
Immunity and Nutrition: The Right Balance in Inflammatory Bowel Disease

 ,
,  ,
,  , ,
, ,  ,
,  and
and
Abstract
1. Introduction
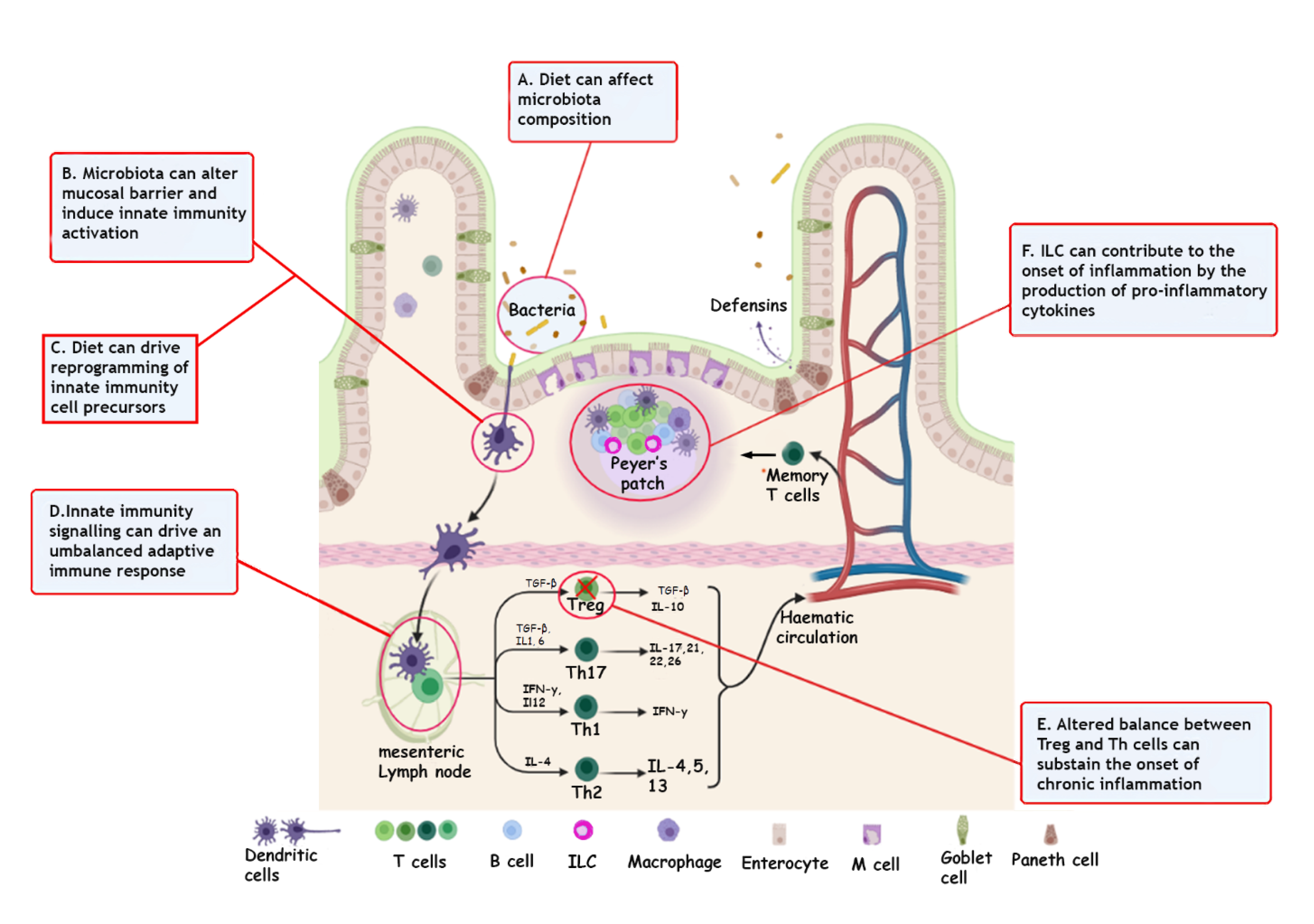
2. Immunity and IBD
3. Nutrition and IBD
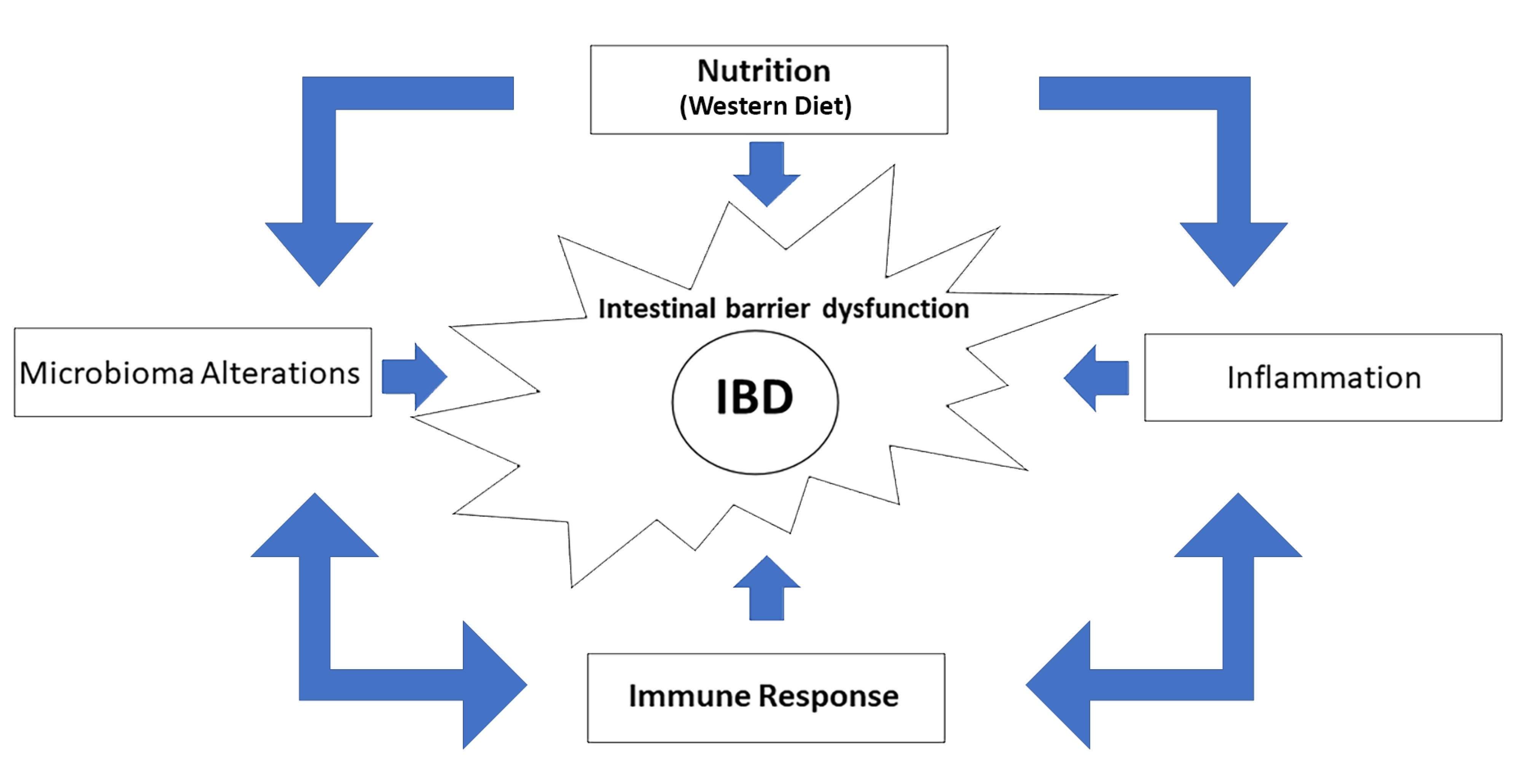
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mak, W.Y.; Zhao, M.; Ng, S.C.; Burisch, J. The epidemiology of inflammatory bowel disease: East meets west. J. Gastroenterol. Hepatol. 2020, 35, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2017, 390, 2769–2778. [Google Scholar] [CrossRef]
- Maloy, K.J.; Powrie, F. Intestinal homeostasis and its breakdown in inflammatory bowel diasease. Nature 2011, 474, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Danese, S.; Fiocchi, C. Ulcerative colitis. N. Engl. J. Med. 2011, 365, 1713–1725. [Google Scholar] [CrossRef]
- Molodecky, N.A.; Soon, S.; Rabi, D.M.; Ghali, W.A.; Ferris, M.; Chernoff, G. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 2012, 42, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Cader, M.Z.; Kaser, A. Recent advances in inflammatory bowel disease: Mucosal immune cells in intestinal inflammatory. Gut 2013, 62, 1653–1664. [Google Scholar] [CrossRef]
- Harlan, W.R., III. Inflammatory Bowel Disease: Epidemiology, Evaluation, Treatment, and Health Maintenance. N. C. Med. J. 2016, 7, 198–201. [Google Scholar] [CrossRef][Green Version]
- Liu, J.Z.; van Sommeren, S.; Huang, H.; Ng, S.C.; Alberts, R.; Takahashi, A.; Ripke, S.; Lee, J.C.; Jostins, L.; Shah, T.; et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 2015, 47, 979–986. [Google Scholar] [CrossRef]
- Venkataraman, G.R.; Rivas, M.A. Rare and common variant discovery in complex disease: The IBD case study. Hum. Mol. Genet. 2019, 28, R162–R169. [Google Scholar] [CrossRef]
- Coleman, O.I.; Haller, D. ER Stress and the UPR in Shaping Intestinal Tissue Homeostasis and Immunity. Front. Immunol. 2019, 10, 2825. [Google Scholar] [CrossRef]
- Brewer, J.W.; Hendershot, L.M. Building an antibody factory: A job for the unfolded protein response. Nat. Immunol. 2005, 6, 23–29. [Google Scholar] [CrossRef]
- De Lange, K.M.; Moutsianas, L.; Lee, J.C.; Lamb, C.A.; Luo, Y.; Kennedy, N.A.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Barkema, H.W.; et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat. Genet. 2017, 49, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Gordon, H.; Trier Moller, F.; Andersen, V.; Harbord, M. Heritability in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2015, 21, 1428–1434. [Google Scholar] [CrossRef]
- Schreiner, P.; Martinho-Grueber, M.; Studerus, D.; Vavricka, S.R.; Tilg, H.; Biedermann, L. Nutrition in Inflammatory Bowel Disease. Digestion 2020, 101, 120–135. [Google Scholar] [CrossRef] [PubMed]
- Wark, G.; Samocha-Bonet, D.; Ghaly, S.; Danta, M. The Role of Diet in the Pathogenesis and Management of Inflammatory Bowel Disease: A Review. Nutrients 2020, 13, 135. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V.; Varum, F.; Bravo, R.; Furrer, E.; Bojic, D.; Basit, A.W. Inflammatory bowel disease: Exploring gut pathophysiology for novel therapeutic targets. Transl. Res. 2016, 176, 38–68. [Google Scholar] [CrossRef] [PubMed]
- Christ, A.; Günther, P.; Lauterbach, M.A.R.; Duewell, P.; Biswas, D.; Pelka, K.; Scholz, C.J.; Oosting, M.; Haendler, K.; Baßler, K.; et al. Western Diet Triggers NLRP3-Dependent Innate Immune Reprogramming. Cell 2018, 172, 162–175.e14. [Google Scholar] [CrossRef] [PubMed]
- Ananthakrishnan, A.N.; Khalili, H.; Pan, A.; Higuchi, L.M.; de Silva, P.; Richter, J.M.; Fuchs, C.S.; Chan, A.T. Association between depressive symptoms and incidence of Crohn’s disease and ulcerative colitis: Results from the Nurses’ Health Study. Clin. Gastroenterol. Hepatol. 2013, 11, 57–62. [Google Scholar] [CrossRef]
- Petronis, A.; Petroniene, R. Epigenetics of inflammatory bowel disease. Gut 2000, 47, 302–306. [Google Scholar] [CrossRef]
- Rengarajan, S.; Vivio, E.E.; Parkes, M.; Peterson, D.A.; Roberson, E.D.O.; Newberry, R.D.; Ciorba, M.A.; Hsieh, C.S. Dynamic immunoglobulin responses to gut bacteria during inflammatory bowel disease. Gut Microbes 2020, 11, 405–420. [Google Scholar] [CrossRef]
- Jiang, W.; Su, J.; Zhang, X.; Cheng, X.; Zhou, J.; Shi, R.; Zhang, H. Elevated levels of Th17 cells and Th17-related cytokines are associated with disease activity in patients with inflammatory bowel disease. Inflamm. Res. 2014, 63, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, K.; Hwang, Y.; Nikolaev, A.; Atreya, R.; Dornhoff, H.; Steiner, S.; Lehr, H.A.; Wirtz, S.; Vieth, M.; Waisman, A.; et al. TH9 cells that express the transcription factor PU.1 drive T cell-mediated colitis via IL-9 receptor signaling in intestinal epithelial cells. Nat. Immunol. 2014, 15, 676–686. [Google Scholar] [CrossRef] [PubMed]
- Nalleweg, N.; Chiriac, M.T.; Podstawa, E.; Lehmann, C.; Rau, T.T.; Atreya, R.; Krauss, E.; Hundorfean, G.; Fichtner-Feigl, S.; Hartmann, A.; et al. IL-9 and its receptor are predominantly involved in the pathogenesis of UC. Gut 2015, 64, 743–755. [Google Scholar] [CrossRef] [PubMed]
- Sartor, R.B. Mechanisms of disease: Pathogenesis of Crohn’s disease and ulcerative colitis. Nat. Clin. Pract. Gastroenterol. Hepatol. 2006, 3, 390–407. [Google Scholar] [CrossRef] [PubMed]
- Geremia, A.; Biancheri, P.; Allan, P.; Corazza, G.R.; Di Sabatino, A. Innate and adaptive immunity in inflammatory bowel disease. Autoimmun. Rev. 2014, 13, 3–10. [Google Scholar] [CrossRef]
- Salim, S.Y.; Söderholm, J.D. Importance of disrupted intestinal barrier in inflammatory bowel diseases. Inflamm. Bowel Dis. 2011, 17, 362–381. [Google Scholar] [CrossRef]
- Uehara, A.; Fujimoto, Y.; Fukase, K.; Takada, H. Various human epithelial cells express functional Toll-like receptors, NOD1 and NOD2 to produce anti-microbial peptides, but not proinflammatory cytokines. Mol. Immunol. 2007, 44, 3100–3111. [Google Scholar] [CrossRef]
- De Souz, H.S.P.; Fiocchi, C. Immunopathogenesis of IBD: Current state of the art. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 13–27. [Google Scholar] [CrossRef]
- Neurath Markus, F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 2014, 14, 329–342. [Google Scholar] [CrossRef]
- Dinarello, C.A. Interleukin-1beta and the autoinflammatory diseases. N. Engl. J. Med. 2009, 360, 2467–2470. [Google Scholar] [CrossRef]
- Pizarro, T.T.; Michie, M.H.; Bentz, M.; Woraratanadharm, J.; Smith, M.F., Jr.; Foley, E.; Moskaluk, C.A.; Bickston, S.J.; Cominelli, F. IL-18, a novel immunoregulatory cytokine, is up-regulated in Crohn’s disease: Expression and localization in intestinal mucosal cells. J. Immunol. 1999, 162, 6829–6835. [Google Scholar] [PubMed]
- Dinarello, C.A.; Novick, D.; Puren, A.J.; Fantuzzi, G.; Shapiro, L.; Mühl, H.; Yoon, D.Y.; Reznikov, L.L.; Kim, S.H.; Rubinstein, M. Overview of interleukin-18, more than an interferon-gamma inducing factor. J. Leukoc. Biol. 1998, 63, 658–664. [Google Scholar] [CrossRef] [PubMed]
- Fiocchi, C. Cytokines and animal models: A combined path to inflammatory bowel disease pathogenesis. Gastroenterology 1993, 104, 1202–1205. [Google Scholar] [CrossRef]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef]
- Li, Y.; de Haar, C.; Chen, M.; Deuring, J.; Gerrits, M.M.; Smits, R.; Xia, B.; Kuipers, E.J.; van der Woude, C.J. Disease-related expression of the IL6/STAT3/SOCS3 signalling pathway in ulcerative colitis and ulcerative colitis-related carcinogenesis. Gut 2010, 59, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Munoz, F.; Dominguez-Lopez, A.; Yamamoto-Furusho, J.K. Role of cytokines in inflammatory bowel disease. World J. Gastroenterol. 2008, 14, 4280–4288. [Google Scholar] [CrossRef]
- Wirtz, S.; Neurath, M.F. Mouse models of inflammatory bowel disease. Adv. Drug Deliv. Rev. 2007, 59, 1073–1083. [Google Scholar] [CrossRef]
- Li, M.C.; He, S.H. IL-10 and its related cytokines for treatment of inflammatory bowel disease. World J. Gastroenterol. 2004, 10, 620–625. [Google Scholar] [CrossRef]
- Marek, A.; Brodzicki, J.; Liberek, A.; Korzon, M. TGF-beta (transforming growth factor-beta) in chronic inflammatory conditions—A new diagnostic and prognostic marker? Med. Sci. Monit. 2002, 8, RA145–RA151. [Google Scholar]
- Lee, S.H.; Kwon, J.E.; Cho, M.L. Immunological pathogenesis of inflammatory bowel disease. Intest. Res. 2018, 16, 26–42. [Google Scholar] [CrossRef]
- Li, M.O.; Flavell, R.A. TGF-beta: A master of all T cell trades. Cell 2008, 134, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Fujino, S.; Andoh, A.; Bamba, S.; Ogawa, A.; Hata, K.; Araki, Y.; Bamba, T.; Fujiyama, Y. Increased expression of interleukin 17 in inflammatory bowel disease. Gut 2003, 52, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Ivanov, I.I.; Spolski, R.; Min, R.; Shenderov, K.; Egawa, T.; Levy, D.E.; Leonard, W.J.; Littman, D.R. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat. Immunol. 2007, 8, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Raza, A.; Shata, M.T. Letter: Pathogenicity of Th17 cells may differ in ulcerative colitis compared with Crohn’s disease. Aliment. Pharmacol. Ther. 2012, 36, 204, author reply 205. [Google Scholar] [CrossRef] [PubMed]
- Geem, D.; Harusato, A.; Flannigan, K.; Denning, T.L. Harnessing regulatory T cells for the treatment of inflammatory bowel disease. Inflamm. Bowel Dis. 2015, 21, 1409–1418. [Google Scholar] [CrossRef] [PubMed]
- Rubtsov, Y.P.; Rasmussen, J.P.; Chi, E.Y.; Fontenot, J.; Castelli, L.; Ye, X.; Treuting, P.; Siewe, L.; Roers, A.; Henderson WRJr Muller, W.; et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity 2008, 28, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Omenetti, S.; Pizarro, T.T. The Treg/Th17 Axis: A Dynamic Balance Regulated by the Gut Microbiome. Front. Immunol. 2015, 6, 639. [Google Scholar] [CrossRef]
- Manichanh, C.; Rigottier-Gois, L.; Bonnaud, E.; Gloux, K.; Pelletier, E.; Frangeul, L.; Nalin, R.; Jarrin, C.; Chardon, P.; Marteau, P.; et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut 2006, 55, 205–211. [Google Scholar] [CrossRef]
- Lepage, P.; Häsler, R.; Spehlmann, M.E.; Rehman, A.; Zvirbliene, A.; Begun, A.; Ott, S.; Kupcinskas, L.; Doré, J.; Raedler, A.; et al. Twin study indicates loss of interaction between microbiota and mucosa of patients with ulcerative colitis. Gastroenterology 2011, 141, 227–236. [Google Scholar] [CrossRef]
- Singh, B.; Read, S.; Asseman, C.; Malmström, V.; Mottet, C.; Stephens, L.A.; Stepankova, R.; Tlaskalova, H.; Powrie, F. Control of intestinal inflammation by regulatory T cells. Immunol. Rev. 2001, 182, 190–200. [Google Scholar] [CrossRef]
- Fantini, M.C.; Becker, C.; Tubbe, I.; Nikolaev, A.; Lehr, H.A.; Galle, P.; Neurath, M.F. Transforming growth factor beta induced FoxP3+ regulatory T cells suppress Th1 mediated experimental colitis. Gut 2006, 55, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.; Dornhoff, H.; Neufert, C.; Fantini, M.C.; Wirtz, S.; Huebner, S.; Nikolaev, A.; Lehr, H.A.; Murphy, A.J.; Valenzuela, D.M.; et al. Cutting edge: IL-23 cross-regulates IL-12 production in T cell-dependent experimental colitis. J. Immunol. 2006, 177, 2760–2764. [Google Scholar] [CrossRef] [PubMed]
- Stolfi, C.; Rizzo, A.; Franzè, E.; Rotondi, A.; Fantini, M.C.; Sarra, M.; Caruso, R.; Monteleone, I.; Sileri, P.; Franceschilli, L.; et al. Involvement of interleukin-21 in the regulation of colitis-associated colon cancer. J. Exp. Med. 2011, 208, 2279–2290. [Google Scholar] [CrossRef] [PubMed]
- Klose, C.S.N.; Flach, M.; Möhle, L.; Rogell, L.; Hoyler, T.; Ebert, K.; Fabiunke, C.; Pfeifer, D.; Sexl, V.; Fonseca-Pereira, D.; et al. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell 2014, 157, 340–356. [Google Scholar] [CrossRef]
- Cortez, V.S.; Robinette, M.L.; Colonna, M. Innate lymphoid cells: New insights into function and development. Curr. Opin. Immunol. 2015, 32, 71–77. [Google Scholar] [CrossRef]
- Li, J.; Glover, S.C. Innate Lymphoid Cells in Inflammatory Bowel Disease. Arch. Immunol. Ther. Exp. 2018, 66, 415–421. [Google Scholar] [CrossRef]
- Spits, H.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.; Mebius, R.E.; et al. Innate lymphoid cells—A proposal for uniform nomenclature. Nat. Rev. Immunol. 2013, 13, 145–149. [Google Scholar] [CrossRef]
- Philip, N.H.; Artis, D. New friendships and old feuds: Relationships between innate lymphoid cells and microbial communities. Immunol. Cell Biol. 2013, 91, 225–231. [Google Scholar] [CrossRef]
- Mjösberg, J.; Spits, H. Human innate lymphoid cells. J. Allergy Clin. Immunol. 2016, 138, 1265–1276. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Knight, R.; Gordon, J.I. The effect of diet on the human gut microbiome: A metagenomic analysis in humanized gnotobiotic mice. Sci. Transl. Med. 2009, 1, 6ra14. [Google Scholar] [CrossRef]
- Faith, J.J.; McNulty, N.P.; Rey, F.E.; Gordon, J.I. Predicting a human gut microbiota’s response to diet in gnotobiotic mice. Science 2011, 333, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Russell, W.R.; Gratz, S.W.; Duncan, S.H.; Holtrop, G.; Ince, J.; Scobbie, L.; Duncan, G.; Johnstone, A.M.; Lobley, G.E.; Wallace, R.J.; et al. High-protein, reduced-carbohydrate weight-loss diets promote metabolite profiles likely to be detrimental to colonic health. Am. J. Clin. Nutr. 2011, 93, 1062–1072. [Google Scholar] [CrossRef] [PubMed]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C. Polyunsaturated fatty acids and inflammation. Biochem. Soc. Trans. 2005, 33, 423–427. [Google Scholar] [CrossRef]
- Lewis, J.D.; Abreu, M.T. Diet as a Trigger or Therapy for Inflammatory Bowel Diseases. Gastroenterology 2017, 152, 398–414.e6. [Google Scholar] [CrossRef] [PubMed]
- Kostic, A.D.; Xavier, R.J.; Gevers, D. The microbiome in inflammatory bowel disease: Current status and the future ahead. Gastroenterology 2014, 146, 1489–1499. [Google Scholar] [CrossRef]
- Patterson, E.; Wall, R.; Fitzgerald, G.F.; Ross, R.P.; Stanton, C. Health implications of high dietary omega-6 polyunsaturated Fatty acids. J. Nutr. Metab. 2012, 2012, 539426. [Google Scholar] [CrossRef]
- Simopoulos, A.P. The importance of the ratio of omega-6/omega-3 essential fatty acids. Biomed. Pharmacother. 2002, 56, 365–379. [Google Scholar] [CrossRef]
- Gutiérrez, S.; Svahn, S.L.; Johansson, M.E. Effects of Omega-3 Fatty Acids on Immune Cells. Int. J. Mol. Sci. 2019, 20, 5028. [Google Scholar] [CrossRef]
- Franzosa, E.A.; Sirota-Madi, A.; Avila-Pacheco, J.; Fornelos, N.; Haiser, H.J.; Reinker, S.; Vatanen, T.; Hall, A.B.; Mallick, H.; McIver, L.J.; et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 2019, 4, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.; Boneh, R.S.; Wine, E. Evolving role of diet in the pathogenesis and treatment of inflammatory bowel diseases. Gut 2018, 67, 1726–1738. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.S.; Seekatz, A.M.; Koropatkin, N.M.; Kamada, N.; Hickey, C.A.; Wolter, M.; Pudlo, N.A.; Kitamoto, S.; Terrapon, N.; Muller, A.; et al. A Dietary Fiber-Deprived Gut Microbiota Degrades the Colonic Mucus Barrier and Enhances Pathogen Susceptibility. Cell 2016, 167, 1339–1353.e21. [Google Scholar] [CrossRef]
- Ma, X.; Torbenson, M.; Hamad, A.R.; Soloski, M.J.; Li, Z. High-fat diet modulates non-CD1d-restricted natural killer T cells and regulatory T cells in mouse colon and exacerbates experimental colitis. Clin. Exp. Immunol. 2008, 151, 130–138. [Google Scholar] [CrossRef]
- Agus, A.; Denizot, J.; Thévenot, J.; Martinez-Medina, M.; Massier, S.; Sauvanet, P.; Bernalier-Donadille, A.; Denis, S.; Hofman, P.; Bonnet, R.; et al. Western diet induces a shift in microbiota composition enhancing susceptibility to Adherent-Invasive E. coli infection and intestinal inflammation. Sci. Rep. 2016, 6, 19032. [Google Scholar] [CrossRef]
- Kim, K.A.; Gu, W.; Lee, I.A.; Joh, E.H.; Kim, D.H. High fat diet-induced gut microbiota exacerbates inflammation and obesity in mice via the TLR4 signaling pathway. PLoS ONE 2012, 7, e47713. [Google Scholar] [CrossRef]
- Liu, Z.; Brooks, R.S.; Ciappio, E.D.; Kim, S.J.; Crott, J.W.; Bennett, G.; Greenberg, A.S.; Mason, J.B. Diet-induced obesity elevates colonic TNF-α in mice and is accompanied by an activation of Wnt signaling: A mechanism for obesity-associated colorectal cancer. J. Nutr. Biochem. 2012, 23, 1207–1213. [Google Scholar] [CrossRef]
- Gonçalves, P.; Araújo, J.R.; Di Santo, J.P. A Cross-Talk Between Microbiota-Derived Short-Chain Fatty Acids and the Host Mucosal Immune System Regulates Intestinal Homeostasis and Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2018, 24, 558–572. [Google Scholar] [CrossRef]
- Castro, F.; de Souza, H.S.P. Dietary Composition and Effects in Inflammatory Bowel Disease. Nutrients 2019, 11, 1398. [Google Scholar] [CrossRef]
- Joossens, M.; Huys, G.; Cnockaert, M.; De Preter, V.; Verbeke, K.; Rutgeerts, P.; Vandamme, P.; Vermeire, S. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut 2011, 60, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.; Leducq, V.; Aschard, H.; Pham, H.P.; Jegou, S.; Landman, C.; Cohen, D.; Liguori, G.; Bourrier, A.; Nion-Larmurier, I.; et al. Fungal microbiota dysbiosis in IBD. Gut 2017, 66, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Pascal, V.; Pozuelo, M.; Borruel, N.; Casellas, F.; Campos, D.; Santiago, A.; Martinez, X.; Varela, E.; Sarrabayrouse, G.; Machiels, K.; et al. A microbial signature for Crohn’s disease. Gut 2017, 66, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Powell, N.; MacDonald, T.T. Recent advances in gut immunology. Parasite Immunol. 2017, 39, e12430. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Oshima, K.; Suda, W.; Nagano, Y.; Nishikawa, H.; Fukuda, S.; Saito, T.; Narushima, S.; Hase, K.; et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 2013, 500, 232–236. [Google Scholar] [CrossRef]
- Sefik, E.; Geva-Zatorsky, N.; Oh, S.; Konnikova, L.; Zemmour, D.; McGuire, A.M.; Burzyn, D.; Ortiz-Lopez, A.; Lobera, M.; Yang, J.; et al. Individual intestinal symbionts induce a distinct population of RORγ⁺ regulatory T cells. Science 2015, 349, 993–997. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Ando, M.; Kamada, N.; Nagano, Y.; Narushima, S.; Suda, W.; Imaoka, A.; Setoyama, H.; Nagamori, T.; et al. Th17 Cell Induction by Adhesion of Microbes to Intestinal Epithelial Cells. Cell 2015, 163, 367–380. [Google Scholar] [CrossRef]
- Tan, T.G.; Sefik, E.; Geva-Zatorsky, N.; Kua, L.; Naskar, D.; Teng, F.; Pasman, L.; Ortiz-Lopez, A.; Jupp, R.; Wu, H.J.; et al. Identifying species of symbiont bacteria from the human gut that, alone, can induce intestinal Th17 cells in mice. Proc. Natl. Acad. Sci. USA 2016, 113, E8141–E8150. [Google Scholar] [CrossRef]
- Russo, G.L. Dietary n-6 and n-3 polyunsaturated fatty acids: From biochemistry to clinical implications in cardiovascular prevention. Biochem. Pharmacol. 2009, 77, 937–946. [Google Scholar] [CrossRef]
- Shoda, R.; Matsueda, K.; Yamato, S.; Umeda, N. Epidemiologic analysis of Crohn disease in Japan: Increased dietary intake of n-6 polyunsaturated fatty acids and animal protein relates to the increased incidence of Crohn disease in Japan. Am. J. Clin. Nutr. 1996, 63, 741–745. [Google Scholar] [CrossRef]
- IBD in EPIC Study Investigators; Tjonneland, A.; Overvad, K.; Bergmann, M.M.; Nagel, G.; Linseisen, J.; Hallmans, G.; Palmqvist, R.; Sjodin, H.; Hagglund, G.; et al. Linoleic acid, a dietary n-6 polyunsaturated fatty acid, and the aetiology of ulcerative colitis: A nested case-control study within a European prospective cohort study. Gut 2009, 58, 1606–1611. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N.; Khalili, H.; Konijeti, G.G.; Higuchi, L.M.; de Silva, P.; Fuchs, C.S.; Willett, W.C.; Richter, J.M.; Chan, A.T. Long-term intake of dietary fat and risk of ulcerative colitis and Crohn’s disease. Gut 2014, 63, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Joffre, C.; Dinel, A.L.; Chataigner, M.; Pallet, V.; Layé, S. n-3 Polyunsaturated Fatty Acids and Their Derivates Reduce Neuroinflammation during Aging. Nutrients 2020, 12, 647. [Google Scholar] [CrossRef] [PubMed]
- Das, U.N. Arachidonic acid and other unsaturated fatty acids and some of their metabolites function as endogenous antimicrobial molecules: A review. J. Adv. Res. 2018, 11, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Al-Khalaifah, H. Modulatory Effect of Dietary Polyunsaturated Fatty Acids on Immunity, Represented by Phagocytic Activity. Front. Vet. Sci. 2020, 7, 569939. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.G.; Contaifer, D.; Madurantakam, P.; Carbone, S.; Price, E.T.; Van Tassell, B.; Brophy, D.F.; Wijesinghe, D.S. Dietary Bioactive Fatty Acids as Modulators of Immune Function: Implications on Human Health. Nutrients 2019, 11, 2974. [Google Scholar] [CrossRef]
- Uchiyama, K.; Nakamura, M.; Odahara, S.; Koido, S.; Katahira, K.; Shiraishi, H.; Ohkusa, T.; Fujise, K.; Tajiri, H. N-3 polyunsaturated fatty acid diet therapy for patients with inflammatory bowel disease. Inflamm. Bowel Dis. 2010, 16, 1696–1707. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Lin, X.; Zhao, Q.; Li, J. Fat intake and risk of ulcerative colitis: Systematic review and dose-response meta-analysis of epidemiological studies. J. Gastroenterol. Hepatol. 2017, 32, 19–27. [Google Scholar] [CrossRef]
- Sakamoto, N.; Kono, S.; Wakai, K.; Fukuda, Y.; Satomi, M.; Shimoyama, T.; Inaba, Y.; Miyake, Y.; Sasaki, S.; Okamoto, K.; et al. Dietary risk factors for inflammatory bowel disease: A multicenter case-control study in Japan. Inflamm. Bowel Dis. 2005, 11, 154–163. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N.; Khalili, H.; Song, M.; Higuchi, L.M.; Lochhead, P.; Richter, J.M.; Chan, A.T. Genetic Polymorphisms in Fatty Acid Metabolism Modify the Association Between Dietary n3, n6 Intake and Risk of Ulcerative Colitis: A Prospective Cohort Study. Inflamm. Bowel Dis. 2017, 23, 1898–1904. [Google Scholar] [CrossRef]
- Le Poul, E.; Loison, C.; Struyf, S.; Springael, J.Y.; Lannoy, V.; Decobecq, M.E.; Brezillon, S.; Dupriez, V.; Vassart, G.; Van Damme, J.; et al. Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J. Biol. Chem. 2003, 278, 25481–25489. [Google Scholar] [CrossRef] [PubMed]
- Aljebab, F.; Choonara, I.; Conroy, S. Systematic Review of the Toxicity of Long-Course Oral Corticosteroids in Children. PLoS ONE 2017, 12, e0170259. [Google Scholar] [CrossRef] [PubMed]
- Rutgeerts, P.J. Review article: The limitations of corticosteroid therapy in Crohn’s disease. Aliment. Pharmacol. Ther. 2001, 15, 1515–1525. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, O.; Cordischi, L.; Cirulli, M.; Paganelli, M.; Labalestra, V.; Uccini, S.; Russo, P.M.; Cucchiara, S. Polymeric diet alone versus corticosteroids in the treatment of active pediatric Crohn’s disease: A randomized controlled open-label trial. Clin. Gastroenterol. Hepatol. 2006, 4, 744–753. [Google Scholar] [CrossRef]
- Hansen, T.; Duerksen, D.R. Enteral Nutrition in the Management of Pediatric and Adult Crohn’s Disease. Nutrients 2018, 10, 537. [Google Scholar] [CrossRef]
- Zachos, M.; Tondeur, M.; Griffiths, A.M. Enteral nutritional therapy for induction of remission in Crohn’s disease. Cochrane Database Syst. Rev. 2007, 4, CD000542. [Google Scholar] [CrossRef]
- Lochs, H.; Dejong, C.; Hammarqvist, F.; Hebuterne, X.; Leon-Sanz, M.; Schütz, T.; van Gemert, W.; van Gossum, A.; Valentini, L.; DGEM (German Society for Nutritional Medicine); et al. ESPEN Guidelines on Enteral Nutrition: Gastroenterology. Clin. Nutr. 2006, 25, 260–274. [Google Scholar] [CrossRef]
- Donnellan, C.F.; Yann, L.H.; Lal, S. Nutritional management of Crohn’s disease. Ther. Adv. Gastroenterol. 2013, 6, 231–242. [Google Scholar] [CrossRef]
- Yamamoto, T.; Nakahigashi, M.; Umegae, S.; Kitagawa, T.; Matsumoto, K. Impact of elemental diet on mucosal inflammation in patients with active Crohn’s disease: Cytokine production and endoscopic and histological findings. Inflamm. Bowel Dis. 2005, 11, 580–588. [Google Scholar] [CrossRef]
- Sanderson, I.R.; Croft, N.M. The anti-inflammatory effects of enteral nutrition. JPEN J. Parenter Enter. Nutr. 2005, 29, S134–S140. [Google Scholar] [CrossRef]
- Svolos, V.; Hansen, R.; Nichols, B.; Quince, C.; Ijaz, U.Z.; Papadopoulou, R.T.; Edwards, C.A.; Watson, D.; Alghamdi, A.; Brejnrod, A.; et al. Treatment of Active Crohn’s Disease With an Ordinary Food-based Diet That Replicates Exclusive Enteral Nutrition. Gastroenterology 2019, 156, 1354–1367.e6. [Google Scholar] [CrossRef] [PubMed]
- Suskind, D.L.; Cohen, S.A.; Brittnacher, M.J.; Wahbeh, G.; Lee, D.; Shaffer, M.L.; Braly, K.; Hayden, H.S.; Klein, J.; Gold, B.; et al. Clinical and Fecal Microbial Changes With Diet Therapy in Active Inflammatory Bowel Disease. J. Clin. Gastroenterol. 2018, 52, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Sigall-Boneh, R.; Pfeffer-Gik, T.; Segal, I.; Zangen, T.; Boaz, M.; Levine, A. Partial enteral nutrition with a Crohn’s disease exclusion diet is effective for induction of remission in children and young adults with Crohn’s disease. Inflamm. Bowel Dis. 2014, 20, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- SigallBoneh, R.; SarbagiliShabat, C.; Yanai, H.; Chermesh, I.; Ben Avraham, S.; Boaz, M.; Levine, A. Dietary Therapy With the Crohn’s Disease Exclusion Diet is a Successful Strategy for Induction of Remission in Children and Adults Failing Biological Therapy. J. Crohn’s Colitis 2017, 11, 1205–1212. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Cytokines, Immune Cells and Microbioma | |||||||
|---|---|---|---|---|---|---|---|
| TNF | IL-1β | IL-6 | M1 Macrophages | Treg | GRAM Negative Bacteria | References | |
| High consumption of fats and sugars | + | + | + | + | − | + | [14,15,17,25,29,30,36,63,66,67] |
| Regular consumption of omega-3 PUFA, fibers and vegetables | − | − | − | − | + | − | [14,15,50,51,65,66,68,69,70] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tamburini, B.; La Manna, M.P.; La Barbera, L.; Mohammadnezhad, L.; Badami, G.D.; Shekarkar Azgomi, M.; Dieli, F.; Caccamo, N. Immunity and Nutrition: The Right Balance in Inflammatory Bowel Disease. Cells 2022, 11, 455. https://doi.org/10.3390/cells11030455
Tamburini B, La Manna MP, La Barbera L, Mohammadnezhad L, Badami GD, Shekarkar Azgomi M, Dieli F, Caccamo N. Immunity and Nutrition: The Right Balance in Inflammatory Bowel Disease. Cells. 2022; 11(3):455. https://doi.org/10.3390/cells11030455
Chicago/Turabian StyleTamburini, Bartolo, Marco Pio La Manna, Lidia La Barbera, Leila Mohammadnezhad, Giusto Davide Badami, Mojtaba Shekarkar Azgomi, Francesco Dieli, and Nadia Caccamo. 2022. "Immunity and Nutrition: The Right Balance in Inflammatory Bowel Disease" Cells 11, no. 3: 455. https://doi.org/10.3390/cells11030455
APA StyleTamburini, B., La Manna, M. P., La Barbera, L., Mohammadnezhad, L., Badami, G. D., Shekarkar Azgomi, M., Dieli, F., & Caccamo, N. (2022). Immunity and Nutrition: The Right Balance in Inflammatory Bowel Disease. Cells, 11(3), 455. https://doi.org/10.3390/cells11030455