
KRAS as a Modulator of the Inflammatory Tumor Microenvironment: Therapeutic Implications

 , , , ,
, , , ,  and
and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
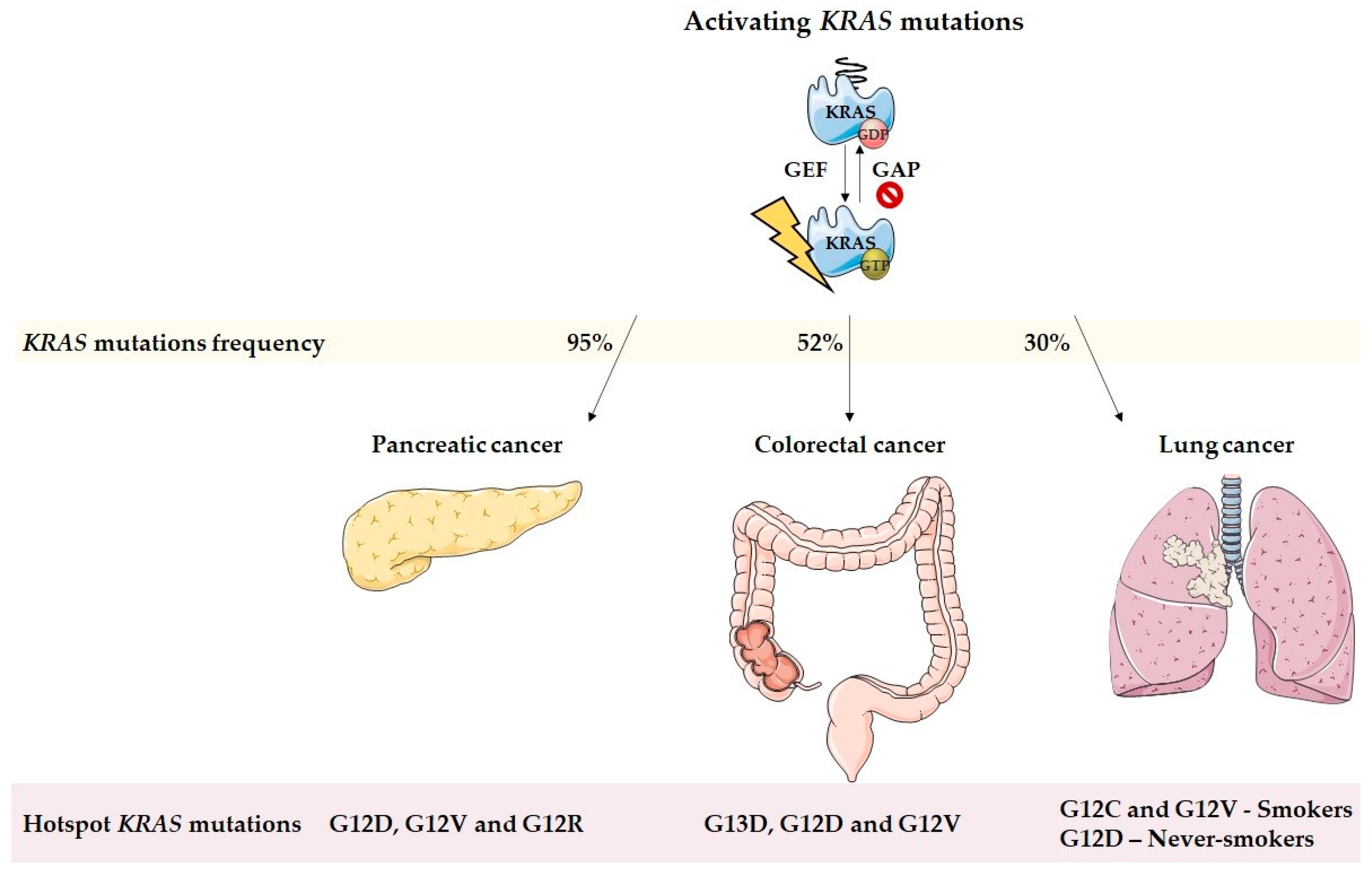
1. Introduction
2. KRAS and the Inflammatory Tumor Microenvironment Modulation
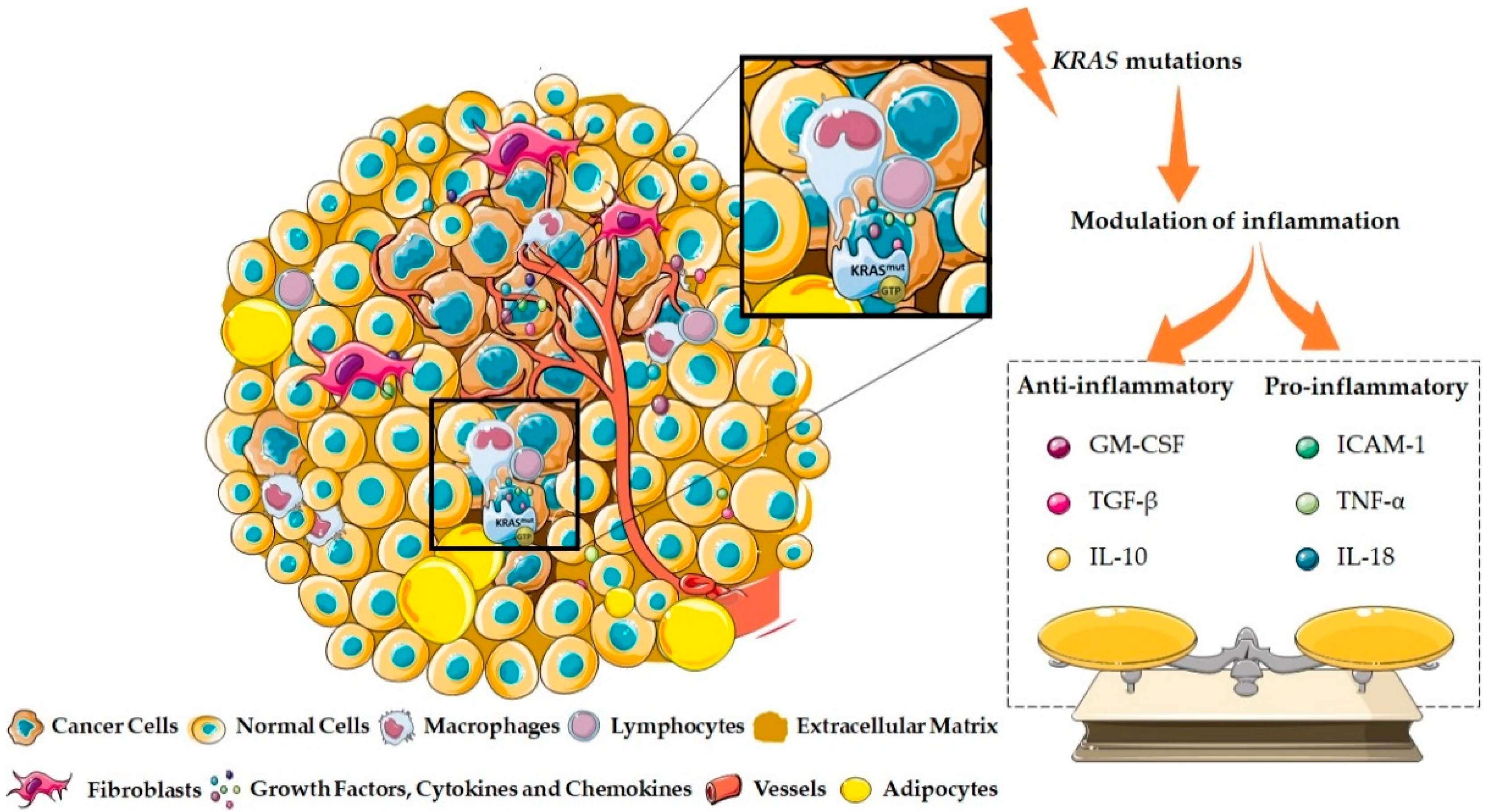
2.1. KRAS as a Pro-Inflammatory Modulator of the Tumor Microenvironment
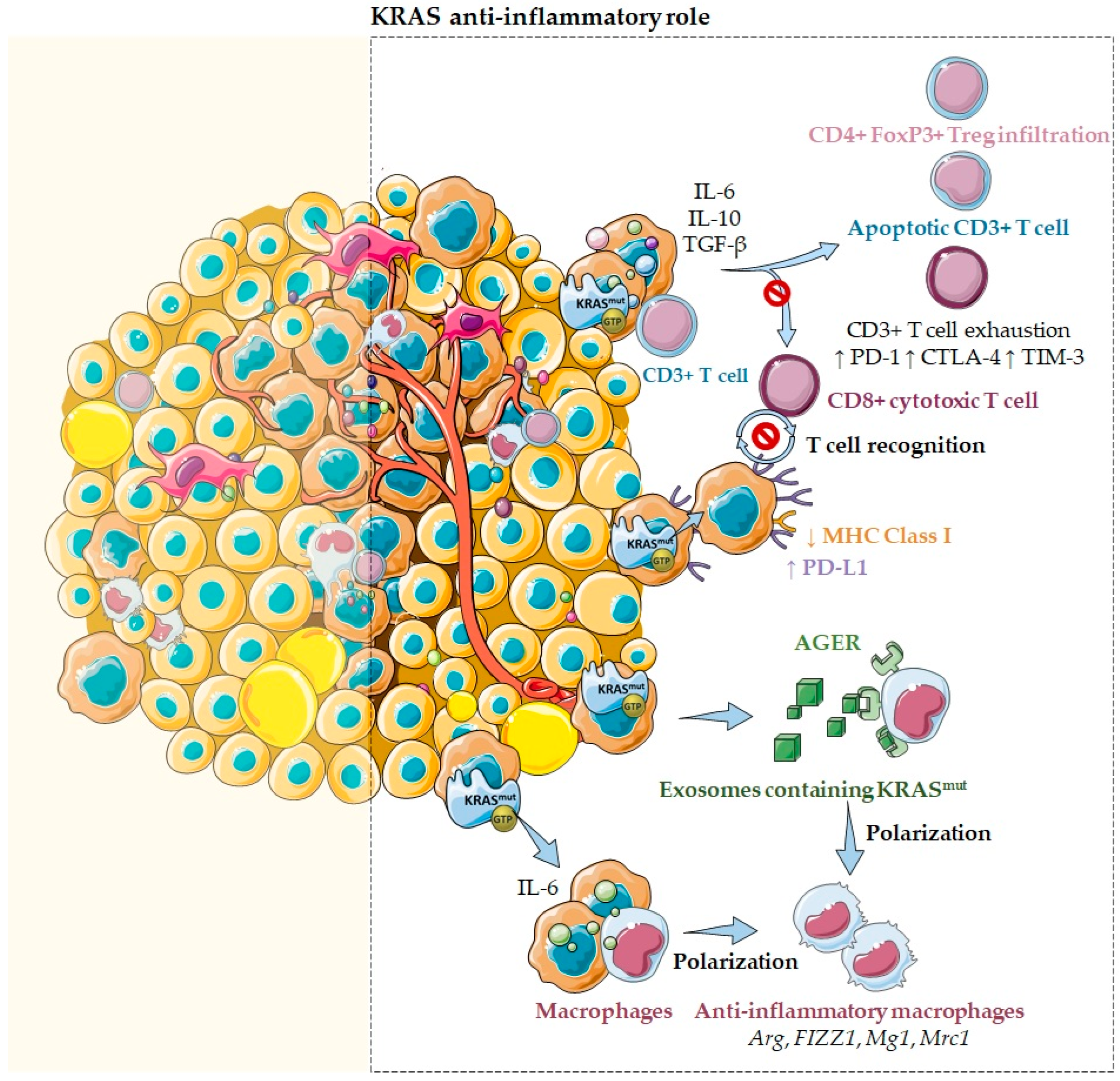
2.2. KRAS as an Anti-Inflammatory Modulator of the Tumor Microenvironment
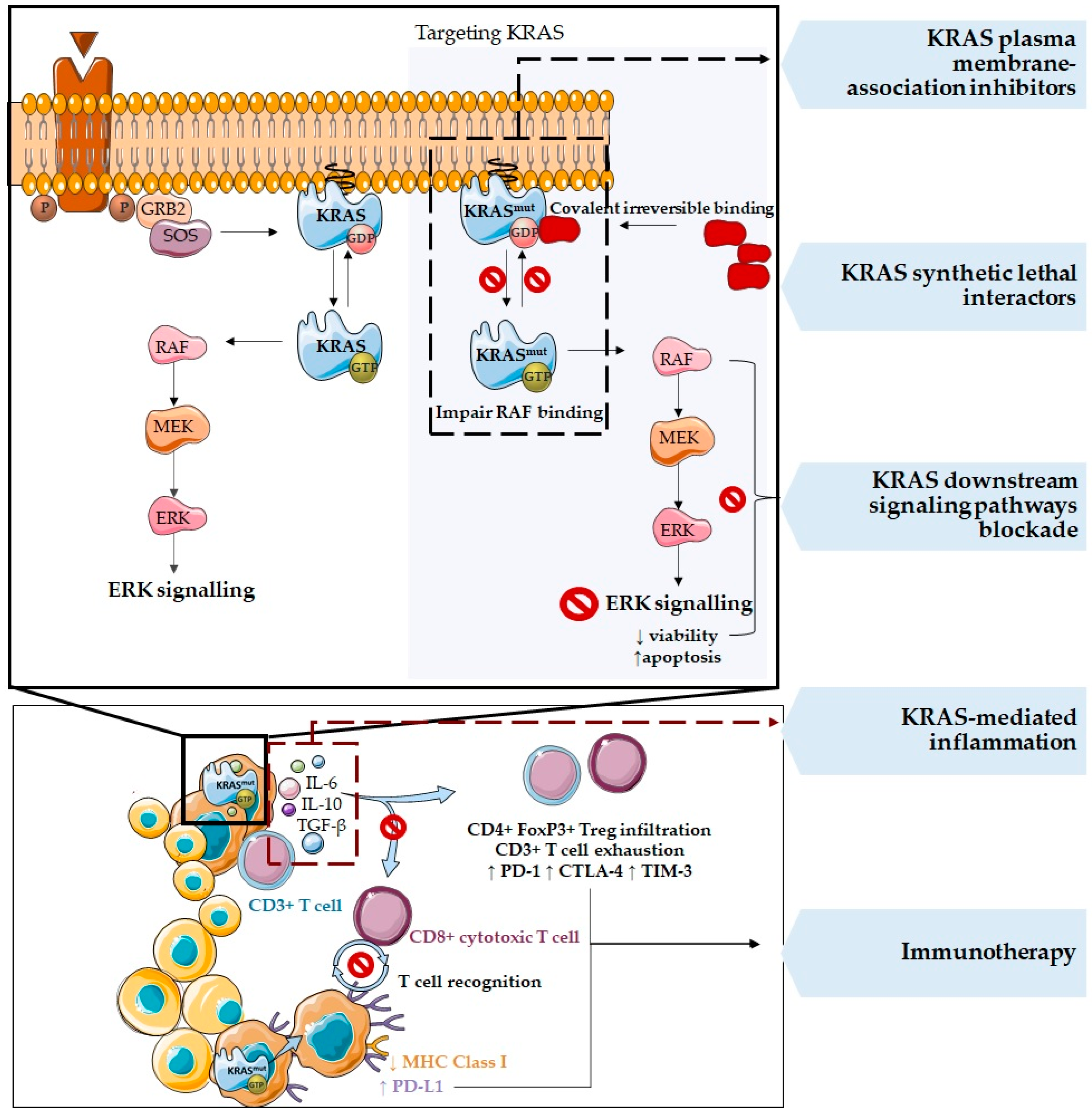
3. Therapies Targeting KRAS Mutations and the Tumor Microenvironment
4. Immunotherapy and Combined Therapeutic Approaches in KRAS Mutated Cancers
5. Final Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hamarsheh, S.; Groß, O.; Brummer, T.; Zeiser, R. Immune modulatory effects of oncogenic KRAS in cancer. Nat. Commun. 2020, 11, 5439. [Google Scholar] [CrossRef] [PubMed]
- Cazzanelli, G.; Pereira, F.; Alves, S.; Francisco, R.; Azevedo, L.; Dias Carvalho, P.; Almeida, A.; Côrte-Real, M.; Oliveira, M.; Lucas, C.; et al. The Yeast Saccharomyces cerevisiae as a Model for Understanding RAS Proteins and their Role in Human Tumorigenesis. Cells 2018, 7, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baines, A.T.; Xu, D.; Der, C.J. Inhibition of Ras for cancer treatment: The search continues. Future Med. Chem. 2011, 3, 1787–1808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Wang, Y.; Li, X. Targeting the untargetable KRAS in cancer therapy. Acta Pharm. Sin. B 2019, 9, 871–879. [Google Scholar] [CrossRef]
- Ancrile, B.; Lim, K.-H.; Counter, C.M. Oncogenic Ras-induced secretion of IL6 is required for tumorigenesis. Genes Dev. 2007, 21, 1714–1719. [Google Scholar] [CrossRef] [Green Version]
- Kratz, C.P.; Schubbert, S.; Bollag, G.; Niemeyer, C.M.; Shannon, K.M.; Zenker, M. Germline Mutations in Components of the Ras Signaling Pathway in Noonan Syndrome and Related Disorders. Cell Cycle 2006, 5, 1607–1611. [Google Scholar] [CrossRef] [Green Version]
- Schubbert, S.; Shannon, K.; Bollag, G. Hyperactive Ras in developmental disorders and cancer. Nat. Rev. Cancer 2007, 7, 295–308. [Google Scholar] [CrossRef]
- Ihle, N. Differential Activity of the KRAS Oncogene by Method of Activation: Implications for Signaling and Therapeutic Intervention. Ph.D. Thesis, The University of Texas, Houston, TX, USA, December 2012. [Google Scholar]
- Salgia, R.; Pharaon, R.; Mambetsariev, I.; Nam, A.; Sattler, M. The improbable targeted therapy: KRAS as an emerging target in non-small cell lung cancer (NSCLC). Cell Rep. Med. 2021, 2, 100186. [Google Scholar] [CrossRef]
- Ghimessy, A.; Radeczky, P.; Laszlo, V.; Hegedus, B.; Renyi-Vamos, F.; Fillinger, J.; Klepetko, W.; Lang, C.; Dome, B.; Megyesfalvi, Z. Current therapy of KRAS-mutant lung cancer. Cancer Metastasis Rev. 2020, 39, 1159–1177. [Google Scholar] [CrossRef]
- Levy, R.; Grafi-Cohen, M.; Kraiem, Z.; Kloog, Y. Galectin-3 Promotes Chronic Activation of K-Ras and Differentiation Block in Malignant Thyroid Carcinomas. Mol. Cancer Ther. 2010, 9, 2208–2219. [Google Scholar] [CrossRef] [Green Version]
- Smith, G.; Bounds, R.; Wolf, H.; Steele, R.J.C.; Carey, F.A.; Wolf, C.R. Activating K-Ras mutations outwith ‘hotspot’ codons in sporadic colorectal tumours–implications for personalised cancer medicine. Br. J. Cancer 2010, 102, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Knickelbein, K.; Zhang, L. Mutant KRAS as a critical determinant of the therapeutic response of colorectal cancer. Genes Dis. 2015, 2, 4–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alves, S.; Castro, L.; Fernandes, M.S.; Francisco, R.; Castro, P.; Priault, M.; Chaves, S.R.; Moyer, M.P.; Oliveira, C.; Seruca, R.; et al. Colorectal cancer-related mutant KRAS alleles function as positive regulators of autophagy. Oncotarget 2015, 6, 30787–30802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Pylayeva-Gupta, Y.; Lee, K.E.; Hajdu, C.H.; Miller, G.; Bar-Sagi, D. Oncogenic Kras-Induced GM-CSF Production Promotes the Development of Pancreatic Neoplasia. Cancer Cell 2012, 21, 836–847. [Google Scholar] [CrossRef] [Green Version]
- Grant, T.J.; Hua, K.; Singh, A. Molecular Pathogenesis of Pancreatic Cancer. Prog. Mol. Biol. Transl. Sci. 2016, 144, 241–275. [Google Scholar]
- Storz, P. The crosstalk between acinar cells with Kras mutations and M1-polarized macrophages leads to initiation of pancreatic precancerous lesions. Oncoimmunology 2015, 4, e1008794. [Google Scholar] [CrossRef] [Green Version]
- Buscail, L.; Bournet, B.; Cordelier, P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 153–168. [Google Scholar] [CrossRef]
- Luo, J. KRAS mutation in pancreatic cancer. Semin. Oncol. 2021, 48, 10–18. [Google Scholar] [CrossRef]
- Hafezi, S.; Saber-Ayad, M.; Abdel-Rahman, W.M. Highlights on the Role of KRAS Mutations in Reshaping the Microenvironment of Pancreatic Adenocarcinoma. Int. J. Mol. Sci. 2021, 22, 10219. [Google Scholar] [CrossRef]
- Lu, S.; Jang, H.; Nussinov, R.; Zhang, J. The Structural Basis of Oncogenic Mutations G12, G13 and Q61 in Small GTPase K-Ras4B. Sci. Rep. 2016, 6, 21949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polidoro, M.A.; Milana, F.; Soldani, C.; Franceschini, B.; Anselmo, A.; Colombo, F.S.; Di Tommaso, L.; Cimino, M.; Carnevale, S.; Lleo, A.; et al. Impact of RAS mutations on the immune infiltrate of colorectal liver metastases: A preliminary study. J. Leukoc. Biol. 2020, 108, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Wang, X.; Duanmu, J.; Li, T.; Jiang, Q. KRAS mutations are negatively correlated with immunity in colon cancer. Aging (Albany. NY) 2021, 13, 750–768. [Google Scholar] [CrossRef] [PubMed]
- Arrington, A.K.; Heinrich, E.L.; Lee, W.; Duldulao, M.; Patel, S.; Sanchez, J.; Garcia-Aguilar, J.; Kim, J. Prognostic and Predictive Roles of KRAS Mutation in Colorectal Cancer. Int. J. Mol. Sci. 2012, 13, 12153–12168. [Google Scholar] [CrossRef] [Green Version]
- Lanfredini, S.; Thapa, A.; O’Neill, E. RAS in pancreatic cancer. Biochem. Soc. Trans. 2019, 47, 961–972. [Google Scholar] [CrossRef]
- Nagasaka, T.; Sasamoto, H.; Notohara, K.; Cullings, H.M.; Takeda, M.; Kimura, K.; Kambara, T.; MacPhee, D.G.; Young, J.; Leggett, B.A.; et al. Colorectal cancer with mutation in BRAF, KRAS, and wild-type with respect to both oncogenes showing different patterns of DNA methylation. J. Clin. Oncol. 2004, 22, 4584–4594. [Google Scholar] [CrossRef]
- Tan, C.; Du, X. KRAS mutation testing in metastatic colorectal cancer. World J. Gastroenterol. 2012, 18, 5171–5180. [Google Scholar] [CrossRef]
- Liu, X.; Jakubowski, M.; Hunt, J.L. KRAS Gene Mutation in Colorectal Cancer Is Correlated With Increased Proliferation and Spontaneous Apoptosis. Am. J. Clin. Pathol. 2011, 135, 245–252. [Google Scholar] [CrossRef]
- Krasinskas, A.M. EGFR Signaling in Colorectal Carcinoma. Patholog. Res. Int. 2011, 2011, 932932. [Google Scholar] [CrossRef]
- Reinacher-Schick, A.; Schulmann, K.; Modest, D.P.; Bruns, N.; Graeven, U.; Jaworska, M.; Greil, R.; Porschen, R.; Arnold, D.; Schmiegel, W.; et al. Effect of KRAS codon13 mutations in patients with advanced colorectal cancer (advanced CRC) under oxaliplatin containing chemotherapy. Results from a translational study of the AIO colorectal study group. BMC Cancer 2012, 12, 349. [Google Scholar] [CrossRef] [PubMed]
- Cullis, J.; Das, S.; Bar-Sagi, D. Kras and Tumor Immunity: Friend or Foe? Cold Spring Harb. Perspect. Med. 2018, 8, a031849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caetano, M.S.; Zhang, H.; Cumpian, A.M.; Gong, L.; Unver, N.; Ostrin, E.J.; Daliri, S.; Chang, S.H.; Ochoa, C.E.; Hanash, S.; et al. IL6 Blockade Reprograms the Lung Tumor Microenvironment to Limit the Development and Progression of K-ras–Mutant Lung Cancer. Cancer Res. 2016, 76, 3189–3199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granville, C.A.; Memmott, R.M.; Balogh, A.; Mariotti, J.; Kawabata, S.; Han, W.; LoPiccolo, J.; Foley, J.; Liewehr, D.J.; Steinberg, S.M.; et al. A Central Role for Foxp3+ Regulatory T Cells in K-Ras-Driven Lung Tumorigenesis. PLoS ONE 2009, 4, e5061. [Google Scholar] [CrossRef] [PubMed]
- Busch, S.E.; Hanke, M.L.; Kargl, J.; Metz, H.E.; MacPherson, D.; Houghton, A.M. Lung Cancer Subtypes Generate Unique Immune Responses. J. Immunol. 2016, 197, 4493–4503. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Zheng, S.; Jin, R.; Wang, X.; Wang, F.; Zang, R.; Xu, H.; Lu, Z.; Huang, J.; Lei, Y.; et al. The superior efficacy of anti-PD-1/PD-L1 immunotherapy in KRAS-mutant non-small cell lung cancer that correlates with an inflammatory phenotype and increased immunogenicity. Cancer Lett. 2020, 470, 95–105. [Google Scholar] [CrossRef]
- Qu, Z.; Sun, F.; Zhou, J.; Li, L.; Shapiro, S.D.; Xiao, G. Interleukin-6 Prevents the Initiation but Enhances the Progression of Lung Cancer. Cancer Res. 2015, 75, 3209–3215. [Google Scholar] [CrossRef] [Green Version]
- Uras, I.Z.; Moll, H.P.; Casanova, E. Targeting KRAS Mutant Non-Small-Cell Lung Cancer: Past, Present and Future. Int. J. Mol. Sci. 2020, 21, 4325. [Google Scholar] [CrossRef]
- Tan, Z.; Xue, H.; Sun, Y.; Zhang, C.; Song, Y.; Qi, Y. The Role of Tumor Inflammatory Microenvironment in Lung Cancer. Front. Pharmacol. 2021, 12, 1168. [Google Scholar] [CrossRef]
- Tsai, M.-J.; Chang, W.-A.; Huang, M.-S.; Kuo, P.-L. Tumor Microenvironment: A New Treatment Target for Cancer. ISRN Biochem. 2014, 2014, 1–8. [Google Scholar] [CrossRef]
- Cheng, H.; Fan, K.; Luo, G.; Fan, Z.; Yang, C.; Huang, Q.; Jin, K.; Xu, J.; Yu, X.; Liu, C. KrasG12D mutation contributes to regulatory T cell conversion through activation of the MEK/ERK pathway in pancreatic cancer. Cancer Lett. 2019, 446, 103–111. [Google Scholar] [CrossRef]
- Pinto, A.T.; Pinto, M.L.; Velho, S.; Pinto, M.T.; Cardoso, A.P.; Figueira, R.; Monteiro, A.; Marques, M.; Seruca, R.; Barbosa, M.A.; et al. Intricate Macrophage-Colorectal Cancer Cell Communication in Response to Radiation. PLoS ONE 2016, 11, e0160891. [Google Scholar] [CrossRef] [Green Version]
- Lyssiotis, C.A.; Kimmelman, A.C. Metabolic Interactions in the Tumor Microenvironment. Trends Cell Biol. 2017, 27, 863–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Q.; Wang, L.; Hei, X. Parameter identification of chaotic systems using artificial raindrop algorithm. J. Comput. Sci. 2015, 8, 20–31. [Google Scholar] [CrossRef]
- Liu, G.; Qin, Q.; Chan, K.W.Y.; Li, Y.; Bulte, J.W.M.; McMahon, M.T.; van Zijl, P.C.M.; Gilad, A.A. Non-invasive temperature mapping using temperature-responsive water saturation shift referencing (T-WASSR) MRI. NMR Biomed. 2014, 27, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Smakman, N.; Veenendaal, L.M.; van Diest, P.; Bos, R.; Offringa, R.; Borel Rinkes, I.H.M.; Kranenburg, O. Dual effect of KrasD12 knockdown on tumorigenesis: Increased immune-mediated tumor clearance and abrogation of tumor malignancy. Oncogene 2005, 24, 8338–8342. [Google Scholar] [CrossRef] [Green Version]
- Fang, C.; Zhang, C.; Zhao, W.-Q.; Hu, W.-W.; Wu, J.; Ji, M. Co-mutations of TP53 and KRAS serve as potential biomarkers for immune checkpoint blockade in squamous-cell non-small cell lung cancer: A case report. BMC Med. Genom. 2019, 12, 136. [Google Scholar] [CrossRef]
- Petanidis, S.; Anestakis, D.; Argyraki, M.; Hadzopoulou-Cladaras, M.; Salifoglou, A. Differential Expression of IL-17, 22 and 23 in the Progression of Colorectal Cancer in Patients with K-ras Mutation: Ras Signal Inhibition and Crosstalk with GM-CSF and IFN-γ. PLoS ONE 2013, 8, e73616. [Google Scholar] [CrossRef]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS oncogenes: Weaving a tumorigenic web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef] [Green Version]
- Dong, Z.-Y.; Zhong, W.-Z.; Zhang, X.-C.; Su, J.; Xie, Z.; Liu, S.-Y.; Tu, H.-Y.; Chen, H.-J.; Sun, Y.-L.; Zhou, Q.; et al. Potential Predictive Value of TP53 and KRAS Mutation Status for Response to PD-1 Blockade Immunotherapy in Lung Adenocarcinoma. Clin. Cancer Res. 2017, 23, 3012–3024. [Google Scholar] [CrossRef] [Green Version]
- Murakami, S.; Shahbazian, D.; Surana, R.; Zhang, W.; Chen, H.; Graham, G.T.; White, S.M.; Weiner, L.M.; Yi, C. Yes-associated protein mediates immune reprogramming in pancreatic ductal adenocarcinoma. Oncogene 2017, 36, 1232–1244. [Google Scholar] [CrossRef] [Green Version]
- Okayama, H.; Saito, M.; Oue, N.; Weiss, J.M.; Stauffer, J.; Takenoshita, S.; Wiltrout, R.H.; Hussain, S.P.; Harris, C.C. NOS2 enhances KRAS-induced lung carcinogenesis, inflammation and microRNA-21 expression. Int. J. Cancer 2013, 132, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Fujiyoshi, K.; Yamamoto, G.; Takahashi, A.; Arai, Y.; Yamada, M.; Kakuta, M.; Yamaguchi, K.; Akagi, Y.; Nishimura, Y.; Sakamoto, H.; et al. High concordance rate of KRAS/BRAF mutations and MSI-H between primary colorectal cancer and corresponding metastases. Oncol. Rep. 2017, 37, 785–792. [Google Scholar] [CrossRef] [Green Version]
- Jo, P.; König, A.; Schirmer, M.; Kitz, J.; Conradi, L.-C.; Azizian, A.; Bernhardt, M.; Wolff, H.A.; Grade, M.; Ghadimi, M.; et al. Heterogeneity of KRAS Mutation Status in Rectal Cancer. PLoS ONE 2016, 11, e0153278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haigis, K.M. KRAS Alleles: The Devil Is in the Detail. Trends Cancer 2017, 3, 686–697. [Google Scholar] [CrossRef]
- Dias Carvalho, P.; Guimarães, C.F.; Cardoso, A.P.; Mendonça, S.; Costa, Â.M.; Oliveira, M.J.; Velho, S. KRAS Oncogenic Signaling Extends beyond Cancer Cells to Orchestrate the Microenvironment. Cancer Res. 2018, 78, 7–14. [Google Scholar] [CrossRef] [Green Version]
- Bellio, H.; Fumet, J.D.; Ghiringhelli, F. Targeting BRAF and RAS in Colorectal Cancer. Cancers 2021, 13, 2201. [Google Scholar] [CrossRef]
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435. [Google Scholar] [CrossRef]
- Chang, S.H.; Mirabolfathinejad, S.G.; Katta, H.; Cumpian, A.M.; Gong, L.; Caetano, M.S.; Moghaddam, S.J.; Dong, C. T helper 17 cells play a critical pathogenic role in lung cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 5664–5669. [Google Scholar] [CrossRef] [Green Version]
- Dias Carvalho, P.; Machado, A.L.; Martins, F.; Seruca, R.; Velho, S. Targeting the Tumor Microenvironment: An Unexplored Strategy for Mutant KRAS Tumors. Cancers 2019, 11, 2010. [Google Scholar] [CrossRef] [Green Version]
- Zeitouni, D.; Pylayeva-Gupta, Y.; Der, C.; Bryant, K. KRAS Mutant Pancreatic Cancer: No Lone Path to an Effective Treatment. Cancers 2016, 8, 45. [Google Scholar] [CrossRef]
- Porru, M.; Pompili, L.; Caruso, C.; Biroccio, A.; Leonetti, C. Targeting KRAS in metastatic colorectal cancer: Current strategies and emerging opportunities. J. Exp. Clin. Cancer Res. 2018, 37, 57. [Google Scholar] [CrossRef] [Green Version]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Van Maldegem, F.; Valand, K.; Cole, M.; Patel, H.; Angelova, M.; Rana, S.; Colliver, E.; Enfield, K.; Bah, N.; Kelly, G.; et al. Characterisation of tumour microenvironment remodelling following oncogene inhibition in preclinical studies with imaging mass cytometry. Nat. Commun. 2021, 12, 5906. [Google Scholar] [CrossRef]
- Xie, Y.-H.; Chen, Y.-X.; Fang, J.-Y. Comprehensive review of targeted therapy for colorectal cancer. Signal. Transduct. Target. Ther. 2020, 5, 22. [Google Scholar] [CrossRef]
- Ferrer, I.; Zugazagoitia, J.; Herbertz, S.; John, W.; Paz-Ares, L.; Schmid-Bindert, G. KRAS-Mutant non-small cell lung cancer: From biology to therapy. Lung Cancer 2018, 124, 53–64. [Google Scholar] [CrossRef] [Green Version]
- Boussios, S.; Ozturk, M.; Moschetta, M.; Karathanasi, A.; Zakynthinakis-Kyriakou, N.; Katsanos, K.; Christodoulou, D.; Pavlidis, N. The Developing Story of Predictive Biomarkers in Colorectal Cancer. J. Pers. Med. 2019, 9, 12. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Gönen, M.; Stadler, Z.K.; Weiser, M.R.; Hechtman, J.F.; Vakiani, E.; Wang, T.; Vyas, M.; Joneja, U.; Al-Bayati, M.; et al. Cellular localization of PD-L1 expression in mismatch-repair-deficient and proficient colorectal carcinomas. Mod. Pathol. 2019, 32, 110–121. [Google Scholar] [CrossRef]
- Gao, G.; Liao, W.; Ma, Q.; Zhang, B.; Chen, Y.; Wang, Y. KRAS G12D mutation predicts lower TMB and drives immune suppression in lung adenocarcinoma. Lung Cancer 2020, 149, 41–45. [Google Scholar] [CrossRef]
- Adderley, H.; Blackhall, F.H.; Lindsay, C.R. KRAS-mutant non-small cell lung cancer: Converging small molecules and immune checkpoint inhibition. EBioMedicine 2019, 41, 711–716. [Google Scholar] [CrossRef] [Green Version]
- Poon, E.; Mullins, S.; Watkins, A.; Williams, G.S.; Koopmann, J.-O.; Di Genova, G.; Cumberbatch, M.; Veldman-Jones, M.; Grosskurth, S.E.; Sah, V.; et al. The MEK inhibitor selumetinib complements CTLA-4 blockade by reprogramming the tumor immune microenvironment. J. Immunother. Cancer 2017, 5, 63. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pereira, F.; Ferreira, A.; Reis, C.A.; Sousa, M.J.; Oliveira, M.J.; Preto, A. KRAS as a Modulator of the Inflammatory Tumor Microenvironment: Therapeutic Implications. Cells 2022, 11, 398. https://doi.org/10.3390/cells11030398
Pereira F, Ferreira A, Reis CA, Sousa MJ, Oliveira MJ, Preto A. KRAS as a Modulator of the Inflammatory Tumor Microenvironment: Therapeutic Implications. Cells. 2022; 11(3):398. https://doi.org/10.3390/cells11030398
Chicago/Turabian StylePereira, Flávia, Anabela Ferreira, Celso Albuquerque Reis, Maria João Sousa, Maria José Oliveira, and Ana Preto. 2022. "KRAS as a Modulator of the Inflammatory Tumor Microenvironment: Therapeutic Implications" Cells 11, no. 3: 398. https://doi.org/10.3390/cells11030398
APA StylePereira, F., Ferreira, A., Reis, C. A., Sousa, M. J., Oliveira, M. J., & Preto, A. (2022). KRAS as a Modulator of the Inflammatory Tumor Microenvironment: Therapeutic Implications. Cells, 11(3), 398. https://doi.org/10.3390/cells11030398