
PCSK9 in Liver Cancers at the Crossroads between Lipid Metabolism and Immunity



Abstract
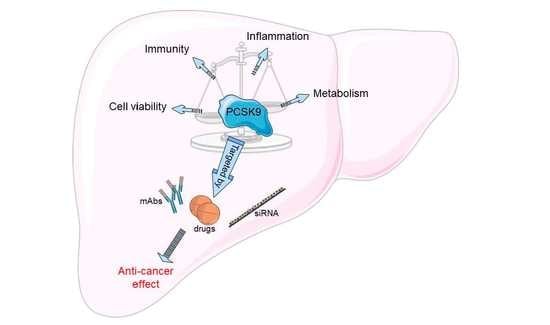
1. Introduction to Liver Cancers
1.1. The Liver, a Multifunctional Organ
1.2. Adult Liver Cancer: Hepatocellular Carcinoma (HCC)
1.3. The Pediatric Liver Cancer: Hepatoblastoma (HB)
2. Hallmarks of Cancer: Deregulated Metabolism and Inflammation
3. PCSK9, a Key Proprotein Convertase
3.1. Background
3.2. Structure
3.3. Function
3.3.1. GOF Mutations of PCSK9
3.3.2. LOF Mutations of PCSK9
4. PCSK9 in Cancers
4.1. Hepatocellular Carcinoma and Hepatoblastoma
4.2. Gastric Cancer
4.3. Lung Cancer
5. PCSK9 Biological Functions
5.1. PCSK9 in Inflammation and Immunity
5.2. PCSK9 in Diabetes
5.3. PCSK9 in Apoptosis
5.4. PCSK9 in Viral Infections
5.5. PCSK9 in Ferroptosis
6. PCSK9 Targeting as a Potential Anti-Cancer Approach
6.1. Blocking PCSK9 Binding to LDLR
6.1.1. PCSK9 Monoclonal Antibodies (mAbs)
6.1.2. Anti-PCSK9 Vaccine
6.1.3. Adnectin
6.1.4. Mimetic Peptides
6.1.5. Pseurotin A
6.1.6. Silibinin A
6.1.7. Orally Active Tricyclic Peptide MK-0616
6.2. Inhibition of PCSK9 Expression
6.2.1. CRISPR/Cas9 Genome-Editing Tool
6.2.2. Antisense Oligonucleotides (ASOs)
6.2.3. siRNA
6.2.4. Pharmaceutical Drugs
6.3. Interfering with PCSK9 Secretion
6.3.1. Sortilin
6.3.2. Sec24a
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bays, H.; Cohen, D.E.; Chalasani, N.; Harrison, S.A. The National Lipid Association’s Statin Safety Task Force, null An Assessment by the Statin Liver Safety Task Force: 2014 Update. J. Clin. Lipidol. 2014, 8, S47–S57. [Google Scholar] [CrossRef] [PubMed]
- Trefts, E.; Gannon, M.; Wasserman, D.H. The Liver. Curr. Biol. CB 2017, 27, R1147–R1151. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular Carcinoma. Nat. Rev. Dis. Primer 2021, 7, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.; Follmann, M.; Malek, N.; Manns, M.P.; Greten, T.F. Critical Appraisal of Clinical Practice Guidelines for Diagnosis and Treatment of Hepatocellular Carcinoma. J. Gastroenterol. Hepatol. 2011, 26, 1779–1786. [Google Scholar] [CrossRef] [PubMed]
- Farazi, P.A.; DePinho, R.A. Hepatocellular Carcinoma Pathogenesis: From Genes to Environment. Nat. Rev. Cancer 2006, 6, 674–687. [Google Scholar] [CrossRef]
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.; Sanyal, A.J. Modeling the Epidemic of Nonalcoholic Fatty Liver Disease Demonstrates an Exponential Increase in Burden of Disease. Hepatol. Baltim. Md 2018, 67, 123–133. [Google Scholar] [CrossRef]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular Carcinoma. Nat. Rev. Dis. Primer 2016, 2, 1–23. [Google Scholar] [CrossRef]
- Bruix, J.; Sherman, M. Management of Hepatocellular Carcinoma. Hepatology 2005, 42, 1208–1236. [Google Scholar] [CrossRef]
- Grandhi, M.S.; Kim, A.K.; Ronnekleiv-Kelly, S.M.; Kamel, I.R.; Ghasebeh, M.A.; Pawlik, T.M. Hepatocellular Carcinoma: From Diagnosis to Treatment. Surg. Oncol. 2016, 25, 74–85. [Google Scholar] [CrossRef]
- Li, D.; Mallory, T.; Satomura, S. AFP-L3: A New Generation of Tumor Marker for Hepatocellular Carcinoma. Clin. Chim. Acta 2001, 313, 15–19. [Google Scholar] [CrossRef]
- International Consensus Group for Hepatocellular Neoplasia. Pathologic Diagnosis of Early Hepatocellular Carcinoma: A Report of the International Consensus Group for Hepatocellular Neoplasia. Hepatology 2009, 49, 658–664. [Google Scholar] [CrossRef]
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular Carcinoma. The Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef]
- Raees, A.; Kamran, M.; Özkan, H.; Jafri, W. Updates on the Diagnosis and Management of Hepatocellular Carcinoma. Euroasian J. Hepato-Gastroenterol. 2021, 11, 32–40. [Google Scholar] [CrossRef]
- Garnier, A.; Ilmer, M.; Kappler, R.; Berger, M. Therapeutic Innovations for Targeting Hepatoblastoma. Anticancer Res. 2016, 36, 5577–5592. [Google Scholar] [CrossRef]
- Litten, J.B.; Tomlinson, G.E. Liver Tumors in Children. Oncologist 2008, 13, 812–820. [Google Scholar] [CrossRef]
- Zhong, S.; Zhao, Y.; Fan, C. Hepatoblastoma with Pure Fetal Epithelial Differentiation in a 10-Year-Old Boy. Medicine (Baltimore) 2018, 97. [Google Scholar] [CrossRef]
- Spector, L.G.; Birch, J. The Epidemiology of Hepatoblastoma. Pediatr. Blood Cancer 2012, 59, 776–779. [Google Scholar] [CrossRef]
- Cairo, S.; Armengol, C.; De Reyniès, A.; Wei, Y.; Thomas, E.; Renard, C.-A.; Goga, A.; Balakrishnan, A.; Semeraro, M.; Gresh, L.; et al. Hepatic Stem-like Phenotype and Interplay of Wnt/β-Catenin and Myc Signaling in Aggressive Childhood Liver Cancer. Cancer Cell 2008, 14, 471–484. [Google Scholar] [CrossRef]
- Buendia, M.A. Unravelling the Genetics of Hepatoblastoma: Few Mutations, What Else? J. Hepatol. 2014, 61, 1202–1204. [Google Scholar] [CrossRef]
- Jia, D.; Dong, R.; Jing, Y.; Xu, D.; Wang, Q.; Chen, L.; Li, Q.; Huang, Y.; Zhang, Y.; Zhang, Z.; et al. Exome Sequencing of Hepatoblastoma Reveals Novel Mutations and Cancer Genes in the Wnt Pathway and Ubiquitin Ligase Complex. Hepatology 2014, 60, 1686–1696. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, G.E.; Kappler, R. Genetics and Epigenetics of Hepatoblastoma. Pediatr. Blood Cancer 2012, 59, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Calvisi, D.F.; Solinas, A. Hepatoblastoma: Current Knowledge and Promises from Preclinical Studies. Transl. Gastroenterol. Hepatol. 2020, 5, 42. [Google Scholar] [CrossRef] [PubMed]
- Baheti, A.D.; Luana Stanescu, A.; Li, N.; Chapman, T. Contrast-Enhanced CT Features of Hepatoblastoma: Can We Predict Histopathology? Clin. Imaging 2017, 44, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Aronson, D.C.; Czauderna, P.; Maibach, R.; Perilongo, G.; Morland, B. The Treatment of Hepatoblastoma: Its Evolution and the Current Status as per the SIOPEL Trials. J. Indian Assoc. Pediatr. Surg. 2014, 19, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Czauderna, P.; Garnier, H. Hepatoblastoma: Current Understanding, Recent Advances, and Controversies. F1000Research 2018, 7. [Google Scholar] [CrossRef]
- Czauderna, P.; Haeberle, B.; Hiyama, E.; Rangaswami, A.; Krailo, M.; Maibach, R.; Rinaldi, E.; Feng, Y.; Aronson, D.; Malogolowkin, M.; et al. The Children’s Hepatic Tumors International Collaboration (CHIC): Novel Global Rare Tumor Database Yields New Prognostic Factors in Hepatoblastoma and Becomes a Research Model. Eur. J. Cancer Oxf. Engl. 1990 2016, 52, 92–101. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Alannan, M.; Fayyad-Kazan, H.; Trézéguet, V.; Merched, A. Targeting Lipid Metabolism in Liver Cancer. Biochemistry 2020, 59, 3951–3964. [Google Scholar] [CrossRef]
- Ali, L.; Schnitzler, J.G.; Kroon, J. Metabolism: The Road to Inflammation and Atherosclerosis. Curr. Opin. Lipidol. 2018, 29, 474–480. [Google Scholar] [CrossRef]
- Zhang, Q.; Lou, Y.; Bai, X.-L.; Liang, T.-B. Immunometabolism: A Novel Perspective of Liver Cancer Microenvironment and Its Influence on Tumor Progression. World J. Gastroenterol. 2018, 24, 3500–3512. [Google Scholar] [CrossRef]
- López-Otín, C.; Overall, C.M. Protease Degradomics: A New Challenge for Proteomics. Nat. Rev. Mol. Cell Biol. 2002, 3, 509–519. [Google Scholar] [CrossRef]
- Seidah, N.G.; Benjannet, S.; Wickham, L.; Marcinkiewicz, J.; Jasmin, S.B.; Stifani, S.; Basak, A.; Prat, A.; Chrétien, M. The Secretory Proprotein Convertase Neural Apoptosis-Regulated Convertase 1 (NARC-1): Liver Regeneration and Neuronal Differentiation. Proc. Natl. Acad. Sci. USA 2003, 100, 928–933. [Google Scholar] [CrossRef]
- Abifadel, M.; Varret, M.; Rabès, J.-P.; Allard, D.; Ouguerram, K.; Devillers, M.; Cruaud, C.; Benjannet, S.; Wickham, L.; Erlich, D.; et al. Mutations in PCSK9 Cause Autosomal Dominant Hypercholesterolemia. Nat. Genet. 2003, 34, 154–156. [Google Scholar] [CrossRef]
- Seidah, N.G.; Prat, A. Precursor Convertases in the Secretory Pathway, Cytosol and Extracellular Milieu. Essays Biochem. 2002, 38, 79–94. [Google Scholar] [CrossRef]
- Wiciński, M.; Żak, J.; Malinowski, B.; Popek, G.; Grześk, G. PCSK9 Signaling Pathways and Their Potential Importance in Clinical Practice. EPMA J. 2017, 8, 391–402. [Google Scholar] [CrossRef]
- Benjannet, S.; Rhainds, D.; Essalmani, R.; Mayne, J.; Wickham, L.; Jin, W.; Asselin, M.-C.; Hamelin, J.; Varret, M.; Allard, D.; et al. NARC-1/PCSK9 and Its Natural Mutants. Zymogen Cleavage and Effects on the Low Density Lipoprotein (LDL) Receptor and LDL Cholesterol. J. Biol. Chem. 2004, 279, 48865–48875. [Google Scholar] [CrossRef]
- Seidah, N.G. The PCSK9 Discovery, an Inactive Protease with Varied Functions in Hypercholesterolemia, Viral Infections, and Cancer. J. Lipid Res. 2021, 62, 100130. [Google Scholar] [CrossRef]
- Cunningham, D.; Danley, D.E.; Geoghegan, K.F.; Griffor, M.C.; Hawkins, J.L.; Subashi, T.A.; Varghese, A.H.; Ammirati, M.J.; Culp, J.S.; Hoth, L.R.; et al. Structural and Biophysical Studies of PCSK9 and Its Mutants Linked to Familial Hypercholesterolemia. Nat. Struct. Mol. Biol. 2007, 14, 413–419. [Google Scholar] [CrossRef]
- Hampton, E.N.; Knuth, M.W.; Li, J.; Harris, J.L.; Lesley, S.A.; Spraggon, G. The Self-Inhibited Structure of Full-Length PCSK9 at 1.9 Å Reveals Structural Homology with Resistin within the C-Terminal Domain. Proc. Natl. Acad. Sci. USA 2007, 104, 14604–14609. [Google Scholar] [CrossRef] [PubMed]
- Lambert, G.; Charlton, F.; Rye, K.-A.; Piper, D.E. Molecular Basis of PCSK9 Function. Atherosclerosis 2009, 203, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Naureckiene, S.; Ma, L.; Sreekumar, K.; Purandare, U.; Frederick Lo, C.; Huang, Y.; Chiang, L.W.; Grenier, J.M.; Ozenberger, B.A.; Steven Jacobsen, J.; et al. Functional Characterization of Narc 1, a Novel Proteinase Related to Proteinase K. Arch. Biochem. Biophys. 2003, 420, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Zhou, A.; Martin, S.; Lipkind, G.; LaMendola, J.; Steiner, D.F. Regulatory Roles of the P Domain of the Subtilisin-like Prohormone Convertases. J. Biol. Chem. 1998, 273, 11107–11114. [Google Scholar] [CrossRef] [PubMed]
- Seidah, N.G.; Chrétien, M. Proprotein and Prohormone Convertases: A Family of Subtilases Generating Diverse Bioactive Polypeptides. Brain Res. 1999, 848, 45–62. [Google Scholar] [CrossRef]
- Seidah, N.G.; Pasquato, A.; Andréo, U. How Do Enveloped Viruses Exploit the Secretory Proprotein Convertases to Regulate Infectivity and Spread? Viruses 2021, 13, 1229. [Google Scholar] [CrossRef]
- Horton, J.D.; Cohen, J.C.; Hobbs, H.H. Molecular Biology of PCSK9: Its Role in LDL Metabolism. Trends Biochem. Sci. 2007, 32, 71–77. [Google Scholar] [CrossRef]
- Kwon, H.J.; Lagace, T.A.; McNutt, M.C.; Horton, J.D.; Deisenhofer, J. Molecular Basis for LDL Receptor Recognition by PCSK9. Proc. Natl. Acad. Sci. USA 2008, 105, 1820–1825. [Google Scholar] [CrossRef]
- Lagace, T.A.; Curtis, D.E.; Garuti, R.; McNutt, M.C.; Park, S.W.; Prather, H.B.; Anderson, N.N.; Ho, Y.K.; Hammer, R.E.; Horton, J.D. Secreted PCSK9 Decreases the Number of LDL Receptors in Hepatocytes and Inlivers of Parabiotic Mice. J. Clin. Investig. 2006, 116, 2995–3005. [Google Scholar] [CrossRef]
- Nassoury, N.; Blasiole, D.A.; Tebon Oler, A.; Benjannet, S.; Hamelin, J.; Poupon, V.; McPherson, P.S.; Attie, A.D.; Prat, A.; Seidah, N.G. The Cellular Trafficking of the Secretory Proprotein Convertase PCSK9 and Its Dependence on the LDLR. Traffic 2007, 8, 718–732. [Google Scholar] [CrossRef]
- Benjannet, S.; Rhainds, D.; Hamelin, J.; Nassoury, N.; Seidah, N.G. The Proprotein Convertase (PC) PCSK9 Is Inactivated by Furin and/or PC5/6A: Functional Consequences of Natural Mutations and Post-Translational Modifications. J. Biol. Chem. 2006, 281, 30561–30572. [Google Scholar] [CrossRef]
- Ouadda, A.B.D.; Gauthier, M.-S.; Susan-Resiga, D.; Girard, E.; Essalmani, R.; Black, M.; Marcinkiewicz, J.; Hamelin, J.; Evagelidis, A.; Ly, K.; et al. Ser-Phosphorylation of PCSK9 by Fam20C-Kinase Enhances Its Ability to Degrade the LDLR. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1996–2013. [Google Scholar] [CrossRef]
- Qian, Y.-W.; Schmidt, R.J.; Zhang, Y.; Chu, S.; Lin, A.; Wang, H.; Wang, X.; Beyer, T.P.; Bensch, W.R.; Li, W.; et al. Secreted PCSK9 Downregulates Low Density Lipoprotein Receptor through Receptor-Mediated Endocytosis. J. Lipid Res. 2007, 48, 1488–1498. [Google Scholar] [CrossRef]
- Park, S.W.; Moon, Y.-A.; Horton, J.D. Post-Transcriptional Regulation of Low Density Lipoprotein Receptor Protein by Proprotein Convertase Subtilisin/Kexin Type 9a in Mouse Liver. J. Biol. Chem. 2004, 279, 50630–50638. [Google Scholar] [CrossRef]
- Poirier, S.; Mayer, G.; Poupon, V.; McPherson, P.S.; Desjardins, R.; Ly, K.; Asselin, M.-C.; Day, R.; Duclos, F.J.; Witmer, M.; et al. Dissection of the Endogenous Cellular Pathways of PCSK9-Induced Low Density Lipoprotein Receptor Degradation. J. Biol. Chem. 2009, 284, 28856–28864. [Google Scholar] [CrossRef]
- Homer, V.M.; Marais, A.D.; Charlton, F.; Laurie, A.D.; Hurndell, N.; Scott, R.; Mangili, F.; Sullivan, D.R.; Barter, P.J.; Rye, K.-A.; et al. Identification and Characterization of Two Non-Secreted PCSK9 Mutants Associated with Familial Hypercholesterolemia in Cohorts from New Zealand and South Africa. Atherosclerosis 2008, 196, 659–666. [Google Scholar] [CrossRef]
- Maxwell, K.N.; Fisher, E.A.; Breslow, J.L. Overexpression of PCSK9 Accelerates the Degradation of the LDLR in a Post-Endoplasmic Reticulum Compartment. Proc. Natl. Acad. Sci. USA 2005, 102, 2069–2074. [Google Scholar] [CrossRef]
- Nishikido, T.; Ray, K.K. Non-Antibody Approaches to Proprotein Convertase Subtilisin Kexin 9 Inhibition: SiRNA, Antisense Oligonucleotides, Adnectins, Vaccination, and New Attempts at Small-Molecule Inhibitors Based on New Discoveries. Front. Cardiovasc. Med. 2019, 5. [Google Scholar] [CrossRef]
- Jang, H.-D.; Lee, S.E.; Yang, J.; Lee, H.-C.; Shin, D.; Lee, H.; Lee, J.; Jin, S.; Kim, S.; Lee, S.J.; et al. Cyclase-Associated Protein 1 Is a Binding Partner of Proprotein Convertase Subtilisin/Kexin Type-9 and Is Required for the Degradation of Low-Density Lipoprotein Receptors by Proprotein Convertase Subtilisin/Kexin Type-9. Eur. Heart J. 2020, 41, 239–252. [Google Scholar] [CrossRef]
- Essalmani, R.; Susan-Resiga, D.; Chamberland, A.; Abifadel, M.; Creemers, J.W.; Boileau, C.; Seidah, N.G.; Prat, A. In Vivo Evidence That Furin from Hepatocytes Inactivates PCSK9. J. Biol. Chem. 2011, 286, 4257–4263. [Google Scholar] [CrossRef]
- Han, B.; Eacho, P.I.; Knierman, M.D.; Troutt, J.S.; Konrad, R.J.; Yu, X.; Schroeder, K.M. Isolation and Characterization of the Circulating Truncated Form of PCSK9. J. Lipid Res. 2014, 55, 1505–1514. [Google Scholar] [CrossRef] [PubMed]
- Pisciotta, L.; Priore Oliva, C.; Cefalù, A.B.; Noto, D.; Bellocchio, A.; Fresa, R.; Cantafora, A.; Patel, D.; Averna, M.; Tarugi, P.; et al. Additive Effect of Mutations in LDLR and PCSK9 Genes on the Phenotype of Familial Hypercholesterolemia. Atherosclerosis 2006, 186, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, K.N.; Breslow, J.L. Adenoviral-Mediated Expression of Pcsk9 in Mice Results in a Low-Density Lipoprotein Receptor Knockout Phenotype. Proc. Natl. Acad. Sci. USA 2004, 101, 7100–7105. [Google Scholar] [CrossRef] [PubMed]
- Timms, K.M.; Wagner, S.; Samuels, M.E.; Forbey, K.; Goldfine, H.; Jammulapati, S.; Skolnick, M.H.; Hopkins, P.N.; Hunt, S.C.; Shattuck, D.M. A Mutation in PCSK9 Causing Autosomal-Dominant Hypercholesterolemia in a Utah Pedigree. Hum. Genet. 2004, 114, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.C.; Boerwinkle, E.; Mosley, T.H.; Hobbs, H.H. Sequence Variations in PCSK9, Low LDL, and Protection against Coronary Heart Disease. N. Engl. J. Med. 2006, 354, 1264–1272. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. Lowering LDL--Not Only How Low, But How Long? Science 2006, 311, 1721–1723. [Google Scholar] [CrossRef]
- Mayne, J.; Dewpura, T.; Raymond, A.; Bernier, L.; Cousins, M.; Ooi, T.C.; Davignon, J.; Seidah, N.G.; Mbikay, M.; Chrétien, M. Novel Loss-of-Function PCSK9 Variant Is Associated with Low Plasma LDL Cholesterol in a French-Canadian Family and with Impaired Processing and Secretion in Cell Culture. Clin. Chem. 2011, 57, 1415–1423. [Google Scholar] [CrossRef]
- Benjannet, S.; Hamelin, J.; Chrétien, M.; Seidah, N.G. Loss- and Gain-of-Function PCSK9 Variants. J. Biol. Chem. 2012, 287, 33745–33755. [Google Scholar] [CrossRef]
- Lebeau, P.F.; Wassef, H.; Byun, J.H.; Platko, K.; Ason, B.; Jackson, S.; Dobroff, J.; Shetterly, S.; Richards, W.G.; Al-Hashimi, A.A.; et al. The Loss-of-Function PCSK9Q152H Variant Increases ER Chaperones GRP78 and GRP94 and Protects against Liver Injury. J. Clin. Investig. 2021, 131, e128650. [Google Scholar] [CrossRef]
- Osono, Y.; Woollett, L.A.; Herz, J.; Dietschy, J.M. Role of the Low Density Lipoprotein Receptor in the Flux of Cholesterol through the Plasma and across the Tissues of the Mouse. J. Clin. Investig. 1995, 95, 1124–1132. [Google Scholar] [CrossRef]
- Glerup, S.; Schulz, R.; Laufs, U.; Schlüter, K.-D. Physiological and Therapeutic Regulation of PCSK9 Activity in Cardiovascular Disease. Basic Res. Cardiol. 2017, 112, 32. [Google Scholar] [CrossRef]
- Poirier, S.; Mayer, G.; Benjannet, S.; Bergeron, E.; Marcinkiewicz, J.; Nassoury, N.; Mayer, H.; Nimpf, J.; Prat, A.; Seidah, N.G. The Proprotein Convertase PCSK9 Induces the Degradation of Low Density Lipoprotein Receptor (LDLR) and Its Closest Family Members VLDLR and ApoER2. J. Biol. Chem. 2008, 283, 2363–2372. [Google Scholar] [CrossRef]
- Canuel, M.; Sun, X.; Asselin, M.-C.; Paramithiotis, E.; Prat, A.; Seidah, N.G. Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) Can Mediate Degradation of the Low Density Lipoprotein Receptor-Related Protein 1 (LRP-1). PLoS ONE 2013, 8, e64145. [Google Scholar] [CrossRef]
- Demers, A.; Samami, S.; Lauzier, B.; Des Rosiers, C.; Sock, E.T.N.; Ong, H.; Mayer, G. PCSK9 Induces CD36 Degradation and Affects Long-Chain Fatty Acid Uptake and Triglyceride Metabolism in Adipocytes and in Mouse Liver. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2517–2525. [Google Scholar] [CrossRef]
- Labonté, P.; Begley, S.; Guévin, C.; Asselin, M.-C.; Nassoury, N.; Mayer, G.; Prat, A.; Seidah, N.G. PCSK9 Impedes Hepatitis C Virus Infection in Vitro and Modulates Liver CD81 Expression. Hepatology 2009, 50, 17–24. [Google Scholar] [CrossRef]
- Levy, E.; Ouadda, A.B.D.; Spahis, S.; Sane, A.T.; Garofalo, C.; Grenier, É.; Emonnot, L.; Yara, S.; Couture, P.; Beaulieu, J.-F.; et al. PCSK9 Plays a Significant Role in Cholesterol Homeostasis and Lipid Transport in Intestinal Epithelial Cells. Atherosclerosis 2013, 227, 297–306. [Google Scholar] [CrossRef]
- Bhat, M.; Skill, N.; Marcus, V.; Deschenes, M.; Tan, X.; Bouteaud, J.; Negi, S.; Awan, Z.; Aikin, R.; Kwan, J.; et al. Decreased PCSK9 Expression in Human Hepatocellular Carcinoma. BMC Gastroenterol. 2015, 15, 1–10. [Google Scholar] [CrossRef]
- Zhang, S.-Z.; Zhu, X.-D.; Feng, L.-H.; Li, X.-L.; Liu, X.-F.; Sun, H.-C.; Tang, Z.-Y. PCSK9 Promotes Tumor Growth by Inhibiting Tumor Cell Apoptosis in Hepatocellular Carcinoma. Exp. Hematol. Oncol. 2021, 10, 25. [Google Scholar] [CrossRef]
- Nagashima, S.; Morishima, K.; Okamoto, H.; Ishibashi, S. Possible Involvement of PCSK9 Overproduction in Hyperlipoproteinemia Associated with Hepatocellular Carcinoma: A Case Report. J. Clin. Lipidol. 2016, 10, 1045–1049. [Google Scholar] [CrossRef]
- Bridge, S.H.; Sheridan, D.A.; Felmlee, D.J.; Crossey, M.M.E.; Fenwick, F.I.; Lanyon, C.V.; Dubuc, G.; Seidah, N.G.; Davignon, J.; Thomas, H.C.; et al. PCSK9, Apolipoprotein E and Lipoviral Particles in Chronic Hepatitis C Genotype 3: Evidence for Genotype-Specific Regulation of Lipoprotein Metabolism. J. Hepatol. 2015, 62, 763–770. [Google Scholar] [CrossRef]
- Fasolato, S.; Pigozzo, S.; Pontisso, P.; Angeli, P.; Ruscica, M.; Savarino, E.; De Martin, S.; Lupo, M.G.; Ferri, N. PCSK9 Levels Are Raised in Chronic HCV Patients with Hepatocellular Carcinoma. J. Clin. Med. 2020, 9, 3134. [Google Scholar] [CrossRef] [PubMed]
- Marimuthu, A.; Subbannayya, Y.; Sahasrabuddhe, N.A.; Balakrishnan, L.; Syed, N.; Sekhar, N.R.; Katte, T.V.; Pinto, S.M.; Srikanth, S.M.; Kumar, P.; et al. SILAC-Based Quantitative Proteomic Analysis of Gastric Cancer Secretome. Proteomics Clin. Appl. 2013, 7, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Li, S.; Fang, Y.; Zou, Y.; Song, D.; Zhang, S.; Cai, Y. Proprotein Convertase Subtilisin/Kexin Type 9 Promotes Gastric Cancer Metastasis and Suppresses Apoptosis by Facilitating MAPK Signaling Pathway Through HSP70 Up-Regulation. Front. Oncol. 2021, 10, 609663. [Google Scholar] [CrossRef] [PubMed]
- Demidyuk, I.V.; Shubin, A.V.; Gasanov, E.V.; Kurinov, A.M.; Demkin, V.V.; Vinogradova, T.V.; Zinovyeva, M.V.; Sass, A.V.; Zborovskaya, I.B.; Kostrov, S.V. Alterations in Gene Expression of Proprotein Convertases in Human Lung Cancer Have a Limited Number of Scenarios. PLoS ONE 2013, 8, e55752. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, A.; Grossi, F.; Carbone, F.; Vecchié, A.; Minetti, S.; Bardi, N.; Elia, E.; Ansaldo, A.M.; Ferrara, D.; Rijavec, E.; et al. Serum PCSK9 Levels at the Second Nivolumab Cycle Predict Overall Survival in Elderly Patients with NSCLC: A Pilot Study. Cancer Immunol. Immunother. 2019, 68, 1351–1358. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, A.; Grossi, F.; Montecucco, F. PCSK9 Is a Promising Prognostic Marker in Patients with Advanced NSCLC. Cancer Immunol. Immunother. 2020, 69, 491–492. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Chowdhury, A.; Chaudhury, K.; Shukla, P.C. Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9): A Potential Multifaceted Player in Cancer. Biochim. Biophys. Acta BBA-Rev. Cancer 2021, 1876, 188581. [Google Scholar] [CrossRef]
- Tang, Z.; Jiang, L.; Peng, J.; Ren, Z.; Wei, D.; Wu, C.; Pan, L.; Jiang, Z.; Liu, L. PCSK9 SiRNA Suppresses the Inflammatory Response Induced by OxLDL through Inhibition of NF-ΚB Activation in THP-1-Derived Macrophages. Int. J. Mol. Med. 2012, 30, 931–938. [Google Scholar] [CrossRef]
- Tang, Z.-H.; Peng, J.; Ren, Z.; Yang, J.; Li, T.-T.; Li, T.-H.; Wang, Z.; Wei, D.-H.; Liu, L.-S.; Zheng, X.-L.; et al. New Role of PCSK9 in Atherosclerotic Inflammation Promotion Involving the TLR4/NF-ΚB Pathway. Atherosclerosis 2017, 262, 113–122. [Google Scholar] [CrossRef]
- Giunzioni, I.; Tavori, H.; Covarrubias, R.; Major, A.S.; Ding, L.; Zhang, Y.; DeVay, R.M.; Hong, L.; Fan, D.; Predazzi, I.M.; et al. Local Effects of Human PCSK9 on the Atherosclerotic Lesion. J. Pathol. 2016, 238, 52–62. [Google Scholar] [CrossRef]
- Ricci, C.; Ruscica, M.; Camera, M.; Rossetti, L.; Macchi, C.; Colciago, A.; Zanotti, I.; Lupo, M.G.; Adorni, M.P.; Cicero, A.F.G.; et al. PCSK9 Induces a Pro-Inflammatory Response in Macrophages. Sci. Rep. 2018, 8, 2267. [Google Scholar] [CrossRef]
- Badimon, L.; Luquero, A.; Crespo, J.; Peña, E.; Borrell-Pages, M. PCSK9 and LRP5 in Macrophage Lipid Internalization and Inflammation. Cardiovasc. Res. 2021, 117, 2054–2068. [Google Scholar] [CrossRef]
- Ruscica, M.; Ricci, C.; Macchi, C.; Magni, P.; Cristofani, R.; Liu, J.; Corsini, A.; Ferri, N. Suppressor of Cytokine Signaling-3 (SOCS-3) Induces Proprotein Convertase Subtilisin Kexin Type 9 (PCSK9) Expression in Hepatic HepG2 Cell Line. J. Biol. Chem. 2016, 291, 3508–3519. [Google Scholar] [CrossRef]
- Liu, X.; Bao, X.; Hu, M.; Chang, H.; Jiao, M.; Cheng, J.; Xie, L.; Huang, Q.; Li, F.; Li, C.-Y. PCSK9 Inhibition Potentiates Cancer Immune Checkpoint Therapy. Nature 2020, 588, 693–698. [Google Scholar] [CrossRef]
- Yuan, J.; Cai, T.; Zheng, X.; Ren, Y.; Qi, J.; Lu, X.; Chen, H.; Lin, H.; Chen, Z.; Liu, M.; et al. Potentiating CD8+ T Cell Antitumor Activity by Inhibiting PCSK9 to Promote LDLR-Mediated TCR Recycling and Signaling. Protein Cell 2021, 12, 240–260. [Google Scholar] [CrossRef]
- Peyot, M.-L.; Roubtsova, A.; Lussier, R.; Chamberland, A.; Essalmani, R.; Murthy Madiraju, S.R.; Seidah, N.G.; Prentki, M.; Prat, A. Substantial PCSK9 Inactivation in β-Cells Does Not Modify Glucose Homeostasis or Insulin Secretion in Mice. Biochim. Biophys. Acta BBA-Mol. Cell Biol. Lipids 2021, 1866, 158968. [Google Scholar] [CrossRef]
- Miao, J.; Manthena, P.V.; Haas, M.E.; Ling, A.V.; Shin, D.-J.; Graham, M.J.; Crooke, R.M.; Liu, J.; Biddinger, S.B. The Role of Insulin in the Regulation of PCSK9. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1589–1596. [Google Scholar] [CrossRef]
- Ai, D.; Chen, C.; Han, S.; Ganda, A.; Murphy, A.J.; Haeusler, R.; Thorp, E.; Accili, D.; Horton, J.D.; Tall, A.R. Regulation of Hepatic LDL Receptors by MTORC1 and PCSK9 in Mice. J. Clin. Investig. 2012, 122, 1262–1270. [Google Scholar] [CrossRef]
- Korsmeyer, S.J. BCL-2 Gene Family and the Regulation of Programmed Cell Death. Cancer Res. 1999, 59, 1693s–1700s. [Google Scholar] [CrossRef]
- Wu, C.-Y.; Tang, Z.-H.; Jiang, L.; Li, X.-F.; Jiang, Z.-S.; Liu, L.-S. PCSK9 SiRNA Inhibits HUVEC Apoptosis Induced by Ox-LDL via Bcl/Bax–Caspase9–Caspase3 Pathway. Mol. Cell. Biochem. 2012, 359, 347–358. [Google Scholar] [CrossRef]
- Kysenius, K.; Muggalla, P.; Mätlik, K.; Arumäe, U.; Huttunen, H.J. PCSK9 Regulates Neuronal Apoptosis by Adjusting ApoER2 Levels and Signaling. Cell. Mol. Life Sci. 2012, 69, 1903–1916. [Google Scholar] [CrossRef] [PubMed]
- Piao, M.-X.; Bai, J.-W.; Zhang, P.-F.; Zhang, Y.-Z. PCSK9 Regulates Apoptosis in Human Neuroglioma U251 Cells via Mitochondrial Signaling Pathways. Int. J. Clin. Exp. Pathol. 2015, 8, 2787–2794. [Google Scholar] [PubMed]
- Xu, X.; Cui, Y.; Cao, L.; Zhang, Y.; Yin, Y.; Hu, X. PCSK9 Regulates Apoptosis in Human Lung Adenocarcinoma A549 Cells via Endoplasmic Reticulum Stress and Mitochondrial Signaling Pathways. Exp. Ther. Med. 2017, 13, 1993–1999. [Google Scholar] [CrossRef] [PubMed]
- Gan, S.-S.; Ye, J.-Q.; Wang, L.; Qu, F.-J.; Chu, C.-M.; Tian, Y.-J.; Yang, W.; Cui, X.-G. Inhibition of PCSK9 Protects against Radiation-Induced Damage of Prostate Cancer Cells. OncoTargets Ther. 2017, 10, 2139–2146. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Essalmani, R.; Day, R.; Khatib, A.M.; Seidah, N.G.; Prat, A. Proprotein Convertase Subtilisin/Kexin Type 9 Deficiency Reduces Melanoma Metastasis in Liver. Neoplasia N. Y. 2012, 14, 1122–1131. [Google Scholar] [CrossRef]
- He, M.; Hou, J.; Wang, L.; Zheng, M.; Fang, T.; Wang, X.; Xia, J. Actinidia Chinensis Planch Root Extract Inhibits Cholesterol Metabolism in Hepatocellular Carcinoma through Upregulation of PCSK9. Oncotarget 2017, 8, 42136–42148. [Google Scholar] [CrossRef]
- Gan, E.S.; Tan, H.C.; Le, D.H.T.; Huynh, T.T.; Wills, B.; Seidah, N.G.; Ooi, E.E.; Yacoub, S. Dengue Virus Induces PCSK9 Expression to Alter Antiviral Responses and Disease Outcomes. J. Clin. Investig. 2020, 130, 5223–5234. [Google Scholar] [CrossRef]
- Seidah, N.G.; Prat, A. The Multifaceted Biology of PCSK9. Endocr. Rev. 2021, bnab035. [Google Scholar] [CrossRef]
- Vuorio, A.; Kovanen, P.T. PCSK9 Inhibitors for COVID-19: An Opportunity to Enhance the Antiviral Action of Interferon in Patients with Hypercholesterolaemia. J. Intern. Med. 2021, 289, 749. [Google Scholar] [CrossRef]
- Hirschhorn, T.; Stockwell, B.R. The Development of the Concept of Ferroptosis. Free Radic. Biol. Med. 2019, 133, 130–143. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Non-Apoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and Function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized Arachidonic/Adrenic Phosphatidylethanolamines Navigate Cells to Ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.; Huang, Z.; Lin, Z.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, Present and Future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Seidah, N.G.; Prat, A. The Biology and Therapeutic Targeting of the Proprotein Convertases. Nat. Rev. Drug Discov. 2012, 11, 367–383. [Google Scholar] [CrossRef]
- Dong, S.; Lu, Y.; Peng, G.; Li, J.; Li, W.; Li, M.; Wang, H.; Liu, L.; Zhao, Q. Furin Inhibits Epithelial Cell Injury and Alleviates Experimental Colitis by Activating the Nrf2-Gpx4 Signaling Pathway. Dig. Liver Dis. 2021, 53, 1276–1285. [Google Scholar] [CrossRef]
- Alannan, M.; Fatrouni, H.; Trézéguet, V.; Dittrich-Domergue, F.; Moreau, P.; Siegfried, G.; Liet, B.; Khatib, A.-M.; Grosset, C.F.; Badran, B.; et al. Targeting PCSK9 in Liver Cancer Cells Triggers Metabolic Ex-Haustion and Cell Death by Ferroptosis. Cells 2022. submitted. [Google Scholar]
- Navarese, E.P.; Kolodziejczak, M.; Schulze, V.; Gurbel, P.A.; Tantry, U.; Lin, Y.; Brockmeyer, M.; Kandzari, D.E.; Kubica, J.M.; D’Agostino, R.B.; et al. Effects of Proprotein Convertase Subtilisin/Kexin Type 9 Antibodies in Adults With Hypercholesterolemia: A Systematic Review and Meta-Analysis. Ann. Intern. Med. 2015, 163, 40. [Google Scholar] [CrossRef]
- Sabatine, M.S.; Giugliano, R.P.; Wiviott, S.D.; Raal, F.J.; Blom, D.J.; Robinson, J.; Ballantyne, C.M.; Somaratne, R.; Legg, J.; Wasserman, S.M.; et al. Efficacy and Safety of Evolocumab in Reducing Lipids and Cardiovascular Events. N. Engl. J. Med. 2015, 372, 1500–1509. [Google Scholar] [CrossRef]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.G.; Farnier, M.; Krempf, M.; Bergeron, J.; Luc, G.; Averna, M.; Stroes, E.S.; Langslet, G.; Raal, F.J.; El Shahawy, M.; et al. Efficacy and Safety of Alirocumab in Reducing Lipids and Cardiovascular Events. N. Engl. J. Med. 2015, 372, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Edelberg, J.M.; Goodman, S.G.; Hanotin, C.; Harrington, R.A.; et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N. Engl. J. Med. 2018, 379, 2097–2107. [Google Scholar] [CrossRef] [PubMed]
- Momtazi-Borojeni, A.A.; Jaafari, M.R.; Badiee, A.; Sahebkar, A. Long-Term Generation of AntiPCSK9 Antibody Using a Nanoliposome-Based Vaccine Delivery System. Atherosclerosis 2019, 283, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Momtazi-Borojeni, A.A.; Jaafari, M.R.; Afshar, M.; Banach, M.; Sahebkar, A. PCSK9 Immunization Using Nanoliposomes: Preventive Efficacy against Hypercholesterolemia and Atherosclerosis. Arch. Med. Sci. 2021, 17, 1365–1377. [Google Scholar] [CrossRef]
- Momtazi-Borojeni, A.A.; Jaafari, M.R.; Badiee, A.; Banach, M.; Sahebkar, A. Therapeutic Effect of Nanoliposomal PCSK9 Vaccine in a Mouse Model of Atherosclerosis. BMC Med. 2019, 17, 223. [Google Scholar] [CrossRef]
- Momtazi-Borojeni, A.A.; Jaafari, M.R.; Banach, M.; Gorabi, A.M.; Sahraei, H.; Sahebkar, A. Pre-Clinical Evaluation of the Nanoliposomal AntiPCSK9 Vaccine in Healthy Non-Human Primates. Vaccines 2021, 9, 749. [Google Scholar] [CrossRef]
- Mitchell, T.; Chao, G.; Sitkoff, D.; Lo, F.; Monshizadegan, H.; Meyers, D.; Low, S.; Russo, K.; DiBella, R.; Denhez, F.; et al. Pharmacologic Profile of the Adnectin BMS-962476, a Small Protein Biologic Alternative to PCSK9 Antibodies for Low-Density Lipoprotein Lowering. J. Pharmacol. Exp. Ther. 2014, 350, 412–424. [Google Scholar] [CrossRef]
- Stein, E.A.; Kasichayanula, S.; Turner, T.; Kranz, T.; Arumugam, U.; Biernat, L.; Lee, J. Ldl Cholesterol Reduction with Bms-962476, an Adnectin Inhibitor of Pcsk9: Results of a Single Ascending Dose Study. J. Am. Coll. Cardiol. 2014, 63, A1372. [Google Scholar] [CrossRef]
- Zhang, Y.; Eigenbrot, C.; Zhou, L.; Shia, S.; Li, W.; Quan, C.; Tom, J.; Moran, P.; Di Lello, P.; Skelton, N.J.; et al. Identification of a Small Peptide That Inhibits PCSK9 Protein Binding to the Low Density Lipoprotein Receptor. J. Biol. Chem. 2014, 289, 942–955. [Google Scholar] [CrossRef]
- Seidah, N.G.; Poirier, S.; Denis, M.; Parker, R.; Miao, B.; Mapelli, C.; Prat, A.; Wassef, H.; Davignon, J.; Hajjar, K.A.; et al. Annexin A2 Is a Natural Extrahepatic Inhibitor of the PCSK9-Induced LDL Receptor Degradation. PLoS ONE 2012, 7, e0041865. [Google Scholar] [CrossRef]
- Abdelwahed, K.S.; Siddique, A.B.; Mohyeldin, M.M.; Qusa, M.H.; Goda, A.A.; Singh, S.S.; Ayoub, N.M.; King, J.A.; Jois, S.D.; El Sayed, K.A. Pseurotin A as a Novel Suppressor of Hormone Dependent Breast Cancer Progression and Recurrence by Inhibiting PCSK9 Secretion and Interaction with LDL Receptor. Pharmacol. Res. 2020, 158, 104847. [Google Scholar] [CrossRef]
- Wellington, K.; Jarvis, B. Silymarin: A Review of Its Clinical Properties in the Management of Hepatic Disorders. BioDrugs 2001, 15, 465–489. [Google Scholar] [CrossRef]
- Dong, Z.; Zhang, W.; Chen, S.; Liu, C. Silibinin A Decreases Statin-induced PCSK9 Expression in Human Hepatoblastoma HepG2 Cells. Mol. Med. Rep. 2019, 20, 1383–1392. [Google Scholar] [CrossRef]
- Tucker, T.J.; Embrey, M.W.; Alleyne, C.; Amin, R.P.; Bass, A.; Bhatt, B.; Bianchi, E.; Branca, D.; Bueters, T.; Buist, N.; et al. A Series of Novel, Highly Potent, and Orally Bioavailable Next-Generation Tricyclic Peptide PCSK9 Inhibitors. J. Med. Chem. 2021, 64, 16770–16800. [Google Scholar] [CrossRef]
- Mullard, A. Merck Readies Oral, Macrocyclic PCSK9 Inhibitor for Phase II Test. Nat. Rev. Drug Discov. 2021, 21, 9. [Google Scholar] [CrossRef]
- Musunuru, K.; Chadwick, A.C.; Mizoguchi, T.; Garcia, S.P.; DeNizio, J.E.; Reiss, C.W.; Wang, K.; Iyer, S.; Dutta, C.; Clendaniel, V.; et al. In Vivo CRISPR Base Editing of PCSK9 Durably Lowers Cholesterol in Primates. Nature 2021, 593, 429–434. [Google Scholar] [CrossRef]
- Rothgangl, T.; Dennis, M.K.; Lin, P.J.C.; Oka, R.; Witzigmann, D.; Villiger, L.; Qi, W.; Hruzova, M.; Kissling, L.; Lenggenhager, D.; et al. In Vivo Adenine Base Editing of PCSK9 in Macaques Reduces LDL Cholesterol Levels. Nat. Biotechnol. 2021, 39, 949–957. [Google Scholar] [CrossRef]
- Graham, M.J.; Lemonidis, K.M.; Whipple, C.P.; Subramaniam, A.; Monia, B.P.; Crooke, S.T.; Crooke, R.M. Antisense Inhibition of Proprotein Convertase Subtilisin/Kexin Type 9 Reduces Serum LDL in Hyperlipidemic Mice. J. Lipid Res. 2007, 48, 763–767. [Google Scholar] [CrossRef]
- Gupta, N.; Fisker, N.; Asselin, M.-C.; Lindholm, M.; Rosenbohm, C.; Ørum, H.; Elmén, J.; Seidah, N.G.; Straarup, E.M. A Locked Nucleic Acid Antisense Oligonucleotide (LNA) Silences PCSK9 and Enhances LDLR Expression In Vitro and In Vivo. PLoS ONE 2010, 5, e0010682. [Google Scholar] [CrossRef]
- van Poelgeest, E.P.; Hodges, M.R.; Moerland, M.; Tessier, Y.; Levin, A.A.; Persson, R.; Lindholm, M.W.; Erichsen, K.D.; Ørum, H.; Cohen, A.F.; et al. Antisense-Mediated Reduction of Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9): A First-in-Human Randomized, Placebo-Controlled Trial. Br. J. Clin. Pharmacol. 2015, 80, 1350–1361. [Google Scholar] [CrossRef] [PubMed]
- van Poelgeest, E.P.; Swart, R.M.; Betjes, M.G.H.; Moerland, M.; Weening, J.J.; Tessier, Y.; Hodges, M.R.; Levin, A.A.; Burggraaf, J. Acute Kidney Injury During Therapy With an Antisense Oligonucleotide Directed Against PCSK9. Am. J. Kidney Dis. 2013, 62, 796–800. [Google Scholar] [CrossRef] [PubMed]
- Novartis Receives EU Approval for Leqvio®* (Inclisiran), a First-in-Class SiRNA to Lower Cholesterol with Two Doses a Year. Available online: https://www.novartis.com/news/media-releases/novartis-receives-eu-approval-leqvio-inclisiran-first-class-sirna-lower-cholesterol-two-doses-year (accessed on 31 August 2021).
- Search the Website. Available online: https://www.ema.europa.eu/en/search/search/type/ema_document (accessed on 31 August 2021).
- Raal, F.J.; Kallend, D.; Ray, K.K.; Turner, T.; Koenig, W.; Wright, R.S.; Wijngaard, P.L.J.; Curcio, D.; Jaros, M.J.; Leiter, L.A.; et al. Inclisiran for the Treatment of Heterozygous Familial Hypercholesterolemia. N. Engl. J. Med. 2020, 382, 1520–1530. [Google Scholar] [CrossRef] [PubMed]
- Novartis Successfully Completes Acquisition of The Medicines Company, Adding a Potentially First-in-Class, Investigational Cholesterol-Lowering Therapy Inclisiran. Available online: https://www.novartis.com/news/media-releases/novartis-successfully-completes-acquisition-medicines-company-adding-potentially-first-class-investigational-cholesterol-lowering-therapy-inclisiran (accessed on 31 August 2021).
- FDA Delays Decision on Novartis Cholesterol Therapy. Available online: https://www.biopharmadive.com/news/novartis-fda-complete-response-inclisiran-inspection/592554/ (accessed on 31 August 2021).
- Petersen, D.N.; Hawkins, J.; Ruangsiriluk, W.; Stevens, K.A.; Maguire, B.A.; O’Connell, T.N.; Rocke, B.N.; Boehm, M.; Ruggeri, R.B.; Rolph, T.; et al. A Small-Molecule Anti-Secretagogue of PCSK9 Targets the 80S Ribosome to Inhibit PCSK9 Protein Translation. Cell Chem. Biol. 2016, 23, 1362–1371. [Google Scholar] [CrossRef][Green Version]
- Li, W.; Ward, F.R.; McClure, K.F.; Tsai-Lan Chang, S.; Montabana, E.; Liras, S.; Dullea, R.G.; Cate, J.H.D. Structural Basis for Selective Stalling of Human Ribosome Nascent Chain Complexes by a Drug-like Molecule. Nat. Struct. Mol. Biol. 2019, 26, 501–509. [Google Scholar] [CrossRef]
- Chan, J.C.Y.; Piper, D.E.; Cao, Q.; Liu, D.; King, C.; Wang, W.; Tang, J.; Liu, Q.; Higbee, J.; Xia, Z.; et al. A Proprotein Convertase Subtilisin/Kexin Type 9 Neutralizing Antibody Reduces Serum Cholesterol in Mice and Nonhuman Primates. Proc. Natl. Acad. Sci. USA 2009, 106, 9820–9825. [Google Scholar] [CrossRef]
- Mahboobnia, K.; Pirro, M.; Marini, E.; Grignani, F.; Bezsonov, E.E.; Jamialahmadi, T.; Sahebkar, A. PCSK9 and Cancer: Rethinking the Link. Biomed. Pharmacother. 2021, 140, 111758. [Google Scholar] [CrossRef]
- Momtazi-Borojeni, A.A.; Nik, M.E.; Jaafari, M.R.; Banach, M.; Sahebkar, A. Effects of Immunization against PCSK9 in an Experimental Model of Breast Cancer. Arch. Med. Sci. AMS 2019, 15, 570–579. [Google Scholar] [CrossRef]
- Momtazi-Borojeni, A.A.; Nik, M.E.; Jaafari, M.R.; Banach, M.; Sahebkar, A. Effects of Immunisation against PCSK9 in Mice Bearing Melanoma. Arch. Med. Sci. AMS 2019, 16, 189–199. [Google Scholar] [CrossRef]
- Lipovšek, D. Adnectins: Engineered Target-Binding Protein Therapeutics. Protein Eng. Des. Sel. 2011, 24, 3–9. [Google Scholar] [CrossRef]
- Craik, D.J.; Fairlie, D.P.; Liras, S.; Price, D. The Future of Peptide-Based Drugs. Chem. Biol. Drug Des. 2013, 81, 136–147. [Google Scholar] [CrossRef]
- Schroeder, C.I.; Swedberg, J.E.; Withka, J.M.; Rosengren, K.J.; Akcan, M.; Clayton, D.J.; Daly, N.L.; Cheneval, O.; Borzilleri, K.A.; Griffor, M.; et al. Design and Synthesis of Truncated EGF-A Peptides That Restore LDL-R Recycling in the Presence of PCSK9 In Vitro. Chem. Biol. 2014, 21, 284–294. [Google Scholar] [CrossRef]
- Luper, S. A Review of Plants Used in the Treatment of Liver Disease: Part 1. Altern. Med. Rev. 1998, 3, 12. [Google Scholar]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome Engineering Using the CRISPR-Cas9 System. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Wang, X.; Raghavan, A.; Chen, T.; Qiao, L.; Zhang, Y.; Ding, Q.; Musunuru, K. CRISPR-Cas9 Targeting of PCSK9 in Human Hepatocytes In Vivo—Brief Report. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 783–786. [Google Scholar] [CrossRef]
- Ding, Q.; Strong, A.; Patel, K.M.; Ng, S.-L.; Gosis, B.S.; Regan, S.N.; Rader, D.J.; Musunuru, K. Permanent Alteration of PCSK9 With In Vivo CRISPR-Cas9 Genome Editing. Circ. Res. 2014, 115, 488–492. [Google Scholar] [CrossRef]
- Rashid, S.; Curtis, D.E.; Garuti, R.; Anderson, N.N.; Bashmakov, Y.; Ho, Y.K.; Hammer, R.E.; Moon, Y.-A.; Horton, J.D. Decreased Plasma Cholesterol and Hypersensitivity to Statins in Mice Lacking Pcsk9. Proc. Natl. Acad. Sci. USA 2005, 102, 5374–5379. [Google Scholar] [CrossRef]
- Bennett, C.F.; Swayze, E.E. RNA Targeting Therapeutics: Molecular Mechanisms of Antisense Oligonucleotides as a Therapeutic Platform. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 259–293. [Google Scholar] [CrossRef]
- Frank-Kamenetsky, M.; Grefhorst, A.; Anderson, N.N.; Racie, T.S.; Bramlage, B.; Akinc, A.; Butler, D.; Charisse, K.; Dorkin, R.; Fan, Y.; et al. Therapeutic RNAi Targeting PCSK9 Acutely Lowers Plasma Cholesterol in Rodents and LDL Cholesterol in Nonhuman Primates. Proc. Natl. Acad. Sci. USA 2008, 105, 11915–11920. [Google Scholar] [CrossRef]
- Susan-Resiga, D.; Girard, E.; Essalmani, R.; Roubtsova, A.; Marcinkiewicz, J.; Derbali, R.M.; Evagelidis, A.; Byun, J.H.; Lebeau, P.F.; Austin, R.C.; et al. Asialoglycoprotein Receptor 1 Is a Novel PCSK9-Independent Ligand of Liver LDLR Cleaved by Furin. J. Biol. Chem. 2021, 297, 101177. [Google Scholar] [CrossRef]
- Lamb, Y.N. Inclisiran: First Approval. Drugs 2021, 81, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Lintner, N.G.; McClure, K.F.; Petersen, D.; Londregan, A.T.; Piotrowski, D.W.; Wei, L.; Xiao, J.; Bolt, M.; Loria, P.M.; Maguire, B.; et al. Selective Stalling of Human Translation through Small-Molecule Engagement of the Ribosome Nascent Chain. PLoS Biol. 2017, 15, e2001882. [Google Scholar] [CrossRef] [PubMed]
- Gustafsen, C.; Kjolby, M.; Nyegaard, M.; Mattheisen, M.; Lundhede, J.; Buttenschøn, H.; Mors, O.; Bentzon, J.F.; Madsen, P.; Nykjaer, A.; et al. The Hypercholesterolemia-Risk Gene SORT1 Facilitates PCSK9 Secretion. Cell Metab. 2014, 19, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Butkinaree, C.; Canuel, M.; Essalmani, R.; Poirier, S.; Benjannet, S.; Asselin, M.-C.; Roubtsova, A.; Hamelin, J.; Marcinkiewicz, J.; Chamberland, A.; et al. Amyloid Precursor-like Protein 2 and Sortilin Do Not Regulate the PCSK9 Convertase-Mediated Low Density Lipoprotein Receptor Degradation but Interact with Each Other. J. Biol. Chem. 2015, 290, 18609–18620. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Targeting Strategy | Drug/Approach | Type of Drug/Approach | Development Stage | Reference |
|---|---|---|---|---|
| Blocking PCSK9 binding to LDLR | Evolocumab (Rephata) | mAb | FDA-approved for treating hypercholesterolemia +/− Statins | [119,120,121] |
| Alirocumab (Praluent) | mAb | FDA-approved for treating hypercholesterolemia +/− Statins | [119,122,123] | |
| L-IFPTA+ | Nanoliposomal vaccine | Tested in vivo on mice Pre-clinically tested in non-human primates | [124,125,126] [127] | |
| BMS-962476 | Adnectin | Pre-clinical trial carried out on cynomolgus monkeys Clinically tested in humans | [128] [129] | |
| Pep2-8 | mimetic peptides | Tested in vitro | [130] | |
| Annexin A2 | mimetic peptides, natural inhibitor | Tested in vivo | [131] | |
| Pseurotin A | Fungal metabolite | Tested in vitro and in vivo | [132] | |
| Silibinin A | TCM: polyphenol | Clinically used for treating liver diseases Tested in vitro +/− Statins | [133] [134] | |
| MK-0616 | Tricyclic peptide | Tested in humans Phase I and II clinical trials | [135] [136] | |
| Inhibiting PCSK9 expression | CRISPR/Cas9 | Genome editing | Tested in vitro and in vivo in cynomolgus monkeys and macaques | [137,138] |
| ISIS 394814/BMS 844421 | ASO | Pre-clinically evaluated in vivo (terminated) | [139] | |
| SPC4061/SPC5001 | ASO | Tested in vivo Clinically tested in humans but caused acute kidney injury (terminated) | [140,141] [142] | |
| Inclisiran (lequivo®) | siRNA | Approved in Europe Passed all three phase III ORION studies 160 Pending FDA approval | [143,144] [145] [146,147] | |
| R-IMPP | Pharmaceutical Molecule | Tested in vitro | [148] | |
| PF-06446846 | Pharmaceutical Molecule | Tested in vivo | [149] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alannan, M.; Seidah, N.G.; Merched, A.J. PCSK9 in Liver Cancers at the Crossroads between Lipid Metabolism and Immunity. Cells 2022, 11, 4132. https://doi.org/10.3390/cells11244132
Alannan M, Seidah NG, Merched AJ. PCSK9 in Liver Cancers at the Crossroads between Lipid Metabolism and Immunity. Cells. 2022; 11(24):4132. https://doi.org/10.3390/cells11244132
Chicago/Turabian StyleAlannan, Malak, Nabil G. Seidah, and Aksam J. Merched. 2022. "PCSK9 in Liver Cancers at the Crossroads between Lipid Metabolism and Immunity" Cells 11, no. 24: 4132. https://doi.org/10.3390/cells11244132
APA StyleAlannan, M., Seidah, N. G., & Merched, A. J. (2022). PCSK9 in Liver Cancers at the Crossroads between Lipid Metabolism and Immunity. Cells, 11(24), 4132. https://doi.org/10.3390/cells11244132