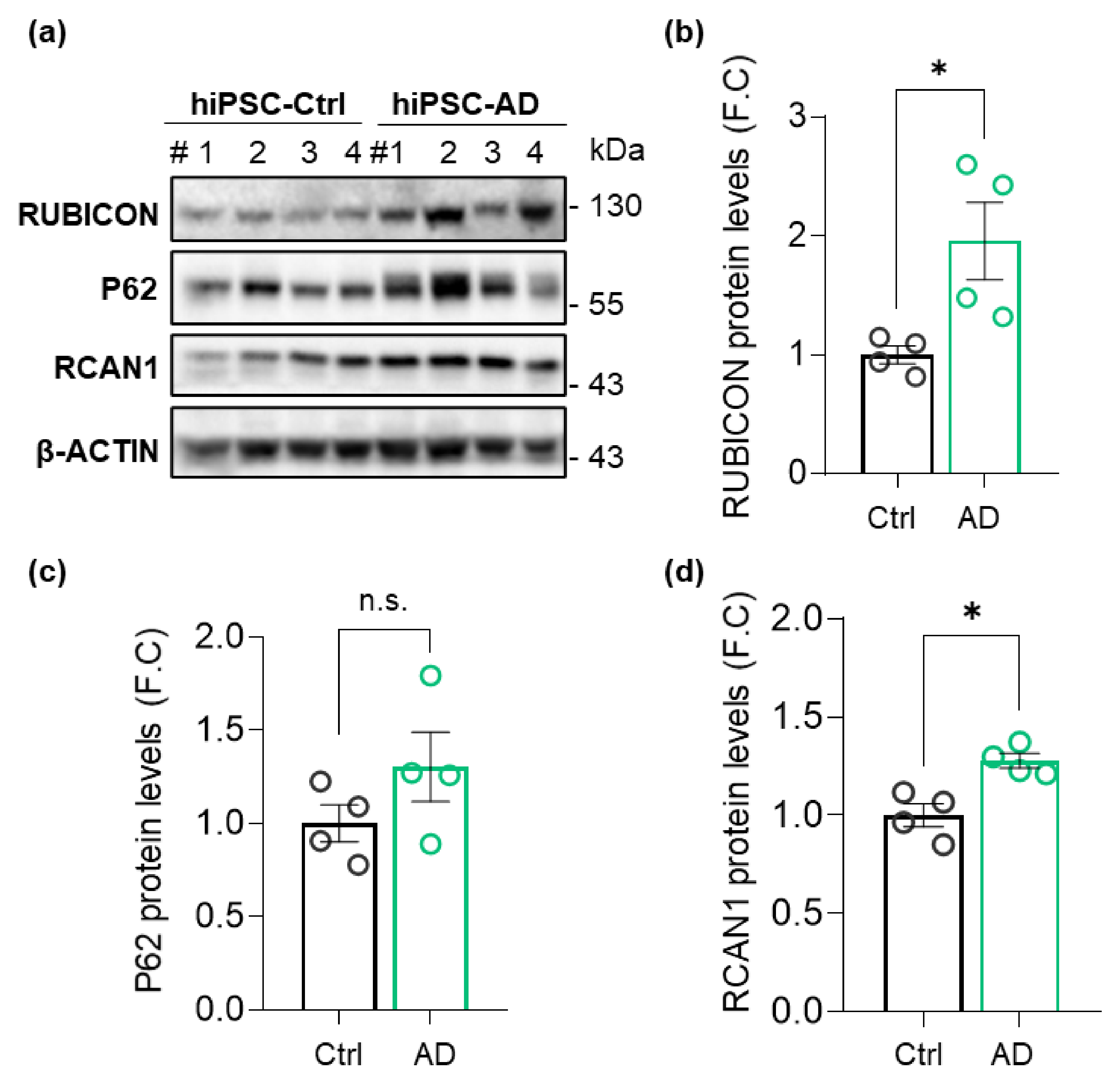
3.1. Levels of Rubicon Are Increased in hiPSCs from an AD Patient from Early-to-Moderate Stage
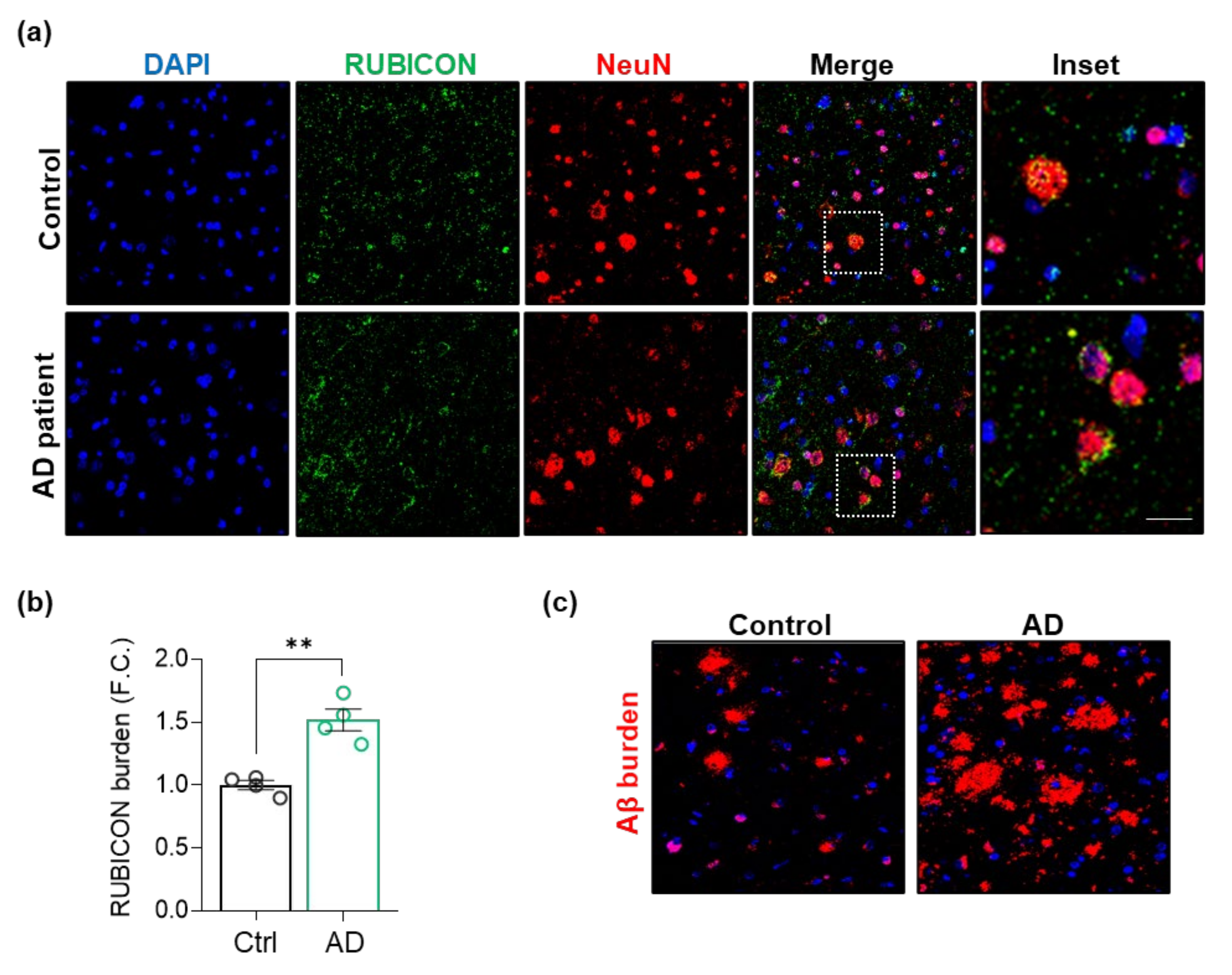
3.2. Rubicon Is Located in Neurons, and Its Levels Are Increased in Postmortem Brain Samples from AD Patients
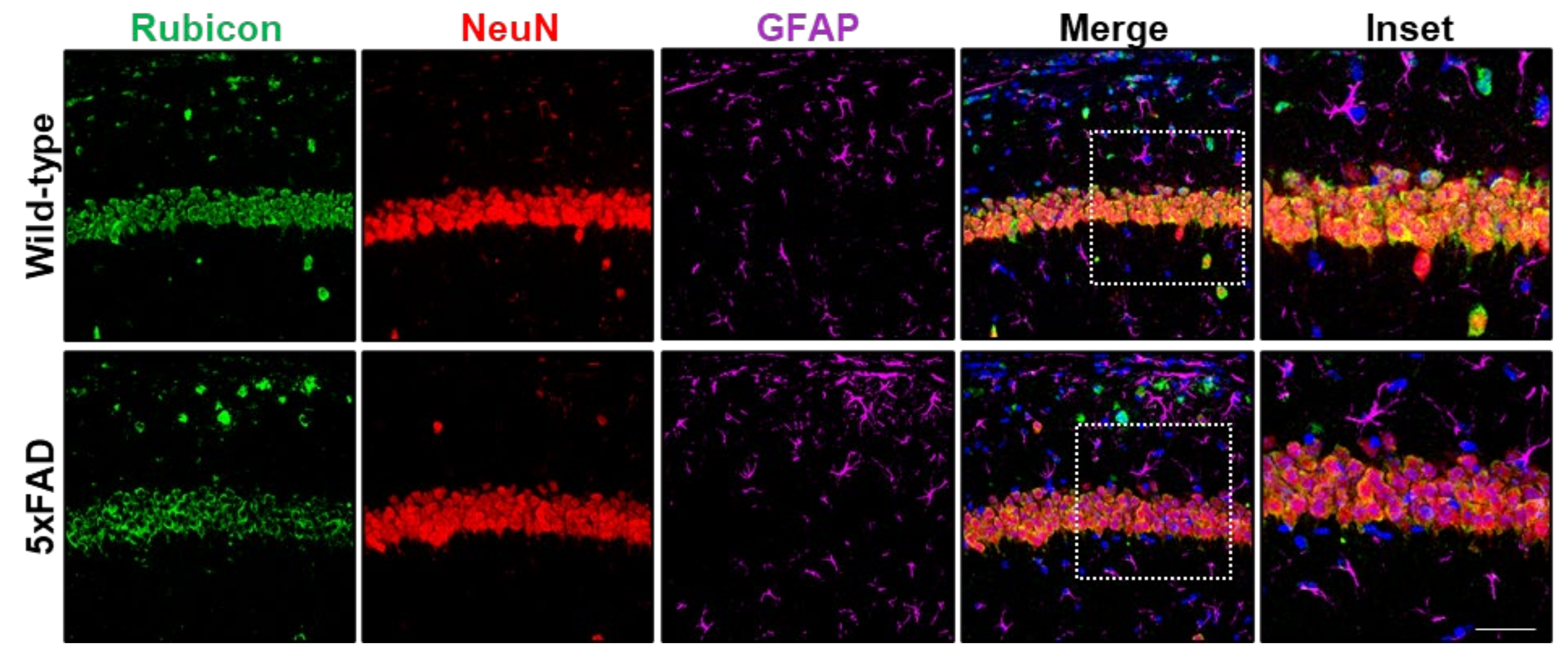
3.3. 5xFAD Mice Lacking Rubicon Present Amyloid β Accumulation in the Hippocampus
3.4. Neuronal Loss, Astrogliosis, and Microglial Activation in 5xFAD Lacking Rubicon
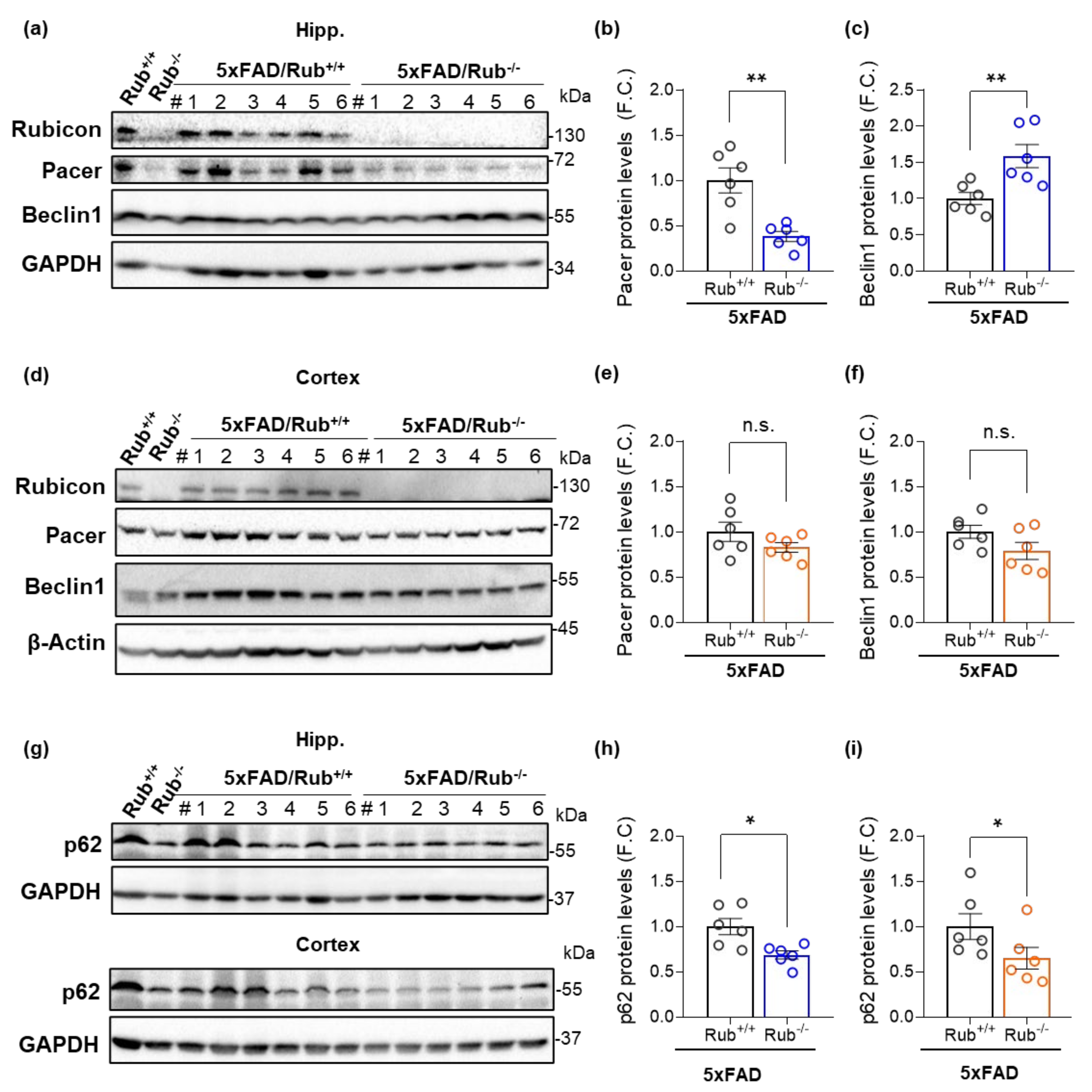
3.5. Autophagy Markers in 5xFAD Lacking Rubicon
3.6. APP-Expressing Neuro2A Cells Deficient in Rubicon Release more APP/Amyloid β
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Patterson, C. World Alzheimer Report 2018—The State of the Art of Dementia Research: New Frontiers; Alzheimer’s Disease International (ADI): London, UK, 2018. [Google Scholar]
- DiBattista, A.M.; Sierra, F.; Masliah, E. NIA Workshop on Senescence in Brain Aging and Alzheimer’s Disease and Its Related Dementias. Geroscience 2020, 42, 389–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caberlotto, L.; Nguyen, T.-P. A systems biology investigation of neurodegenerative dementia reveals a pivotal role of autophagy. BMC Syst. Biol. 2014, 8, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caberlotto, L.; Nguyen, T.-P.; Lauria, M.; Priami, C.; Rimondini, R.; Maioli, S.; Cedazo-Minguez, A.; Sita, G.; Morroni, F.; Corsi, M.; et al. Cross-disease analysis of Alzheimer’s disease and type-2 Diabetes highlights the role of autophagy in the pathophysiology of two highly comorbid diseases. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, A.; Irie, T.; Suzuki, R.; Maemura, T.; Nasser, H.; Uriu, K.; Kosugi, Y.; Shirakawa, K.; Sadamasu, K.; Kimura, I.; et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2010, 107, 14164–14169. [Google Scholar] [CrossRef]
- Raj, T.; Li, Y.I.; Wong, G.; Humphrey, J.; Wang, M.; Ramdhani, S.; Wang, Y.-C.; Ng, B.; Gupta, I.; Haroutunian, V.; et al. Integrative transcriptome analyses of the aging brain implicate altered splicing in Alzheimer’s disease susceptibility. Nat. Genet. 2018, 50, 1584–1592. [Google Scholar] [CrossRef]
- Gao, S.; Casey, A.; Sargeant, T.; Mäkinen, V.-P. Genetic variation within endolysosomal system is associated with late-onset Alzheimer’s disease. Brain 2018, 141, 2711–2720. [Google Scholar] [CrossRef]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef]
- Kuo, C.-C.; Chiang, A.W.T.; Baghdassarian, H.M.; Lewis, N.E. Dysregulation of the secretory pathway connects Alzheimer’s disease genetics to aggregate formation. Cell Syst. 2021, 12, 873–884.e4. [Google Scholar] [CrossRef]
- Cai, C.-Z.; Yang, C.; Zhuang, X.-X.; Yuan, N.-N.; Wu, M.-Y.; Tan, J.-Q.; Song, J.-X.; Cheung, K.-H.; Su, H.; Wang, Y.-T.; et al. NRBF2 is a RAB7 effector required for autophagosome maturation and mediates the association of APP-CTFs with active form of RAB7 for degradation. Autophagy 2021, 17, 1112–1130. [Google Scholar] [CrossRef]
- Burrinha, T.; Martinsson, I.; Gomes, R.; Terrasso, A.P.; Gouras, G.K.; Almeida, C.G. Upregulation of APP endocytosis by neuronal aging drives amyloid-dependent synapse loss. J. Cell Sci. 2021, 134. [Google Scholar] [CrossRef]
- Lauritzen, I.; Pardossi-Piquard, R.; Bourgeois, A.; Pagnotta, S.; Biferi, M.G.; Barkats, M.; Lacor, P.; Klein, W.; Bauer, C.; Checler, F. Intraneuronal aggregation of the β-CTF fragment of APP (C99) induces Aβ-independent lysosomal-autophagic pathology. Acta Neuropathol. 2016, 132, 257–276. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Sato, Y.; Im, E.; Berg, M.; Bordi, M.; Darji, S.; Kumar, A.; Mohan, P.S.; Bandyopadhyay, U.; Diaz, A.; et al. Lysosomal dysfunction in down syndrome is app-dependent and mediated by APP-βCTF (c99). J. Neurosci. 2019, 39, 5255–5268. [Google Scholar] [CrossRef]
- Haung, Y.W.; Cuervo, A.M.; Kumar, A.; Peterhoff, C.M.; Schmidt, S.D.; Lee, J.H.; Mohan, P.S.; Mercken, M.; Farmery, M.R.; Tjernberg, L.O.; et al. Macroautophagy—A novel β-amyloid peptide-generating pathway activated in Alzheimer’s disease. J. Cell Biol. 2005, 171, 87–98. [Google Scholar]
- Mizushima, N. A brief history of autophagy from cell biology to physiology and disease. Nat. Cell Biol. 2018, 20, 521–527. [Google Scholar] [CrossRef]
- Yim, W.W.-Y.; Mizushima, N. Lysosome biology in autophagy. Cell Discov. 2020, 6, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Bourdenx, M.; Martín-Segura, A.; Scrivo, A.; Rodriguez-Navarro, J.A.; Kaushik, S.; Tasset, I.; Diaz, A.; Storm, N.J.; Xin, Q.; Juste, Y.R.; et al. Chaperone-mediated autophagy prevents collapse of the neuronal metastable proteome. Cell 2021, 184, 2696–2714.e25. [Google Scholar] [CrossRef]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.-I.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef]
- Maday, S.; Holzbaur, E.L.F. Compartment-specific regulation of autophagy in primary neurons. J. Neurosci. 2016, 36, 5933–5945. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Ma, X.; Ding, X.; Li, L.; Jiang, X.; Shen, Z.; Chen, S.; Liu, W.; Gong, W.; Sun, Q. Pacer Mediates the Function of Class III PI3K and HOPS Complexes in Autophagosome Maturation by Engaging Stx17. Mol. Cell 2017, 65, 1029–1043.e5. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Y.; Wang, Q.; Li, X.; Yan, Y.; Backer, J.M.; Chait, B.T.; Heintz, N.; Yue, Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1–phosphatidylinositol-3-kinase complex. Nat. Cell Biol. 2009, 11, 468–476. [Google Scholar] [CrossRef]
- Matsunaga, K.; Saitoh, T.; Tabata, K.; Omori, H.; Satoh, T.; Kurotori, N.; Maejima, I.; Shirahama-Noda, K.; Ichimura, T.; Isobe, T.; et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 2009, 11, 385–396. [Google Scholar] [CrossRef]
- McEwan, D.G.; Popovic, D.; Gubas, A.; Terawaki, S.; Suzuki, H.; Stadel, D.; Coxon, F.P.; de Stegmann, D.M.; Bhogaraju, S.; Maddi, K.; et al. PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol. Cell 2015, 57, 39–54. [Google Scholar] [CrossRef] [Green Version]
- Tsukada, M.; Ohsumi, Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993, 333, 169–174. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Westphal, W.; Wong, K.N.; Tan, I.; Zhong, Q. Rubicon controls endosome maturation as a Rab7 effector. Proc. Natl. Acad. Sci. USA 2010, 107, 19338–19343. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, S.; Oba, M.; Suzuki, M.; Takahashi, A.; Yamamuro, T.; Fujiwara, M.; Ikenaka, K.; Minami, S.; Tabata, N.; Yamamoto, K.; et al. Suppression of autophagic activity by Rubicon is a signature of aging. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef]
- Yamamoto-Imoto, H.; Minami, S.; Shioda, T.; Yamashita, Y.; Sakai, S.; Maeda, S.; Yamamoto, T.; Oki, S.; Takashima, M.; Yamamuro, T.; et al. Age-associated decline of MondoA drives cellular senescence through impaired autophagy and mitochondrial homeostasis. Cell Rep. 2022, 38, 110444. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.; Malireddi, R.; Lu, Q.; Cunha, L.D.; Pelletier, S.; Gingras, S.; Orchard, R.; Guan, J.-L.; Tan, H.; Peng, J.; et al. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat. Cell Biol. 2015, 17, 893–906. [Google Scholar] [CrossRef] [Green Version]
- Heckmann, B.; Teubner, B.J.; Tummers, B.; Boada-Romero, E.; Harris, L.; Yang, M.; Guy, C.S.; Zakharenko, S.S.; Green, D.R. LC3-Associated Endocytosis Facilitates β-Amyloid Clearance and Mitigates Neurodegeneration in Murine Alzheimer’s Disease. Cell 2019, 178, 536–551.e14. [Google Scholar] [CrossRef]
- Muniz-Feliciano, L.; Doggett, T.A.; Zhou, Z.; Ferguson, T.A. RUBCN/rubicon and EGFR regulate lysosomal degradative processes in the retinal pigment epithelium (RPE) of the eye. Autophagy 2017, 13, 2072–2085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leyton, E.; Matus, D.; Espinoza, S.; Benitez, J.M.; Cortés, B.I.; Gomez, W.; Arévalo, N.B.; Murgas, P.; Manque, P.; Woehlbier, U.; et al. DEF8 and Autophagy-Associated Genes Are Altered in Mild Cognitive Impairment, Probable Alzheimer’s Disease Patients, and a Transgenic Model of the Disease. J. Alzheimer's Dis. 2021, 82, S163–S178. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer's Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Morris, J.C. The clinical dementia rating (cdr): Current version and scoring rules. Neurology 1993, 43, 2412–2414. [Google Scholar] [CrossRef]
- Okita, K.; Matsumura, Y.; Sato, Y.; Okada, A.; Morizane, A.; Okamoto, S.; Hong, H.; Nakagawa, M.; Tanabe, K.; Tezuka, K.-I.; et al. A more efficient method to generate integration-free human iPS cells. Nat. Methods 2011, 8, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Parra, V.; Altamirano, F.; Hernández-Fuentes, C.P.; Tong, D.; Kyrychenko, V.; Rotter, D.; Pedrozo, Z.; Hill, J.A.; Eisner, V.; Lavandero, S.; et al. Down syndrome critical region 1 gene, rcan1, helps maintain a more fused mitochondrial network. Circ. Res. 2018, 122, e20–e33. [Google Scholar] [CrossRef]
- Franklin, E.E.; Perrin, R.J.; Vincent, B.; Baxter, M.; Morris, J.C.; Cairns, N.J. Brain collection, standardized neuropathologic assessment, and comorbidity in Alzheimer’s Disease Neuroimaging Initiative 2 participants. Alzheimer’s Dement. 2015, 7, 815–822. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef]
- Jawhar, S.; Trawicka, A.; Jenneckens, C.; Bayer, T.A.; Wirths, O. Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal Aβ aggregation in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol. Aging 2012, 33, 196.e29–196.e40. [Google Scholar] [CrossRef]
- Beltrán, S.; Nassif, M.; Vicencio, E.; Arcos, J.; Labrador, L.; Cortes, B.I.; Cortez, C.; Bergmann, C.A.; Espinoza, S.; Hernandez, M.F.; et al. Network approach identifies Pacer as an autophagy protein involved in ALS pathogenesis. Mol. Neurodegener. 2019, 14, 14. [Google Scholar] [CrossRef]
- Prabhu, Y.; Burgos, P.V.; Schindler, C.; Farías, G.G.; Magadán, J.G.; Bonifacino, J.S. Adaptor protein 2–mediated endocytosis of the β-secretase BACE1 is dispensable for amyloid precursor protein processing. Mol. Biol. Cell 2012, 23, 2339–2351. [Google Scholar] [CrossRef] [PubMed]
- García-Huerta, P.; Troncoso-Escudero, P.; Wu, D.; Thiruvalluvan, A.; Cisternas-Olmedo, M.; Henríquez, D.R.; Plate, L.; Chana-Cuevas, P.; Saquel, C.; Thielen, P.; et al. Insulin-like growth factor 2 (IGF2) protects against Huntington’s disease through the extracellular disposal of protein aggregates. Acta Neuropathol. 2020, 140, 737–764. [Google Scholar] [CrossRef]
- Kondo, T.; Asai, M.; Tsukita, K.; Kutoku, Y.; Ohsawa, Y.; Sunada, Y.; Imamura, K.; Egawa, N.; Yahata, N.; Okita, K.; et al. Modeling Alzheimer’s Disease with iPSCs Reveals Stress Phenotypes Associated with Intracellular Aβ and Differential Drug Responsiveness. Cell Stem Cell 2013, 12, 487–496. [Google Scholar] [CrossRef] [Green Version]
- Seranova, E.; Palhegyi, A.M.; Verma, S.; Dimova, S.; Lasry, R.; Naama, M.; Sun, C.; Barrett, T.; Rosenstock, T.R.; Kumar, D.; et al. Human Induced Pluripotent Stem Cell Models of Neurodegenerative Disorders for Studying the Biomedical Implications of Autophagy. J. Mol. Biol. 2020, 432, 2754–2798. [Google Scholar] [CrossRef]
- Cervo, P.R.D.V.; Besusso, D.; Conforti, P.; Cattaneo, E. hiPSCs for predictive modelling of neurodegenerative diseases: Dreaming the possible. Nat. Rev. Neurol. 2021, 17, 381–392. [Google Scholar] [CrossRef]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.-A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [Green Version]
- Harris, C.D.; Ermak, G.; Davies, K.J.A. RCAN1-1L is overexpressed in neurons of Alzheimer's disease patients. FEBS J. 2007, 274, 1715–1724. [Google Scholar] [CrossRef]
- Assoum, M.; Salih, M.; Drouot, N.; Hnia, K.; Martelli, A.; Koenig, M. The Salih Ataxia Mutation Impairs Rubicon Endosomal Localization. Cerebellum 2013, 12, 835–840. [Google Scholar] [CrossRef]
- Assoum, M.; Salih, M.A.; Drouot, N.; Brahim, R.H.-B.; Lagier-Tourenne, C.; AlDrees, A.; Elmalik, S.A.; Ahmed, T.S.; Seidahmed, M.Z.; Kabiraj, M.M.; et al. Rundataxin, a novel protein with RUN and diacylglycerol binding domains, is mutant in a new recessive ataxia. Brain 2010, 133, 2439–2447. [Google Scholar] [CrossRef] [Green Version]
- Ba, L.; Chen, X.-H.; Chen, Y.-L.; Nie, Q.; Li, Z.-J.; Ding, F.-F.; Zhang, M. Distinct Rab7-related endosomal-autophagic-lysosomal dysregulation observed in cortex and hippocampus in APPswe/PSEN1dE9 mouse model of Alzheimer’s disease. Chin. Med. J. 2017, 130, 2941–2950. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Joh, T.H. Microglia, major player in the brain inflammation: Their roles in the pathogenesis of Parkinson’s disease. Exp. Mol. Med. 2006, 4, 333–347. [Google Scholar] [CrossRef] [Green Version]
- Kenkhuis, B.; Somarakis, A.; de Haan, L.; Dzyubachyk, O.; Ijsselsteijn, M.E.; de Miranda, N.F.C.C.; Lelieveldt, B.P.F.; Dijkstra, J.; van Roon-Mom, W.M.C.; Höllt, T.; et al. Iron loading is a prominent feature of activated microglia in Alzheimer’s disease patients. Acta Neuropathol. Commun. 2021, 9, 27. [Google Scholar] [CrossRef]
- Hoogland, I.C.M.; Houbolt, C.; van Westerloo, D.J.; van Gool, W.A.; van de Beek, D. Systemic inflammation and microglial activation: Systematic review of animal experiments. J. Neuroinflammation 2015, 12, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, X.; Ma, X.; Zhu, Q.; Song, D.; Ding, X.; Li, L.; Jiang, X.; Wang, X.; Tian, R.; Su, H.; et al. Pacer Is a Mediator of mTORC1 and GSK3-TIP60 Signaling in Regulation of Autophagosome Maturation and Lipid Metabolism. Mol. Cell 2019, 73, 788–802.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B.; et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J. Clin. Invest. 2008, 118, 2190–2199. [Google Scholar] [CrossRef] [Green Version]
- Ostrowski, M.; Carmo, N.B.; Krumeich, S.; Fanget, I.; Raposo, G.; Savina, A.; Moita, C.F.; Schauer, K.; Hume, A.N.; Freitas, R.P.; et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat. Cell Biol. 2010, 12, 19–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Zhang, S.; An, L.; Wu, J.; Hu, X.; Lai, S.; Mazhar, H.; Zou, Y.; He, L.; Zhu, H. Loss of Rubicon ameliorates doxorubicin-induced cardiotoxicity through enhancement of mitochondrial quality. Int. J. Cardiol. 2019, 296, 129–135. [Google Scholar] [CrossRef]
- Zi, Z.; Song, Z.; Zhang, S.; Ye, Y.; Li, C.; Xu, M.; Zou, Y.; He, L.; Zhu, H. Rubicon deficiency enhances cardiac autophagy and protects mice from lipopolysaccharide-induced lethality and reduction in stroke volume. J. Cardiovasc. Pharmacol. 2015, 65, 252–261. [Google Scholar] [CrossRef]
- Tanaka, S.; Hikita, H.; Tatsumi, T.; Sakamori, R.; Nozaki, Y.; Sakane, S.; Shiode, Y.; Nakabori, T.; Saito, Y.; Hiramatsu, N.; et al. Rubicon inhibits autophagy and accelerates hepatocyte apoptosis and lipid accumulation in nonalcoholic fatty liver disease in mice. Hepatology 2016, 64, 1994–2014. [Google Scholar] [CrossRef]
- Yamamuro, T.; Nakamura, S.; Yanagawa, K.; Tokumura, A.; Kawabata, T.; Fukuhara, A.; Teranishi, H.; Hamasaki, M.; Shimomura, I.; Yoshimori, T. Loss of RUBCN/rubicon in adipocytes mediates the upregulation of autophagy to promote the fasting response. Autophagy 2022, 14, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Yamamuro, T.; Kawabata, T.; Fukuhara, A.; Saita, S.; Nakamura, S.; Takeshita, H.; Fujiwara, M.; Enokidani, Y.; Yoshida, G.; Tabata, K.; et al. Age-dependent loss of adipose Rubicon promotes metabolic disorders via excess autophagy. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Ando, S.; Hashida, N.; Yamashita, D.; Kawabata, T.; Asao, K.; Kawasaki, S.; Sakurai, K.; Yoshimori, T.; Nishida, K. Rubicon regulates A2E-induced autophagy impairment in the retinal pigment epithelium implicated in the pathology of age-related macular degeneration. Biochem. Biophys. Res. Commun. 2021, 551, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Minami, S.; Nakamura, S.; Yoshimori, T. Rubicon in Metabolic Diseases and Ageing. Front. Cell Dev. Biol. 2022, 9, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.W.; Llop, J.R.; Farrell, S.F.; Yuan, J.; Stolzenburg, L.R.; Samuelson, A.v. The Caenorhabditis elegans Myc-Mondo/Mad complexes integrate diverse longevity signals. PLoS Genet. 2014, 10, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, S.; Karalay, Ö.; Jäger, P.S.; Horikawa, M.; Klein, C.; Nakamura, K.; Latza, C.; Templer, S.E.; Dieterich, C.; Antebi, A. Mondo Complexes Regulate TFEB via TOR Inhibition to Promote Longevity in Response to Gonadal Signals. Nat. Commun. 2016, 7. Available online: https://pubmed.ncbi.nlm.nih.gov/27001890/ (accessed on 25 April 2022).
- Stoltzman, C.A.; Peterson, C.W.; Breen, K.T.; Muoio, D.M.; Billin, A.N.; Ayer, D.E. Glucose sensing by MondoA:Mlx complexes: A role for hexokinases and direct regulation of thioredoxin-interacting protein expression. Proc. Natl. Acad. Sci. USA 2008, 105, 6912–6917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKnight, N.; Zhong, Y.; Wold, M.S.; Gong, S.; Phillips, G.R.; Dou, Z.; Zhao, Y.; Heintz, N.; Zong, W.-X.; Yue, Z. Beclin 1 Is Required for Neuron Viability and Regulates Endosome Pathways via the UVRAG-VPS34 Complex. PLoS Genet. 2014, 10, e1004626. [Google Scholar] [CrossRef] [Green Version]
- Glatigny, M.; Moriceau, S.; Rivagorda, M.; Ramos-Brossier, M.; Nascimbeni, A.C.; Lanté, F.; Shanley, M.R.; Boudarene, N.; Rousseaud, A.; Friedman, A.K.; et al. Autophagy Is Required for Memory Formation and Reverses Age-Related Memory Decline. Curr. Biol. 2019, 29, 435–448.e8. [Google Scholar] [CrossRef] [Green Version]
- Brashear, H.R.; Godec, M.S.; Carlsen, J. The distribution of neuritic plaques and acetylcholinesterase staining in the amygdala in alzheimer’s disease. Neurology 1988, 38, 1694–1698. [Google Scholar] [CrossRef]
- Nelson, P.T.; Abner, E.L.; Patel, E.; Anderson, S.; Wilcock, D.M.; Kryscio, R.J.; Van Eldik, L.J.; Jicha, G.A.; Gal, Z.; Nelson, R.S.; et al. The Amygdala as a Locus of Pathologic Misfolding in Neurodegenerative Diseases. J. Neuropathol. Exp. Neurol. 2018, 77, 2–20. [Google Scholar] [CrossRef]
- Scott, S.A.; DeKosky, S.T.; Scheff, S.W. Volumetrie atrophy of the amygdala in Alzheimer’s disease: Quantitative serial reconstruction. Neurology 1991, 41, 351–356. [Google Scholar] [CrossRef]
- Swart, C.; Khoza, A.; Khan, K.; Le Roux, S.; Du Plessis, A.; Loos, B. Investigating Basal Autophagic Activity in Brain Regions Associated with Neurodegeneration using In Vivo and Ex Vivo Models. J. Alzheimer’s Dis. Park. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Heckmann, B.L.; Teubner, B.J.W.; Boada-Romero, E.; Tummers, B.; Guy, C.; Fitzgerald, P.; Mayer, U.; Carding, S.; Zakharenko, S.S.; Wileman, T.; et al. Noncanonical function of an autophagy protein prevents spontaneous Alzheimer’s disease. Sci. Adv. 2020, 6, 1–12. [Google Scholar] [CrossRef]
- Pensalfini, A.; Kim, S.; Subbanna, S.; Bleiwas, C.; Goulbourne, C.N.; Stavrides, P.H.; Jiang, Y.; Lee, J.-H.; Darji, S.; Pawlik, M.; et al. Endosomal Dysfunction Induced by Directly Overactivating Rab5 Recapitulates Prodromal and Neurodegenerative Features of Alzheimer’s Disease. Cell Rep. 2020, 33, 108420. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Donor | Gender | Disease Duration (Years) | CDR | Braak Stage |
|---|---|---|---|---|
| C1 | Male | - | 0 | I |
| C2 | Female | - | 0 | II |
| C3 | Female | - | 0 | II |
| C4 | Female | - | 0 | II |
| AD1 | Female | 6 | 3 | IV |
| AD2 | Female | 6 | 3 | IV |
| AD3 | Male | 6 | 3 | IV |
| AD4 | Male | 10 | 3 | IV |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Espinoza, S.; Grunenwald, F.; Gomez, W.; García, F.; Abarzúa-Catalan, L.; Oyarce-Pezoa, S.; Hernandez, M.F.; Cortés, B.I.; Uhrig, M.; Ponce, D.P.; et al. Neuronal Rubicon Represses Extracellular APP/Amyloid β Deposition in Alzheimer’s Disease. Cells 2022, 11, 1860. https://doi.org/10.3390/cells11121860
Espinoza S, Grunenwald F, Gomez W, García F, Abarzúa-Catalan L, Oyarce-Pezoa S, Hernandez MF, Cortés BI, Uhrig M, Ponce DP, et al. Neuronal Rubicon Represses Extracellular APP/Amyloid β Deposition in Alzheimer’s Disease. Cells. 2022; 11(12):1860. https://doi.org/10.3390/cells11121860
Chicago/Turabian StyleEspinoza, Sandra, Felipe Grunenwald, Wileidy Gomez, Felipe García, Lorena Abarzúa-Catalan, Sebastián Oyarce-Pezoa, Maria Fernanda Hernandez, Bastián I. Cortés, Markus Uhrig, Daniela P. Ponce, and et al. 2022. "Neuronal Rubicon Represses Extracellular APP/Amyloid β Deposition in Alzheimer’s Disease" Cells 11, no. 12: 1860. https://doi.org/10.3390/cells11121860
APA StyleEspinoza, S., Grunenwald, F., Gomez, W., García, F., Abarzúa-Catalan, L., Oyarce-Pezoa, S., Hernandez, M. F., Cortés, B. I., Uhrig, M., Ponce, D. P., Durán-Aniotz, C., Hetz, C., SanMartín, C. D., Cornejo, V. H., Ezquer, F., Parra, V., Behrens, M. I., Manque, P. A., Rojas-Rivera, D., ... Nassif, M. (2022). Neuronal Rubicon Represses Extracellular APP/Amyloid β Deposition in Alzheimer’s Disease. Cells, 11(12), 1860. https://doi.org/10.3390/cells11121860