
Influence of the Metabolism on Myeloid Cell Functions in Cancers: Clinical Perspectives


Abstract
1. Introduction
2. Metabolism of TAMs and MDSCs, the Major Components of the Tumor Myeloid Landscape
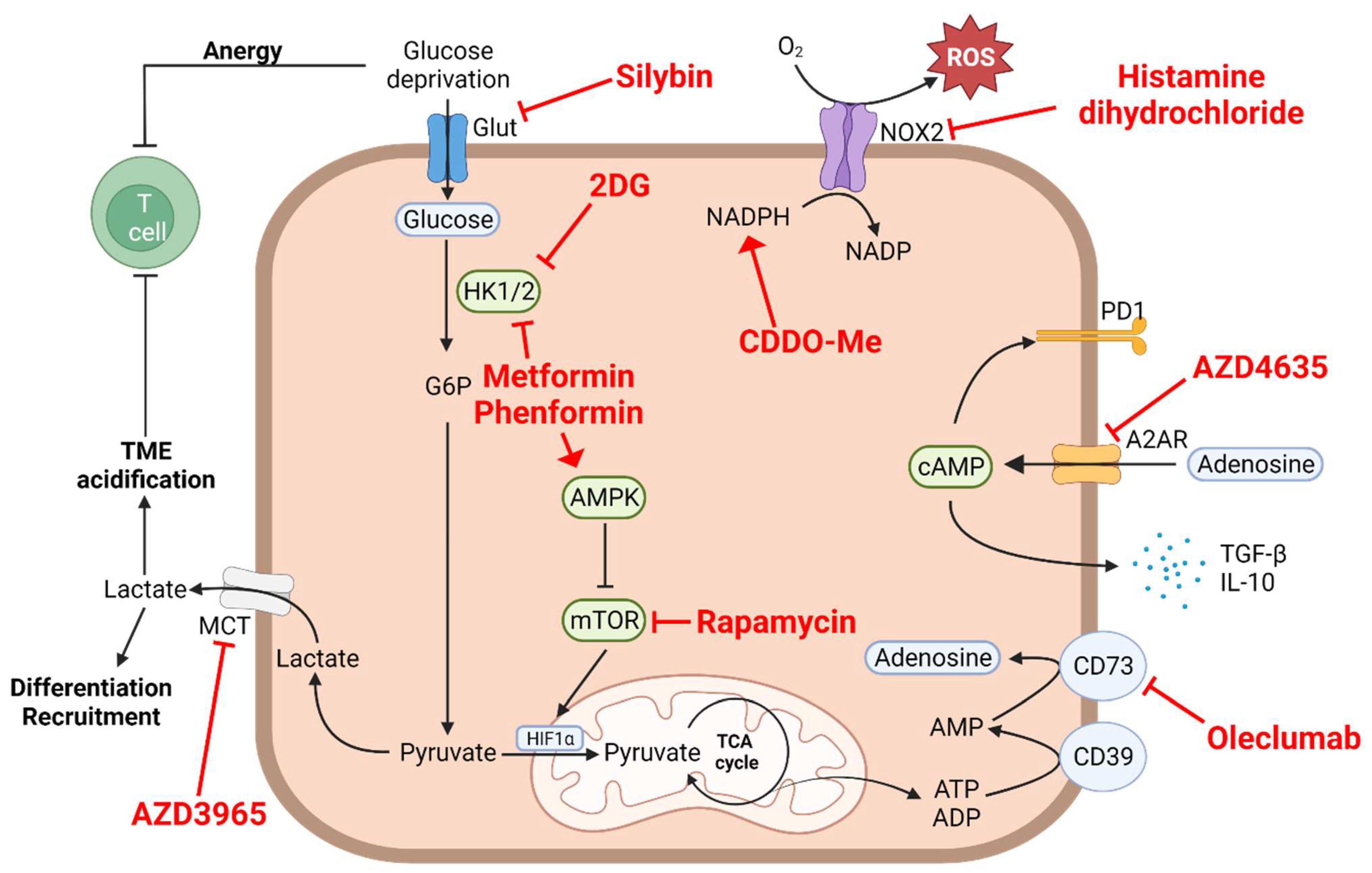
3. Aerobic Glycolysis, Lactate Accumulation and Acidification of the Medium
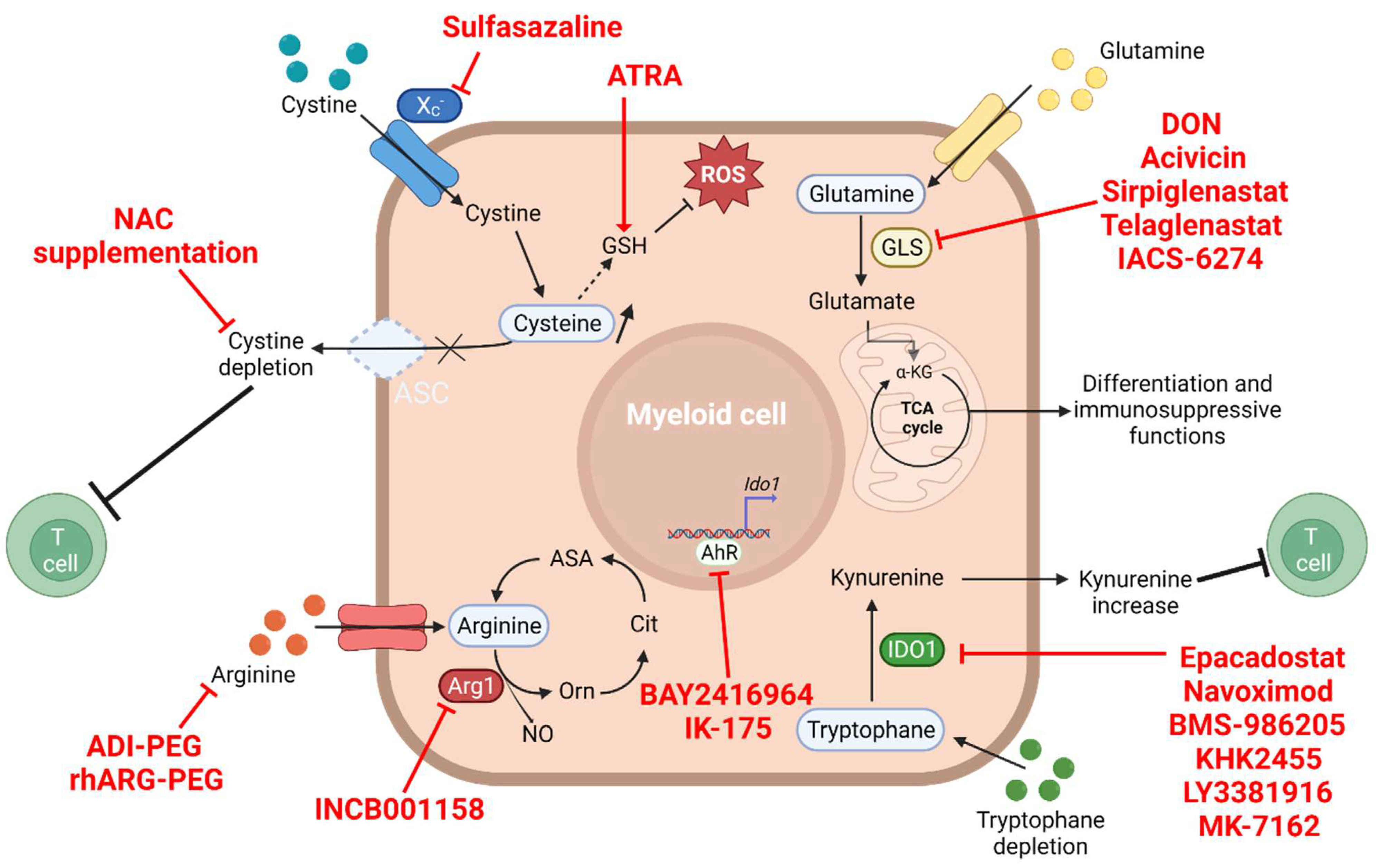
4. The Amino Acid Metabolism
4.1. Glutamine and Glutaminolysis
4.2. Arginine Depletion
4.3. Tryptophan–Kynurenine Pathway
4.4. Cysteine Depletion
5. Extracellular Adenosine
6. The Oxidative Stress
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Arora, S.; Velichinskii, R.; Lesh, R.W.; Ali, U.; Kubiak, M.; Bansal, P.; Borghaei, H.; Edelman, M.J.; Boumber, Y. Existing and Emerging Biomarkers for Immune Checkpoint Immunotherapy in Solid Tumors. Adv. Ther. 2019, 36, 2638–2678. [Google Scholar] [CrossRef]
- Postow, M.A.; Sidlow, R.; Hellmann, M.D. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. N. Engl. J. Med. 2018, 378, 158–168. [Google Scholar] [CrossRef]
- Schoenfeld, A.J.; Hellmann, M.D. Acquired Resistance to Immune Checkpoint Inhibitors. Cancer Cell 2020, 37, 443–455. [Google Scholar] [CrossRef]
- Bai, R.; Chen, N.; Li, L.; Du, N.; Bai, L.; Lv, Z.; Tian, H.; Cui, J. Mechanisms of Cancer Resistance to Immunotherapy. Front. Oncol. 2020, 10, 1290. [Google Scholar] [CrossRef]
- Bettonville, M.; D’Aria, S.; Braun, M.Y. Metabolic Programming in Chronically Stimulated T Cells: Lessons from Cancer and Viral Infections. Eur. J. Immunol. 2016, 46, 1574–1582. [Google Scholar] [CrossRef]
- Sieow, J.L.; Gun, S.Y.; Wong, S.C. The Sweet Surrender: How Myeloid Cell Metabolic Plasticity Shapes the Tumor Microenvironment. Front. Cell Dev. Biol. 2018, 6, 168. [Google Scholar] [CrossRef]
- Labani-Motlagh, A.; Ashja-Mahdavi, M.; Loskog, A. The Tumor Microenvironment: A Milieu Hindering and Obstructing Antitumor Immune Responses. Front. Immunol. 2020, 11, 940. [Google Scholar] [CrossRef]
- Seoane, S.; Martinez-Ordoñez, A.; Eiro, N.; Cabezas-Sainz, P.; Garcia-Caballero, L.; Gonzalez, L.O.; Macia, M.; Sanchez, L.; Vizoso, F.; Perez-Fernandez, R. POU1F1 Transcription Factor Promotes Breast Cancer Metastasis via Recruitment and Polarization of Macrophages. J. Pathol. 2019, 249, 381–394. [Google Scholar] [CrossRef]
- Bieniasz-Krzywiec, P.; Martín-Pérez, R.; Ehling, M.; García-Caballero, M.; Pinioti, S.; Pretto, S.; Kroes, R.; Aldeni, C.; Di Matteo, M.; Prenen, H.; et al. Podoplanin-Expressing Macrophages Promote Lymphangiogenesis and Lymphoinvasion in Breast Cancer. Cell Metab. 2019, 30, 917–936.e10. [Google Scholar] [CrossRef]
- Chen, X.W.; Yu, T.J.; Zhang, J.; Li, Y.; Chen, H.L.; Yang, G.F.; Yu, W.; Liu, Y.Z.; Liu, X.X.; Duan, C.F.; et al. CYP4A in Tumor-Associated Macrophages Promotes Pre-Metastatic Niche Formation and Metastasis. Oncogene 2017, 36, 5045–5057. [Google Scholar] [CrossRef]
- Eisenblaetter, M.; Flores-Borja, F.; Lee, J.J.; Wefers, C.; Smith, H.; Hueting, R.; Cooper, M.S.; Blower, P.J.; Patel, D.; Rodriguez-Justo, M.; et al. Visualization of Tumor-Immune Interaction - Target-Specific Imaging of S100A8/A9 Reveals Pre-Metastatic Niche Establishment. Theranostics 2017, 7, 2392–2401. [Google Scholar] [CrossRef] [PubMed]
- Grover, A.; Sanseviero, E.; Timosenko, E.; Gabrilovich, D.I. Myeloid-Derived Suppressor Cells: A Propitious Road to Clinic. Cancer Discov. 2021, 11, 2693–2706. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Jaillon, S.; Garlanda, C.; Allavena, P. Tumor-Associated Myeloid Cells: Diversity and Therapeutic Targeting. Cell Mol. Immunol. 2021, 18, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11, 3151. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Hawkins, G.A.; Wudel, L.; Chou, P.; Forbes, E.; Pullikuth, A.K.; Liu, L.; Jin, G.; Craddock, L.; Topaloglu, U.; et al. Dissecting Intratumoral Myeloid Cell Plasticity by Single Cell RNA-seq. Cancer Med. 2019, 8, 3072–3085. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Edwards, J.P. Exploring the Full Spectrum of Macrophage Activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Domblides, C.; Lartigue, L.; Faustin, B. Control of the Antitumor Immune Response by Cancer Metabolism. Cells 2019, 8, 104. [Google Scholar] [CrossRef] [PubMed]
- Goffaux, G.; Hammami, I.; Jolicoeur, M. A Dynamic Metabolic Flux Analysis of Myeloid-Derived Suppressor Cells Confirms Immunosuppression-Related Metabolic Plasticity. Sci. Rep. 2017, 7, 9850. [Google Scholar] [CrossRef]
- Jian, S.-L.; Chen, W.-W.; Su, Y.-C.; Su, Y.-W.; Chuang, T.-H.; Hsu, S.-C.; Huang, L.-R. Glycolysis Regulates the Expansion of Myeloid-Derived Suppressor Cells in Tumor-Bearing Hosts through Prevention of ROS-Mediated Apoptosis. Cell Death Dis. 2017, 8, e2779. [Google Scholar] [CrossRef]
- Hossain, F.; Al-Khami, A.A.; Wyczechowska, D.; Hernandez, C.; Zheng, L.; Reiss, K.; Valle, L.D.; Trillo-Tinoco, J.; Maj, T.; Zou, W.; et al. Inhibition of Fatty Acid Oxidation Modulates Immunosuppressive Functions of Myeloid-Derived Suppressor Cells and Enhances Cancer Therapies. Cancer Immunol. Res. 2015, 3, 1236–1247. [Google Scholar] [CrossRef]
- Hu, C.; Pang, B.; Lin, G.; Zhen, Y.; Yi, H. Energy Metabolism Manipulates the Fate and Function of Tumour Myeloid-Derived Suppressor Cells. Br. J. Cancer 2020, 122, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Domblides, C.; Lartigue, L.; Faustin, B. Metabolic Stress in the Immune Function of T Cells, Macrophages and Dendritic Cells. Cells 2018, 7, 68. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.C.-C.; Smith, A.M.; Everts, B.; Colonna, M.; Pearce, E.L.; Schilling, J.D.; Pearce, E.J. Metabolic Reprogramming Mediated by the MTORC2-IRF4 Signaling Axis Is Essential for Macrophage Alternative Activation. Immunity 2016, 45, 817–830. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Allavena, P.; Mantovani, A. Tumor-Associated Macrophages: Functional Diversity, Clinical Significance, and Open Questions. Semin. Immunopathol. 2013, 35, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, C.; Muthana, M.; Coffelt, S.B.; Lewis, C.E. The Role of Myeloid Cells in the Promotion of Tumour Angiogenesis. Nat. Rev. Cancer 2008, 8, 618–631. [Google Scholar] [CrossRef]
- Biswas, S.K. Metabolic Reprogramming of Immune Cells in Cancer Progression. Immunity 2015, 43, 435–449. [Google Scholar] [CrossRef]
- Halaby, M.J.; Hezaveh, K.; Lamorte, S.; Ciudad, M.T.; Kloetgen, A.; MacLeod, B.L.; Guo, M.; Chakravarthy, A.; Medina, T.D.S.; Ugel, S.; et al. GCN2 Drives Macrophage and MDSC Function and Immunosuppression in the Tumor Microenvironment. Sci. Immunol. 2019, 4, eaax8189. [Google Scholar] [CrossRef]
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-Derived Lactate Modifies Antitumor Immune Response: Effect on Myeloid-Derived Suppressor Cells and NK Cells. J. Immunol. 2013, 191, 1486–1495. [Google Scholar] [CrossRef]
- Fukumura, D.; Xu, L.; Chen, Y.; Gohongi, T.; Seed, B.; Jain, R.K. Hypoxia and Acidosis Independently Up-Regulate Vascular Endothelial Growth Factor Transcription in Brain Tumors in Vivo. Cancer Res. 2001, 61, 6020–6024. [Google Scholar]
- Ohashi, T.; Akazawa, T.; Aoki, M.; Kuze, B.; Mizuta, K.; Ito, Y.; Inoue, N. Dichloroacetate Improves Immune Dysfunction Caused by Tumor-Secreted Lactic Acid and Increases Antitumor Immunoreactivity. Int. J. Cancer 2013, 133, 1107–1118. [Google Scholar] [CrossRef]
- Janeway, C.A.; Travers, P.; Walport, M.; Shlomchik, M.J.; Jr, C.A.J.; Travers, P.; Walport, M.; Shlomchik, M.J. Immunobiology; 5th ed.; Garland Science: New York, NY, USA, 2001; ISBN 978-0-8153-3642-6. [Google Scholar]
- Liu, P.-S.; Wang, H.; Li, X.; Chao, T.; Teav, T.; Christen, S.; Di Conza, G.; Cheng, W.-C.; Chou, C.-H.; Vavakova, M.; et al. α-Ketoglutarate Orchestrates Macrophage Activation through Metabolic and Epigenetic Reprogramming. Nat. Immunol. 2017, 18, 985–994. [Google Scholar] [CrossRef]
- Oh, M.-H.; Sun, I.-H.; Zhao, L.; Leone, R.D.; Sun, I.-M.; Xu, W.; Collins, S.L.; Tam, A.J.; Blosser, R.L.; Patel, C.H.; et al. Targeting Glutamine Metabolism Enhances Tumor-Specific Immunity by Modulating Suppressive Myeloid Cells. J. Clin. Invest. 2020, 130, 3865–3884. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A Nutrient and Energy Sensor That Maintains Energy Homeostasis. Nat. Rev. Mol. Cell Biol 2012, 13, 251–262. [Google Scholar] [CrossRef]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- Dimmer, K.-S.; Friedrich, B.; Lang, F.; Deitmer, J.W.; Bröer, S. The Low-Affinity Monocarboxylate Transporter MCT4 Is Adapted to the Export of Lactate in Highly Glycolytic Cells. Biochem. J. 2000, 350, 219–227. [Google Scholar] [CrossRef]
- Fox, C.J.; Hammerman, P.S.; Thompson, C.B. Fuel Feeds Function: Energy Metabolism and the T-Cell Response. Nat. Rev. Immunol. 2005, 5, 844–852. [Google Scholar] [CrossRef]
- Renner, K.; Singer, K.; Koehl, G.E.; Geissler, E.K.; Peter, K.; Siska, P.J.; Kreutz, M. Metabolic Hallmarks of Tumor and Immune Cells in the Tumor Microenvironment. Front. Immunol. 2017, 8, 248. [Google Scholar] [CrossRef]
- Calcinotto, A.; Filipazzi, P.; Grioni, M.; Iero, M.; Milito, A.D.; Ricupito, A.; Cova, A.; Canese, R.; Jachetti, E.; Rossetti, M.; et al. Modulation of Microenvironment Acidity Reverses Anergy in Human and Murine Tumor-Infiltrating T Lymphocytes. Cancer Res. 2012, 72, 2746–2756. [Google Scholar] [CrossRef]
- Flaig, T.W.; Gustafson, D.L.; Su, L.-J.; Zirrolli, J.A.; Crighton, F.; Harrison, G.S.; Pierson, A.S.; Agarwal, R.; Glodé, L.M. A Phase I and Pharmacokinetic Study of Silybin-Phytosome in Prostate Cancer Patients. Invest. New Drugs 2007, 25, 139–146. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, K.; Shi, L.; Xiang, F.; Tao, K.; Wang, G. Prognostic Significance of the Metabolic Marker Hexokinase-2 in Various Solid Tumors: A Meta-Analysis. PLOS ONE 2016, 11, e0166230. [Google Scholar] [CrossRef] [PubMed]
- Sottnik, J.L.; Lori, J.C.; Rose, B.J.; Thamm, D.H. Glycolysis Inhibition by 2-Deoxy-D-Glucose Reverts the Metastatic Phenotype in Vitro and in Vivo. Clin. Exp. Metastasis 2011, 28, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Eikawa, S.; Nishida, M.; Mizukami, S.; Yamazaki, C.; Nakayama, E.; Udono, H. Immune-Mediated Antitumor Effect by Type 2 Diabetes Drug, Metformin. PNAS 2015. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.-H.; Yang, W.-H.; Xia, W.; Wei, Y.; Chan, L.-C.; Lim, S.-O.; Li, C.-W.; Kim, T.; Chang, S.-S.; Lee, H.-H.; et al. Metformin Promotes Antitumor Immunity via Endoplasmic-Reticulum-Associated Degradation of PD-L1. Mol. Cell 2018, 71, 606–620.e7. [Google Scholar] [CrossRef]
- Kim, S.H.; Li, M.; Trousil, S.; Zhang, Y.; Pasca di Magliano, M.; Swanson, K.D.; Zheng, B. Phenformin Inhibits Myeloid-Derived Suppressor Cells and Enhances the Anti-Tumor Activity of PD-1 Blockade in Melanoma. J. Invest. Derm. 2017, 137, 1740–1748. [Google Scholar] [CrossRef]
- Qin, G.; Lian, J.; Huang, L.; Zhao, Q.; Liu, S.; Zhang, Z.; Chen, X.; Yue, D.; Li, L.; Li, F.; et al. Metformin Blocks Myeloid-Derived Suppressor Cell Accumulation through AMPK-DACH1-CXCL1 Axis. Oncoimmunology 2018, 7, e1442167. [Google Scholar] [CrossRef]
- Xu, P.; Yin, K.; Tang, X.; Tian, J.; Zhang, Y.; Ma, J.; Xu, H.; Xu, Q.; Wang, S. Metformin Inhibits the Function of Granulocytic Myeloid-Derived Suppressor Cells in Tumor-Bearing Mice. Biomed. Pharm. 2019, 120, 109458. [Google Scholar] [CrossRef]
- Mercalli, A.; Calavita, I.; Dugnani, E.; Citro, A.; Cantarelli, E.; Nano, R.; Melzi, R.; Maffi, P.; Secchi, A.; Sordi, V.; et al. Rapamycin Unbalances the Polarization of Human Macrophages to M1. Immunology 2013, 140, 179–190. [Google Scholar] [CrossRef]
- Deng, Y.; Yang, J.; Luo, F.; Qian, J.; Liu, R.; Zhang, D.; Yu, H.; Chu, Y. MTOR-Mediated Glycolysis Contributes to the Enhanced Suppressive Function of Murine Tumor-Infiltrating Monocytic Myeloid-Derived Suppressor Cells. Cancer Immunol. Immunother. 2018, 67, 1355–1364. [Google Scholar] [CrossRef]
- Ma, P.; Xing, M.; Han, L.; Gan, S.; Ma, J.; Wu, F.; Huang, Y.; Chen, Y.; Tian, W.; An, C.; et al. High PD-L1 Expression Drives Glycolysis via an Akt/MTOR/HIF-1α Axis in Acute Myeloid Leukemia. Oncol. Rep. 2020, 43, 999–1009. [Google Scholar] [CrossRef]
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.W.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef]
- Wagner, J.; Rapsomaniki, M.A.; Chevrier, S.; Anzeneder, T.; Langwieder, C.; Dykgers, A.; Rees, M.; Ramaswamy, A.; Muenst, S.; Soysal, S.D.; et al. A Single-Cell Atlas of the Tumor and Immune Ecosystem of Human Breast Cancer. Cell 2019, 177, 1330–1345.e18. [Google Scholar] [CrossRef] [PubMed]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 Alters T-Cell Metabolic Reprogramming by Inhibiting Glycolysis and Promoting Lipolysis and Fatty Acid Oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 Is a Novel Direct Target of HIF-1α, and Its Blockade under Hypoxia Enhanced MDSC-Mediated T Cell Activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Puri, S.; Juvale, K. Monocarboxylate Transporter 1 and 4 Inhibitors as Potential Therapeutics for Treating Solid Tumours: A Review with Structure-Activity Relationship Insights. Eur. J. Med. Chem. 2020, 199, 112393. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Morris, M.E. In Vitro and In Vivo Efficacy of AZD3965 and Alpha-Cyano-4-Hydroxycinnamic Acid in the Murine 4T1 Breast Tumor Model. AAPS J 2020, 22, 84. [Google Scholar] [CrossRef] [PubMed]
- Halford, S.E.R.; Walter, H.; McKay, P.; Townsend, W.; Linton, K.; Heinzmann, K.; Dragoni, I.; Brotherton, L.; Veal, G.; Siskos, A.; et al. Phase I Expansion Study of the First-in-Class Monocarboxylate Transporter 1 (MCT1) Inhibitor AZD3965 in Patients with Diffuse Large B-Cell Lymphoma (DLBCL) and Burkitt Lymphoma (BL). JCO 2021, 39, 3115. [Google Scholar] [CrossRef]
- McNeillis, R.; Greystoke, A.; Walton, J.; Bacon, C.; Keun, H.; Siskos, A.; Petrides, G.; Leech, N.; Jenkinson, F.; Bowron, A.; et al. A Case of Malignant Hyperlactaemic Acidosis Appearing upon Treatment with the Mono-Carboxylase Transporter 1 Inhibitor AZD3965. Br. J. Cancer 2020, 122, 1141–1145. [Google Scholar] [CrossRef]
- Metzler, B.; Gfeller, P.; Guinet, E. Restricting Glutamine or Glutamine-Dependent Purine and Pyrimidine Syntheses Promotes Human T Cells with High FOXP3 Expression and Regulatory Properties. J. Immunol. 2016, 196, 3618–3630. [Google Scholar] [CrossRef]
- Lemberg, K.M.; Vornov, J.J.; Rais, R.; Slusher, B.S. We’re Not “DON” Yet: Optimal Dosing and Prodrug Delivery of 6-Diazo-5-Oxo-L-Norleucine. Mol. Cancer Ther. 2018, 17, 1824–1832. [Google Scholar] [CrossRef]
- Sun, H.-W.; Wu, W.-C.; Chen, H.-T.; Xu, Y.-T.; Yang, Y.-Y.; Chen, J.; Yu, X.-J.; Wang, Z.; Shuang, Z.-Y.; Zheng, L. Glutamine Deprivation Promotes the Generation and Mobilization of MDSCs by Enhancing Expression of G-CSF and GM-CSF. Front. Immunol. 2020, 11, 616367. [Google Scholar] [CrossRef] [PubMed]
- Leone, R.D.; Zhao, L.; Englert, J.M.; Sun, I.-M.; Oh, M.-H.; Sun, I.-H.; Arwood, M.L.; Bettencourt, I.A.; Patel, C.H.; Wen, J.; et al. Glutamine Blockade Induces Divergent Metabolic Programs to Overcome Tumor Immune Evasion. Science 2019, 366, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Masisi, B.K.; El Ansari, R.; Alfarsi, L.; Rakha, E.A.; Green, A.R.; Craze, M.L. The Role of Glutaminase in Cancer. Histopathology 2020, 76, 498–508. [Google Scholar] [CrossRef]
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor Activity of the Glutaminase Inhibitor CB-839 in Triple-Negative Breast Cancer. Mol. Cancer Ther. 2014, 13, 890–901. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D.N.; Ngwa, V.M.; Raybuck, A.L.; Wang, S.; Hwang, Y.; Kim, L.C.; Cho, S.H.; Paik, Y.; Wang, Q.; Zhang, S.; et al. Selective Glutamine Metabolism Inhibition in Tumor Cells Improves Antitumor T Lymphocyte Activity in Triple-Negative Breast Cancer. J. Clin. Invest. 2021, 131, 140100. [Google Scholar] [CrossRef] [PubMed]
- Tannir, N.M.; Agarwal, N.; Porta, C.; Lawrence, N.J.; Motzer, R.J.; Lee, R.J.; Jain, R.K.; Davis, N.B.; Appleman, L.J.; Goodman, O.B.; et al. CANTATA: Primary Analysis of a Global, Randomized, Placebo (Pbo)-Controlled, Double-Blind Trial of Telaglenastat (CB-839) + Cabozantinib versus Pbo + Cabozantinib in Advanced/Metastatic Renal Cell Carcinoma (MRCC) Patients (Pts) Who Progressed on Immune Checkpoint Inhibitor (ICI) or Anti-Angiogenic Therapies. JCO 2021, 39, 4501. [Google Scholar] [CrossRef]
- Yap, T.A.; Dumbrava, E.E.; Rodon Ahnert, J.; Hong, D.S.; Pant, S.; Karp, D.D.; Piha-Paul, S.A.A.; Subbiah, V.; Tsimberidou, A.M.; Fu, S.; et al. First-in-Human Biomarker-Driven Phase I Trial of the Potent and Selective Glutaminase-1 (GLS1) Inhibitor IACS-6274 (IPN60090) in Patients (Pts) with Molecularly Selected Advanced Solid Tumors. JCO 2021, 39, 3001. [Google Scholar] [CrossRef]
- Byun, J.-K.; Park, M.; Lee, S.; Yun, J.W.; Lee, J.; Kim, J.S.; Cho, S.J.; Jeon, H.-J.; Lee, I.-K.; Choi, Y.-K.; et al. Inhibition of Glutamine Utilization Synergizes with Immune Checkpoint Inhibitor to Promote Antitumor Immunity. Mol. Cell 2020, 80, 592–606.e8. [Google Scholar] [CrossRef]
- Ekici, S.; Nye, J.A.; Neill, S.G.; Allen, J.W.; Shu, H.-K.; Fleischer, C.C. Glutamine Imaging: A New Avenue for Glioma Management. AJNR Am. J. Neuroradiol. 2021. [Google Scholar] [CrossRef]
- Chen, C.-L.; Hsu, S.-C.; Ann, D.K.; Yen, Y.; Kung, H.-J. Arginine Signaling and Cancer Metabolism. Cancers 2021, 13, 3541. [Google Scholar] [CrossRef]
- Patil, M.D.; Bhaumik, J.; Babykutty, S.; Banerjee, U.C.; Fukumura, D. Arginine Dependence of Tumor Cells: Targeting a Chink in Cancer’s Armor. Oncogene 2016, 35, 4957–4972. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Lian, J.; Yang, M.; Wuyang, J.; Zhao, C.; Chen, W.; Liu, C.; Zhao, Q.; Lou, C.; Han, J.; et al. Overexpression of Arginase-1 Is an Indicator of Poor Prognosis in Patients with Colorectal Cancer. Pathol. Res. Pract. 2019, 215. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Zanovello, P. Regulation of Immune Responses by L-Arginine Metabolism. Nat. Rev. Immunol. 2005, 5, 641–654. [Google Scholar] [CrossRef] [PubMed]
- Grzywa, T.M.; Sosnowska, A.; Matryba, P.; Rydzynska, Z.; Jasinski, M.; Nowis, D.; Golab, J. Myeloid Cell-Derived Arginase in Cancer Immune Response. Front. Immunol. 2020, 11, 938. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.C.; Ernstoff, M.S.; Hernandez, C.; Atkins, M.; Zabaleta, J.; Sierra, R.; Ochoa, A.C. Arginase I-Producing Myeloid-Derived Suppressor Cells in Renal Cell Carcinoma Are a Subpopulation of Activated Granulocytes. Cancer Res. 2009, 69, 1553–1560. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.C.; Quiceno, D.G.; Zabaleta, J.; Ortiz, B.; Zea, A.H.; Piazuelo, M.B.; Delgado, A.; Correa, P.; Brayer, J.; Sotomayor, E.M.; et al. Arginase I Production in the Tumor Microenvironment by Mature Myeloid Cells Inhibits T-Cell Receptor Expression and Antigen-Specific T-Cell Responses. Cancer Res. 2004, 64, 5839–5849. [Google Scholar] [CrossRef]
- Hoechst, B.; Ormandy, L.A.; Ballmaier, M.; Lehner, F.; Krüger, C.; Manns, M.P.; Greten, T.F.; Korangy, F. A New Population of Myeloid-Derived Suppressor Cells in Hepatocellular Carcinoma Patients Induces CD4+CD25+Foxp3+ T Cells. Gastroenterology 2008, 135, 234–243. [Google Scholar] [CrossRef]
- Mazzoni, A.; Bronte, V.; Visintin, A.; Spitzer, J.H.; Apolloni, E.; Serafini, P.; Zanovello, P.; Segal, D.M. Myeloid Suppressor Lines Inhibit T Cell Responses by an NO-Dependent Mechanism. J. Immunol. 2002, 168, 689–695. [Google Scholar] [CrossRef]
- Molon, B.; Ugel, S.; Del Pozzo, F.; Soldani, C.; Zilio, S.; Avella, D.; De Palma, A.; Mauri, P.; Monegal, A.; Rescigno, M.; et al. Chemokine Nitration Prevents Intratumoral Infiltration of Antigen-Specific T Cells. J. Exp. Med. 2011, 208, 1949–1962. [Google Scholar] [CrossRef]
- Ekmekcioglu, S.; Grimm, E.A.; Roszik, J. Targeting INOS to Increase Efficacy of Immunotherapies. Hum. Vaccin. Immunother. 2017, 13, 1105–1108. [Google Scholar] [CrossRef]
- Serafini, P.; Meckel, K.; Kelso, M.; Noonan, K.; Califano, J.; Koch, W.; Dolcetti, L.; Bronte, V.; Borrello, I. Phosphodiesterase-5 Inhibition Augments Endogenous Antitumor Immunity by Reducing Myeloid-Derived Suppressor Cell Function. J. Exp. Med. 2006, 203, 2691–2702. [Google Scholar] [CrossRef]
- Miret, J.J.; Kirschmeier, P.; Koyama, S.; Zhu, M.; Li, Y.Y.; Naito, Y.; Wu, M.; Malladi, V.S.; Huang, W.; Walker, W.; et al. Suppression of Myeloid Cell Arginase Activity Leads to Therapeutic Response in a NSCLC Mouse Model by Activating Anti-Tumor Immunity. J. Immunother. Cancer 2019, 7, 32. [Google Scholar] [CrossRef]
- Steggerda, S.M.; Bennett, M.K.; Chen, J.; Emberley, E.; Huang, T.; Janes, J.R.; Li, W.; MacKinnon, A.L.; Makkouk, A.; Marguier, G.; et al. Inhibition of Arginase by CB-1158 Blocks Myeloid Cell-Mediated Immune Suppression in the Tumor Microenvironment. J. Immunother. Cancer 2017, 5, 101. [Google Scholar] [CrossRef]
- Naing, A.; Bauer, T.; Papadopoulos, K.P.; Rahma, O.; Tsai, F.; Garralda, E.; Naidoo, J.; Pai, S.; Gibson, M.K.; Rybkin, I.; et al. Phase I Study of the Arginase Inhibitor INCB001158 (1158) Alone and in Combination with Pembrolizumab (PEM) in Patients (Pts) with Advanced/Metastatic (Adv/Met) Solid Tumours. Ann. Oncol. 2019, 30, v160. [Google Scholar] [CrossRef]
- O’Neil, B.H.; Wallmark, J.M.; Lorente, D.; Elez, E.; Raimbourg, J.; Gomez-Roca, C.; Ejadi, S.; Piha-Paul, S.A.; Stein, M.N.; Abdul Razak, A.R.; et al. Safety and Antitumor Activity of the Anti-PD-1 Antibody Pembrolizumab in Patients with Advanced Colorectal Carcinoma. PLoS ONE 2017, 12, e0189848. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.M.; Bridgewater, J.A.; Gbolahan, O.B.; Jungels, C.; Cho, M.T.; Papadopoulos, K.P.; Thistlethwaite, F.C.; Canon, J.-L.R.; Cheng, L.; Ioannidis, S.; et al. A Phase I/II Study of Safety and Efficacy of the Arginase Inhibitor INCB001158 plus Chemotherapy in Patients (Pts) with Advanced Biliary Tract Cancers. JCO 2021, 39, 311. [Google Scholar] [CrossRef]
- Valle, J.; Wasan, H.; Palmer, D.H.; Cunningham, D.; Anthoney, A.; Maraveyas, A.; Madhusudan, S.; Iveson, T.; Hughes, S.; Pereira, S.P.; et al. Cisplatin plus Gemcitabine versus Gemcitabine for Biliary Tract Cancer. N. Engl. J. Med. 2010, 362, 1273–1281. [Google Scholar] [CrossRef]
- Fletcher, M.; Ramirez, M.E.; Sierra, R.A.; Raber, P.; Thevenot, P.; Al-Khami, A.A.; Sanchez-Pino, D.; Hernandez, C.; Wyczechowska, D.D.; Ochoa, A.C.; et al. L-Arginine Depletion Blunts Anti-Tumor T Cell Responses by Inducing Myeloid-Derived Suppressor Cells. Cancer Res. 2015, 75, 275–283. [Google Scholar] [CrossRef]
- Aaboe Jørgensen, M.; Ugel, S.; Linder Hübbe, M.; Carretta, M.; Perez-Penco, M.; Weis-Banke, S.E.; Martinenaite, E.; Kopp, K.; Chapellier, M.; Adamo, A.; et al. Arginase 1-Based Immune Modulatory Vaccines Induce Anticancer Immunity and Synergize with Anti-PD-1 Checkpoint Blockade. Cancer Immunol. Res. 2021, 9, 1316–1326. [Google Scholar] [CrossRef]
- Canale, F.P.; Basso, C.; Antonini, G.; Perotti, M.; Li, N.; Sokolovska, A.; Neumann, J.; James, M.J.; Geiger, S.; Jin, W.; et al. Metabolic Modulation of Tumours with Engineered Bacteria for Immunotherapy. Nature 2021, 598, 662–666. [Google Scholar] [CrossRef]
- De Sanctis, F.; Lamolinara, A.; Boschi, F.; Musiu, C.; Caligola, S.; Trovato, R.; Fiore, A.; Frusteri, C.; Anselmi, C.; Poffe, O.; et al. Interrupting the Nitrosative Stress Fuels Tumor-Specific Cytotoxic T Lymphocytes in Pancreatic Cancer. J. Immunother. Cancer 2022, 10, e003549. [Google Scholar] [CrossRef] [PubMed]
- Pudlo, M.; Demougeot, C.; Girard-Thernier, C. Arginase Inhibitors: A Rational Approach Over One Century. Med. Res. Rev. 2017, 37, 475–513. [Google Scholar] [CrossRef] [PubMed]
- Aune, T.M.; Pogue, S.L. Inhibition of Tumor Cell Growth by Interferon-Gamma Is Mediated by Two Distinct Mechanisms Dependent upon Oxygen Tension: Induction of Tryptophan Degradation and Depletion of Intracellular Nicotinamide Adenine Dinucleotide. J. Clin. Invest. 1989, 84, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Mándi, Y.; Vécsei, L. The Kynurenine System and Immunoregulation. J. Neural. Transm. 2012, 119, 197–209. [Google Scholar] [CrossRef]
- Halaby, M.J.; McGaha, T.L. Amino Acid Transport and Metabolism in Myeloid Function. Front. Immunol. 2021, 12, 695238. [Google Scholar] [CrossRef]
- Mondanelli, G.; Bianchi, R.; Pallotta, M.T.; Orabona, C.; Albini, E.; Iacono, A.; Belladonna, M.L.; Vacca, C.; Fallarino, F.; Macchiarulo, A.; et al. A Relay Pathway between Arginine and Tryptophan Metabolism Confers Immunosuppressive Properties on Dendritic Cells. Immunity 2017, 46, 233–244. [Google Scholar] [CrossRef]
- Wang, S.; Wu, J.; Shen, H.; Wang, J. The Prognostic Value of IDO Expression in Solid Tumors: A Systematic Review and Meta-Analysis. BMC Cancer 2020, 20, 471. [Google Scholar] [CrossRef]
- Munn, D.H.; Mellor, A.L. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol. 2016, 37, 193–207. [Google Scholar] [CrossRef]
- Holmgaard, R.B.; Zamarin, D.; Munn, D.H.; Wolchok, J.D.; Allison, J.P. Indoleamine 2,3-Dioxygenase Is a Critical Resistance Mechanism in Antitumor T Cell Immunotherapy Targeting CTLA-4. J. Exp. Med. 2013, 210, 1389–1402. [Google Scholar] [CrossRef]
- Pallotta, M.T.; Orabona, C.; Volpi, C.; Vacca, C.; Belladonna, M.L.; Bianchi, R.; Servillo, G.; Brunacci, C.; Calvitti, M.; Bicciato, S.; et al. Indoleamine 2,3-Dioxygenase Is a Signaling Protein in Long-Term Tolerance by Dendritic Cells. Nat. Immunol. 2011, 12, 870–878. [Google Scholar] [CrossRef]
- Fallarino, F.; Grohmann, U.; You, S.; McGrath, B.C.; Cavener, D.R.; Vacca, C.; Orabona, C.; Bianchi, R.; Belladonna, M.L.; Volpi, C.; et al. The Combined Effects of Tryptophan Starvation and Tryptophan Catabolites Down-Regulate T Cell Receptor Zeta-Chain and Induce a Regulatory Phenotype in Naive T Cells. J. Immunol. 2006, 176, 6752–6761. [Google Scholar] [CrossRef] [PubMed]
- Hippen, K.L.; O’Connor, R.S.; Lemire, A.M.; Saha, A.; Hanse, E.A.; Tennis, N.C.; Merkel, S.C.; Kelekar, A.; Riley, J.L.; Levine, B.L.; et al. In Vitro Induction of Human Regulatory T Cells Using Conditions of Low Tryptophan Plus Kynurenines. Am. J. Transpl. 2017, 17, 3098–3113. [Google Scholar] [CrossRef] [PubMed]
- Kiyozumi, Y.; Baba, Y.; Okadome, K.; Yagi, T.; Ishimoto, T.; Iwatsuki, M.; Miyamoto, Y.; Yoshida, N.; Watanabe, M.; Komohara, Y.; et al. IDO1 Expression Is Associated With Immune Tolerance and Poor Prognosis in Patients With Surgically Resected Esophageal Cancer. Ann. Surg. 2019, 269, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Sun, J.; Wang, S.E.; Li, H.; Cao, S.; Cong, Y.; Liu, J.; Ren, X. Upregulated Expression of Indoleamine 2, 3-Dioxygenase in Primary Breast Cancer Correlates with Increase of Infiltrated Regulatory T Cells In Situ and Lymph Node Metastasis. Clin. Dev. Immunol. 2011, 2011, e469135. [Google Scholar] [CrossRef]
- Ananieva, E. Targeting Amino Acid Metabolism in Cancer Growth and Anti-Tumor Immune Response. World J. Biol. Chem. 2015, 6, 281–289. [Google Scholar] [CrossRef]
- Heng, B.; Lim, C.K.; Lovejoy, D.B.; Bessede, A.; Gluch, L.; Guillemin, G.J. Understanding the Role of the Kynurenine Pathway in Human Breast Cancer Immunobiology. Oncotarget 2015, 7, 6506–6520. [Google Scholar] [CrossRef]
- Smith, C.; Chang, M.Y.; Parker, K.H.; Beury, D.W.; DuHadaway, J.B.; Flick, H.E.; Boulden, J.; Sutanto-Ward, E.; Soler, A.P.; Laury-Kleintop, L.D.; et al. IDO Is a Nodal Pathogenic Driver of Lung Cancer and Metastasis Development. Cancer Discov. 2012, 2, 722–735. [Google Scholar] [CrossRef]
- Lewis-Ballester, A.; Pham, K.N.; Batabyal, D.; Karkashon, S.; Bonanno, J.B.; Poulos, T.L.; Yeh, S.-R. Structural Insights into Substrate and Inhibitor Binding Sites in Human Indoleamine 2,3-Dioxygenase 1. Nat. Commun. 2017, 8, 1693. [Google Scholar] [CrossRef]
- Jochems, C.; Fantini, M.; Fernando, R.I.; Kwilas, A.R.; Donahue, R.N.; Lepone, L.M.; Grenga, I.; Kim, Y.-S.; Brechbiel, M.W.; Gulley, J.L.; et al. The IDO1 Selective Inhibitor Epacadostat Enhances Dendritic Cell Immunogenicity and Lytic Ability of Tumor Antigen-Specific T Cells. Oncotarget 2016, 7, 37762–37772. [Google Scholar] [CrossRef]
- Liu, M.; Wang, X.; Wang, L.; Ma, X.; Gong, Z.; Zhang, S.; Li, Y. Targeting the IDO1 Pathway in Cancer: From Bench to Bedside. J. Hematol. Oncol. 2018, 11, 100. [Google Scholar] [CrossRef]
- Li, A.; Barsoumian, H.B.; Schoenhals, J.E.; Caetano, M.S.; Wang, X.; Menon, H.; Valdecanas, D.R.; Niknam, S.; Younes, A.I.; Cortez, M.A.; et al. IDO1 Inhibition Overcomes Radiation-Induced “Rebound Immune Suppression” by Reducing Numbers of IDO1-Expressing Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Int. J. Radiat. Oncol. Biol. Phys. 2019, 104, 903–912. [Google Scholar] [CrossRef]
- Komiya, T.; Huang, C.H. Updates in the Clinical Development of Epacadostat and Other Indoleamine 2,3-Dioxygenase 1 Inhibitors (IDO1) for Human Cancers. Front. Oncol. 2018, 8, 423. [Google Scholar] [CrossRef]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.-J.; Kim, T.M.; et al. Epacadostat plus Pembrolizumab versus Placebo plus Pembrolizumab in Patients with Unresectable or Metastatic Melanoma (ECHO-301/KEYNOTE-252): A Phase 3, Randomised, Double-Blind Study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef]
- Blair, A.B.; Kleponis, J.; Thomas, D.L.; Muth, S.T.; Murphy, A.G.; Kim, V.; Zheng, L. IDO1 Inhibition Potentiates Vaccine-Induced Immunity against Pancreatic Adenocarcinoma. J. Clin. Invest. 2019, 129, 1742–1755. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.J.; DuHadaway, J.B.; Donover, P.S.; Sutanto-Ward, E.; Prendergast, G.C. Inhibition of Indoleamine 2,3-Dioxygenase, an Immunoregulatory Target of the Cancer Suppression Gene Bin1, Potentiates Cancer Chemotherapy. Nat. Med. 2005, 11, 312–319. [Google Scholar] [CrossRef]
- Platten, M.; Nollen, E.A.A.; Röhrig, U.F.; Fallarino, F.; Opitz, C.A. Tryptophan Metabolism as a Common Therapeutic Target in Cancer, Neurodegeneration and Beyond. Nat. Rev. Drug Discov. 2019, 18, 379–401. [Google Scholar] [CrossRef]
- Kjeldsen, J.W.; Lorentzen, C.L.; Martinenaite, E.; Ellebaek, E.; Donia, M.; Holmstroem, R.B.; Klausen, T.W.; Madsen, C.O.; Ahmed, S.M.; Weis-Banke, S.E.; et al. A Phase 1/2 Trial of an Immune-Modulatory Vaccine against IDO/PD-L1 in Combination with Nivolumab in Metastatic Melanoma. Nat. Med. 2021, 27, 2212–2223. [Google Scholar] [CrossRef]
- Campesato, L.F.; Budhu, S.; Tchaicha, J.; Weng, C.-H.; Gigoux, M.; Cohen, I.J.; Redmond, D.; Mangarin, L.; Pourpe, S.; Liu, C.; et al. Blockade of the AHR Restricts a Treg-Macrophage Suppressive Axis Induced by L-Kynurenine. Nat. Commun. 2020, 11, 4011. [Google Scholar] [CrossRef]
- Bonifácio, V.D.B.; Pereira, S.A.; Serpa, J.; Vicente, J.B. Cysteine Metabolic Circuitries: Druggable Targets in Cancer. Br. J. Cancer 2021, 124, 862–879. [Google Scholar] [CrossRef]
- Angelini, G.; Gardella, S.; Ardy, M.; Ciriolo, M.R.; Filomeni, G.; Di Trapani, G.; Clarke, F.; Sitia, R.; Rubartelli, A. Antigen-Presenting Dendritic Cells Provide the Reducing Extracellular Microenvironment Required for T Lymphocyte Activation. Proc. Natl. Acad. Sci. 2002, 99, 1491–1496. [Google Scholar] [CrossRef]
- Srivastava, M.K.; Sinha, P.; Clements, V.K.; Rodriguez, P.; Ostrand-Rosenberg, S. Myeloid-Derived Suppressor Cells Inhibit T-Cell Activation by Depleting Cystine and Cysteine. Cancer Res. 2010, 70, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Scheffel, M.J.; Scurti, G.; Simms, P.; Garrett-Mayer, E.; Mehrotra, S.; Nishimura, M.I.; Voelkel-Johnson, C. Efficacy of Adoptive T-Cell Therapy Is Improved by Treatment with the Antioxidant N-Acetyl Cysteine, Which Limits Activation-Induced T-Cell Death. Cancer Res. 2016, 76, 6006–6016. [Google Scholar] [CrossRef]
- Scheffel, M.J.; Scurti, G.; Wyatt, M.M.; Garrett-Mayer, E.; Paulos, C.M.; Nishimura, M.I.; Voelkel-Johnson, C. N-Acetyl Cysteine Protects Anti-Melanoma Cytotoxic T Cells from Exhaustion Induced by Rapid Expansion via the Downmodulation of Foxo1 in an Akt-Dependent Manner. Cancer Immunol. Immunother. 2018, 67, 691–702. [Google Scholar] [CrossRef]
- Bansal, A.; Simon, M.C. Glutathione Metabolism in Cancer Progression and Treatment Resistance. J. Cell Biol. 2018, 217, 2291–2298. [Google Scholar] [CrossRef]
- Lin, W.; Wang, C.; Liu, G.; Bi, C.; Wang, X.; Zhou, Q.; Jin, H. SLC7A11/XCT in Cancer: Biological Functions and Therapeutic Implications. Am. J. Cancer Res. 2020, 10, 3106–3126. [Google Scholar]
- Ge, C.; Cao, B.; Feng, D.; Zhou, F.; Zhang, J.; Yang, N.; Feng, S.; Wang, G.; Aa, J. The Down-Regulation of SLC7A11 Enhances ROS Induced P-Gp over-Expression and Drug Resistance in MCF-7 Breast Cancer Cells. Sci. Rep. 2017, 7, 3791. [Google Scholar] [CrossRef]
- Wangpaichitr, M.; Wu, C.; Li, Y.Y.; Nguyen, D.J.M.; Kandemir, H.; Shah, S.; Chen, S.; Feun, L.G.; Prince, J.S.; Kuo, M.T.; et al. Exploiting ROS and Metabolic Differences to Kill Cisplatin Resistant Lung Cancer. Oncotarget 2017, 8, 49275–49292. [Google Scholar] [CrossRef]
- Ogihara, K.; Kikuchi, E.; Okazaki, S.; Hagiwara, M.; Takeda, T.; Matsumoto, K.; Kosaka, T.; Mikami, S.; Saya, H.; Oya, M. Sulfasalazine Could Modulate the CD44v9-XCT System and Enhance Cisplatin-Induced Cytotoxic Effects in Metastatic Bladder Cancer. Cancer Sci. 2019, 110, 1431–1441. [Google Scholar] [CrossRef]
- Leone, R.D.; Emens, L.A. Targeting Adenosine for Cancer Immunotherapy. J. Immunother. Cancer 2018, 6, 57. [Google Scholar] [CrossRef]
- Chiarella, A.M.; Ryu, Y.K.; Manji, G.A.; Rustgi, A.K. Extracellular ATP and Adenosine in Cancer Pathogenesis and Treatment. Trends Cancer 2021, 7, 731–750. [Google Scholar] [CrossRef]
- Linnemann, C.; Schildberg, F.A.; Schurich, A.; Diehl, L.; Hegenbarth, S.I.; Endl, E.; Lacher, S.; Müller, C.E.; Frey, J.; Simeoni, L.; et al. Adenosine Regulates CD8 T-Cell Priming by Inhibition of Membrane-Proximal T-Cell Receptor Signalling. Immunology 2009, 128, e728–e737. [Google Scholar] [CrossRef] [PubMed]
- Ohta, A.; Sitkovsky, M. Extracellular Adenosine-Mediated Modulation of Regulatory T Cells. Front. Immunol. 2014, 5, 304. [Google Scholar] [CrossRef]
- Morello, S.; Pinto, A.; Blandizzi, C.; Antonioli, L. Myeloid Cells in the Tumor Microenvironment: Role of Adenosine. Oncoimmunology 2015, 5, e1108515. [Google Scholar] [CrossRef]
- Chen, L.; Diao, L.; Yang, Y.; Yi, X.; Rodriguez, B.L.; Li, Y.; Villalobos, P.A.; Cascone, T.; Liu, X.; Tan, L.; et al. CD38-Mediated Immunosuppression as a Mechanism of Tumor Cell Escape from PD-1/PD-L1 Blockade. Cancer Discov. 2018, 8, 1156–1175. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Marti, A.; Majem, M.; Barlesi, F.; Carcereny Costa, E.; Chu, Q.; Monnet, I.; Sanchez, A.; Dahkil, S.; Camidge, D.R.; He, P.; et al. COAST: An Open-Label, Randomised, Phase II Platform Study of Durvalumab Alone or in Combination with Novel Agents in Patients with Locally Advanced, Unresectable, Stage III NSCLC. Ann. Oncol. 2021, 32. [Google Scholar] [CrossRef]
- Bendell, J.C.; LoRusso, P.; Overman, M.J.; Noonan, A.M.; Kim, D.-W.; Strickler, J.; Kim, S.-W.; Clarke, S.J.; George, T.J.; Grimison, P.S.; et al. Safety and Efficacy of the Anti-CD73 Monoclonal Antibody (MAb) Oleclumab ± Durvalumab in Patients (Pts) with Advanced Colorectal Cancer (CRC), Pancreatic Ductal Adenocarcinoma (PDAC), or EGFR-Mutant Non-Small Cell Lung Cancer (EGFRm NSCLC). JCO 2021, 39, 9047. [Google Scholar] [CrossRef]
- Lim, E.A.; Bauer, T.M.; Patel, M.R.; Falchook, G.S.; Karlix, J.L.; Choe, J.H.; George, D.J.; Mugundu, G.M.; Pilling, E.; Chen, H.; et al. A Phase I, Open-Label, Multicenter Study to Assess the Safety, Pharmacokinetics, and Preliminary Antitumor Activity of AZD4635 Both as Monotherapy and in Combination in Patients with Advanced Solid Malignancies: Results from Prostate Cancer Patients (NCT02740985). JCO 2020, 38, 5518. [Google Scholar] [CrossRef]
- Fong, L.; Hotson, A.; Powderly, J.D.; Sznol, M.; Heist, R.S.; Choueiri, T.K.; George, S.; Hughes, B.G.M.; Hellmann, M.D.; Shepard, D.R.; et al. Adenosine 2A Receptor Blockade as an Immunotherapy for Treatment-Refractory Renal Cell Cancer. Cancer Discov. 2020, 10, 40–53. [Google Scholar] [CrossRef]
- Surikova, E.I.; Goroshinskaja, I.A.; Nerodo, G.A.; Frantsiyants, E.M.; Malejko, M.L.; Shalashnaja, E.V.; Kachesova, P.S.; Nemashkalova, L.A.; Leonova, A.V. The Activity of Redox-Regulatory Systems in the Tumor and Its Surrounding Tissues in Various Histological Types of Tumor. Biomed. Khimiya. 2016, 62, 187–192. [Google Scholar] [CrossRef][Green Version]
- Corzo, C.A.; Cotter, M.J.; Cheng, P.; Cheng, F.; Kusmartsev, S.; Sotomayor, E.; Padhya, T.; McCaffrey, T.V.; McCaffrey, J.C.; Gabrilovich, D.I. Mechanism Regulating Reactive Oxygen Species in Tumor-Induced Myeloid-Derived Suppressor Cells. J. Immunol. 2009, 182, 5693–5701. [Google Scholar] [CrossRef]
- Zhang, Y.; Choksi, S.; Chen, K.; Pobezinskaya, Y.; Linnoila, I.; Liu, Z.-G. ROS Play a Critical Role in the Differentiation of Alternatively Activated Macrophages and the Occurrence of Tumor-Associated Macrophages. Cell Res. 2013, 23, 898–914. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Yu, S.; Kappes, J.; Wang, J.; Grizzle, W.E.; Zinn, K.R.; Zhang, H.-G. Expansion of Spleen Myeloid Suppressor Cells Represses NK Cell Cytotoxicity in Tumor-Bearing Host. Blood 2007, 109, 4336–4342. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, S.; Youn, J.-I.; Weber, H.; Iclozan, C.; Lu, L.; Cotter, M.J.; Meyer, C.; Becerra, C.R.; Fishman, M.; Antonia, S.; et al. Anti-Inflammatory Triterpenoid Blocks Immune Suppressive Function of MDSCs and Improves Immune Response in Cancer. Clin. Cancer Res. 2010, 16, 1812–1823. [Google Scholar] [CrossRef]
- Nefedova, Y.; Fishman, M.; Sherman, S.; Wang, X.; Beg, A.A.; Gabrilovich, D.I. Mechanism of All-Trans Retinoic Acid Effect on Tumor-Associated Myeloid-Derived Suppressor Cells. Cancer Res. 2007, 67, 11021–11028. [Google Scholar] [CrossRef]
- Tobin, R.P.; Jordan, K.R.; Robinson, W.A.; Davis, D.; Borges, V.F.; Gonzalez, R.; Lewis, K.D.; McCarter, M.D. Targeting Myeloid-Derived Suppressor Cells Using All-Trans Retinoic Acid in Melanoma Patients Treated with Ipilimumab. Int. Immunopharmacol. 2018, 63, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Grauers Wiktorin, H.; Nilsson, M.S.; Kiffin, R.; Sander, F.E.; Lenox, B.; Rydström, A.; Hellstrand, K.; Martner, A. Histamine Targets Myeloid-Derived Suppressor Cells and Improves the Anti-Tumor Efficacy of PD-1/PD-L1 Checkpoint Blockade. Cancer Immunol. Immunother. 2019, 68, 163–174. [Google Scholar] [CrossRef]
- Amin, M.; Desai, M.D.; Sorscher, S.; Lim, K.-H.; Wang-Gillam, A.; Tan, B.R.; Picus, J.; Highkin, M.; Lears, K.; Lockhart, A.C. Phase II Trial of Levocetirizine with Capecitabine and Bevacizumab to Overcome the Resistance of Antiangiogenic Therapies in Refractory Metastatic Colorectal Cancer. JCO 2015, 33, 763. [Google Scholar] [CrossRef]
- Chamoto, K.; Chowdhury, P.S.; Kumar, A.; Sonomura, K.; Matsuda, F.; Fagarasan, S.; Honjo, T. Mitochondrial Activation Chemicals Synergize with Surface Receptor PD-1 Blockade for T Cell-Dependent Antitumor Activity. PNAS 2017, 114, E761–E770. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Aerobic Glycolysis, Lactate Accumulation and Acidification of the Medium | |||||
|---|---|---|---|---|---|
| Target | Drugs | Phases | Tumor Types | NCT Number | Enrollment |
| GLUT4 | Silybin | I | Advanced hepatocellular carcinoma | NCT01129570 | 3 |
| II | Prostate cancer | NCT00487721 | 12 | ||
| HK2 | 2DG | I | Lung, breast, pancreatic, head and neck, gastric cancer | NCT00096707 | 50 |
| I/II | Advanced prostate cancer | NCT00633087 | 12 | ||
| HK1/2 | Metformin | II | Non-small cell lung cancer (NSCLC) | NCT03048500 | 17 |
| II | Small cell lung cancer (SCLC) | NCT03800602 | 24 | ||
| II | Small cell lung cancer (SCLC) | NCT03994744 | 68 | ||
| II | Solid tumor | NCT04114136 | 108 | ||
| MCT1 | AZD3965 | I | Adult solid tumor | Diffuse large B cell lymphoma | Burkitt lymphoma | NCT01791595 | 53 |
| mTOR | rapamycin | ||||
| Glutamine and Glutaminolysis | |||||
| Target | Drugs | Phases | Tumor Types | NCT Number | Enrollment |
| Glutamine | DRP-104 (Sirpiglenastat) | I/II | Solid tumor | NCT04471415 | 246 |
| Glutaminase (GLS) | Teleglenastat (CD-839) | I | Ovarian cancer | NCT03944902 | 33 |
| I | Multiple myeloma | NCT03798678 | 36 | ||
| I | Non-small cell lung cancer (NSCLC) | NCT03831932 | 18 | ||
| I | Non-small cell lung cancer (NSCLC) | NCT04250545 | 85 | ||
| I | Hematological tumors | NCT02071888 | 25 | ||
| I | Solid tumor | NCT02071862 | 210 | ||
| I | Anaplastic or diffuse astrocytoma | NCT03528642 | 40 | ||
| I | Acute myeloid leukemia (AML)|Acute lymphocytic leukemia (ALL) | NCT02071927 | 43 | ||
| I/II | Advanced myelodysplastic syndrome | NCT03047993 | 40 | ||
| I/II | Colorectal cancer | NCT03263429 | 40 | ||
| I/II | Clear cell renal cell carcinoma (ccRCC)|Melanoma|Non-small cell lung cancer (NSCLC) | NCT02771626 | 118 | ||
| I/II | Solid tumor | NCT03875313 | 33 | ||
| I/II | Solid tumor | NCT03965845 | 53 | ||
| II | Triple-negative breast cancer (TNBC) | NCT03057600 | 52 | ||
| II | Prostate cancer metastatic | NCT04824937 | 30 | ||
| II | Solid tumor | NCT03872427 | 108 | ||
| II | Non-squamous non-small cell lung cancer (NSCLC) | NCT04265534 | 120 | ||
| II | Renal cell carcinoma | NCT03428217 | 445 | ||
| II | Renal cell carcinoma | NCT03163667 | 63 | ||
| IACS-6274 | I | Solid tumor | NCT05039801 | 36 | |
| Arginine | |||||
| Target | Drugs | Phases | Tumor Types | NCT Number | Enrollment |
| Arginase 1/2 inhibitors | INCB001158 | I | Solid tumor | NCT03910530 | 18 |
| I/II | Solid tumor | NCT03361228 | 5 | ||
| I/II | Solid tumor | NCT03314935 | 149 | ||
| I/II | Solid tumor | NCT02903914 | 260 | ||
| I/II | Multiple myeloma | NCT03837509 | 12 | ||
| Arginine depletion | ADI-PEG | I | HER2-negative breast cancer | NCT01948843 | 15 |
| I | Solid tumors|Prostate cancer | NCT01497925 | 43 | ||
| I | Advanced pancreatic cancer | NCT02101580 | 21 | ||
| I | Advanced solid cancers | NCT03254732 | 47 | ||
| I | Solid tumor | NCT01665183 | 8 | ||
| I | Solid tumor | NCT02029690 | 85 | ||
| I | Metastatic melanoma | NCT00029900 | 15 | ||
| I | Hepatocellular carcinoma | NCT02101593 | 8 | ||
| I | Uveal melanoma | NCT03922880 | 9 | ||
| I | Acute myeloid leukemia | NCT05001828 | 60 | ||
| I | Acute myeloid leukemia | NCT02875093 | 23 | ||
| I | Glioblastoma | NCT04587830 | 32 | ||
| I/II | Advanced gastrointestinal (GI) malignancies|Hepatocellular carcinoma|Gastric cancer|Colorectal cancer | NCT02102022 | 140 | ||
| I/II | Metastatic melanoma|Skin cancer|Neoplasm | NCT00520299 | 31 | ||
| II | Hepatocellular carcinoma | NCT00056992 | 34 | ||
| II | Melanoma (skin) | NCT00450372 | 38 | ||
| II | Soft tissue sarcoma | NCT03449901 | 98 | ||
| II | Re-sectable hepatocellular carcinoma | NCT04965714 | 10 | ||
| II | Acute myeloid leukemia | NCT01910012 | 43 | ||
| II | Small cell lung cancer | NCT01266018 | 22 | ||
| II | Hepatocellular carcinoma | NCT02006030 | 30 | ||
| II | Non-Hodgkin’s lymphoma | NCT01910025 | 18 | ||
| II/III | Mesothelioma | NCT02709512 | 386 | ||
| III | Hepatocellular carcinoma | NCT01287585 | 636 | ||
| Tryptophane Kynurenine | |||||
| Target | Drugs | Phases | Tumor Types | NCT Number | Enrollment |
| IDO inhibitors | Epacadostat (INCB024360) | I | Rectal cancer | NCT03516708 | 39 |
| I | Solid tumors and hematologic malignancy | NCT01195311 | 52 | ||
| I | Ovarian cancer|Fallopian tube carcinoma|Primary peritoneal carcinoma | NCT02118285 | 2 | ||
| I | NSCLC (non-small cell lung carcinoma)|UC (urothelial cancer) | NCT02298153 | 29 | ||
| I | Advanced solid tumor|Non-small cell lung cancer (NSCLC) | NCT03217669 | 22 | ||
| I | Solid tumors | NCT02559492 | 142 | ||
| I | Unresectable or Metastatic Solid Tumors | NCT03589651 | 83 | ||
| I | Neoplasms|Non-small-cell lung carcinoma | NCT02862457 | 34 | ||
| I | Glioblastoma|Glioblastoma multiforme | NCT03707457 | 3 | ||
| I | Solid tumor | NCT03471286 | 2 | ||
| I | Fallopian and ovarian cancer | NCT02042430 | 17 | ||
| I/II | Melanoma | NCT01604889 | 136 | ||
| I/II | Fallopian and ovarian cancer | NCT02166905 | 40 | ||
| I/II | Advanced solid tumors|Lymphoma | NCT03322384 | 20 | ||
| I/II | Solid tumors | NCT02178722 | 444 | ||
| I/II | B-cell malignancies|Colorectal cancer (CRC)|Head and neck cancer|Lung cancer|Lymphoma|Melanoma|Ovarian cancer|Glioblastoma | NCT02327078 | 307 | ||
| I/II | Epithelial ovarian cancer|Tube cancer|Peritoneal cancer | NCT02785250 | 85 | ||
| I/II | Solid tumor | NCT03361228 | 5 | ||
| I/II | Solid tumor | NCT02959437 | 70 | ||
| I/II | Breast cancer | NCT03328026 | 60 | ||
| I/II | Platinum-resistant ovarian cancer|Platinum-resistant fallopian cancer|Platinum-resistant peritoneal cancer | NCT02575807 | 35 | ||
| I/II | Solid tumor | NCT03085914 | 70 | ||
| I/II | Solid tumors|Head and neck cancer|Lung cancer|UC (urothelial cancer) | NCT02318277 | 176 | ||
| I/II | Solid tumor | NCT03347123 | 11 | ||
| I/II | Solid tumors | NCT03277352 | 10 | ||
| II | Gastrointestinal stromal tumors | NCT03291054 | 1 | ||
| II | Metastatic pancreatic adenocarcinoma | NCT03006302 | 44 | ||
| II | Ovarian cancer|Genitourinary (GU) tumors | NCT01685255 | 83 | ||
| II | Lung cancer | NCT03322566 | 233 | ||
| II | Head and neck carcinoma | NCT03463161 | 2 | ||
| II | Lung cancer | NCT03322540 | 154 | ||
| II | Colorectal cancer | NCT03196232 | 3 | ||
| II | Thymic carcinoma|Thymus neoplasms|Thymus cancer | NCT02364076 | 45 | ||
| II | Head and neck carcinoma | NCT03823131 | 14 | ||
| II | Melanoma | NCT01961115 | 11 | ||
| II | Endometrial cancer | NCT04463771 | 220 | ||
| II | Malignant ovarian clear cell tumor|Recurrent ovarian carcinoma | NCT03602586 | 14 | ||
| II | Sarcoma | NCT03414229 | 30 | ||
| II | Urothelial carcinoma | NCT04586244 | 45 | ||
| II | Glioma|Glioblastoma | NCT03532295 | 55 | ||
| II | Myelodysplastic syndromes | NCT01822691 | 15 | ||
| III | Lung cancer | NCT03348904 | 2 | ||
| III | UC (urothelial cancer) | NCT03361865 | 93 | ||
| III | UC (urothelial cancer) | NCT03374488 | 84 | ||
| III | Head and neck cancer | NCT03358472 | 89 | ||
| III | Renal cell carcinoma (RCC) | NCT03260894 | 129 | ||
| III | Melanoma | NCT02752074 | 706 | ||
| Linrodostat (BMS-986205) | II | Bladder cancer|Bladder tumors|Bladder neoplasms | NCT03519256 | 69 | |
| I/II | Advanced cancer | NCT03792750 | 17 | ||
| I | Advanced cancer | NCT03192943 | 11 | ||
| III | Bladder cancer|Muscle-invasive bladder cancer|BMS-986205 | NCT03661320 | 1200 | ||
| I/II | Advanced cancer|Melanoma|Non-small cell lung cancer | NCT02658890 | 630 | ||
| II | Endometrial adenocarcinoma|Endometrial carcinosarcoma | NCT04106414 | 50 | ||
| I/II | Advanced cancer | NCT03459222 | 184 | ||
| III | Melanoma|Skin cancer | NCT03329846 | 20 | ||
| I | Multiple malignancies | NCT03346837 | 53 | ||
| I | Cancer | NCT03247283 | 9 | ||
| I/II | Hepatocellular carcinoma | NCT03695250 | 8 | ||
| II | Advanced cancer | NCT02750514 | 295 | ||
| II | Head and neck carcinoma | NCT03854032 | 48 | ||
| II | Solid tumors | NCT02996110 | 200 | ||
| II | Advanced gastric cancer | NCT02935634 | 186 | ||
| I | Solid tumors | NCT03335540 | 50 | ||
| I | Glioblastoma | NCT04047706 | 30 | ||
| KHK2455 | I | Solid tumors | NCT02867007 | 36 | |
| I | Urothelial carcinoma | NCT03915405 | 50 | ||
| LY3381916 | I | Solid tumors | NCT03343613 | 60 | |
| AhR inhibitors | BAY 2416964 | I | Solid tumors | NCT04999202 | 78 |
| I | Solid tumors | NCT04069026 | 141 | ||
| IK-175 | I | Solid tumors, urothelial carcinoma | NCT04200963 | 93 | |
| Cysteine | |||||
| Target | Drugs | Phases | Tumor Types | NCT Number | Enrollment |
| Cysteine supplementation | N-acetylcysteine (NAC) | I | Breast cancer | NCT01878695 | 13 |
| I | Brain tumors | NCT00238173 | 2 | ||
| I | Lymphoma | NCT05081479 | 32 | ||
| I/II | Peritoneal cancer|Mucinous adenocarcinoma | NCT03976973 | 100 | ||
| I/II | Ovarian cancer | NCT04520139 | 102 | ||
| II | Ovarian cancer | NCT02569957 | 1 | ||
| II | Melanoma | NCT00003346 | 80 | ||
| Xc- antiporter | Sulfasalazine | II | Breast cancer | NCT03847311 | 40 |
| Adenosine | |||||
| Target | Drugs | Phases | Tumor Types | NCT Number | Enrollment |
| CD73 inhibitors | Oleclumab | I | Bladder cancer | NCT03773666 | 24 |
| I | Solid tumors | NCT04261075 | 57 | ||
| I | Solid tumors | NCT03736473 | 6 | ||
| I | Solid tumors | NCT02503774 | 192 | ||
| I | Non-small cell lung cancer (NSCLC) | NCT03819465 | 212 | ||
| I/II | Triple-negative breast cancer (TNBC) | NCT03742102 | 203 | ||
| I/II | Triple-negative breast cancer (TNBC) | NCT03616886 | 129 | ||
| I/II | Pancreatic adenocarcinoma | NCT03611556 | 208 | ||
| I/II | Colorectal cancer | NCT04068610 | 60 | ||
| II | Non-small cell lung cancer (NSCLC) | NCT05061550 | 140 | ||
| II | Breast cancer luminal B | NCT03875573 | 147 | ||
| II | Pancreatic cancer | NCT04940286 | 30 | ||
| II | Non-small cell lung cancer (NSCLC) | NCT03822351 | 189 | ||
| II | Non-small cell lung cancer (NSCLC) | NCT03794544 | 84 | ||
| II | Non-small cell lung cancer (NSCLC) | NCT03334617 | 420 | ||
| II | Non-small cell lung cancer (NSCLC) | NCT03833440 | 120 | ||
| II | Sarcoma | NCT04668300 | 75 | ||
| AZD3965 | I | Solid tumors | NCT03980821 | 10 | |
| II | Prostate cancer | NCT04495179 | 30 | ||
| Oleclumab, AZD4635 | I | Solid tumors, non-small cell lung cancer (NSCLC), prostate cancer, colorectal cancer | NCT02740985 | 313 | |
| I/II | Non-small cell lung cancer (NSCLC) | NCT03381274 | 43 | ||
| II | Prostate cancer | NCT04089553 | 59 | ||
| Oxidative Stress | |||||
| Target | Drugs | Phases | Tumor Types | NCT Number | Enrollment |
| NAD(P)H | CDDO-Me | I | Advanced solid tumors|Lymphoid malignancies | NCT00529438 | 47 |
| I | Lymphoid malignancies|Solid tumors | NCT00508807 | 21 | ||
| I/II | Pancreatic neoplasms|Pancreatic cancer | NCT00529113 | 33 | ||
| Glutathione | ATRA | II | Advanced melanoma | NCT02403778 | 10 |
| NOX2 | Histamine dihydrochloride | II | Colorectal neoplasms | NCT01722162 | 47 |
| III | Leukemia | NCT00003991 | 360 | ||
| IV | Acute myeloid leukemia | NCT01347996 | 84 | ||
| Acute myeloid leukemia | NCT01770158 | 8 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boyer, T.; Blaye, C.; Larmonier, N.; Domblides, C. Influence of the Metabolism on Myeloid Cell Functions in Cancers: Clinical Perspectives. Cells 2022, 11, 554. https://doi.org/10.3390/cells11030554
Boyer T, Blaye C, Larmonier N, Domblides C. Influence of the Metabolism on Myeloid Cell Functions in Cancers: Clinical Perspectives. Cells. 2022; 11(3):554. https://doi.org/10.3390/cells11030554
Chicago/Turabian StyleBoyer, Thomas, Céline Blaye, Nicolas Larmonier, and Charlotte Domblides. 2022. "Influence of the Metabolism on Myeloid Cell Functions in Cancers: Clinical Perspectives" Cells 11, no. 3: 554. https://doi.org/10.3390/cells11030554
APA StyleBoyer, T., Blaye, C., Larmonier, N., & Domblides, C. (2022). Influence of the Metabolism on Myeloid Cell Functions in Cancers: Clinical Perspectives. Cells, 11(3), 554. https://doi.org/10.3390/cells11030554