
Neuron–Microglia Contact-Dependent Mechanisms Attenuate Methamphetamine-Induced Microglia Reactivity and Enhance Neuronal Plasticity

 ,
,  ,
,  , , , ,
, , , , 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Neuronal Cell Culture
2.3. Microglia and Astrocyte Purified Cultures Obtained from Mixed Glial Cell Cultures
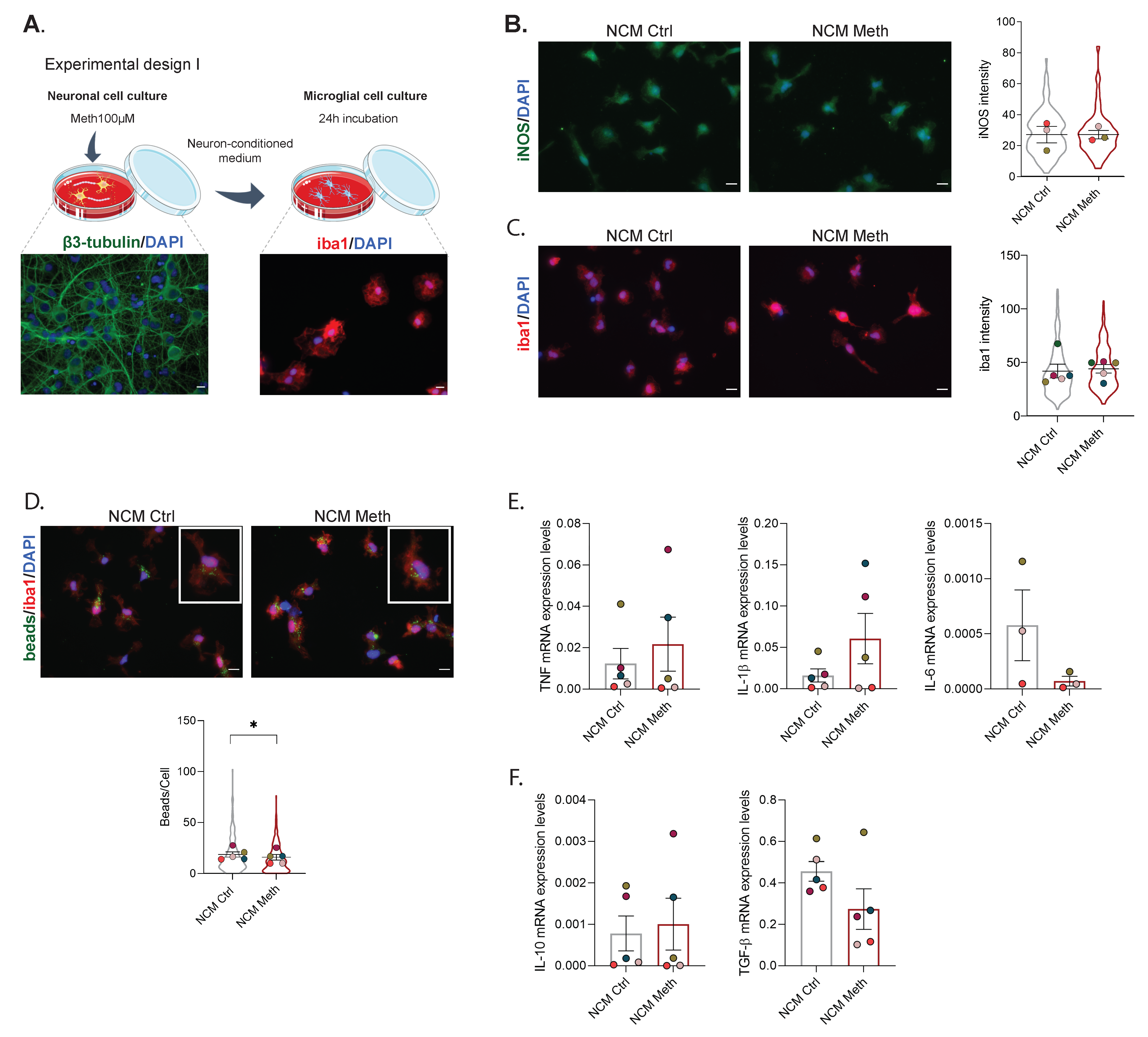
2.4. Microglia Incubation with Neuron-Conditioned Medium
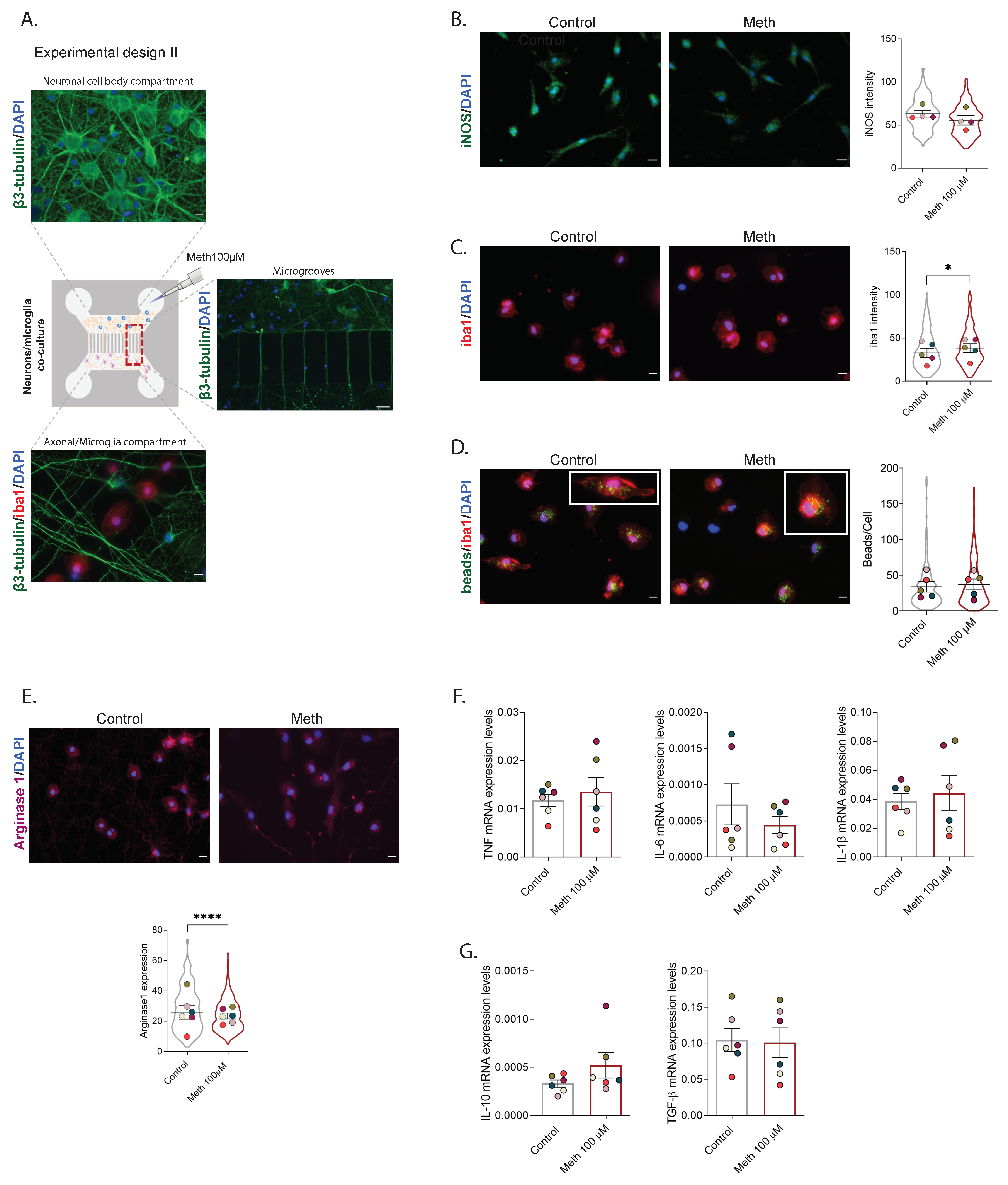
2.5. Co-Cultures of Neurons and Microglia in Microfluidic Devices
2.6. Astrocytic-Conditioned Medium
2.7. Treatment of Neuron–Microglia Co-Cultures
2.8. Immunocytochemistry and Image Acquisition
2.9. Phagocytic Assay
2.10. Fluorescence Intensity Quantification
2.11. RNA Extraction, cDNA Synthesis, and qRT-PCR
2.12. Flow Cytometry of Microglia
2.13. Quantification of Synaptic Proteins
2.14. Statistical Analysis
3. Results
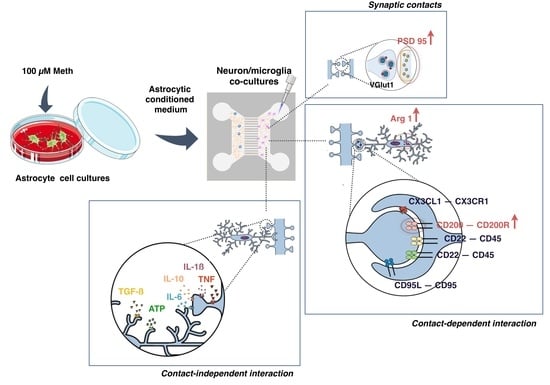
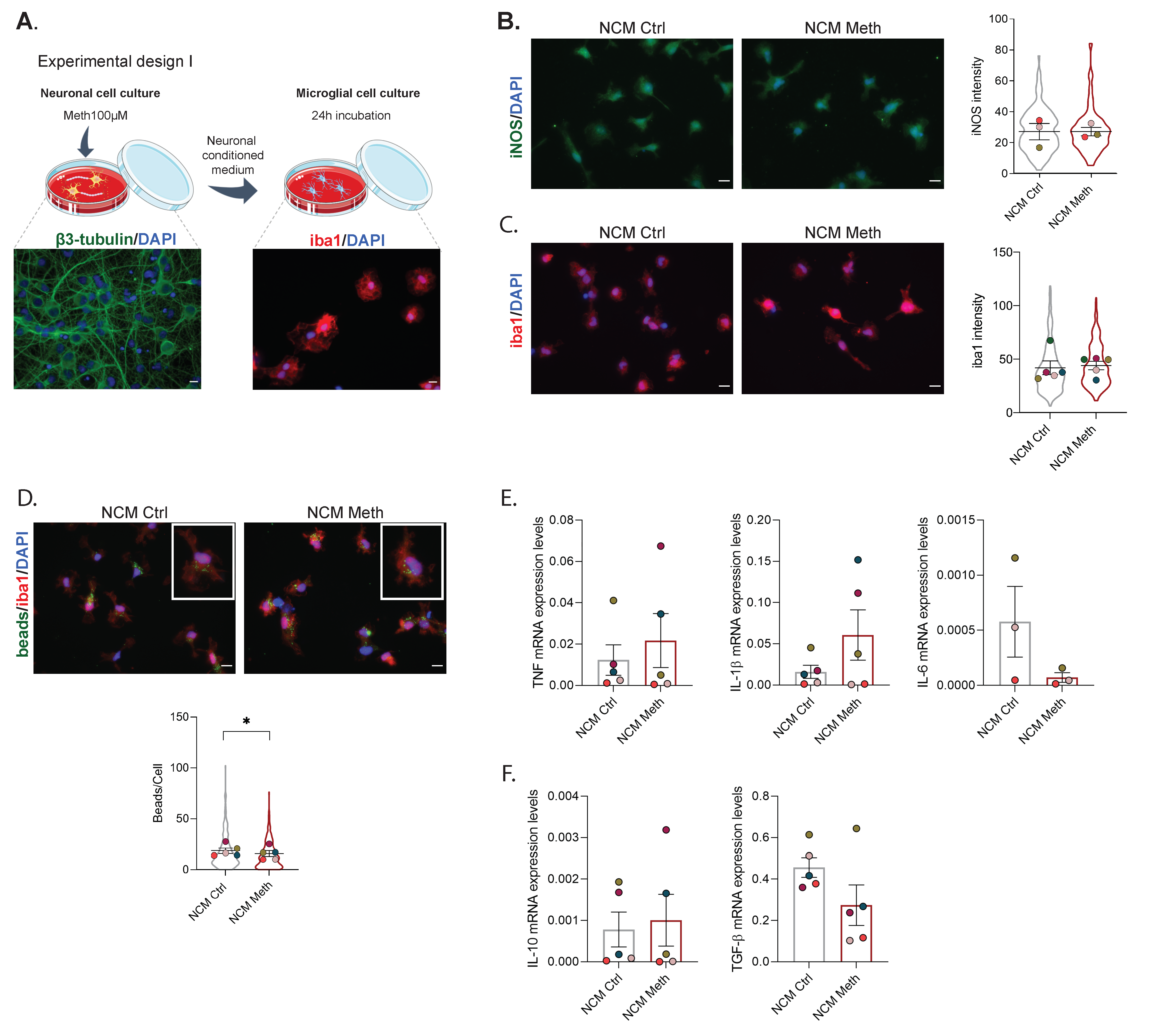
3.1. Neurons Exposed to Meth Do Not Promote Microglial Activation through Contact-Dependent or -Independent Mechanisms
3.2. Microglia Activation Induced by Meth-Exposed Astrocytes Is Partially Prevented by Neurons
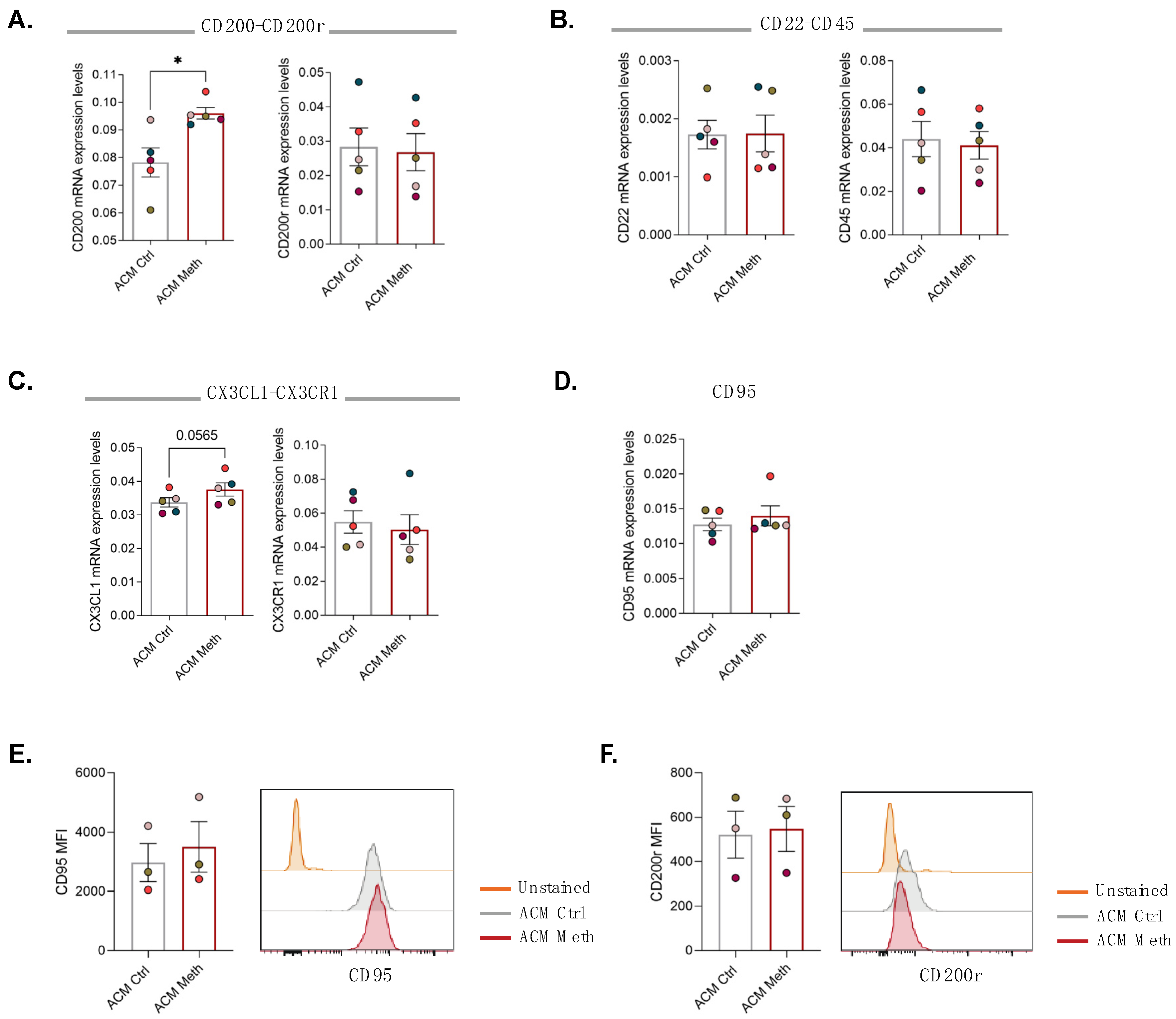
3.3. Neurons Increased Self-Protection from Meth-Induced Activation of Microglia through Contact-Dependent Mechanisms
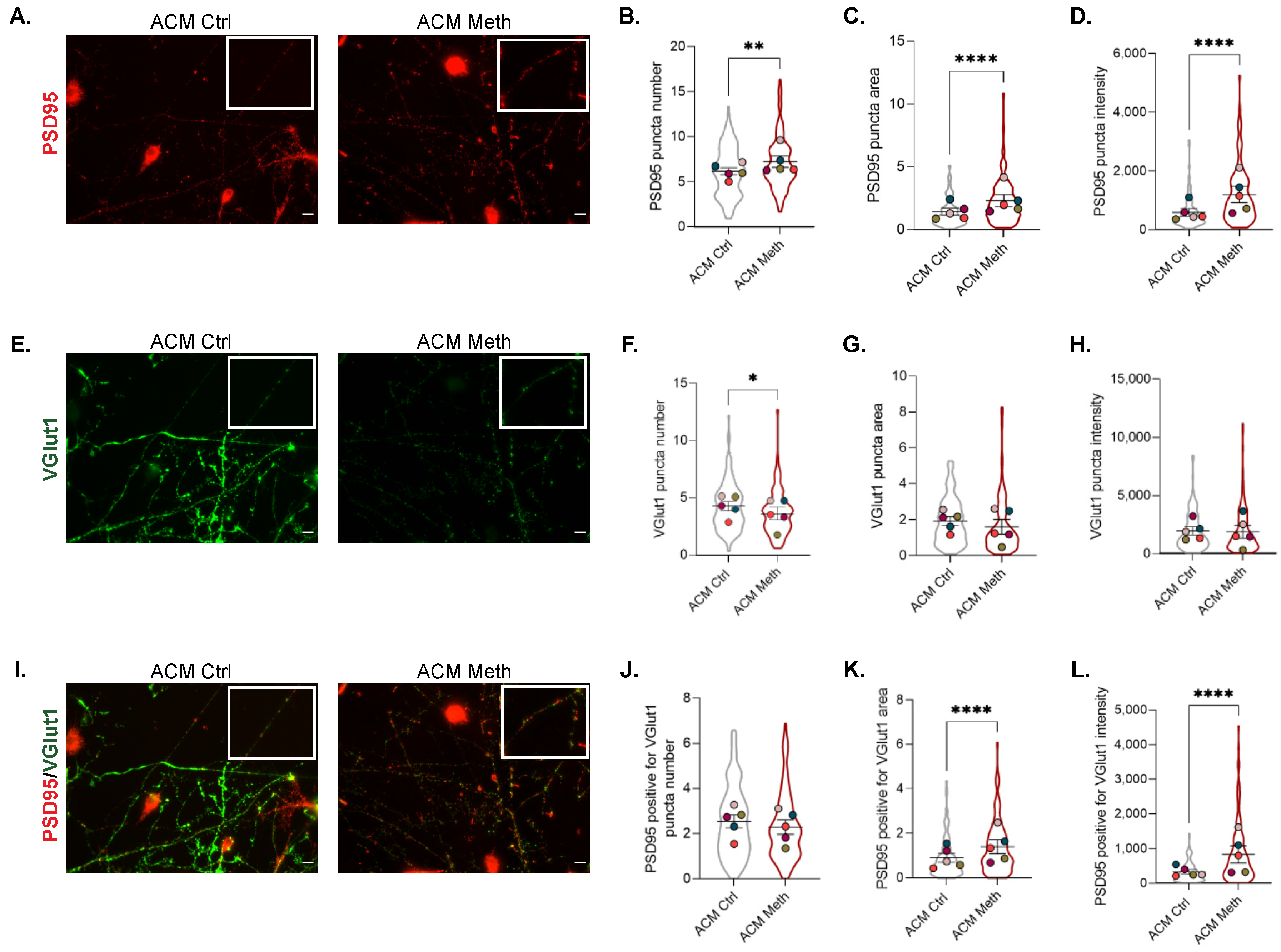
3.4. Synaptic Proteins Expression Increases in Co-Cultures of Neurons and ACM Meth-Microglia
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nestler, E.J.; Luscher, C. The Molecular Basis of Drug Addiction: Linking Epigenetic to Synaptic and Circuit Mechanisms. Neuron 2019, 102, 48–59. [Google Scholar] [CrossRef] [Green Version]
- Krasnova, I.N.; Cadet, J.L. Methamphetamine toxicity and messengers of death. Brain Res. Rev. 2009, 60, 379–407. [Google Scholar] [CrossRef] [Green Version]
- Jayanthi, S.; Daiwile, A.P.; Cadet, J.L. Neurotoxicity of methamphetamine: Main effects and mechanisms. Exp. Neurol. 2021, 344, 113795. [Google Scholar] [CrossRef] [PubMed]
- Tata, D.A.; Yamamoto, B.K. Interactions between methamphetamine and environmental stress: Role of oxidative stress, glutamate and mitochondrial dysfunction. Addiction 2007, 102 (Suppl. 1), 49–60. [Google Scholar] [CrossRef]
- Simões, P.F.; Silva, A.P.; Pereira, F.C.; Marques, E.; Grade, S.; Milhazes, N.; Borges, F.; Ribeiro, C.F.; Macedo, T.R. Methamphetamine induces alterations on hippocampal NMDA and AMPA receptor subunit levels and impairs spatial working memory. Neuroscience 2007, 150, 433–441. [Google Scholar] [CrossRef]
- Avila, J.A.; Memos, N.; Aslan, A.; Andrejewski, T.; Luine, V.N.; Serrano, P.A. Voluntary oral methamphetamine increases memory deficits and contextual sensitization during abstinence associated with decreased PKMζ and increased κOR in the hippocampus of female mice. J. Psychopharmacol. 2021, 35, 1240–1252. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.R.; Bu, M.; Gopinath, A.; Martinez, L.R.; Khoshbouei, H. Methamphetamine Dysregulation of the Central Nervous System and Peripheral Immunity. J. Pharmacol. Exp. Ther. 2021, 379, 372. [Google Scholar] [CrossRef]
- Cadet, J.L.; Bisagno, V. Glial-neuronal ensembles: Partners in drug addiction-associated synaptic plasticity. Front. Pharmacol. 2014, 5, 204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miguel-Hidalgo, J.J. The role of glial cells in drug abuse. Curr. Drug Abuse Rev. 2009, 2, 76–82. [Google Scholar] [CrossRef] [Green Version]
- Dang, J.; Tiwari, S.K.; Agrawal, K.; Hui, H.; Qin, Y.; Rana, T.M. Glial cell diversity and methamphetamine-induced neuroinflammation in human cerebral organoids. Mol. Psychiatry 2021, 26, 1194–1207. [Google Scholar] [CrossRef] [Green Version]
- Frank, M.G.; Adhikary, S.; Sobesky, J.L.; Weber, M.D.; Watkins, L.R.; Maier, S.F. The danger-associated molecular pattern HMGB1 mediates the neuroinflammatory effects of methamphetamine. Brain Behav. Immun. 2016, 51, 99–108. [Google Scholar] [CrossRef] [Green Version]
- Canedo, T.; Portugal, C.C.; Socodato, R.; Almeida, T.O.; Terceiro, A.F.; Bravo, J.; Silva, A.I.; Magalhaes, J.D.; Guerra-Gomes, S.; Oliveira, J.F.; et al. Astrocyte-derived TNF and glutamate critically modulate microglia activation by methamphetamine. Neuropsychopharmacology 2021, 46, 2358–2370. [Google Scholar] [CrossRef] [PubMed]
- Chavarria, A.; Cardenas, G. Neuronal influence behind the central nervous system regulation of the immune cells. Front. Integr. Neurosci. 2013, 7, 64. [Google Scholar] [CrossRef] [Green Version]
- Tian, L.; Rauvala, H.; Gahmberg, C.G. Neuronal regulation of immune responses in the central nervous system. Trends Immunol. 2009, 30, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Brás, J.P.; Bravo, J.; Freitas, J.; Barbosa, M.A.; Santos, S.G.; Summavielle, T.; Almeida, M.I. TNF-alpha-induced microglia activation requires miR-342: Impact on NF-kB signaling and neurotoxicity. Cell Death Dis. 2020, 11, 415. [Google Scholar] [CrossRef]
- Biber, K.; Neumann, H.; Inoue, K.; Boddeke, H.W. Neuronal ‘On’ and ‘Off’ signals control microglia. Trends Neurosci. 2007, 30, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Hernangomez, M.; Klusakova, I.; Joukal, M.; Hradilova-Svizenska, I.; Guaza, C.; Dubovy, P. CD200R1 agonist attenuates glial activation, inflammatory reactions, and hypersensitivity immediately after its intrathecal application in a rat neuropathic pain model. J. Neuroinflammation 2016, 13, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; He, X.; Tao, X.; Hou, T.; Chen, M.; He, M.; Liao, H. The CD200/CD200R signaling pathway contributes to spontaneous functional recovery by enhancing synaptic plasticity after stroke. J. Neuroinflammation 2020, 17, 171. [Google Scholar] [CrossRef]
- Denieffe, S.; Kelly, R.J.; McDonald, C.; Lyons, A.; Lynch, M.A. Classical activation of microglia in CD200-deficient mice is a consequence of blood brain barrier permeability and infiltration of peripheral cells. Brain Behav. Immun. 2013, 34, 86–97. [Google Scholar] [CrossRef] [Green Version]
- Feng, D.; Huang, A.; Yan, W.; Chen, D. CD200 dysfunction in neuron contributes to synaptic deficits and cognitive impairment. Biochem. Biophys. Res. Commun. 2019, 516, 1053–1059. [Google Scholar] [CrossRef]
- Braz, S.O.; Cruz, A.; Lobo, A.; Bravo, J.; Moreira-Ribeiro, J.; Pereira-Castro, I.; Freitas, J.; Relvas, J.B.; Summavielle, T.; Moreira, A. Expression of Rac1 alternative 3′ UTRs is a cell specific mechanism with a function in dendrite outgrowth in cortical neurons. Biochim. Biophys. Acta Gene Regul Mech. 2017, 1860, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Balasubramaniyan, V.; Peng, J.; Hurlock, E.C.; Tallquist, M.; Li, J.; Lu, Q.R. Isolation and culture of rat and mouse oligodendrocyte precursor cells. Nat. Protoc. 2007, 2, 1044–1051. [Google Scholar] [CrossRef] [PubMed]
- Socodato, R.; Portugal, C.C.; Domith, I.; Oliveira, N.A.; Coreixas, V.S.; Loiola, E.C.; Martins, T.; Santiago, A.R.; Paes-de-Carvalho, R.; Ambrosio, A.F.; et al. c-Src function is necessary and sufficient for triggering microglial cell activation. Glia 2015, 63, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Bortell, N.; Basova, L.; Semenova, S.; Fox, H.S.; Ravasi, T.; Marcondes, M.C. Astrocyte-specific overexpressed gene signatures in response to methamphetamine exposure in vitro. J. Neuroinflammation 2017, 14, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, C.D.; Gomes, C.P.; Neto, E.; Sampaio, P.; Aguiar, P.; Pego, A.P. Microfluidic-based platform to mimic the in vivo peripheral administration of neurotropic nanoparticles. Nanomedicine (Lond) 2016, 11, 3205–3221. [Google Scholar] [CrossRef] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Andrade, E.B.; Magalhaes, A.; Puga, A.; Costa, M.; Bravo, J.; Portugal, C.C.; Ribeiro, A.; Correia-Neves, M.; Faustino, A.; Firon, A.; et al. A mouse model reproducing the pathophysiology of neonatal group B streptococcal infection. Nat. Commun. 2018, 9, 3138. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.D.; Iuliano, O.; Ribeiro, L.; Veran, J.; Ferreira, J.S.; Rio, P.; Mulle, C.; Duarte, C.B.; Carvalho, A.L. Contactin-associated protein 1 (Caspr1) regulates the traffic and synaptic content of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)-type glutamate receptors. J. Biol. Chem. 2012, 287, 6868–6877. [Google Scholar] [CrossRef] [Green Version]
- Verstraelen, P.; Garcia-Diaz Barriga, G.; Verschuuren, M.; Asselbergh, B.; Nuydens, R.; Larsen, P.H.; Timmermans, J.P.; De Vos, W.H. Systematic Quantification of Synapses in Primary Neuronal Culture. iScience 2020, 23, 101542. [Google Scholar] [CrossRef]
- McLeod, F.; Marzo, A.; Podpolny, M.; Galli, S.; Salinas, P. Evaluation of Synapse Density in Hippocampal Rodent Brain Slices. J. Vis. Exp. 2017, 128, 56153. [Google Scholar] [CrossRef] [Green Version]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J. Neuroinflammation 2014, 11, 98. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Zhu, L.; Shen, Q.; Bai, X.; Di, X. Recent advances in methamphetamine neurotoxicity mechanisms and its molecular pathophysiology. Behav. Neurol. 2015, 2015, 103969. [Google Scholar] [CrossRef] [Green Version]
- Shin, E.J.; Dang, D.K.; Tran, T.V.; Tran, H.Q.; Jeong, J.H.; Nah, S.Y.; Jang, C.G.; Yamada, K.; Nabeshima, T.; Kim, H.C. Current understanding of methamphetamine-associated dopaminergic neurodegeneration and psychotoxic behaviors. Arch. Pharm. Res. 2017, 40, 403–428. [Google Scholar] [CrossRef]
- Krystofova, J.; Pathipati, P.; Russ, J.; Sheldon, A.; Ferriero, D. The Arginase Pathway in Neonatal Brain Hypoxia-Ischemia. Dev. Neurosci. 2018, 40, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.C.; Hudson, P.; Wilson, B.; Liu, B.; Abel, H.; Hemperly, J.; Hong, J.S. Immune modulatory effects of neural cell adhesion molecules on lipopolysaccharide-induced nitric oxide production by cultured glia. Brain Res. Mol. Brain Res. 2000, 81, 197–201. [Google Scholar] [CrossRef]
- Hoek, R.M.; Ruuls, S.R.; Murphy, C.A.; Wright, G.J.; Goddard, R.; Zurawski, S.M.; Blom, B.; Homola, M.E.; Streit, W.J.; Brown, M.H.; et al. Down-regulation of the macrophage lineage through interaction with OX2 (CD200). Science 2000, 290, 1768–1771. [Google Scholar] [CrossRef]
- Mott, R.T.; Ait-Ghezala, G.; Town, T.; Mori, T.; Vendrame, M.; Zeng, J.; Ehrhart, J.; Mullan, M.; Tan, J. Neuronal expression of CD22: Novel mechanism for inhibiting microglial proinflammatory cytokine production. Glia 2004, 46, 369–379. [Google Scholar] [CrossRef]
- Beier, C.P.; Wischhusen, J.; Gleichmann, M.; Gerhardt, E.; Pekanovic, A.; Krueger, A.; Taylor, V.; Suter, U.; Krammer, P.H.; Endres, M.; et al. FasL (CD95L/APO-1L) resistance of neurons mediated by phosphatidylinositol 3-kinase-Akt/protein kinase B-dependent expression of lifeguard/neuronal membrane protein 35. J. Neurosci. 2005, 25, 6765–6774. [Google Scholar] [CrossRef]
- Medana, I.; Li, Z.; Flugel, A.; Tschopp, J.; Wekerle, H.; Neumann, H. Fas ligand (CD95L) protects neurons against perforin-mediated T lymphocyte cytotoxicity. J. Immunol. 2001, 167, 674–681. [Google Scholar] [CrossRef] [Green Version]
- Shi, S.; Chen, T.; Zhao, M. The Crosstalk Between Neurons and Glia in Methamphetamine-Induced Neuroinflammation. Neurochem. Res. 2022. [Google Scholar] [CrossRef]
- Taft, C.E.; Turrigiano, G.G. PSD-95 promotes the stabilization of young synaptic contacts. Philos Trans. R Soc. Lond B Biol Sci 2014, 369, 20130134. [Google Scholar] [CrossRef] [Green Version]
- El-Husseini, A.E.; Schnell, E.; Chetkovich, D.M.; Nicoll, R.A.; Bredt, D.S. PSD-95 involvement in maturation of excitatory synapses. Science 2000, 290, 1364–1368. [Google Scholar] [CrossRef] [PubMed]
- Berry, K.P.; Nedivi, E. Spine Dynamics: Are They All the Same? Neuron 2017, 96, 43–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mark, K.A.; Quinton, M.S.; Russek, S.J.; Yamamoto, B.K. Dynamic changes in vesicular glutamate transporter 1 function and expression related to methamphetamine-induced glutamate release. J. Neurosci. 2007, 27, 6823–6831. [Google Scholar] [CrossRef] [PubMed]
- Dietz, D.M.; Dietz, K.C.; Nestler, E.J.; Russo, S.J. Molecular mechanisms of psychostimulant-induced structural plasticity. Pharmacopsychiatry 2009, 42 (Suppl. 1), S69–S78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutlu, M.G.; Gould, T.J. Effects of drugs of abuse on hippocampal plasticity and hippocampus-dependent learning and memory: Contributions to development and maintenance of addiction. Learn. Mem. 2016, 23, 515–533. [Google Scholar] [CrossRef] [Green Version]
- Nakahata, Y.; Yasuda, R. Plasticity of Spine Structure: Local Signaling, Translation and Cytoskeletal Reorganization. Front. Synaptic Neurosci. 2018, 10, 29. [Google Scholar] [CrossRef] [Green Version]
- Murakoshi, H.; Wang, H.; Yasuda, R. Local, persistent activation of Rho GTPases during plasticity of single dendritic spines. Nature 2011, 472, 100–104. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bravo, J.; Ribeiro, I.; Terceiro, A.F.; Andrade, E.B.; Portugal, C.C.; Lopes, I.M.; Azevedo, M.M.; Sousa, M.; Lopes, C.D.F.; Lobo, A.C.; et al. Neuron–Microglia Contact-Dependent Mechanisms Attenuate Methamphetamine-Induced Microglia Reactivity and Enhance Neuronal Plasticity. Cells 2022, 11, 355. https://doi.org/10.3390/cells11030355
Bravo J, Ribeiro I, Terceiro AF, Andrade EB, Portugal CC, Lopes IM, Azevedo MM, Sousa M, Lopes CDF, Lobo AC, et al. Neuron–Microglia Contact-Dependent Mechanisms Attenuate Methamphetamine-Induced Microglia Reactivity and Enhance Neuronal Plasticity. Cells. 2022; 11(3):355. https://doi.org/10.3390/cells11030355
Chicago/Turabian StyleBravo, Joana, Inês Ribeiro, Ana Filipa Terceiro, Elva B. Andrade, Camila Cabral Portugal, Igor M. Lopes, Maria M. Azevedo, Mafalda Sousa, Cátia D. F. Lopes, Andrea C. Lobo, and et al. 2022. "Neuron–Microglia Contact-Dependent Mechanisms Attenuate Methamphetamine-Induced Microglia Reactivity and Enhance Neuronal Plasticity" Cells 11, no. 3: 355. https://doi.org/10.3390/cells11030355
APA StyleBravo, J., Ribeiro, I., Terceiro, A. F., Andrade, E. B., Portugal, C. C., Lopes, I. M., Azevedo, M. M., Sousa, M., Lopes, C. D. F., Lobo, A. C., Canedo, T., Relvas, J. B., & Summavielle, T. (2022). Neuron–Microglia Contact-Dependent Mechanisms Attenuate Methamphetamine-Induced Microglia Reactivity and Enhance Neuronal Plasticity. Cells, 11(3), 355. https://doi.org/10.3390/cells11030355