
Inhibition of Nigral Microglial Activation Reduces Age-Related Loss of Dopaminergic Neurons and Motor Deficits


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Treadmill Running Training
2.3. Ibuprofen Supplement
2.4. Ki20227 and Lipopolysaccharide Administration
2.5. Beam Traversal Test
2.6. Rotarod Test
2.7. Preparing Brain Tissue
2.8. Immunohistochemistry
2.9. Counting Cells
2.10. Quantifying the Microglial Area
2.11. Quantifying DA Fibers in the Striatum
2.12. Transmission Electron Microscopy
2.13. Western Blotting
2.14. Quantifying TNF and IL-6 in the SN
2.15. Preparing TrkB shRNA and Gene-Expressing Lentivirus
2.16. Delivering shTrkB to the Striatum
2.17. Statistical Analysis
3. Results
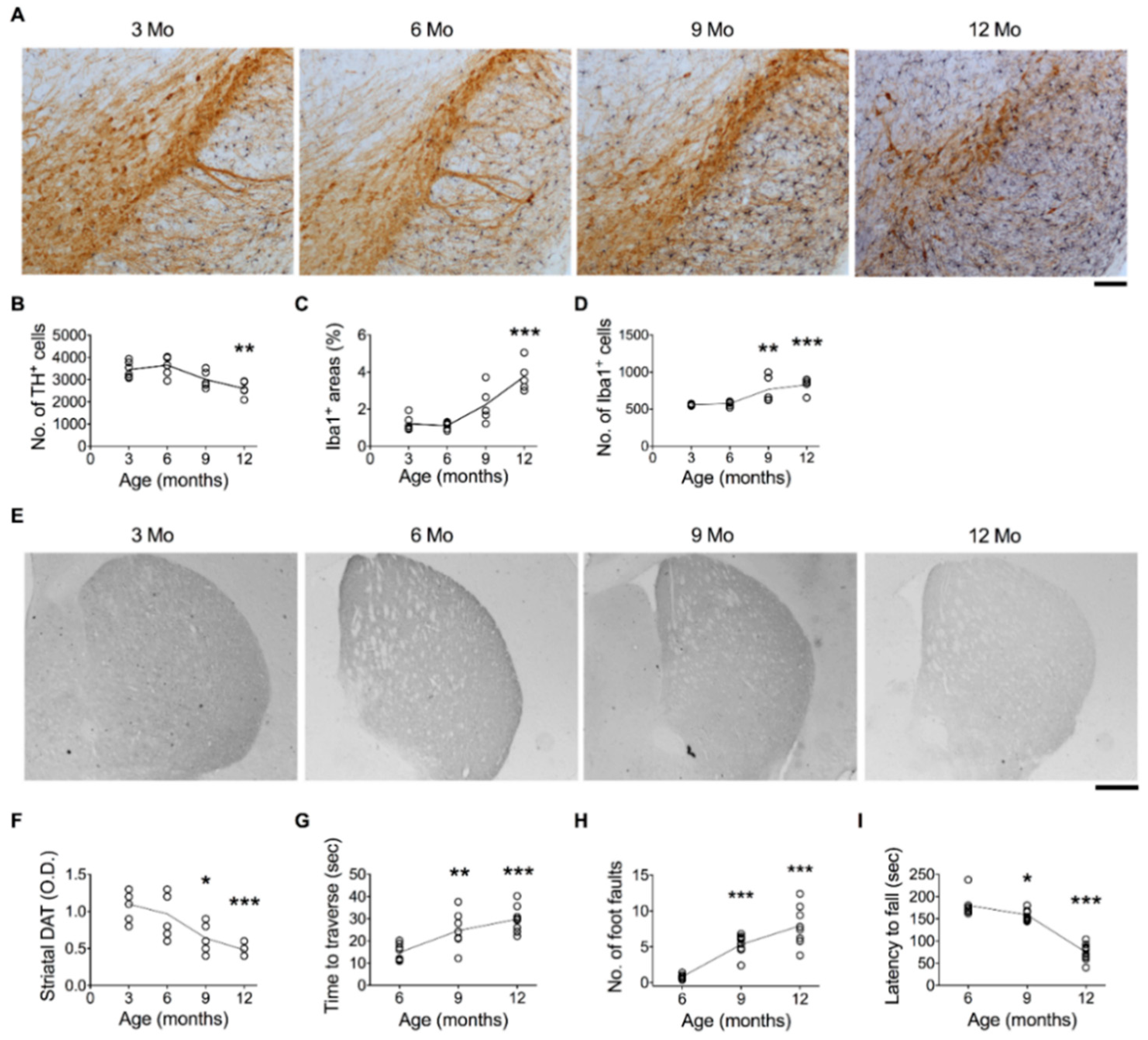
3.1. Age-Related DA Neuron Loss in the SN Is Associated with Microglial Activation
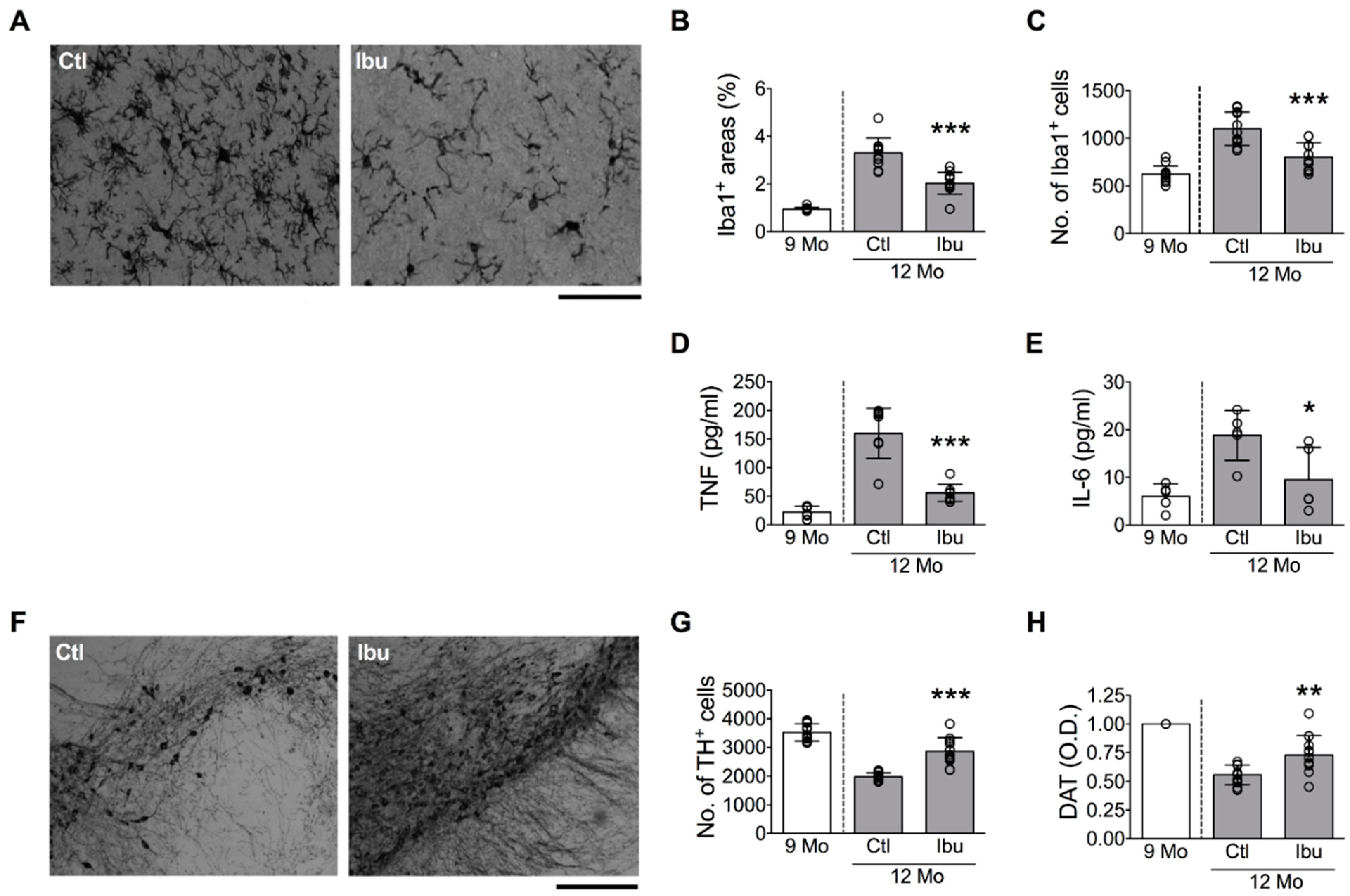
3.2. Blocking Microglial Activation Suppresses Age-Related and Lipopolysaccharide-Induced DA Neuron Loss in the SN
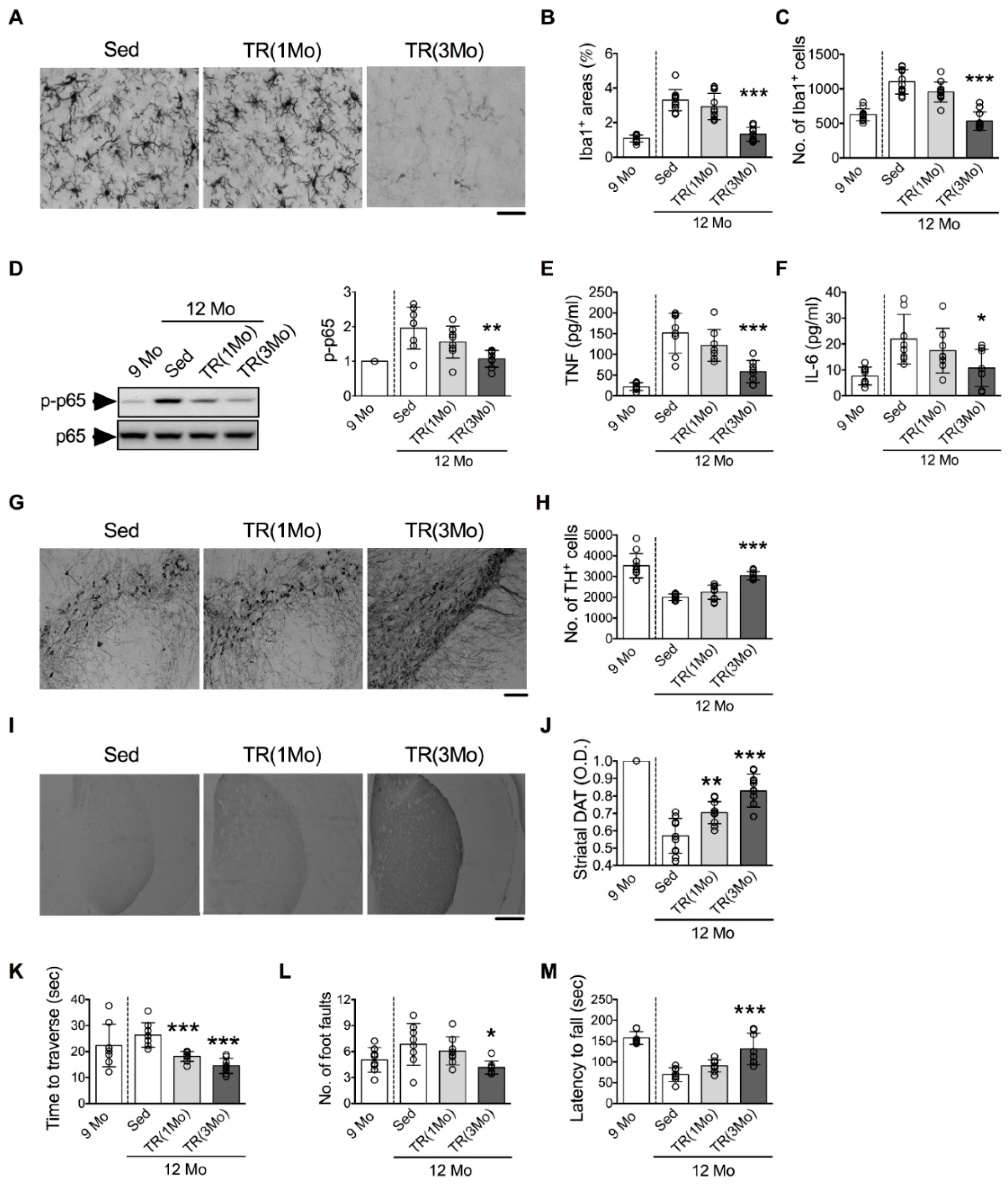
3.3. Running Exercise Ameliorates Age-Related Microglial Activation and DA Neuron
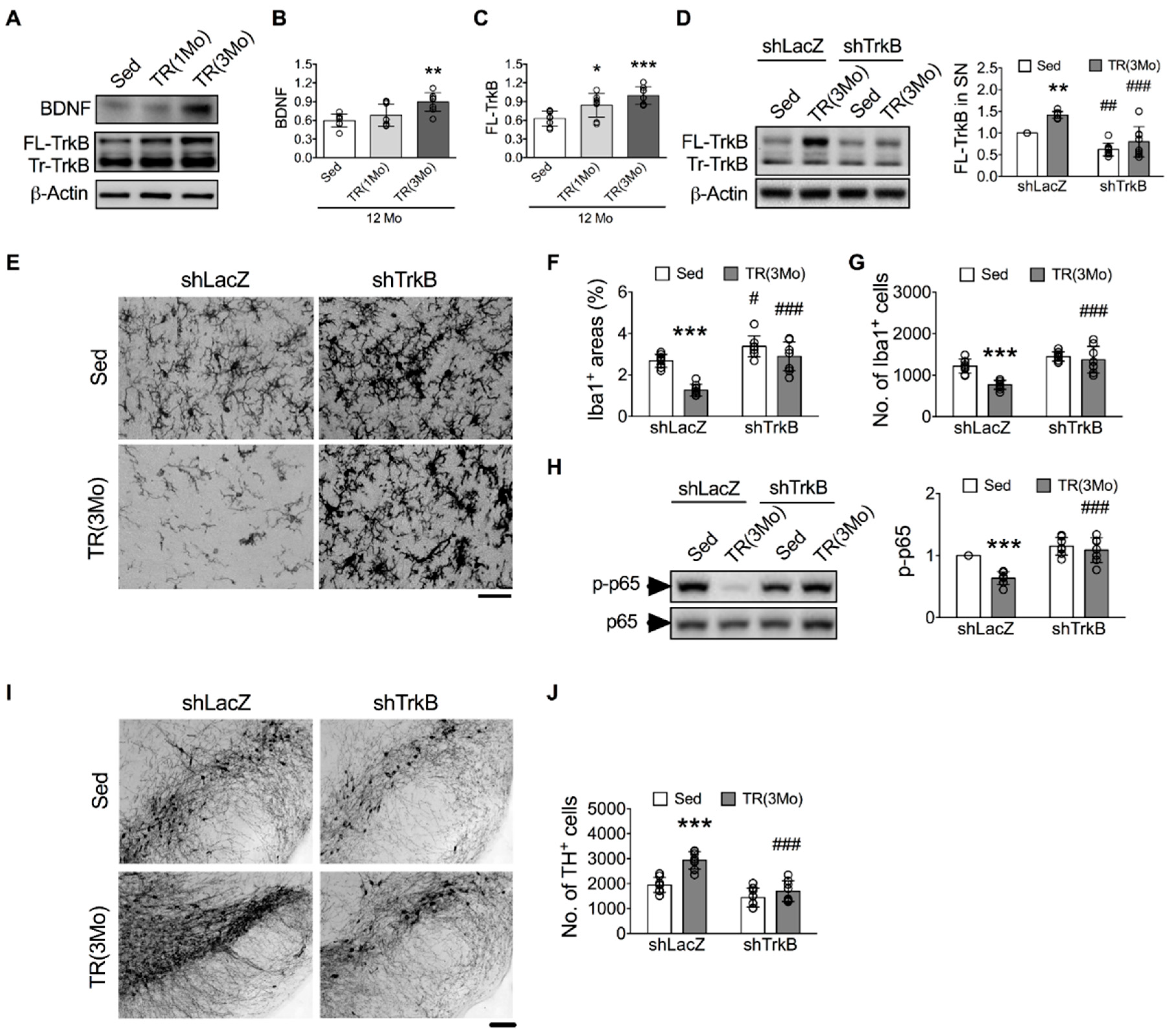
3.4. Inhibition of BDNF-TrkB Signaling Blocked the TR-Induced Protection against Age-Related Microglial Activation, DA Neuron Loss, and Motor Deficit
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Collier, T.J.; Kanaan, N.M.; Kordower, J.H. Ageing as a primary risk factor for Parkinson’s disease: Evidence from studies of non-human primates. Nat. Rev. Neurosci. 2011, 12, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Hong, J.S. Microglia and inflammation-mediated neurodegeneration: Multiple triggers with a common mechanism. Prog. Neurobiol. 2005, 76, 77–98. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- Ouchi, Y.; Yoshikawa, E.; Sekine, Y.; Futatsubashi, M.; Kanno, T.; Ogusu, T.; Torizuka, T. Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann. Neurol. 2005, 57, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Lull, M.E.; Block, M.L. Microglial activation and chronic neurodegeneration. Neurotherapeutics 2010, 7, 354–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, V.H.; Holmes, C. Microglial priming in neurodegenerative disease. Nat. Rev. Neurol. 2014, 10, 217–224. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [Green Version]
- Hart, A.D.; Wyttenbach, A.; Perry, V.H.; Teeling, J.L. Age related changes in microglial phenotype vary between CNS regions: Grey versus white matter differences. Brain Behav. Immun. 2012, 26, 754–765. [Google Scholar] [CrossRef] [Green Version]
- Norden, D.M.; Muccigrosso, M.M.; Godbout, J.P. Microglial priming and enhanced reactivity to secondary insult in aging, and traumatic CNS injury, and neurodegenerative disease. Neuropharmacology 2015, 96, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.F.; Tsai, S.F.; Zhao, Z.W.; Shih, M.M.; Wang, C.Y.; Yang, T.T.; Kuo, Y.M. Exercise-induced increases of corticosterone contribute to exercise-enhanced adult hippocampal neurogenesis in mice. Chin. J. Physiol. 2021, 64, 186–193. [Google Scholar] [CrossRef]
- Wu, C.W.; Chang, Y.T.; Yu, L.; Chen, H.I.; Jen, C.J.; Wu, S.Y.; Lo, C.P.; Kuo, Y.M. Exercise enhances the proliferation of neural stem cells and neurite growth and survival of neuronal progenitor cells in dentate gyrus of middle-aged mice. J. Appl. Physiol. 2008, 105, 1585–1594. [Google Scholar] [CrossRef]
- Prakash, R.S.; Voss, M.W.; Erickson, K.I.; Kramer, A.F. Physical activity and cognitive vitality. Annu. Rev. Psychol. 2015, 66, 769–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotman, C.W.; Berchtold, N.C. Exercise: A behavioral intervention to enhance brain health and plasticity. Trends Neurosci. 2002, 25, 295–301. [Google Scholar] [CrossRef]
- Tillerson, J.L.; Caudle, W.M.; Reveron, M.E.; Miller, G.W. Exercise induces behavioral recovery and attenuates neurochemical deficits in rodent models of Parkinson’s disease. Neuroscience 2003, 119, 899–911. [Google Scholar] [CrossRef]
- Gobbo, O.L.; O’Mara, S.M. Exercise, but not environmental enrichment, improves learning after kainic acid-induced hippocampal neurodegeneration in association with an increase in brain-derived neurotrophic factor. Behav. Brain Res. 2005, 159, 21–26. [Google Scholar] [CrossRef] [PubMed]
- van Praag, H. Exercise and the brain: Something to chew on. Trends Neurosci. 2009, 32, 283–290. [Google Scholar] [CrossRef] [Green Version]
- Gleeson, M.; Bishop, N.C.; Stensel, D.J.; Lindley, M.R.; Mastana, S.S.; Nimmo, M.A. The anti-inflammatory effects of exercise: Mechanisms and implications for the prevention and treatment of disease. Nat. Rev. Immunol. 2011, 11, 607–615. [Google Scholar] [CrossRef]
- He, X.F.; Liu, D.X.; Zhang, Q.; Liang, F.Y.; Dai, G.Y.; Zeng, J.S.; Pei, Z.; Xu, G.Q.; Lan, Y. Voluntary Exercise Promotes Glymphatic Clearance of Amyloid Beta and Reduces the Activation of Astrocytes and Microglia in Aged Mice. Front. Mol. Neurosci. 2017, 10, 144. [Google Scholar] [CrossRef] [Green Version]
- van Praag, H.; Christie, B.R.; Sejnowski, T.J.; Gage, F.H. Running enhances neurogenesis, learning, and long-term potentiation in mice. Proc. Natl. Acad. Sci. USA 1999, 96, 13427–13431. [Google Scholar] [CrossRef] [Green Version]
- Scheffer, D.D.L.; Latini, A. Exercise-induced immune system response: Anti-inflammatory status on peripheral and central organs. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165823. [Google Scholar] [CrossRef]
- Hojman, P. Exercise protects from cancer through regulation of immune function and inflammation. Biochem. Soc. Trans. 2017, 45, 905–911. [Google Scholar] [CrossRef]
- Faherty, C.J.; Raviie Shepherd, K.; Herasimtschuk, A.; Smeyne, R.J. Environmental enrichment in adulthood eliminates neuronal death in experimental Parkinsonism. Brain Res. Mol. Brain Res. 2005, 134, 170–179. [Google Scholar] [CrossRef]
- Tajiri, N.; Yasuhara, T.; Shingo, T.; Kondo, A.; Yuan, W.; Kadota, T.; Wang, F.; Baba, T.; Tayra, J.T.; Morimoto, T.; et al. Exercise exerts neuroprotective effects on Parkinson’s disease model of rats. Brain Res. 2010, 1310, 200–207. [Google Scholar] [CrossRef] [Green Version]
- Gerecke, K.M.; Jiao, Y.; Pani, A.; Pagala, V.; Smeyne, R.J. Exercise protects against MPTP-induced neurotoxicity in mice. Brain Res. 2010, 1341, 72–83. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.F.; Chen, H.I.; Yu, L.; Kuo, Y.M.; Wu, F.S.; Chuang, J.I.; Liao, P.C.; Jen, C.J. Upregulation of hippocampal TrkB and synaptotagmin is involved in treadmill exercise-enhanced aversive memory in mice. Neurobiol. Learn. Mem. 2008, 90, 81–89. [Google Scholar] [CrossRef]
- Conner, J.M.; Lauterborn, J.C.; Yan, Q.; Gall, C.M.; Varon, S. Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: Evidence for anterograde axonal transport. J. Neurosci. 1997, 17, 2295–2313. [Google Scholar] [CrossRef] [Green Version]
- Tapia-Arancibia, L.; Aliaga, E.; Silhol, M.; Arancibia, S. New insights into brain BDNF function in normal aging and Alzheimer disease. Brain Res. Rev. 2008, 59, 201–220. [Google Scholar] [CrossRef]
- Lu, B.; Nagappan, G.; Lu, Y. BDNF and synaptic plasticity, cognitive function, and dysfunction. Handb. Exp. Pharmacol. 2014, 220, 223–250. [Google Scholar] [CrossRef]
- Zuccato, C.; Cattaneo, E. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat. Rev. Neurol. 2009, 5, 311–322. [Google Scholar] [CrossRef]
- Allen, S.J.; Watson, J.J.; Shoemark, D.K.; Barua, N.U.; Patel, N.K. GDNF, NGF and BDNF as therapeutic options for neurodegeneration. Pharmacol. Ther. 2013, 138, 155–175. [Google Scholar] [CrossRef]
- Palasz, E.; Niewiadomski, W.; Gasiorowska, A.; Mietelska-Porowska, A.; Niewiadomska, G. Neuroplasticity and Neuroprotective Effect of Treadmill Training in the Chronic Mouse Model of Parkinson’s Disease. Neural Plast. 2019, 2019, 8215017. [Google Scholar] [CrossRef] [Green Version]
- Hsueh, S.C.; Chen, K.Y.; Lai, J.H.; Wu, C.C.; Yu, Y.W.; Luo, Y.; Hsieh, T.H.; Chiang, Y.H. Voluntary Physical Exercise Improves Subsequent Motor and Cognitive Impairments in a Rat Model of Parkinson’s Disease. Int. J. Mol. Sci. 2018, 19, 508. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.Y.; Wang, T.F.; Yu, L.; Jen, C.J.; Chuang, J.I.; Wu, F.S.; Wu, C.W.; Kuo, Y.M. Running exercise protects the substantia nigra dopaminergic neurons against inflammation-induced degeneration via the activation of BDNF signaling pathway. Brain Behav. Immun. 2011, 25, 135–146. [Google Scholar] [CrossRef]
- Palasz, E.; Wysocka, A.; Gasiorowska, A.; Chalimoniuk, M.; Niewiadomski, W.; Niewiadomska, G. BDNF as a Promising Therapeutic Agent in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 1170. [Google Scholar] [CrossRef] [Green Version]
- Elmore, M.R.; Najafi, A.R.; Koike, M.A.; Dagher, N.N.; Spangenberg, E.E.; Rice, R.A.; Kitazawa, M.; Matusow, B.; Nguyen, H.; West, B.L.; et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 2014, 82, 380–397. [Google Scholar] [CrossRef] [Green Version]
- Fleming, S.M.; Salcedo, J.; Hutson, C.B.; Rockenstein, E.; Masliah, E.; Levine, M.S.; Chesselet, M.F. Behavioral effects of dopaminergic agonists in transgenic mice overexpressing human wildtype alpha-synuclein. Neuroscience 2006, 142, 1245–1253. [Google Scholar] [CrossRef] [Green Version]
- Hancock, M.B. Visualization of peptide-immunoreactive processes on serotonin-immunoreactive cells using two-color immunoperoxidase staining. J. Histochem. Cytochem. 1984, 32, 311–314. [Google Scholar] [CrossRef]
- Sui, Y.; Stanic, D.; Tomas, D.; Jarrott, B.; Horne, M.K. Meloxicam reduces lipopolysaccharide-induced degeneration of dopaminergic neurons in the rat substantia nigra pars compacta. Neurosci. Lett. 2009, 460, 121–125. [Google Scholar] [CrossRef]
- Wu, S.Y.; Chen, Y.W.; Tsai, S.F.; Wu, S.N.; Shih, Y.H.; Jiang-Shieh, Y.F.; Yang, T.T.; Kuo, Y.M. Estrogen ameliorates microglial activation by inhibiting the Kir2.1 inward-rectifier K(+) channel. Sci. Rep. 2016, 6, 22864. [Google Scholar] [CrossRef] [Green Version]
- Franklin, K.; Paxinos, G. The Mouse Brain in Stereotaxic Coordinates, Compact: The Coronal Plates and Diagrams, 3rd ed.; Academic Press: Cambridge, MA, USA, 2008. [Google Scholar]
- Yang, T.T.; Lin, C.; Hsu, C.T.; Wang, T.F.; Ke, F.Y.; Kuo, Y.M. Differential distribution and activation of microglia in the brain of male C57BL/6J mice. Brain Struct. Funct. 2013, 218, 1051–1060. [Google Scholar] [CrossRef]
- Wu, S.Y.; Pan, B.S.; Tsai, S.F.; Chiang, Y.T.; Huang, B.M.; Mo, F.E.; Kuo, Y.M. BDNF reverses aging-related microglial activation. J. Neuroinflamm. 2020, 17, 210. [Google Scholar] [CrossRef]
- Zhao, X.; van Praag, H. Steps towards standardized quantification of adult neurogenesis. Nat. Commun. 2020, 11, 4275. [Google Scholar] [CrossRef]
- Beggs, S.; Salter, M.W. Stereological and somatotopic analysis of the spinal microglial response to peripheral nerve injury. Brain Behav. Immun. 2007, 21, 624–633. [Google Scholar] [CrossRef] [Green Version]
- Li, M.Y.; Lai, F.J.; Hsu, L.J.; Lo, C.P.; Cheng, C.L.; Lin, S.R.; Lee, M.H.; Chang, J.Y.; Subhan, D.; Tsai, M.S.; et al. Dramatic co-activation of WWOX/WOX1 with CREB and NF-kappaB in delayed loss of small dorsal root ganglion neurons upon sciatic nerve transection in rats. PLoS ONE 2009, 4, e7820. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Ho, D.H.; Suk, J.E.; You, S.; Michael, S.; Kang, J.; Joong Lee, S.; Masliah, E.; Hwang, D.; Lee, H.J.; et al. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 2013, 4, 1562. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.W.; Bang, M.S.; Han, T.R.; Ko, Y.J.; Yoon, B.W.; Kim, J.H.; Kang, L.M.; Lee, K.M.; Kim, M.H. Exercise increased BDNF and trkB in the contralateral hemisphere of the ischemic rat brain. Brain Res. 2005, 1052, 16–21. [Google Scholar] [CrossRef]
- Teema, A.M.; Zaitone, S.A.; Moustafa, Y.M. Ibuprofen or piroxicam protects nigral neurons and delays the development of l-dopa induced dyskinesia in rats with experimental Parkinsonism: Influence on angiogenesis. Neuropharmacology 2016, 107, 432–450. [Google Scholar] [CrossRef]
- Casper, D.; Yaparpalvi, U.; Rempel, N.; Werner, P. Ibuprofen protects dopaminergic neurons against glutamate toxicity in vitro. Neurosci. Lett. 2000, 289, 201–204. [Google Scholar] [CrossRef]
- Poly, T.N.; Islam, M.M.R.; Yang, H.C.; Li, Y.J. Non-steroidal anti-inflammatory drugs and risk of Parkinson’s disease in the elderly population: A meta-analysis. Eur. J. Clin. Pharmacol. 2019, 75, 99–108. [Google Scholar] [CrossRef]
- Brunk, U.T.; Terman, A. Lipofuscin: Mechanisms of age-related accumulation and influence on cell function. Free Radic. Biol. Med. 2002, 33, 611–619. [Google Scholar] [CrossRef]
- Sulzer, D.; Bogulavsky, J.; Larsen, K.E.; Behr, G.; Karatekin, E.; Kleinman, M.H.; Turro, N.; Krantz, D.; Edwards, R.H.; Greene, L.A.; et al. Neuromelanin biosynthesis is driven by excess cytosolic catecholamines not accumulated by synaptic vesicles. Proc. Natl. Acad. Sci. USA 2000, 97, 11869–11874. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Phillips, K.; Wielgus, A.R.; Liu, J.; Albertini, A.; Zucca, F.A.; Faust, R.; Qian, S.Y.; Miller, D.S.; Chignell, C.F.; et al. Neuromelanin activates microglia and induces degeneration of dopaminergic neurons: Implications for progression of Parkinson’s disease. Neurotox. Res. 2011, 19, 63–72. [Google Scholar] [CrossRef] [Green Version]
- Buchman, A.S.; Shulman, J.M.; Nag, S.; Leurgans, S.E.; Arnold, S.E.; Morris, M.C.; Schneider, J.A.; Bennett, D.A. Nigral pathology and parkinsonian signs in elders without Parkinson disease. Ann. Neurol. 2012, 71, 258–266. [Google Scholar] [CrossRef] [Green Version]
- Reeve, A.; Simcox, E.; Turnbull, D. Ageing and Parkinson’s disease: Why is advancing age the biggest risk factor? Ageing Res. Rev. 2014, 14, 19–30. [Google Scholar] [CrossRef]
- Sulzer, D. Multiple hit hypotheses for dopamine neuron loss in Parkinson’s disease. Trends Neurosci. 2007, 30, 244–250. [Google Scholar] [CrossRef]
- Goldberg, J.A.; Guzman, J.N.; Estep, C.M.; Ilijic, E.; Kondapalli, J.; Sanchez-Padilla, J.; Surmeier, D.J. Calcium entry induces mitochondrial oxidant stress in vagal neurons at risk in Parkinson’s disease. Nat. Neurosci. 2012, 15, 1414–1421. [Google Scholar] [CrossRef]
- Jiang, T.; Sun, Q.; Chen, S. Oxidative stress: A major pathogenesis and potential therapeutic target of antioxidative agents in Parkinson’s disease and Alzheimer’s disease. Prog. Neurobiol. 2016, 147, 1–19. [Google Scholar] [CrossRef]
- Castelli, V.; Benedetti, E.; Antonosante, A.; Catanesi, M.; Pitari, G.; Ippoliti, R.; Cimini, A.; d’Angelo, M. Neuronal Cells Rearrangement During Aging and Neurodegenerative Disease: Metabolism, Oxidative Stress and Organelles Dynamic. Front. Mol. Neurosci. 2019, 12, 132. [Google Scholar] [CrossRef] [Green Version]
- Noda, S.; Sato, S.; Fukuda, T.; Tada, N.; Uchiyama, Y.; Tanaka, K.; Hattori, N. Loss of Parkin contributes to mitochondrial turnover and dopaminergic neuronal loss in aged mice. Neurobiol. Dis. 2020, 136, 104717. [Google Scholar] [CrossRef]
- Greenamyre, J.T.; Hastings, T.G. Biomedicine. Parkinson’s–divergent causes, convergent mechanisms. Science 2004, 304, 1120–1122. [Google Scholar] [CrossRef]
- Giannoccaro, M.P.; La Morgia, C.; Rizzo, G.; Carelli, V. Mitochondrial DNA and primary mitochondrial dysfunction in Parkinson’s disease. Mov. Disord. 2017, 32, 346–363. [Google Scholar] [CrossRef]
- Truban, D.; Hou, X.; Caulfield, T.R.; Fiesel, F.C.; Springer, W. PINK1, Parkin, and Mitochondrial Quality Control: What can we Learn about Parkinson’s Disease Pathobiology? J. Parkinsons Dis. 2017, 7, 13–29. [Google Scholar] [CrossRef] [Green Version]
- Segura-Aguilar, J.; Paris, I.; Muñoz, P.; Ferrari, E.; Zecca, L.; Zucca, F.A. Protective and toxic roles of dopamine in Parkinson’s disease. J. Neurochem. 2014, 129, 898–915. [Google Scholar] [CrossRef]
- Cai, Y.; Arikkath, J.; Yang, L.; Guo, M.L.; Periyasamy, P.; Buch, S. Interplay of endoplasmic reticulum stress and autophagy in neurodegenerative disorders. Autophagy 2016, 12, 225–244. [Google Scholar] [CrossRef]
- Mercado, G.; Castillo, V.; Soto, P.; Sidhu, A. ER stress and Parkinson’s disease: Pathological inputs that converge into the secretory pathway. Brain Res. 2016, 1648, 626–632. [Google Scholar] [CrossRef]
- Masato, A.; Plotegher, N.; Boassa, D.; Bubacco, L. Impaired dopamine metabolism in Parkinson’s disease pathogenesis. Mol. Neurodegener. 2019, 14, 35. [Google Scholar] [CrossRef] [Green Version]
- Joshi, N.; Singh, S. Updates on immunity and inflammation in Parkinson disease pathology. J. Neurosci. Res. 2018, 96, 379–390. [Google Scholar] [CrossRef]
- Dilger, R.N.; Johnson, R.W. Aging, microglial cell priming, and the discordant central inflammatory response to signals from the peripheral immune system. J. Leukoc. Biol. 2008, 84, 932–939. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, C. Microglia and neurodegeneration: The role of systemic inflammation. Glia 2013, 61, 71–90. [Google Scholar] [CrossRef]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef]
- Tatton, W.G.; Greenwood, C.E.; Verrier, M.C.; Holland, D.P.; Kwan, M.M.; Biddle, F.E. Different rates of age-related loss for four murine monoaminergic neuronal populations. Neurobiol. Aging 1991, 12, 543–556. [Google Scholar] [CrossRef]
- Brandt, M.D.; Krüger-Gerlach, D.; Hermann, A.; Meyer, A.K.; Kim, K.S.; Storch, A. Early Postnatal but Not Late Adult Neurogenesis Is Impaired in the Pitx3-Mutant Animal Model of Parkinson’s Disease. Front. Neurosci. 2017, 11, 471. [Google Scholar] [CrossRef] [Green Version]
- Shaerzadeh, F.; Phan, L.; Miller, D.; Dacquel, M.; Hachmeister, W.; Hansen, C.; Bechtle, A.; Tu, D.; Martcheva, M.; Foster, T.C.; et al. Microglia senescence occurs in both substantia nigra and ventral tegmental area. Glia 2020, 68, 2228–2245. [Google Scholar] [CrossRef]
- Gür, E.; Duyan, Y.A.; Arkan, S.; Karson, A.; Balcı, F. Interval timing deficits and their neurobiological correlates in aging mice. Neurobiol. Aging 2020, 90, 33–42. [Google Scholar] [CrossRef]
- Osterburg, H.H.; Donahue, H.G.; Severson, J.A.; Finch, C.E. Catecholamine levels and turnover during aging in brain regions of male C57BL/6J mice. Brain Res. 1981, 224, 337–352. [Google Scholar] [CrossRef]
- Yue, M.; Hinkle, K.M.; Davies, P.; Trushina, E.; Fiesel, F.C.; Christenson, T.A.; Schroeder, A.S.; Zhang, L.; Bowles, E.; Behrouz, B.; et al. Progressive dopaminergic alterations and mitochondrial abnormalities in LRRK2 G2019S knock-in mice. Neurobiol. Dis. 2015, 78, 172–195. [Google Scholar] [CrossRef] [Green Version]
- Noda, S.; Sato, S.; Fukuda, T.; Tada, N.; Hattori, N. Aging-related motor function and dopaminergic neuronal loss in C57BL/6 mice. Mol. Brain 2020, 13, 46. [Google Scholar] [CrossRef]
- Howell, R.D.; Dominguez-Lopez, S.; Ocañas, S.R.; Freeman, W.M.; Beckstead, M.J. Female mice are resilient to age-related decline of substantia nigra dopamine neuron firing parameters. Neurobiol. Aging 2020, 95, 195–204. [Google Scholar] [CrossRef]
- Ji, K.A.; Eu, M.Y.; Kang, S.H.; Gwag, B.J.; Jou, I.; Joe, E.H. Differential neutrophil infiltration contributes to regional differences in brain inflammation in the substantia nigra pars compacta and cortex. Glia 2008, 56, 1039–1047. [Google Scholar] [CrossRef]
- Qin, L.; Wu, X.; Block, M.L.; Liu, Y.; Breese, G.R.; Hong, J.S.; Knapp, D.J.; Crews, F.T. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 2007, 55, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Jiang, C.; Wang, Z.N.; Kang, Y.C.; Chen, Y.; Lu, W.X.; Ren, H.J.; Hou, B.R. Ki20227 aggravates apoptosis, inflammatory response, and oxidative stress after focal cerebral ischemia injury. Neural. Regen. Res. 2022, 17, 137–143. [Google Scholar] [CrossRef]
- Neumann, H.; Kotter, M.R.; Franklin, R.J. Debris clearance by microglia: An essential link between degeneration and regeneration. Brain 2009, 132, 288–295. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Perry, V.H. Microglial physiology: Unique stimuli, specialized responses. Annu. Rev. Immunol. 2009, 27, 119–145. [Google Scholar] [CrossRef]
- Jin, X.; Yamashita, T. Microglia in central nervous system repair after injury. J. Biochem. 2016, 159, 491–496. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Park, Y.; Huang, X.; Hollenbeck, A.; Blair, A.; Schatzkin, A.; Chen, H. Physical activities and future risk of Parkinson disease. Neurology 2010, 75, 341–348. [Google Scholar] [CrossRef]
- Jiang, Y.; Wei, N.; Lu, T.; Zhu, J.; Xu, G.; Liu, X. Intranasal brain-derived neurotrophic factor protects brain from ischemic insult via modulating local inflammation in rats. Neuroscience 2011, 172, 398–405. [Google Scholar] [CrossRef]
- Makar, T.K.; Trisler, D.; Sura, K.T.; Sultana, S.; Patel, N.; Bever, C.T. Brain derived neurotrophic factor treatment reduces inflammation and apoptosis in experimental allergic encephalomyelitis. J. Neurol. Sci. 2008, 270, 70–76. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, T.-F.; Wu, S.-Y.; Pan, B.-S.; Tsai, S.-F.; Kuo, Y.-M. Inhibition of Nigral Microglial Activation Reduces Age-Related Loss of Dopaminergic Neurons and Motor Deficits. Cells 2022, 11, 481. https://doi.org/10.3390/cells11030481
Wang T-F, Wu S-Y, Pan B-S, Tsai S-F, Kuo Y-M. Inhibition of Nigral Microglial Activation Reduces Age-Related Loss of Dopaminergic Neurons and Motor Deficits. Cells. 2022; 11(3):481. https://doi.org/10.3390/cells11030481
Chicago/Turabian StyleWang, Tzu-Feng, Shih-Ying Wu, Bo-Syong Pan, Sheng-Feng Tsai, and Yu-Min Kuo. 2022. "Inhibition of Nigral Microglial Activation Reduces Age-Related Loss of Dopaminergic Neurons and Motor Deficits" Cells 11, no. 3: 481. https://doi.org/10.3390/cells11030481
APA StyleWang, T.-F., Wu, S.-Y., Pan, B.-S., Tsai, S.-F., & Kuo, Y.-M. (2022). Inhibition of Nigral Microglial Activation Reduces Age-Related Loss of Dopaminergic Neurons and Motor Deficits. Cells, 11(3), 481. https://doi.org/10.3390/cells11030481