
New Insights into the Mechanisms of Chaperon-Mediated Autophagy and Implications for Kidney Diseases


Abstract
1. Introduction
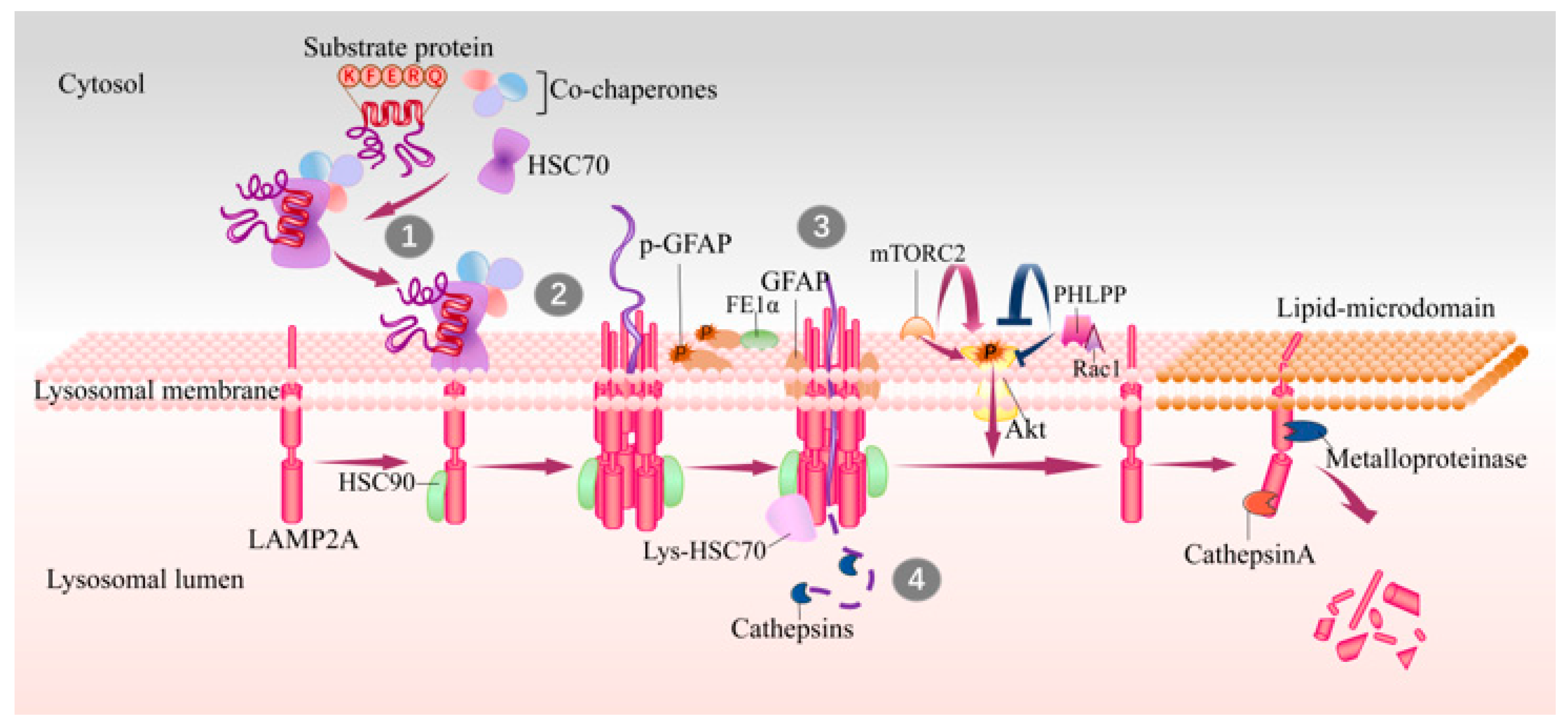
2. Biology of Chaperon-Mediated Autophagy
3. Regulation of CMA
3.1. Regulation of LAMP2A
3.2. Regulation of Chaperones
3.3. Regulation of Lysosomal PH
3.4. Regulate Molecules of CMA
3.5. Starvation
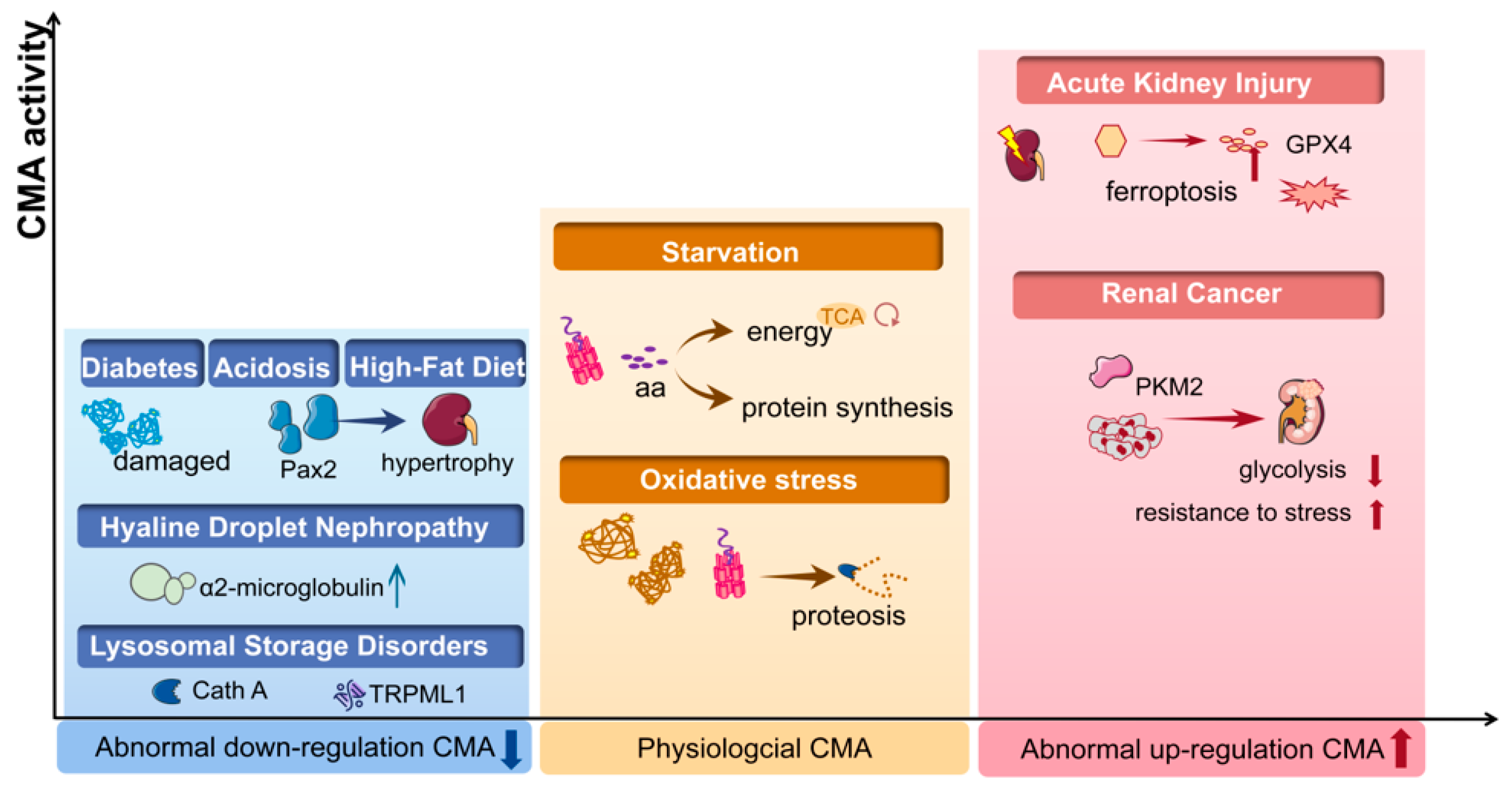
4. Implication of CMA in the Kidney Diseases
4.1. Diabetic Renal Hypertrophy
4.2. Lysosomal Storage Disorders (LSDs)
4.3. Hyaline Droplet Nephropathy
4.4. Chronic Kidney Disease (CKD) and Kidney Aging
4.5. Acute Kidney Injury (AKI)
4.6. HFD-Mediated Kidney Injury
4.7. Renal Cancer
4.8. Other Kidney Diseases
5. Conclusions Remarks and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Debnath, J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 2015, 16, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Oku, M.; Sakai, Y. Three Distinct Types of Microautophagy Based on Membrane Dynamics and Molecular Ma-chineries. Bioessays 2018, 40, e1800008. [Google Scholar] [CrossRef] [PubMed]
- Sahu, R.; Kaushik, S.; Clement, C.C.; Cannizzo, E.S.; Scharf, B.; Follenzi, A.; Potolicchio, I.; Nieves, E.; Cuervo, A.M.; Santambrogio, L. Microautophagy of cytosolic proteins by late endosomes. Dev Cell. 2011, 20, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Massey, A.C.; Kaushik, S.; Sovak, G.; Kiffin, R.; Cuervo, A.M. Consequences of the selective blockage of chaperone-mediated autophagy. Proc. Natl. Acad. Sci. USA 2006, 103, 5805–5810. [Google Scholar] [CrossRef]
- Schneider, J.L.; Villarroya, J.; Diaz-Carretero, A.; Patel, B.; Urbanska, A.M.; Thi, M.M.; Villarroya, F.; Santambrogio, L.; Cuervo, A.M. Loss of hepatic chaperone-mediated autophagy accelerates proteostasis failure in aging. Aging Cell 2015, 14, 249–264. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Knecht, E.; Terlecky, S.R.; Dice, J.F. Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. Am. J. Physiol. Physiol. 1995, 269, C1200–C1208. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Dice, J.F. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem. Sci. 1990, 15, 305–309. [Google Scholar] [CrossRef]
- Tekirdag, K.; Cuervo, A.M. Chaperone-mediated autophagy and endosomal microautophagy: Joint by a chaper-one. J. Biol. Chem. 2018, 293, 5414–5424. [Google Scholar] [CrossRef]
- Dice, J.F.; Terlecky, S.R. Targeting of cytosolic proteins to lysosomes for degradation. Crit. Rev. Ther. Drug Carr. Syst. 1990, 7, 211–233. [Google Scholar]
- Liu, S.; Hartleben, B.; Kretz, O.; Wiech, T.; Igarashi, P.; Mizushima, N.; Walz, G.; Huber, T.B. Autophagy plays a critical role in kidney tubule maintenance, aging and ischemia-reperfusion injury. Autophagy 2012, 8, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Franch, H.A. Chaperone-Mediated Autophagy in the Kidney: The Road More Traveled. Semin. Nephrol. 2014, 34, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, R.; Osaki, M.; Sasaki, R.; Ishikawa, M.; Yumioka, T.; Yamaguchi, N.; Iwamoto, H.; Honda, M.; Kabuta, T.; Takenaka, A.; et al. Splice variants of lyso-some-associated membrane proteins 2A and 2B are involved in sunitinib resistance in human renal cell carcinoma cells. Oncol. Rep. 2020, 44, 1810–1820. [Google Scholar]
- Cuervo, A.M.; Dice, J.F. Age-related Decline in Chaperone-mediated Autophagy. J. Biol. Chem. 2000, 275, 31505–31513. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Cuervo, A.M. Restoration of chaperone-mediated autophagy in aging liver improves cellular mainte-nance and hepatic function. Nat. Med. 2008, 14, 959–965. [Google Scholar] [CrossRef]
- Kiffin, R.; Christian, C.; Knecht, E.; Cuervo, A.M. Activation of Chaperone-mediated Autophagy during Oxidative Stress. Mol. Biol. Cell 2004, 15, 4829–4840. [Google Scholar] [CrossRef]
- Liu, C.; Gidlund, E.-K.; Witasp, A.; Qureshi, A.R.; Söderberg, M.; Thorell, A.; Nader, G.A.; Barany, P.; Stenvinkel, P.; Von Walden, F. Reduced skeletal muscle expression of mitochondrial-derived peptides humanin and MOTS-C and Nrf2 in chronic kidney disease. Am. J. Physiol. Physiol. 2019, 317, F1122–F1131. [Google Scholar] [CrossRef]
- Sooparb, S.; Price, S.R.; Shaoguang, J.; Franch, H.A. Suppression of chaperone-mediated autophagy in the renal cor-tex during acute diabetes mellitus. Kidney Int. 2004, 65, 2135–2144. [Google Scholar] [CrossRef]
- Kirchner, P.; Bourdenx, M.; Madrigal-Matute, J.; Tiano, S.; Diaz, A.; Bartholdy, B.A.; Will, B.; Cuervo, A.M. Proteome-wide analysis of chaperone-mediated autophagy targeting motifs. PLoS Biol. 2019, 17, e3000301. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Dice, J.F. A Receptor for the Selective Uptake and Degradation of Proteins by Lysosomes. Science 1996, 273, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, U.; Kaushik, S.; Varticovski, L.; Cuervo, A.M. The chaperone-mediated autophagy receptor organiz-es in dynamic protein complexes at the lysosomal membrane. Mol. Cell. Biol. 2008, 28, 5747–5763. [Google Scholar] [CrossRef]
- Gong, Z.; Tasset, I.; Diaz, A.; Anguiano, J.; Tas, E.; Cui, L.; Kuliawat, R.; Liu, H.; Kühn, B.; Cuervo, A.M.; et al. Humanin is an endogenous activator of chaperone-mediated autophagy. J. Cell Biol. 2017, 217, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.; Dice, J. Regulation of Lamp2a Levels in the Lysosomal Membrane. Traffic 2000, 1, 570–583. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, U.; Sridhar, S.; Kaushik, S.; Kiffin, R.; Cuervo, A.M. Identification of Regulators of Chaperone-Mediated Autophagy. Mol. Cell 2010, 39, 535–547. [Google Scholar] [CrossRef] [PubMed]
- Arias, E.; Koga, H.; Diaz, A.; Mocholi, E.; Patel, B.; Cuervo, A.M. Lysosomal mTORC2/PHLPP1/Akt Regulate Chaperone-Mediated Autophagy. Mol. Cell 2015, 59, 270–284. [Google Scholar] [CrossRef]
- Kaushik, S.; Massey, A.C.; Cuervo, A.M. Lysosome membrane lipid microdomains: Novel regulators of chaperone-mediated autophagy. EMBO J. 2006, 25, 3921–3933. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Mann, L.; Bonten, E.J.; D’Azzo, A.; Dice, J.F. Cathepsin A regulates chaperone-mediated autophagy through cleavage of the lysosomal receptor. EMBO J. 2003, 22, 47–59. [Google Scholar] [CrossRef]
- Pajares, M.; Rojo, A.I.; Arias, E.; Díaz-Carretero, A.; Cuervo, A.M.; Cuadrado, A. Transcription factor NFE2L2/NRF2 mod-ulates chaperone-mediated autophagy through the regulation of LAMP2A. Autophagy 2018, 14, 1310–1322. [Google Scholar] [CrossRef]
- Park, C.; Suh, Y.; Cuervo, A.M. Regulated degradation of Chk1 by chaperone-mediated autophagy in response to DNA damage. Nat. Commun. 2015, 6, 1–14. [Google Scholar] [CrossRef]
- Valdor, R.; Mocholi, E.; Botbol, Y.; Guerrero-Ros, I.; Chandra, D.; Koga, H.; Gravekamp, C.; Cuervo, A.M.; Macian, F. Chaperone-mediated autophagy regu-lates T cell responses through targeted degradation of negative regulators of T cell activation. Nat. Immunol. 2014, 15, 1046–1054. [Google Scholar] [CrossRef]
- Anguiano, J.; Garner, T.P.; Mahalingam, M.; Das, B.C.; Gavathiotis, E.; Cuervo, A.M. Chemical modulation of chaperone-mediated autophagy by retinoic acid derivatives. Nat. Chem. Biol. 2013, 9, 374–382. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Dice, J.F.; Knecht, E. A population of rat liver lysosomes responsible for the selective uptake and deg-radation of cytosolic proteins. J. Biol. Chem. 1997, 272, 5606–5615. [Google Scholar] [CrossRef] [PubMed]
- Agarraberes, F.A.; Terlecky, S.R.; Dice, J.F. An intralysosomal hsp70 is required for a selective pathway of lysosomal protein degradation. J. Cell. Biol. 1997, 137, 825–834. [Google Scholar] [CrossRef]
- Agarraberes, F.A.; Dice, J.F. A molecular chaperone complex at the lysosomal membrane is required for protein translocation. J. Cell Sci. 2001, 114 Pt 13, 2491–2499. [Google Scholar] [CrossRef]
- Sautin, Y.Y.; Lu, M.; Gaugler, A.; Zhang, L.; Gluck, S.L. Phosphatidylinositol 3-Kinase-Mediated Effects of Glucose on Vacuolar H+-ATPase Assembly, Translocation, and Acidification of Intracellular Compartments in Renal Epithelial Cells. Mol. Cell. Biol. 2005, 25, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Kohio, H.P.; Adamson, A.L. Glycolytic control of vacuolar-type ATPase activity: A mechanism to regulate influenza viral infection. Virology 2013, 444, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Forgac, M.; McGuire, C.; Stransky, L.; Cotter, K. Regulation of V-ATPase activity. Front. Biosci. 2017, 22, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Franch, H.A.; Preisig, P.A. NH4Cl-induced hypertrophy is mediated by weak base effects and is independent of cell cycle processes. Am. J. Physiol. Physiol. 1996, 270, C932–C938. [Google Scholar] [CrossRef]
- Ling, H.; Vamvakas, S.; Gekle, M.; Schaefer, L.; Teschner, M.; Schaefer, R.M.; Heidland, A. Role of lysosomal cathepsin activities in cell hypertrophy induced by NH4Cl in cultured renal proximal tubule cells. J. Am. Soc. Nephrol. 1996, 7, 73–80. [Google Scholar] [CrossRef]
- Franch, H.A.; Sooparb, S.; Du, J.; Brown, N.S. A Mechanism Regulating Proteolysis of Specific Proteins during Renal Tubular Cell Growth. J. Biol. Chem. 2001, 276, 19126–19131. [Google Scholar] [CrossRef]
- Porter, K.; Nallathambi, J.; Lin, Y.; Liton, P.B. Lysosomal basification and decreased autophagic flux in oxidatively stressed trabecular meshwork cells: Implications for glaucoma pathogenesis. Autophagy 2013, 9, 581–594. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Murashige, D.S.; Humphrey, S.; James, D.E. A Positive Feedback Loop between Akt and mTORC2 via SIN1 Phosphorylation. Cell Rep. 2015, 12, 937–943. [Google Scholar] [CrossRef]
- Endicott, S.J.; Ziemba, Z.J.; Beckmann, L.J.; Boynton, D.N.; Miller, R.A. Inhibition of class I PI3K enhances chaperone-mediated autophagy. J. Cell Biol. 2020, 219, e202001031. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhu, J.; Dou, J.; She, H.; Tao, K.; Xu, H.; Yang, Q.; Mao, Z. Phosphorylation of LAMP2A by p38 MAPK couples ER stress to chaperone-mediated autophagy. Nat. Commun. 2017, 8, 1763. [Google Scholar] [CrossRef]
- Obayashi, H.; Nagano, Y.; Takahashi, T.; Seki, T.; Tanaka, S.; Sakai, N.; Matsumoto, M.; Maruyama, H. Histone deacetylase 10 knockout activates chaperone-mediated autophagy and accelerates the decomposition of its substrate. Biochem. Biophys. Res. Commun. 2020, 523, 246–252. [Google Scholar] [CrossRef]
- Sato, M.; Ueda, E.; Konno, A.; Hirai, H.; Kurauchi, Y.; Hisatsune, A.; Katsuki, H.; Seki, T. Glucocorticoids negatively regulates chaper-one mediated autophagy and microautophagy. Biochem. Biophys. Res. Commun. 2020, 528, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Liu, J.; Shen, S.; Tong, Q.; Ma, X.; Lin, L. SIRT3 promotes lipophagy and chaperon-mediated autophagy to protect hepatocytes against lipotoxicity. Cell Death Differ. 2020, 27, 329–344. [Google Scholar] [CrossRef]
- Wang, Y.; Chang, J.; Wang, Z.Q.; Li, Y. Sirt3 promotes the autophagy of HK-2 human proximal tubular epithelial cells via the inhibition of Notch-1/Hes-1 signaling. Mol. Med. Rep. 2021, 24, 634. [Google Scholar] [CrossRef]
- Massey, A.; Kiffin, R.; Cuervo, A.M. Pathophysiology of chaperone-mediated autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2420–2434. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Hu, W.; Lim, B.; Dice, J.F. IkappaB is a substrate for a selective pathway of lysosomal proteolysis. Mol. Biol. Cell. 1998, 9, 1995–2010. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat. Cell Biol. 2015, 17, 759–770. [Google Scholar] [CrossRef]
- Finn, P.F.; Dice, J.F.; Chiu, P.C.N.; Chung, M.-K.; Tsang, H.-Y.; Koistinen, R.; Koistinen, H.; Seppala, M.; Lee, K.-F.; Yeung, W.S. Ketone Bodies Stimulate Chaperone-mediatedAutophagy. J. Biol. Chem. 2005, 280, 25864–25870. [Google Scholar] [CrossRef]
- Finn, P.F.; Dice, J.F. Proteolytic and lipolytic responses to starvation. Nutrition 2006, 22, 830–844. [Google Scholar] [CrossRef]
- Napolitano, G.; Johnson, J.L.; He, J.; Rocca, C.; Monfregola, J.; Pestonjamasp, K.; Cherqui, S.; Catz, S.D. Impairment of chaperone-mediated autophagy leads to selective lysosomal degradation defects in the lysosomal storage disease cystinosis. EMBO Mol. Med. 2015, 7, 158–174. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, B.; Mesires, N.T.; Kennedy, J.C.; Curcio-Morelli, C.; Laplante, J.M.; Dice, J.F.; Slaugenhaupt, S.A. Chaperone-mediated au-tophagy is defective in mucolipidosis type IV. J. Cell. Physiol. 2009, 219, 344–353. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Hildebrand, H.; Bomhard, E.M.; Dice, J.F. Direct lysosomal uptake of α2-microglobulin contributes to chemically induced nephropathy. Kidney Int. 1999, 55, 529–545. [Google Scholar] [CrossRef][Green Version]
- Hard, G.C.; Rodgers, I.S.; Baetcke, K.P.; Richards, W.L.; McGaughy, R.E.; Valcovic, L.R. Hazard Evaluation of Chemicals That Cause Accumulation of α 2u -Globulin, Hyaline Droplet Nephropathy, and Tubule Neoplasia in the Kidneys of Male Rats. Environ. Health Perspect. 1993, 99, 313. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Geng, Y.; Lu, X.; Shi, Y.; Wu, G.; Zhang, M.; Shan, B.; Pan, H.; Yuan, J. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc. Natl. Acad. Sci. USA 2019, 116, 2996–3005. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wang, D.; Yu, Y.; Zhao, T.; Min, N.; Wu, Y.; Kang, L.; Zhao, Y.; Du, L.; Zhang, M.; et al. Legumain promotes tubular ferroptosis by facilitating chaper-one-mediated autophagy of GPX4 in AKI. Cell Death Dis. 2021, 12, 65. [Google Scholar] [CrossRef]
- Xiao, S.; Xu, G.; Wang, Z.; Chong, T. Chaperon-mediated autophagy can promote proliferation and invasion of renal carcinoma cells and inhibit apoptosis through PKM. Oncol. Rep. 2021, 46, 1–9. [Google Scholar] [CrossRef]
- Luo, Y.; Wu, M.-Y.; Deng, B.-Q.; Huang, J.; Hwang, S.H.; Li, M.-Y.; Zhou, C.-Y.; Zhang, Q.-Y.; Yu, H.-B.; Zhao, D.-K.; et al. Inhibition of soluble epoxide hydrolase attenuates a high-fat diet-mediated renal injury by activating PAX2 and AMPK. Proc. Natl. Acad. Sci. USA 2019, 116, 5154–5159. [Google Scholar] [CrossRef]
- Macri, C.; Tasset, I.; Schall, N.; Page, N.; Briand, J.-P.; Cuervo, A.M.; Muller, S.; Wang, F. Modulation of deregulated chaperone-mediated autophagy by a phosphopeptide. Autophagy 2015, 11, 472–486. [Google Scholar] [CrossRef]
- Belibi, F.; Zafar, I.; Ravichandran, K.; Segvic, A.B.; Jani, A.; Ljubanovic, D.G.; Edelstein, C.L. Hypoxia-inducible factor-1α (HIF-1α) and autophagy in polycystic kidney disease (PKD). Am. J. Physiol.-Ren. Physiol. 2011, 300, F1235–F1243. [Google Scholar] [CrossRef]
- Belibi, F.; Ravichandran, K.; Zafar, I.; He, Z.; Edelstein, C.L. mTORC1/2 and rapamycin in female Han:SPRD rats with polycystic kidney disease. Am. J. Physiol. Renal. Physiol. 2011, 300, F236–F244. [Google Scholar] [CrossRef]
- Belfiore, F.; Rabuazzo, A.; Iannello, S.; Campione, R.; Vasta, D. Anabolic response of some tissues to diabetes. Biochem. Med. Metab. Biol. 1986, 35, 149–155. [Google Scholar] [CrossRef]
- Franch, H.A. Pathways of proteolysis affecting renal cell growth. Curr. Opin. Nephrol. Hypertens. 2002, 11, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Hammerman, M.R.; O’Shea, M.; Miller, S.B. Role of growth factors in regulation of renal growth. Annu. Rev. Physiol. 1993, 55, 305–321. [Google Scholar] [CrossRef] [PubMed]
- Franch, H.A.; Curtis, P.V.; Mitch, W.E. Mechanisms of renal tubular cell hypertrophy: Mitogen-induced suppression of proteolysis. Am. J. Physiol. Content 1997, 273, C843–C851. [Google Scholar] [CrossRef] [PubMed]
- Franch, H.A.; Wang, X.; Sooparb, S.; Brown, N.S.; Du, J. Phosphatidylinositol 3-kinase activity is required for epidermal growth factor to suppress proteolysis. J. Am. Soc. Nephrol. 2002, 13, 903–909. [Google Scholar] [CrossRef]
- Shen, W.; Brown, N.S.; Finn, P.F.; Dice, J.F.; Franch, H.A. Akt and Mammalian target of rapamycin regulate separate sys-tems of proteolysis in renal tubular cells. J. Am. Soc. Nephrol. 2006, 17, 2414–2423. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, A.P.; Puertollano, R.; Raben, N.; Slaugenhaupt, S.; Walkley, S.U.; Ballabio, A. Autophagy in lysosomal stor-age disorders. Autophagy 2012, 8, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Johnson, J.L.; He, J.; Napolitano, G.; Ramadass, M.; Rocca, C.; Kiosses, W.B.; Bucci, C.; Xin, Q.; Gavathiotis, E.; et al. Cystinosin, the small GTPase Rab11, and the Rab7 effector RILP regulate intracellular trafficking of the chaperone-mediated autophagy receptor LAMP2A. J. Biol. Chem. 2017, 292, 10328–10346. [Google Scholar] [CrossRef]
- Zhang, J.; He, J.; Johnson, J.L.; Rahman, F.; Gavathiotis, E.; Cuervo, A.M.; Catz, S.D. Chaperone-Mediated Autophagy Upregu-lation Rescues Megalin Expression and Localization in Cystinotic Proximal Tubule Cells. Front. Endocrinol. 2019, 10, 21. [Google Scholar] [CrossRef]
- Brott, D.A.; Bentley, P.; Nadella, M.V.; Thurman, D.; Fikes, J.; Cheatham, L.; McGrath, F.; Luo, W.; Kinter, L.B. Renal biomarker changes associated with hyaline droplet nephropathy in rats are time and potentially compound dependent. Toxicology 2013, 303, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Read, N.G. The role of lysosomes in hyaline droplet nephropathy induced by a variety of pharmacological agents in the male rat. Histochem. J. 1991, 23, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Coresh, J.; Selvin, E.; Stevens, L.A.; Manzi, J.; Kusek, J.W.; Eggers, P.; Van Lente, F.; Levey, A.S. Prevalence of Chronic Kidney Disease in the United States. J. Am. Med. Assoc. 2007, 298, 2038–2047. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.J.; Foley, R.N.; Herzog, C.; Chavers, B.; Gilbertson, D.; Ishani, A.; Kasiske, B.; Liu, J.; Mau, L.-W.; McBean, M.; et al. US Renal Data System 2010 Annual Data Report. Am. J. Kidney Dis. 2011, 57, A8-526. [Google Scholar] [CrossRef]
- Cai, H.; Liu, Y.; Men, H.; Zheng, Y. Protective Mechanism of Humanin Against Oxidative Stress in Aging-Related Car-diovascular Diseases. Front. Endocrinol. 2021, 12, 683151. [Google Scholar] [CrossRef]
- Rodriguez-Navarro, J.A.; Kaushik, S.; Koga, H.; Dall’Armi, C.; Shui, G.; Wenk, M.R.; Di Paolo, G.; Cuervo, A.M. Inhibitory effect of dietary lipids on chaperone-mediated autophagy. Proc. Natl. Acad. Sci. USA 2012, 109, E705–E714. [Google Scholar] [CrossRef]
- Kiffin, R.; Kaushik, S.; Zeng, M.; Bandyopadhyay, U.; Zhang, C.; Massey, A.C.; Martinez-Vicente, M.; Cuervo, A.M. Altered dynamics of the lysosomal receptor for chaperone-mediated autophagy with age. J. Cell Sci. 2007, 120 Pt 5, 782–791. [Google Scholar] [CrossRef]
- Rajawat, Y.S.; Hilioti, Z.; Bossis, I. Aging: Central role for autophagy and the lysosomal degradative system. Ageing Res. Rev. 2009, 8, 199–213. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A. Autophagy as a cell-repair mechanism: Activation of chaperone-mediated autophagy during oxidative stress. Mol. Asp. Med. 2006, 27, 444–454. [Google Scholar] [CrossRef]
- Diceglie, C.; Anelli, G.M.; Martelli, C.; Serati, A.; Lo Dico, A.; Lisso, F.; Parisi, F.; Novielli, C.; Paleari, R.; Cetin, I.; et al. Placental Antioxidant Defenses and Autopha-gy-Related Genes in Maternal Obesity and Gestational Diabetes Mellitus. Nutrients 2021, 13, 1303. [Google Scholar] [CrossRef]
- Kon, M.; Kiffin, R.; Koga, H.; Chapochnick, J.; Macian, F.; Varticovski, L.; Cuervo, A.M. Chaperone-mediated autophagy is re-quired for tumor growth. Sci. Transl. Med. 2011, 3, 109ra17. [Google Scholar] [CrossRef] [PubMed]
- Arias, E.; Cuervo, A.M. Pros and Cons of Chaperone-Mediated Autophagy in Cancer Biology. Trends Endocrinol. Metab. 2020, 31, 53–66. [Google Scholar] [CrossRef]
- Li, Y.-H.; Li, X.-F.; Liu, J.-T.; Wang, H.; Fan, L.-L.; Li, J.; Sun, G.-P. PKM2, a potential target for regulating cancer. Gene 2018, 668, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Liu, W.; Li, F.; Sun, Y.; Li, L.; Wang, X.; He, D. MP92-15 suppression of chaperone-mediated autophagy: A novel mechanism of action of silibimin against bladder and renal cancer. J. Urol. 2016, 195, e1167. [Google Scholar] [CrossRef]
- Blazer, A.D.; Clancy, R.M. ApoL1 and the Immune Response of Patients with Systemic Lupus Erythematosus. Curr. Rheumatol. Rep. 2017, 19, 13. [Google Scholar] [CrossRef]
- Freedman, B.I.; Langefeld, C.D.; Andringa, K.K.; Croker, J.A.; Williams, A.H.; Garner, N.E.; Birmingham, D.J.; Hebert, L.A.; Hicks, P.J.; Segal, M.S.; et al. End-stage renal disease in African Americans with lupus nephritis is associated with APOL1. Arthritis Rheumatol. 2014, 66, 390–396. [Google Scholar] [CrossRef]
- Qi, Y.-Y.; Zhou, X.-J.; Cheng, F.-J.; Hou, P.; Ren, Y.-L.; Wang, S.-X.; Zhao, M.-H.; Yang, L.; Martinez, J.; Zhang, H. Increased autophagy is cytoprotective against podocyte injury induced by antibody and interferon-α in lupus nephritis. Ann. Rheum. Dis. 2018, 77, 1799–1809. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Yue, Y.; Dong, C.; Shi, Y.; Xiong, S. Blockade of macrophage autophagy ameliorates activated lymphocytes-derived DNA induced murine lupus possibly via inhibition of proinflammatory cytokine production. Clin. Exp. Rheumatol. 2014, 32, 705–714. [Google Scholar] [PubMed]
- Clarke, A.J.; Ellinghaus, U.; Cortini, A.; Stranks, A.; Simon, A.K.; Botto, M.; Vyse, T.J. Autophagy is activated in systemic lupus erythematosus and required for plasmablast development. Ann. Rheum. Dis. 2015, 74, 912–920. [Google Scholar] [CrossRef] [PubMed]
- Weindel, C.G.; Richey, L.J.; Bolland, S.; Mehta, A.J.; Kearney, J.F.; Huber, B.T. B cell autophagy mediates TLR7-dependent autoimmunity and inflammation. Autophagy 2015, 11, 1010–1024. [Google Scholar] [CrossRef]
- Muller, S.; Monneaux, F.; Schall, N.; Rashkov, R.K.; Oparanov, B.A.; Wiesel, P.; Geiger, J.M.; Zimmer, R. Spliceosomal peptide P140 for im-munotherapy of systemic lupus erythematosus: Results of an early phase II clinical trial. Arthritis Rheum. 2008, 58, 3873–3883. [Google Scholar] [CrossRef]
- Zimmer, R.; Scherbarth, H.R.; Rillo, O.L.; Gomez-Reino, J.J.; Muller, S. Lupuzor/P140 peptide in patients with systemic lupus erythematosus: A randomised, double-blind, placebo-controlled phase IIb clinical trial. Ann. Rheum. Dis. 2012, 72, 1830–1835. [Google Scholar] [CrossRef]
- Monneaux, F.; Lozano, J.M.; Patarroyo, M.E.; Briand, J.P.; Muller, S. T cell recognition and therapeutic effect of a phos-phorylated synthetic peptide of the 70K snRNP protein administered in MR/lpr mice. Eur. J. Immunol. 2003, 33, 287–296. [Google Scholar] [CrossRef]
- Page, N.; Gros, F.; Schall, N.; Décossas, M.; Bagnard, D.; Briand, J.-P.; Muller, S. HSC70 blockade by the therapeutic peptide P140 affects autophagic processes and endogenous MHCII presentation in murine lupus. Ann. Rheum. Dis. 2011, 70, 837–843. [Google Scholar] [CrossRef] [PubMed]
- Wilson, P.D. Polycystic kidney disease. N. Engl. J. Med. 2004, 350, 151–164. [Google Scholar] [CrossRef]
- Shi, W.; Xu, D.; Gu, J.; Xue, C.; Yang, B.; Fu, L.; Song, S.; Liu, D.; Zhou, W.; Lv, J.; et al. Saikosaponin-d inhibits proliferation by up-regulating autophagy via the CaMKKβ-AMPK-mTOR pathway in ADPKD cells. Mol. Cell. Biochem. 2018, 449, 219–226. [Google Scholar] [CrossRef]
- Sun, L.; Hu, C.; Zhang, X. Histone Deacetylase Inhibitors Reduce Cysts by Activating Autophagy in Polycystic Kidney Disease. Kidney Dis. 2019, 5, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Sieben, C.J.; Xu, X.; Harris, P.C.; Lin, X. Autophagy activators suppress cystogenesis in an autosomal dominant polycystic kidney disease model. Hum. Mol. Genet. 2016, 26, 158–172. [Google Scholar] [CrossRef]
- Chou, L.-F.; Cheng, Y.-L.; Hsieh, C.-Y.; Lin, C.-Y.; Yang, H.-Y.; Chen, Y.-C.; Hung, C.-C.; Tian, Y.-C.; Yang, C.-W.; Chang, M.-Y. Effect of Trehalose Supplementation on Autophagy and Cystogenesis in a Mouse Model of Polycystic Kidney Disease. Nutrients 2018, 11, 42. [Google Scholar] [CrossRef]
- Hubbi, M.E.; Hu, H.; Kshitiz Ahmed, I.; Levchenko, A.; Semenza, G.L. Chaperone-mediated autophagy targets hypoxia-inducible factor-1α (HIF-1α) for lysosomal degradation. J. Biol. Chem. 2013, 288, 10703–10714. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Jin, J.; Gong, J.; Lin, B.; Li, Y.; He, Q. How many podocyte autophagosomes are there in immunoglobulin A nephropathy and idiopathic membranous nephropathy? Int. Urol. Nephrol. 2016, 48, 2109–2114. [Google Scholar] [CrossRef]
- Mao, X.; Xu, Z.; Xu, X.; Zeng, M.; Zhao, Z.; Zhang, Z.; Ding, X.; Wu, H. TGF-β1 inhibits the autophagy of podocytes by activating mTORC1 in IgA nephropathy. Exp. Cell Res. 2019, 385, 111670. [Google Scholar] [CrossRef]
- Xia, M.; Liu, D.; Tang, X.; Liu, Y.; Liu, H.; Liu, Y.; Chen, G.; Liu, H. Dihydroartemisinin inhibits the proliferation of IgAN mesangial cells through the mTOR signaling pathway. Int. Immunopharmacol. 2020, 80, 106125. [Google Scholar] [CrossRef] [PubMed]
- Hartleben, B.; Gödel, M.; Meyer-Schwesinger, C.; Liu, S.; Ulrich, T.; Köbler, S.; Wiech, T.; Grahammer, F.; Arnold, S.J.; Lindenmeyer, M.T.; et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J. Clin. Investig. 2010, 120, 1084–1096. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Disease | Substrates | CMA Activity | Roles of CMA | Reference |
|---|---|---|---|---|
| Diabetes | PKM2, GAPDH | ↓ | The excessive PKM2, GAPDH may provide more energy for kidney hypertrophy by upregulating the glycolysis activity | [20] |
| Pax2 | ↓ | The decreased degradation of Pax2 results in kidney hypertrophy | ||
| Cystinosis | N/A | ↓ | LAMP2A is mislocalized and CMA is impaired | [56] |
| Mucolipidosis Type IV | GAPDH, RNase A | ↓ | The mutation in the TRPML1 leads to MLIV and decreases CMA activity | [57] |
| Hyaline droplet nephropathy | α2-microglobulin | ↑ | Elevated proportion of direct uptake α2-microglobulin into lysosomes | [58,59] |
| Acute kidney injury | GPX4 | ↑ | Ferroptosis is enchanced and increased-CMA promotes the degradation of GPX4 | [60,61] |
| Aging | RNase A, GAPDH | ↓ | The decreased CMA leads to the accumulation of oxidized substrates | [16,17,18] |
| Cancer | PKM2 | ↑ | CMA promotes the proliferation and invasion of renal carcinoma cells through PKM2 | [62] |
| Starvation | nonessential proteins (e.g., glycolytic enzymes) | ↑ | CMA provides amino acids for the synthesis of essential proteins | [51] |
| PLIN2, PLIN3 | ↑ | Lipolysis of LD generates free fatty acids to provide energy and CMA promotes degradation of LD-associated proteins. | [53] | |
| Acidosis | Pax2, GAPDH | ↓ | The decreased degradation of Pax2 results in kidney hypertrophy. | [42] |
| High-fat diet/Obesity | Pax2 | ↓ | High-fat diet leads to decrease of Pax2 and AMPK and an increase of renal sEH, mediating the renal injury | [63] |
| Lupus nephritis | GAPDH | ↑ | Interfere with the endogenous autoantigen processing and loading to MHCII and lead to lower activation of autoreactive T cells. | [64] |
| Polycystic kidney disease | N/A | ↓? | Impaired CMA may be associated with increased apoptosis and cyst growth. | [65,66] |
| IgAN/ Glomerulonephritis | N/A | ? | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, Z.; Wang, S.; Tan, X.; Wang, D. New Insights into the Mechanisms of Chaperon-Mediated Autophagy and Implications for Kidney Diseases. Cells 2022, 11, 406. https://doi.org/10.3390/cells11030406
Yuan Z, Wang S, Tan X, Wang D. New Insights into the Mechanisms of Chaperon-Mediated Autophagy and Implications for Kidney Diseases. Cells. 2022; 11(3):406. https://doi.org/10.3390/cells11030406
Chicago/Turabian StyleYuan, Zhen, Shuyuan Wang, Xiaoyue Tan, and Dekun Wang. 2022. "New Insights into the Mechanisms of Chaperon-Mediated Autophagy and Implications for Kidney Diseases" Cells 11, no. 3: 406. https://doi.org/10.3390/cells11030406
APA StyleYuan, Z., Wang, S., Tan, X., & Wang, D. (2022). New Insights into the Mechanisms of Chaperon-Mediated Autophagy and Implications for Kidney Diseases. Cells, 11(3), 406. https://doi.org/10.3390/cells11030406