

Vascular Smooth Muscle Cells Phenotypic Switching in Cardiovascular Diseases


 ,
,
Abstract
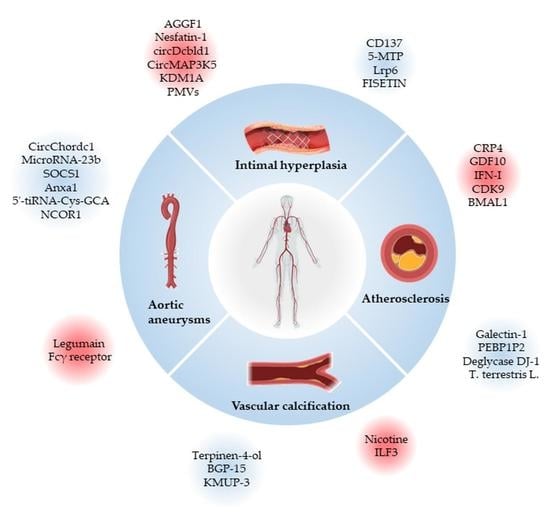
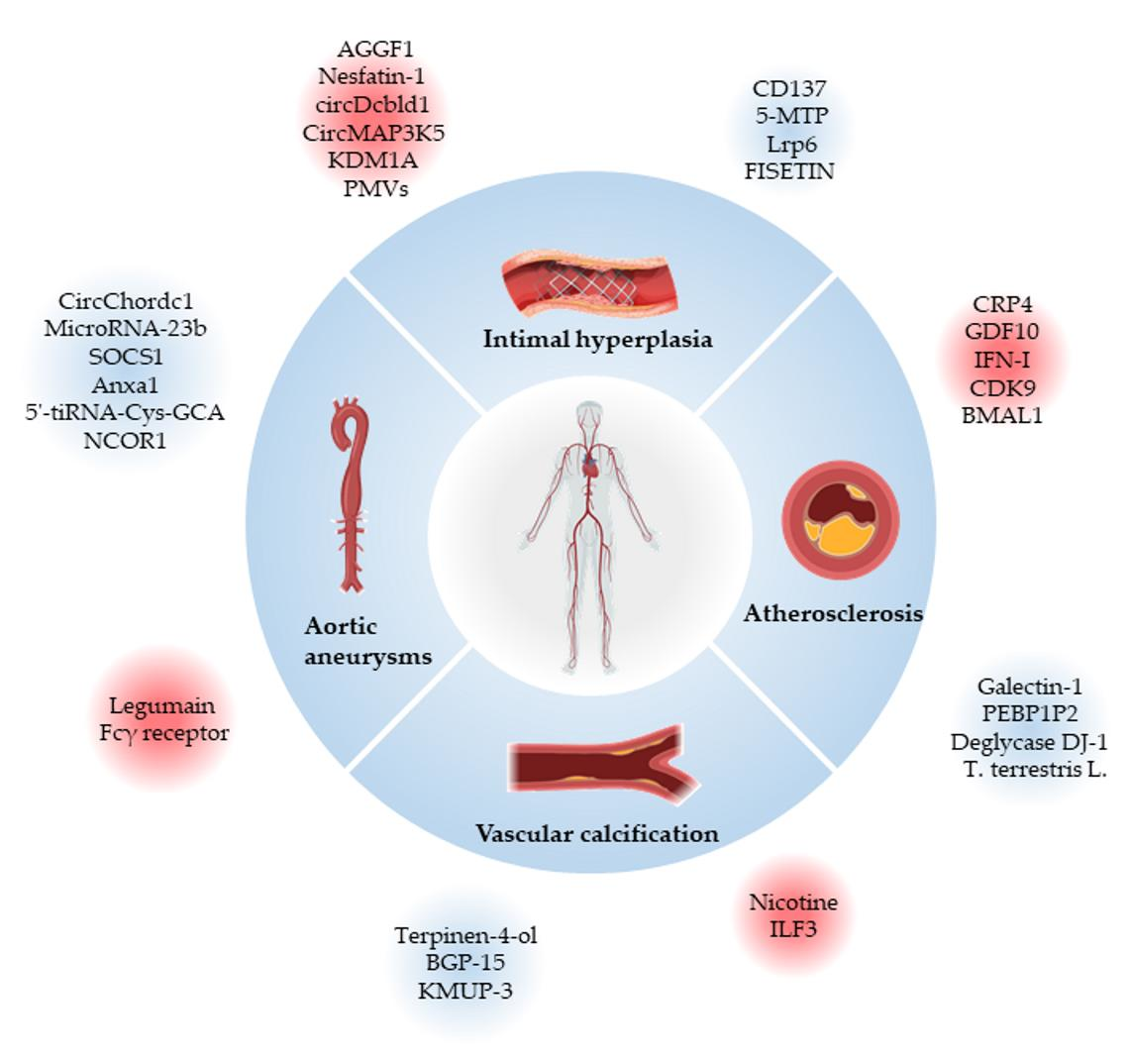
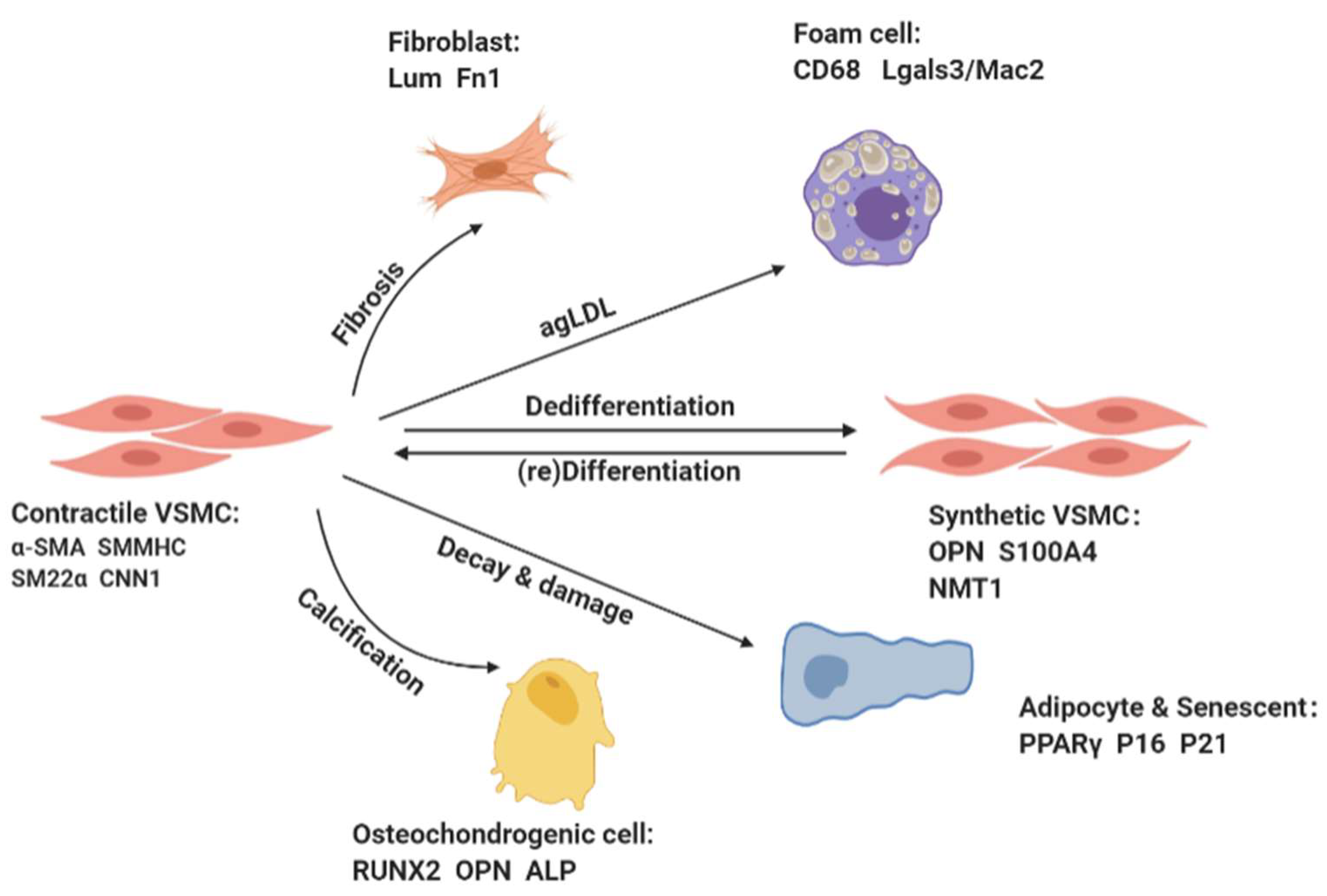
1. Vascular Smooth Muscle Cells
2. Cardiovascular Diseases Associated with VSMCs Phenotypic Switching
2.1. Atherosclerosis
2.2. Intimal Hyperplasia and Restenosis
2.3. Aortic Aneurysms
2.4. Vascular Calcification
3. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- He, X.; Deng, J.; Yu, X.J.; Yang, S.; Yang, Y.; Zang, W.J. Activation of M3AChR (Type 3 Muscarinic Acetylcholine Receptor) and Nrf2 (Nuclear Factor Erythroid 2-Related Factor 2) Signaling by Choline Alleviates Vascular Smooth Muscle Cell Phenotypic Switching and Vascular Remodeling. Arter. Thromb. Vasc. Biol. 2020, 40, 2649–2664. [Google Scholar] [CrossRef] [PubMed]
- Farina, F.M.; Hall, I.F.; Serio, S.; Zani, S.; Climent, M.; Salvarani, N.; Carullo, P.; Civilini, E.; Condorelli, G.; Elia, L.; et al. miR-128-3p Is a Novel Regulator of Vascular Smooth Muscle Cell Phenotypic Switch and Vascular Diseases. Circ. Res. 2020, 126, e120–e135. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Son, M.; Park, C.H.; Jang, J.T.; Son, K.H.; Byun, K. Pyrogallol-Phloroglucinol-6,6-Bieckolon Attenuates Vascular Smooth Muscle Cell Proliferation and Phenotype Switching in Hyperlipidemia through Modulation of Chemokine Receptor 5. Mar. Drugs 2020, 18, 393. [Google Scholar] [CrossRef] [PubMed]
- Tierney, J.W.; Evans, B.C.; Cheung-Flynn, J.; Wang, B.; Colazo, J.M.; Polcz, M.E.; Cook, R.S.; Brophy, C.M.; Duvall, C.L. Therapeutic MK2 inhibition blocks pathological vascular smooth muscle cell phenotype switch. JCI Insight 2021, 6, 142339. [Google Scholar] [CrossRef]
- He, X.; Lian, Z.; Yang, Y.; Wang, Z.; Fu, X.; Liu, Y.; Li, M.; Tian, J.; Yu, T.; Xin, H. Long Non-coding RNA PEBP1P2 Suppresses Proliferative VSMCs Phenotypic Switching and Proliferation in Atherosclerosis. Mol. Ther. Nucleic Acids 2020, 22, 84–98. [Google Scholar] [CrossRef]
- Zhang, C.Y.; Hu, Y.C.; Zhang, Y.; Ma, W.D.; Song, Y.F.; Quan, X.H.; Guo, X.; Wang, C.X. Glutamine switches vascular smooth muscle cells to synthetic phenotype through inhibiting miR-143 expression and upregulating THY1 expression. Life Sci. 2021, 277, 119365. [Google Scholar] [CrossRef]
- Zhang, X.; Huang, T.; Zhai, H.; Peng, W.; Zhou, Y.; Li, Q.; Yang, H. Inhibition of lysine-specific demethylase 1A suppresses neointimal hyperplasia by targeting bone morphogenetic protein 2 and mediating vascular smooth muscle cell phenotype. Cell Prolif. 2020, 53, e12711. [Google Scholar] [CrossRef]
- Shanahan, C.M.; Crouthamel, M.H.; Kapustin, A.; Giachelli, C.M. Arterial calcification in chronic kidney disease: Key roles for calcium and phosphate. Circ. Res. 2011, 109, 697–711. [Google Scholar] [CrossRef]
- Frismantiene, A.; Philippova, M.; Erne, P.; Resink, T.J. Smooth muscle cell-driven vascular diseases and molecular mechanisms of VSMC plasticity. Cell Signal. 2018, 52, 48–64. [Google Scholar] [CrossRef]
- Allahverdian, S.; Chaabane, C.; Boukais, K.; Francis, G.A.; Bochaton-Piallat, M.L. Smooth muscle cell fate and plasticity in atherosclerosis. Cardiovasc. Res. 2018, 114, 540–550. [Google Scholar] [CrossRef]
- Minty, A.; Kedes, L. Upstream regions of the human cardiac actin gene that modulate its transcription in muscle cells: Presence of an evolutionarily conserved repeated motif. Mol. Cell Biol. 1986, 6, 2125–2136. [Google Scholar] [CrossRef] [PubMed]
- Treisman, R. Identification of a protein-binding site that mediates transcriptional response of the c-fos gene to serum factors. Cell 1986, 46, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Wamhoff, B.R.; Hoofnagle, M.H.; Burns, A.; Sinha, S.; McDonald, O.G.; Owens, G.K. A G/C element mediates repression of the SM22alpha promoter within phenotypically modulated smooth muscle cells in experimental atherosclerosis. Circ. Res. 2004, 95, 981–988. [Google Scholar] [CrossRef]
- Van Der Heide, L.P.; Hoekman, M.F.; Smidt, M.P. The ins and outs of FoxO shuttling: Mechanisms of FoxO translocation and transcriptional regulation. Biochem. J. 2004, 380, 297–309. [Google Scholar] [CrossRef]
- Tang, Y.; Yu, S.; Liu, Y.; Zhang, J.; Han, L.; Xu, Z. MicroRNA-124 controls human vascular smooth muscle cell phenotypic switch via Sp1. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H641–H649. [Google Scholar] [CrossRef]
- Torella, D.; Iaconetti, C.; Catalucci, D.; Ellison, G.M.; Leone, A.; Waring, C.D.; Bochicchio, A.; Vicinanza, C.; Aquila, I.; Curcio, A.; et al. MicroRNA-133 controls vascular smooth muscle cell phenotypic switch in vitro and vascular remodeling in vivo. Circ. Res. 2011, 109, 880–893. [Google Scholar] [CrossRef]
- Sun, H.; Cai, S.; Zhang, M.; Zhao, J.; Wei, S.; Luo, Y.; Meng, X.; Zhou, X.; Li, Y.; Zhang, W. MicroRNA-206 regulates vascular smooth muscle cell phenotypic switch and vascular neointimal formation. Cell Biol. Int. 2017, 41, 739–748. [Google Scholar] [CrossRef]
- Liu, X.; Cheng, Y.; Zhang, S.; Lin, Y.; Yang, J.; Zhang, C. A necessary role of miR-221 and miR-222 in vascular smooth muscle cell proliferation and neointimal hyperplasia. Circ. Res. 2009, 104, 476–487. [Google Scholar] [CrossRef]
- Zeng, Z.; Xia, L.; Fan, S.; Zheng, J.; Qin, J.; Fan, X.; Liu, Y.; Tao, J.; Liu, Y.; Li, K.; et al. Circular RNA CircMAP3K5 Acts as a MicroRNA-22-3p Sponge to Promote Resolution of Intimal Hyperplasia Via TET2-Mediated Smooth Muscle Cell Differentiation. Circulation 2021, 143, 354–371. [Google Scholar] [CrossRef]
- Rong, Z.H.; Chang, N.B.; Yao, Q.P.; Li, T.; Zhu, X.L.; Cao, Y.; Jiang, M.J.; Cheng, Y.S.; Jiang, R.; Jiang, J. Suppression of circDcbld1 Alleviates Intimal Hyperplasia in Rat Carotid Artery by Targeting miR-145-3p/Neuropilin-1. Mol. Ther. Nucleic Acids 2019, 18, 999–1008. [Google Scholar] [CrossRef]
- Hall, I.F.; Climent, M.; Quintavalle, M.; Farina, F.M.; Schorn, T.; Zani, S.; Carullo, P.; Kunderfranco, P.; Civilini, E.; Condorelli, G.; et al. Circ_Lrp6, a Circular RNA Enriched in Vascular Smooth Muscle Cells, Acts as a Sponge Regulating miRNA-145 Function. Circ. Res. 2019, 124, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Lauf, P.K.; Sharma, N.; Adragna, N.C. Kinetic studies of K-Cl cotransport in cultured rat vascular smooth muscle cells. Am. J. Physiol. Cell Physiol. 2019, 316, C274–C284. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Wang, Z.Y.; Cheng, J.; Liu, B.; Xie, F.; Li, C.L.; Qiao, W.; Lu, Q.H.; Wang, Y.; Zhang, M.X. Protein deglycase DJ-1 deficiency induces phenotypic switching in vascular smooth muscle cells and exacerbates atherosclerotic plaque instability. J. Cell Mol. Med. 2021, 25, 2816–2827. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Much, A.A.; Maor, E.; Asher, E.; Younis, A.; Xu, Y.; Lu, Y.; Liu, X.; Shu, J.; Bragazzi, N.L. Global, regional, and national burden of ischemic heart disease and its attributable risk factors, 1990–2017: Results from the global Burden of Disease Study 2017. Eur. Heart J. Qual. Care Clin. Outcomes 2020, 8, 50–60. [Google Scholar] [CrossRef]
- Grootaert, M.O.J.; Bennett, M.R. Vascular smooth muscle cells in atherosclerosis: Time for a re-assessment. Cardiovasc. Res. 2021, 117, 2326–2339. [Google Scholar] [CrossRef]
- Arora, S.; Stouffer, G.A.; Kucharska-Newton, A.M.; Qamar, A.; Vaduganathan, M.; Pandey, A.; Porterfield, D.; Blankstein, R.; Rosamond, W.D.; Bhatt, D.L.; et al. Twenty Year Trends and Sex Differences in Young Adults Hospitalized With Acute Myocardial Infarction. Circulation 2019, 139, 1047–1056. [Google Scholar] [CrossRef]
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2021 Update: A Report From the American Heart Association. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and challenges in translating the biology of atherosclerosis. Nature 2011, 473, 317–325. [Google Scholar] [CrossRef]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular Smooth Muscle Cells in Atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. A century of cholesterol and coronaries: From plaques to genes to statins. Cell 2015, 161, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Chin, D.D.; Poon, C.; Wang, J.; Joo, J.; Ong, V.; Jiang, Z.; Cheng, K.; Plotkin, A.; Magee, G.A.; Chung, E.J. miR-145 micelles mitigate atherosclerosis by modulating vascular smooth muscle cell phenotype. Biomaterials 2021, 273, 120810. [Google Scholar] [CrossRef] [PubMed]
- Rykaczewska, U.; Suur, B.E.; Röhl, S.; Razuvaev, A.; Lengquist, M.; Sabater-Lleal, M.; van der Laan, S.W.; Miller, C.L.; Wirka, R.C.; Kronqvist, M.; et al. PCSK6 Is a Key Protease in the Control of Smooth Muscle Cell Function in Vascular Remodeling. Circ. Res. 2020, 126, 571–585. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Gao, K.; Huang, P.; Tang, Z.; Nie, F.; Jia, S.; Guo, R. Bazedoxifene inhibits PDGF-BB induced VSMC phenotypic switch via regulating the autophagy level. Life Sci. 2020, 259, 118397. [Google Scholar] [CrossRef] [PubMed]
- Swiatlowska, P.; Sit, B.; Feng, Z.; Marhuenda, E.; Xanthis, I.; Zingaro, S.; Ward, M.; Zhou, X.; Xiao, Q.; Shanahan, C.; et al. Pressure and stiffness sensing together regulate vascular smooth muscle cell phenotype switching. Sci. Adv. 2022, 8, eabm3471. [Google Scholar] [CrossRef]
- Roldán-Montero, R.; Pérez-Sáez, J.M.; Cerro-Pardo, I.; Oller, J.; Martinez-Lopez, D.; Nuñez, E.; Maller, S.M.; Gutierrez-Muñoz, C.; Mendez-Barbero, N.; Escola-Gil, J.C.; et al. Galectin-1 prevents pathological vascular remodeling in atherosclerosis and abdominal aortic aneurysm. Sci. Adv. 2022, 8, eabm7322. [Google Scholar] [CrossRef]
- Längst, N.; Adler, J.; Kuret, A.; Peter, A.; Ruth, P.; Boldt, K.; Lukowski, R. Cysteine-Rich LIM-Only Protein 4 (CRP4) Promotes Atherogenesis in the ApoE−/− Mouse Model. Cells 2022, 11, 1364. [Google Scholar] [CrossRef]
- Brandt, K.J.; Burger, F.; Baptista, D.; Roth, A.; Fernandes da Silva, R.; Montecucco, F.; Mach, F.; Miteva, K. Single-Cell Analysis Uncovers Osteoblast Factor Growth Differentiation Factor 10 as Mediator of Vascular Smooth Muscle Cell Phenotypic Modulation Associated with Plaque Rupture in Human Carotid Artery Disease. Int. J. Mol. Sci. 2022, 23, 1796. [Google Scholar] [CrossRef]
- Bi, X.; Du, C.; Wang, X.; Wang, X.Y.; Han, W.; Wang, Y.; Qiao, Y.; Zhu, Y.; Ran, L.; Liu, Y.; et al. Mitochondrial Damage-Induced Innate Immune Activation in Vascular Smooth Muscle Cells Promotes Chronic Kidney Disease-Associated Plaque Vulnerability. Adv. Sci. (Weinh) 2021, 8, 2002738. [Google Scholar] [CrossRef]
- Huang, S.; Luo, W.; Wu, G.; Shen, Q.; Zhuang, Z.; Yang, D.; Qian, J.; Hu, X.; Cai, Y.; Chattipakorn, N.; et al. Inhibition of CDK9 attenuates atherosclerosis by inhibiting inflammation and phenotypic switching of vascular smooth muscle cells. Aging (Albany N. Y.) 2021, 13, 14892–14909. [Google Scholar] [CrossRef]
- Shen, Y.; Xu, L.R.; Yan, D.; Zhou, M.; Han, T.L.; Lu, C.; Tang, X.; Lin, C.P.; Qian, R.Z.; Guo, D.Q. BMAL1 modulates smooth muscle cells phenotypic switch towards fibroblast-like cells and stabilizes atherosclerotic plaques by upregulating YAP1. Biochim. Biophys. Acta Mol. Basis Dis. 2022, 1868, 166450. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhao, W.R.; Shi, W.T.; Tan, J.J.; Zhang, K.Y.; Tang, J.Y.; Chen, X.L.; Zhou, Z.Y. Tribulus terrestris L. extract ameliorates atherosclerosis by inhibition of vascular smooth muscle cell proliferation in ApoE−/− mice and A7r5 cells via suppression of Akt/MEK/ERK signaling. J. Ethnopharmacol. 2022, 297, 115547. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Zhang, Y.; Hsu, C.; Korshunov, V.A.; Long, X.; Knight, P.A.; Berk, B.C.; Yan, C. Role of PDE10A in vascular smooth muscle cell hyperplasia and pathological vascular remodeling. Cardiovasc. Res. 2021, 118, 2703–2717. [Google Scholar] [CrossRef]
- Scott, R.A.; Panitch, A. Macromolecular approaches to prevent thrombosis and intimal hyperplasia following percutaneous coronary intervention. Biomacromolecules 2014, 15, 2825–2832. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Pi, S.; Xiong, M.; Liu, Z.; Huang, X.; An, R.; Zhang, T.; Yuan, B. WD Repeat Domain 1 Deficiency Inhibits Neointima Formation in Mice Carotid Artery by Modulation of Smooth Muscle Cell Migration and Proliferation. Mol. Cells 2020, 43, 749–762. [Google Scholar] [CrossRef]
- Liu, J.T.; Bao, H.; Fan, Y.J.; Li, Z.T.; Yao, Q.P.; Han, Y.; Zhang, M.L.; Jiang, Z.L.; Qi, Y.X. Platelet-Derived Microvesicles Promote VSMC Dedifferentiation After Intimal Injury via Src/Lamtor1/mTORC1 Signaling. Front. Cell Dev. Biol. 2021, 9, 744320. [Google Scholar] [CrossRef]
- Gao, X.F.; Chen, A.Q.; Wang, Z.M.; Wang, F.; Luo, S.; Chen, S.Y.; Gu, Y.; Kong, X.Q.; Zuo, G.F.; Chen, Y.; et al. Single-Cell RNA Sequencing of the Rat Carotid Arteries Uncovers Potential Cellular Targets of Neointimal Hyperplasia. Front. Cardiovasc. Med. 2021, 8, 751525. [Google Scholar] [CrossRef]
- Zhu, X.L.; Li, T.; Cao, Y.; Yao, Q.P.; Liu, X.; Li, Y.; Guan, Y.Y.; Deng, J.J.; Jiang, R.; Jiang, J. tRNA-derived fragments tRF(GlnCTG) induced by arterial injury promote vascular smooth muscle cell proliferation. Mol. Ther. Nucleic Acids 2021, 23, 603–613. [Google Scholar] [CrossRef]
- Qiu, L.; Hu, L.; Liu, X.; Li, W.; Zhang, X.; Xia, H.; Zhang, C. Physalin B inhibits PDGF-BB-induced VSMC proliferation, migration and phenotypic transformation by activating the Nrf2 pathway. Food Funct. 2021, 12, 10950–10966. [Google Scholar] [CrossRef]
- Ye, B.; Wu, Z.H.; Tsui, T.Y.; Zhang, B.F.; Su, X.; Qiu, Y.H.; Zheng, X.T. lncRNA KCNQ1OT1 Suppresses the Inflammation and Proliferation of Vascular Smooth Muscle Cells through IκBa in Intimal Hyperplasia. Mol. Ther. Nucleic Acids 2020, 20, 62–72. [Google Scholar] [CrossRef]
- Ran, R.; Cai, D.; King, S.D.; Que, X.; Bath, J.M.; Chen, S.Y. Surfactant Protein A, a Novel Regulator for Smooth Muscle Phenotypic Modulation and Vascular Remodeling-Brief Report. Arter. Thromb. Vasc. Biol. 2021, 41, 808–814. [Google Scholar] [CrossRef] [PubMed]
- Long, F.; Yang, D.; Wang, J.; Wang, Q.; Ni, T.; Wei, G.; Zhu, Y.; Liu, X. SMYD3-PARP16 axis accelerates unfolded protein response and mediates neointima formation. Acta Pharm. Sin. B 2021, 11, 1261–1273. [Google Scholar] [CrossRef] [PubMed]
- Han, J.H.; Heo, K.S.; Myung, C.S. Cytokine-induced apoptosis inhibitor 1 (CIAPIN1) accelerates vascular remodelling via p53 and JAK2-STAT3 regulation in vascular smooth muscle cells. Br. J. Pharmacol. 2021, 178, 4533–4551. [Google Scholar] [CrossRef] [PubMed]
- Riascos-Bernal, D.F. Perking Up Strategies to Control Restenosis. JACC Basic Transl. Sci. 2020, 5, 264–266. [Google Scholar] [CrossRef]
- Pei, F.; Pei, H.; Su, C.; Du, L.; Wang, J.; Xie, F.; Yin, Q.; Gao, Z. Fisetin Alleviates Neointimal Hyperplasia via PPARγ/PON2 Antioxidative Pathway in SHR Rat Artery Injury Model. Oxid. Med. Cell Longev. 2021, 2021, 6625517. [Google Scholar] [CrossRef]
- Jain, M.; Dhanesha, N.; Doddapattar, P.; Chorawala, M.R.; Nayak, M.K.; Cornelissen, A.; Guo, L.; Finn, A.V.; Lentz, S.R.; Chauhan, A.K. Smooth muscle cell–specific fibronectin-EDA mediates phenotypic switching and neointimal hyperplasia. J. Clin. Investig. 2019, 130, 295–314. [Google Scholar] [CrossRef]
- Ji, Z.; Li, J.; Wang, J. Jujuboside B Inhibits Neointimal Hyperplasia and Prevents Vascular Smooth Muscle Cell Dedifferentiation, Proliferation, and Migration via Activation of AMPK/PPAR-γ Signaling. Front. Pharmacol. 2021, 12, 672150. [Google Scholar] [CrossRef]
- Ding, X.; Yan, Y.; Zhang, C.; Xu, X.; Yang, F.; Liu, Y.; Wang, G.; Qin, Y. OCT4 regulated neointimal formation in injured mouse arteries by matrix metalloproteinase 2-mediated smooth muscle cells proliferation and migration. J. Cell Physiol. 2021, 236, 5421–5431. [Google Scholar] [CrossRef]
- Kural, M.H.; Wang, J.; Gui, L.; Yuan, Y.; Li, G.; Leiby, K.L.; Quijano, E.; Tellides, G.; Saltzman, W.M.; Niklason, L.E. Fas ligand and nitric oxide combination to control smooth muscle growth while sparing endothelium. Biomaterials 2019, 212, 28–38. [Google Scholar] [CrossRef]
- Yu, Y.; Li, Y.; Peng, H.; Song, Q.; Da, X.; Li, H.; He, Z.; Ren, X.; Xu, C.; Yao, Y.; et al. Angiogenic factor AGGF1 blocks neointimal formation after vascular injury via interaction with integrin α7 on vascular smooth muscle cells. J. Biol. Chem. 2022, 298, 101759. [Google Scholar] [CrossRef]
- Zhang, J.R.; Lu, Q.B.; Feng, W.B.; Wang, H.P.; Tang, Z.H.; Cheng, H.; Du, Q.; Wang, Y.B.; Li, K.X.; Sun, H.J. Nesfatin-1 promotes VSMC migration and neointimal hyperplasia by upregulating matrix metalloproteinases and downregulating PPARγ. Biomed. Pharm. 2018, 102, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zang, G.; Zhong, W.; Chen, R.; Zhang, Y.; Yang, P.; Yan, J. Activation of CD137 signaling promotes neointimal formation by attenuating TET2 and transferrring from endothelial cell-derived exosomes to vascular smooth muscle cells. Biomed. Pharm. 2020, 121, 109593. [Google Scholar] [CrossRef]
- Chen, C.H.; Ho, Y.C.; Ho, H.H.; Liang, L.Y.; Jiang, W.C.; Lee, G.L.; Lee, J.K.; Hsu, Y.J.; Kuo, C.C.; Wu, K.K.; et al. Tryptophan metabolite 5-methoxytryptophan ameliorates arterial denudation-induced intimal hyperplasia via opposing effects on vascular endothelial and smooth muscle cells. Aging (Albany N. Y.) 2019, 11, 8604–8622. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Chen, Q.; He, S.; Yang, M.; Maguire, E.M.; An, W.; Afzal, T.A.; Luong, L.A.; Zhang, L.; Xiao, Q. miR-22 Is a Novel Mediator of Vascular Smooth Muscle Cell Phenotypic Modulation and Neointima Formation. Circulation 2018, 137, 1824–1841. [Google Scholar] [CrossRef] [PubMed]
- Rombouts, K.B.; van Merrienboer, T.A.R.; Ket, J.C.F.; Bogunovic, N.; van der Velden, J.; Yeung, K.K. The role of vascular smooth muscle cells in the development of aortic aneurysms and dissections. Eur. J. Clin. Investig. 2021, 52, e13697. [Google Scholar] [CrossRef]
- Yang, K.; Ren, J.; Li, X.; Wang, Z.; Xue, L.; Cui, S.; Sang, W.; Xu, T.; Zhang, J.; Yu, J.; et al. Prevention of aortic dissection and aneurysm via an ALDH2-mediated switch in vascular smooth muscle cell phenotype. Eur. Heart J. 2020, 41, 2442–2453. [Google Scholar] [CrossRef]
- Oliver-Williams, C.; Sweeting, M.J.; Turton, G.; Parkin, D.; Cooper, D.; Rodd, C.; Thompson, S.G.; Earnshaw, J.J. Lessons learned about prevalence and growth rates of abdominal aortic aneurysms from a 25-year ultrasound population screening programme. Br. J. Surg. 2018, 105, 68–74. [Google Scholar] [CrossRef]
- Si, X.; Chen, Q.; Zhang, J.; Zhou, W.; Chen, L.; Chen, J.; Deng, N.; Li, W.; Liu, D.; Wang, L.; et al. MicroRNA-23b prevents aortic aneurysm formation by inhibiting smooth muscle cell phenotypic switching via FoxO4 suppression. Life Sci. 2021, 288, 119092. [Google Scholar] [CrossRef]
- Ailawadi, G.; Moehle, C.W.; Pei, H.; Walton, S.P.; Yang, Z.; Kron, I.L.; Lau, C.L.; Owens, G.K. Smooth muscle phenotypic modulation is an early event in aortic aneurysms. J. Thorac. Cardiovasc. Surg. 2009, 138, 1392–1399. [Google Scholar] [CrossRef]
- Lu, W.; Zhou, Y.; Zeng, S.; Zhong, L.; Zhou, S.; Song, H.; Ding, R.; Zhong, G.; Li, Q.; Hu, Y.; et al. Loss of FoxO3a prevents aortic aneurysm formation through maintenance of VSMC homeostasis. Cell Death Dis. 2021, 12, 378. [Google Scholar] [CrossRef]
- Pan, L.; Bai, P.; Weng, X.; Liu, J.; Chen, Y.; Chen, S.; Ma, X.; Hu, K.; Sun, A.; Ge, J. Legumain Is an Endogenous Modulator of Integrin αvβ3 Triggering Vascular Degeneration, Dissection, and Rupture. Circulation 2022, 145, 659–674. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Li, X.; Han, Y.; Chen, G.; Xu, T.; Cai, D.; Sun, Y.; Wang, S.; Lai, Y.; Teng, Z.; et al. CircRNA Chordc1 protects mice from abdominal aortic aneurysm by contributing to the phenotype and growth of vascular smooth muscle cells. Mol. Ther. Nucleic Acids 2022, 27, 81–98. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Sanz, L.; Bernal, S.; Jimenez-Castilla, L.; Prieto, I.; La Manna, S.; Gomez-Lopez, S.; Blanco-Colio, L.M.; Egido, J.; Martin-Ventura, J.L.; Gomez-Guerrero, C. Fcγ receptor activation mediates vascular inflammation and abdominal aortic aneurysm development. Clin. Transl. Med. 2021, 11, e463. [Google Scholar] [CrossRef] [PubMed]
- Bernal, S.; Lopez-Sanz, L.; Jimenez-Castilla, L.; Prieto, I.; Melgar, A.; La Manna, S.; Martin-Ventura, J.L.; Blanco-Colio, L.M.; Egido, J.; Gomez-Guerrero, C. Protective effect of suppressor of cytokine signalling 1-based therapy in experimental abdominal aortic aneurysm. Br. J. Pharmacol. 2021, 178, 564–581. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Lin, Z.; Cao, H.; Chen, Y.; Li, J.; Zhuang, X.; Ma, D.; Ji, L.; Li, W.; Xu, S.; et al. Anxa1 in smooth muscle cells protects against acute aortic dissection. Cardiovasc. Res. 2021, 118, 1564–1582. [Google Scholar] [CrossRef] [PubMed]
- Zong, T.; Yang, Y.; Lin, X.; Jiang, S.; Zhao, H.; Liu, M.; Meng, Y.; Li, Y.; Zhao, L.; Tang, G.; et al. 5’-tiRNA-Cys-GCA regulates VSMC proliferation and phenotypic transition by targeting STAT4 in aortic dissection. Mol. Ther. Nucleic Acids 2021, 26, 295–306. [Google Scholar] [CrossRef]
- Du, L.J.; Sun, J.Y.; Zhang, W.C.; Liu, Y.; Liu, Y.; Lin, W.Z.; Liu, T.; Zhu, H.; Wang, Y.L.; Shao, S.; et al. NCOR1 maintains the homeostasis of vascular smooth muscle cells and protects against aortic aneurysm. Cell Death Differ. 2022. [Google Scholar] [CrossRef]
- Zheng, H.; Qiu, Z.; Chai, T.; He, J.; Zhang, Y.; Wang, C.; Ye, J.; Wu, X.; Li, Y.; Zhang, L.; et al. Insulin Resistance Promotes the Formation of Aortic Dissection by Inducing the Phenotypic Switch of Vascular Smooth Muscle Cells. Front. Cardiovasc. Med. 2021, 8, 732122. [Google Scholar] [CrossRef]
- Petsophonsakul, P.; Furmanik, M.; Forsythe, R.; Dweck, M.; Schurink, G.W.; Natour, E.; Reutelingsperger, C.; Jacobs, M.; Mees, B.; Schurgers, L. Role of Vascular Smooth Muscle Cell Phenotypic Switching and Calcification in Aortic Aneurysm Formation. Arter. Thromb. Vasc. Biol. 2019, 39, 1351–1368. [Google Scholar] [CrossRef]
- Lacolley, P.; Regnault, V.; Segers, P.; Laurent, S. Vas.s.scular Smooth Muscle Cells and Arterial Stiffening: Relevance in Development, Aging, and Disease. Physiol. Rev. 2017, 97, 1555–1617. [Google Scholar] [CrossRef]
- Durham, A.L.; Speer, M.Y.; Scatena, M.; Giachelli, C.M.; Shanahan, C.M. Role of smooth muscle cells in vascular calcification: Implications in atherosclerosis and arterial stiffness. Cardiovasc. Res. 2018, 114, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Schurgers, L.J.; Akbulut, A.C.; Kaczor, D.M.; Halder, M.; Koenen, R.R.; Kramann, R. Initiation and Propagation of Vascular Calcification Is Regulated by a Concert of Platelet- and Smooth Muscle Cell-Derived Extracellular Vesicles. Front. Cardiovasc. Med. 2018, 5, 36. [Google Scholar] [CrossRef] [PubMed]
- Jaminon, A.; Reesink, K.; Kroon, A.; Schurgers, L. The Role of Vascular Smooth Muscle Cells in Arterial Remodeling: Focus on Calcification-Related Processes. Int. J. Mol. Sci. 2019, 20, 5694. [Google Scholar] [CrossRef] [PubMed]
- Neven, E.; D’Haese, P.C. Vascular calcification in chronic renal failure: What have we learned from animal studies? Circ. Res. 2011, 108, 249–264. [Google Scholar] [CrossRef]
- Pawade, T.A.; Newby, D.E.; Dweck, M.R. Calcification in Aortic Stenosis: The Skeleton Key. J. Am. Coll. Cardiol. 2015, 66, 561–577. [Google Scholar] [CrossRef]
- Goettsch, C.; Hutcheson, J.D.; Aikawa, M.; Iwata, H.; Pham, T.; Nykjaer, A.; Kjolby, M.; Rogers, M.; Michel, T.; Shibasaki, M.; et al. Sortilin mediates vascular calcification via its recruitment into extracellular vesicles. J. Clin. Invest. 2016, 126, 1323–1336. [Google Scholar] [CrossRef]
- Carlo, A.S.; Nykjaer, A.; Willnow, T.E. Sorting receptor sortilin-a culprit in cardiovascular and neurological diseases. J. Mol. Med. (Berl.) 2014, 92, 905–911. [Google Scholar] [CrossRef]
- Petsophonsakul, P.; Burgmaier, M.; Willems, B.; Heeneman, S.; Stadler, N.; Gremse, F.; Reith, S.; Burgmaier, K.; Kahles, F.; Marx, N.; et al. Nicotine promotes vascular calcification via intracellular Ca2+-mediated, Nox5-induced oxidative stress and extracellular vesicle release in vascular smooth muscle cells. Cardiovasc. Res. 2021, 118, 2196–2210. [Google Scholar] [CrossRef]
- Zhang, Y.; He, L.; Tu, M.; Huang, M.; Chen, Y.; Pan, D.; Peng, J.; Shen, X. The ameliorative effect of terpinen-4-ol on ER stress-induced vascular calcification depends on SIRT1-mediated regulation of PERK acetylation. Pharmacol. Res. 2021, 170, 105629. [Google Scholar] [CrossRef]
- Xie, F.; Cui, Q.K.; Wang, Z.Y.; Liu, B.; Qiao, W.; Li, N.; Cheng, J.; Hou, Y.M.; Dong, X.Y.; Wang, Y.; et al. ILF3 is responsible for hyperlipidemia-induced arteriosclerotic calcification by mediating BMP2 and STAT1 transcription. J. Mol. Cell Cardiol. 2021, 161, 39–52. [Google Scholar] [CrossRef]
- Nagy, A.; Pethő, D.; Gesztelyi, R.; Juhász, B.; Balla, G.; Szilvássy, Z.; Balla, J.; Gáll, T. BGP-15 Inhibits Hyperglycemia-Aggravated VSMC Calcification Induced by High Phosphate. Int. J. Mol. Sci. 2021, 22, 9263. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Zhang, J.; Huang, S.; Cheng, N.; Zhang, C.; Li, Y.; Wang, X.; Liu, J.; You, B.; Du, J. MicroRNA-223-3p inhibits vascular calcification and the osteogenic switch of vascular smooth muscle cells. J. Biol. Chem. 2021, 296, 100483. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.H.; Chang, C.W.; Lee, F.T.; Kuo, C.H.; Hsu, J.H.; Liu, C.P.; Wu, H.L.; Yeh, J.L. Targeting vascular smooth muscle cell dysfunction with xanthine derivative KMUP-3 inhibits abdominal aortic aneurysm in mice. Atherosclerosis 2020, 297, 16–24. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Protein/RNA | Animal Model | Downstream Molecule/ Signaling Pathway | Promotes (+)/Inhibits (−) VSMCs Phenotypic Switching | VSMCs Differentiation Markers | VSMCs Dedifferentiation/ Transdifferentiation Markers | Reference | |
|---|---|---|---|---|---|---|---|
| Galectin-1 | Mice | Severe atherosclerosis induced by pAAV/D377Y-mPCSK9 adenovirus | - | α-SMA | [36] | ||
| Cysteine-rich LIM-only protein 4 (CRP4) | Mice | ApoE−/− mice fed with Western diet | + | α-SMA | [37] | ||
| Growth Differentiation Factor 10 (GDF10) | Mice | ApoE−/− mice fed with high cholesterol diet (HCD) | + | α-SMA SMMHC | Alkaline Phosphatase RUNX2 | [38] | |
| Type-I-interferon (IFN-I) | Mice | ApoE−/− mice | + | α-SMA SMMHC CNN1 | [39] | ||
| Cyclin-dependent kinases 9 (CDK9) | Mice | ApoE−/− mice fed with high fat diet (HFD) | NF-κB | + | α-SMA | Vimentin OPN | [40] |
| PEBP1P2 | Rats | Rats carotid artery injury models | CDK9 | − | α-SMA SMMHC CNN1 | [5] | |
| Deglycase DJ-1 | Mice | ApoE−/− mice fed with western diet | KLF4 | − | α-SMA CNN1 | Vimentin OPN | [24] |
| BMAL1 | Mice | Severe atherosclerosis induced by pAAV/D377Y-mPCSK9 adenovirus, western diet | YAP1 | + | SMMHC | [41] | |
| Furostanol saponins enriched extract (FSEE) of T. terrestris L. | Mice | ApoE−/− mice fed with high fat diet (HFD) | Akt/MEK/ERK | − | α-SMA | OPN | [42] |
| Protein/RNA | Animal Model | Downstream Molecule/ Signaling Pathway | Promotes (+)/Inhibits (−) VSMCs Phenotypic Switching | VSMCs Differentiation Markers | VSMCs Dedifferentiation/ Transdifferentiation Markers | Reference | |
|---|---|---|---|---|---|---|---|
| Angiogenic factor AGGF1 | Mice | Mice carotid artery wire injury model | Integrin α7 | + | α-SMA SMMHC SM22α | [60] | |
| Nesfatin-1 | Rats | Rats carotid artery injury | MMP2/MMP-9 PPARγ | + | α-SMA SMMHC | OPN | [61] |
| CD137 | Mice | Mice carotid artery wire injury model | TET2 | − | α-SMA SMMHC CNN1 | Vimentin | [62] |
| Tryptophan metabolite 5-methoxytryptophan (5-MTP) | Mice | Mice femoral artery denudation injury model | p38 MAPK NFκB-p65 | − | α-SMA | [63] | |
| CircRNA_009723 (circDcbld1) | Mice | Common carotid artery (CCA) intima hyperplasia model | miR-145-3p/Nrp1 | + | α-SMA SMMHC CNN1 | [20] | |
| Lrp6 (lipoprotein receptor 6) | Mice | ApoE−/− mice placed in perivascular collar | miR-145 | − | α-SMA SM22α | [21] | |
| Circular mitogen-activated protein kinase 5 (CircMAP3K5) | Mice | Mice femoral artery wire denudation model | miR-22-3p/TET2 | + | [19] | ||
| Lysine (K)-specific demethylase 1A (KDM1A) | Rats | Rats carotid artery balloon injury model | BMP-2 | + | α-SMA | OPN | [7] |
| Platelet-derived microvesicles (PMVs) | Mice | Mice carotid wire injury model | Src/Lamtor1/mTORC1 | + | α-SMA SM22α CNN1 | [46] | |
| FISETIN | Rats | Rats carotid artery balloon injury model | PPARγ/PON2 | − | α-SMA | OPN | [55] |
| Protein/RNA | Animal Model | Downstream Molecule/ Signaling Pathway | Promotes (+)/ Inhibits (−) VSMCs Phenotypic Switching | VSMCs Differentiation Markers | VSMCs Dedifferentiation/ Transdifferentiation Markers | Reference | |
|---|---|---|---|---|---|---|---|
| Legumain | Mice | Mice model of BAPN-induced TAD | Integrin αvβ3 | + | SM22α SMMHC CNN1 | [71] | |
| Cysteine and histidine-rich domain containing 1 (CircChordc1) | Mice | Mice model of angiotensin (Ang) II- and CaCl2-induced AAA | Annexin A2 and glycogen synthase kinase 3 beta (GSK3β) | − | SM22α CNN1 | Vimentin OPN | [72] |
| MicroRNA-23b | Mice | Mice model of AngII-induced AAA | FoxO4 | − | α-SMA SM22α CNN1 | [68] | |
| Fcγ receptor | Mice | Mice model of aortic perfusion of elastase from porcine pancreas induced AAA | + | α-SMA SM22α | KLF4 | [73] | |
| Cytokine signaling-1 (SOCS1) | Mice | Mice model of elastase-induced AAA | JAK/STAT | − | α-SMA SM22α CNN1 | KLF4 | [74] |
| Anxa1 | Mice | Mice model of AngII-induced AAD | JunB/MYL9 | − | α-SMA SMMHC SM22α CNN1 | [75] | |
| 5’-tiRNA-Cys-GCA | Mice | Mice model of intraperitoneal injection of AngII combined with intraperitoneal injection of BAPN induced AAA | STAT4 | − | α-SMA SMMHC CNN1 | [76] | |
| Nuclear receptor corepressor1 (NCOR1) | Mice | Mice model of angiotensin (Ang) II | FoxO3a NFAT5 ATF3 | − | α-SMA SMMHC CNN1 | [77] | |
| Protein/RNA | Animal Model | Downstream Molecule/ Signaling Pathway | Promotes (+)/Inhibits (−) VSMCs Phenotypic Switching | VSMCs Differentiation Markers | VSMCs Dedifferentiation/ Transdifferentiation Markers | Reference | |
|---|---|---|---|---|---|---|---|
| Nicotine | Nox5 | + | α-SMA SM22α CNN1 | S100A4 KLF4 | [88] | ||
| Terpinen-4-ol | Mice | Mice fed with a high phosphorus diet supplemented with adenine | Sirtuin 1 (SIRT1) | − | α-SMA | RUNX2 ALP BMP2 | [89] |
| Interleukin enhancer Binding factor 3 (ILF3) | Mice | ApoE−/− mice fed with high fat diet (HFD) | BMP2 STAT1 | + | α-SMA | BMP2 RUNX2 STAT1 | [90] |
| BGP-15 | Annexin A2 | − | α-SMA | KLF-5 Msx-2 Sp7 BMP-2 | [91] | ||
| KMUP-3 | Mice | Mice model of AngII-induced AAA | AMPK | − | α-SMA | RUNX2 | [93] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, H.-Y.; Chen, A.-Q.; Zhang, H.; Gao, X.-F.; Kong, X.-Q.; Zhang, J.-J. Vascular Smooth Muscle Cells Phenotypic Switching in Cardiovascular Diseases. Cells 2022, 11, 4060. https://doi.org/10.3390/cells11244060
Tang H-Y, Chen A-Q, Zhang H, Gao X-F, Kong X-Q, Zhang J-J. Vascular Smooth Muscle Cells Phenotypic Switching in Cardiovascular Diseases. Cells. 2022; 11(24):4060. https://doi.org/10.3390/cells11244060
Chicago/Turabian StyleTang, Hao-Yue, Ai-Qun Chen, Huan Zhang, Xiao-Fei Gao, Xiang-Quan Kong, and Jun-Jie Zhang. 2022. "Vascular Smooth Muscle Cells Phenotypic Switching in Cardiovascular Diseases" Cells 11, no. 24: 4060. https://doi.org/10.3390/cells11244060
APA StyleTang, H.-Y., Chen, A.-Q., Zhang, H., Gao, X.-F., Kong, X.-Q., & Zhang, J.-J. (2022). Vascular Smooth Muscle Cells Phenotypic Switching in Cardiovascular Diseases. Cells, 11(24), 4060. https://doi.org/10.3390/cells11244060