
Subviral Dense Bodies of Human Cytomegalovirus Induce an Antiviral Type I Interferon Response

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Purification of DBs and the UV-Inactivation
2.3. Application of the Virus Supernatants and DBs to the Cells
2.4. Plasmids
2.5. Antibodies
2.6. Reagents and Kits
2.7. Generation of the HFF-Knockout Cells Using CRISPR/Cas9-Mediated Genome Editing
2.8. siRNA Knockdown
2.9. Immunoblot Analysis
2.10. Extraction of the Viral DNA
2.11. Quantitative Real-Time Polymerase Chain Reaction (TaqMan qPCR) and Quantification of the Viral DNA
2.12. Mass Spectrometry-Based Proteomics
2.13. Statistical Analysis
3. Results
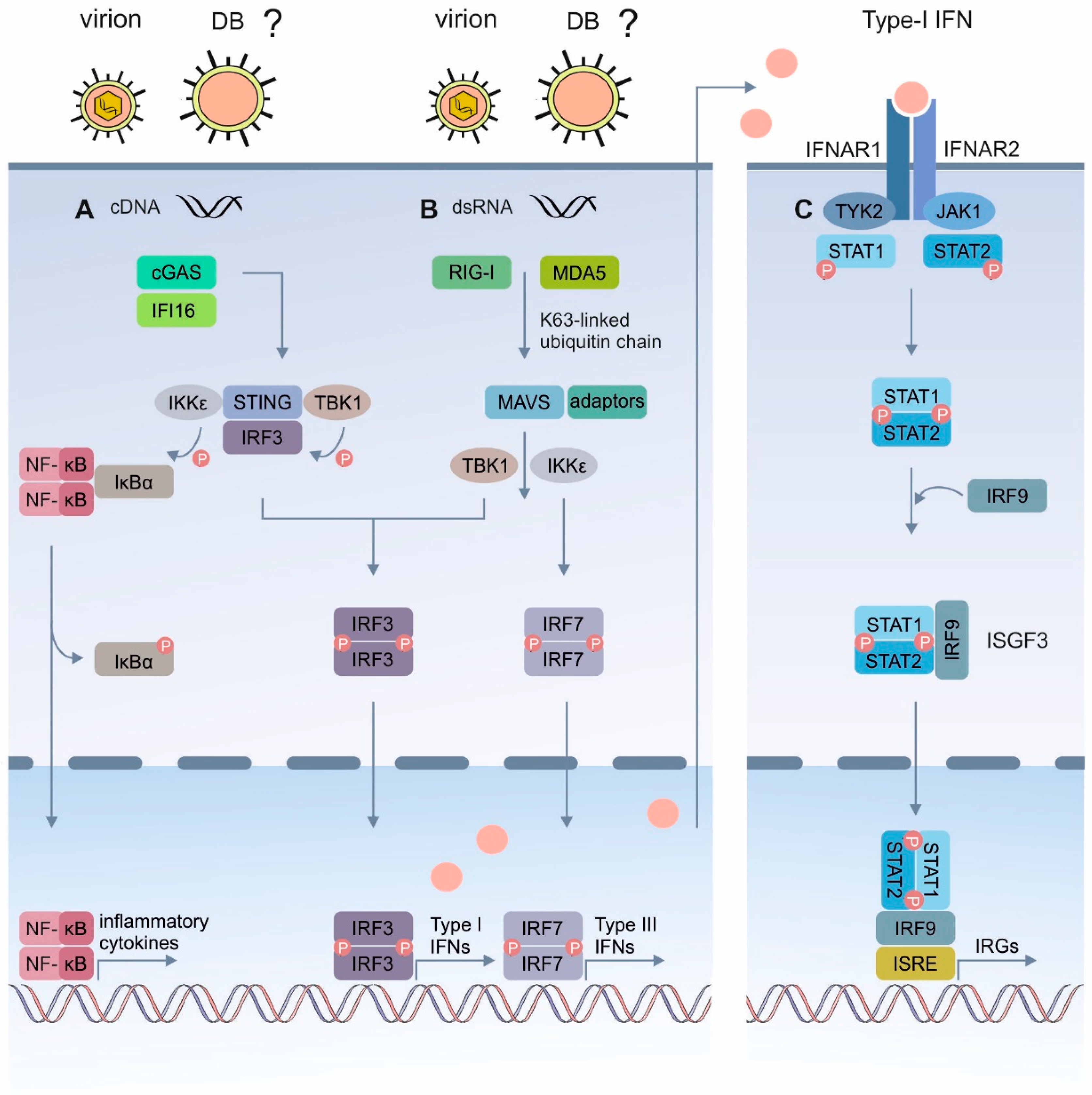
3.1. DBs Induce Antiviral IFN-Type I Responses
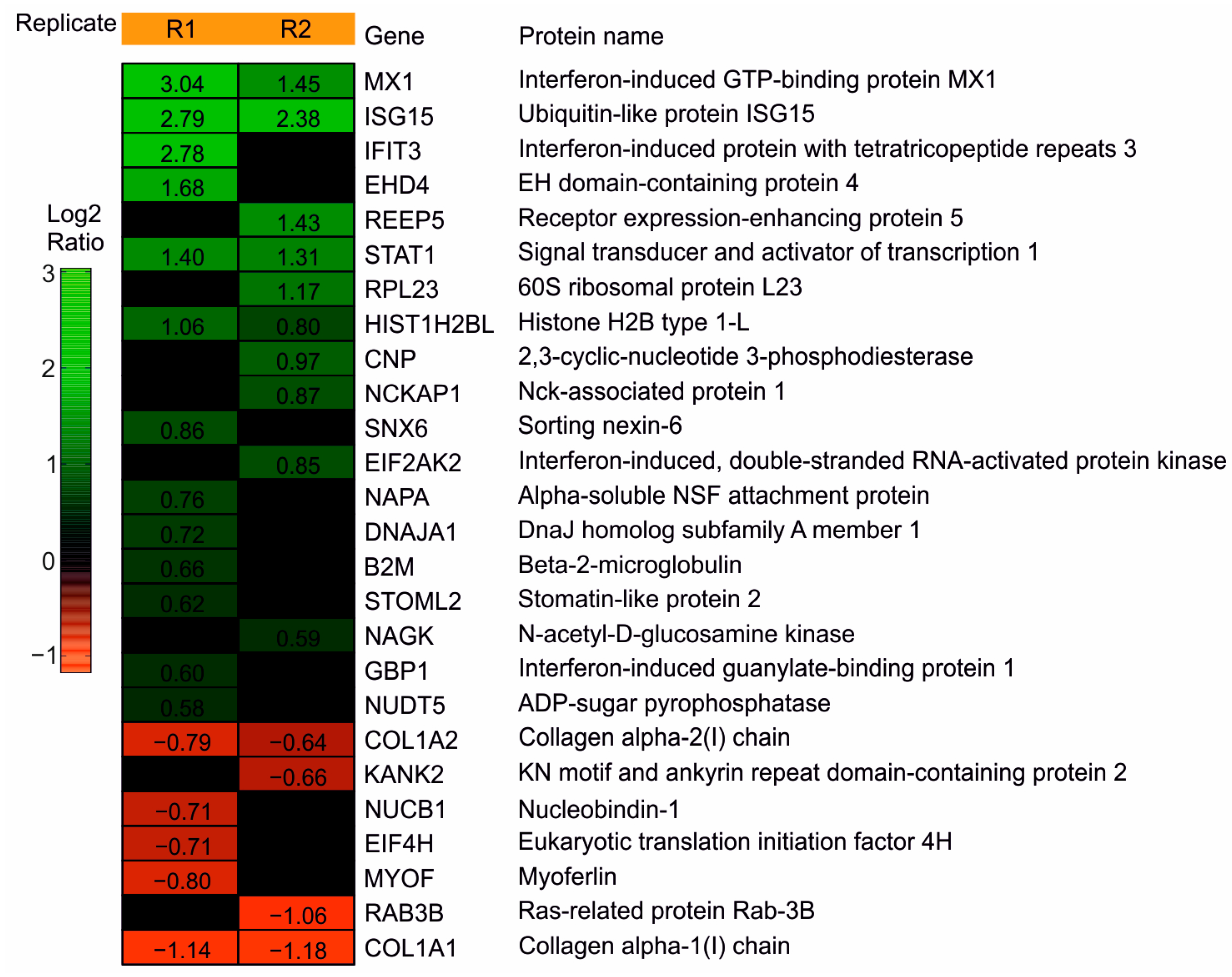
3.1.1. Mass Spec Analysis Reveals the IRGs to Be Differentially Regulated following the DB Exposure of HFFs
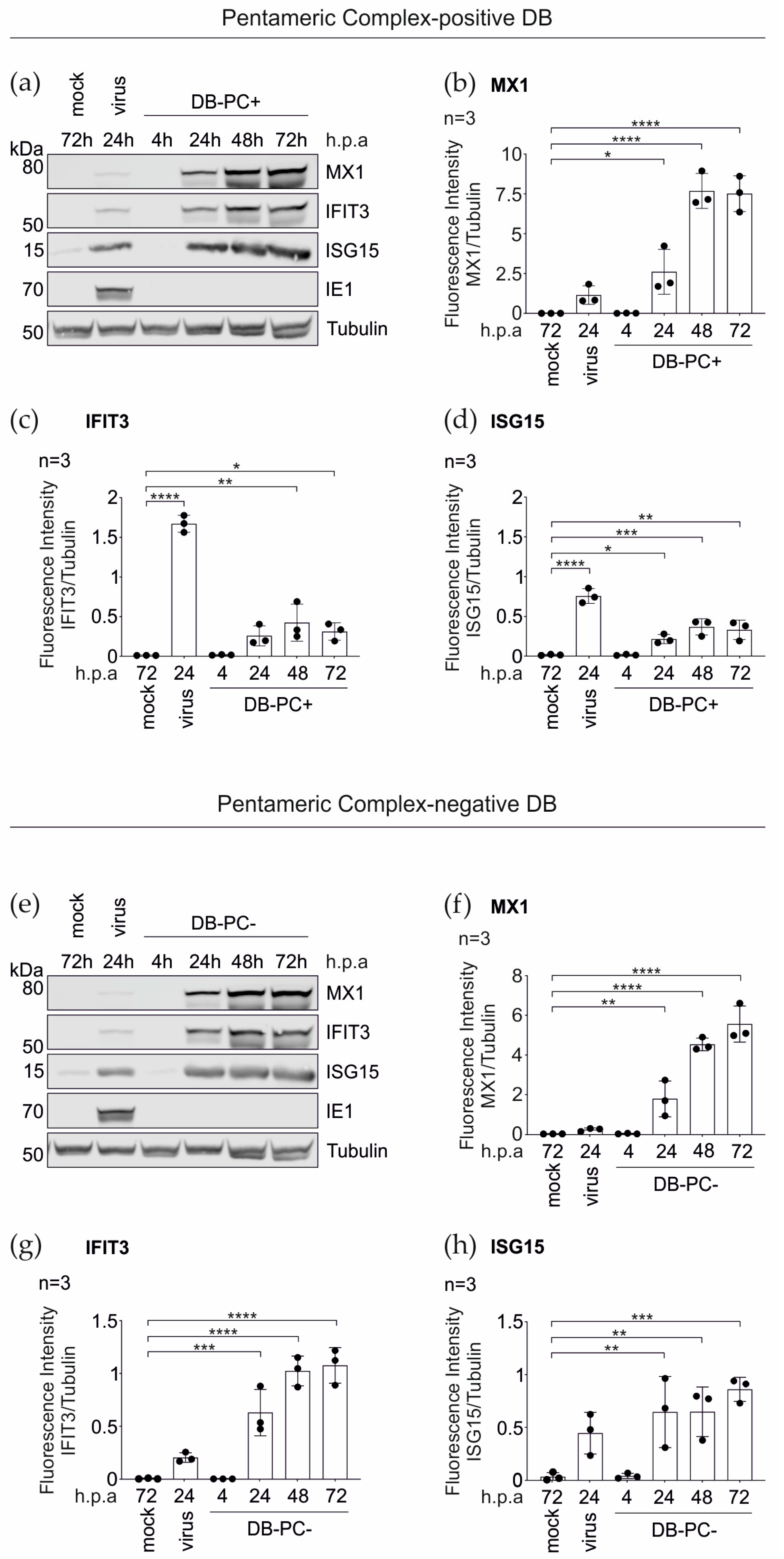
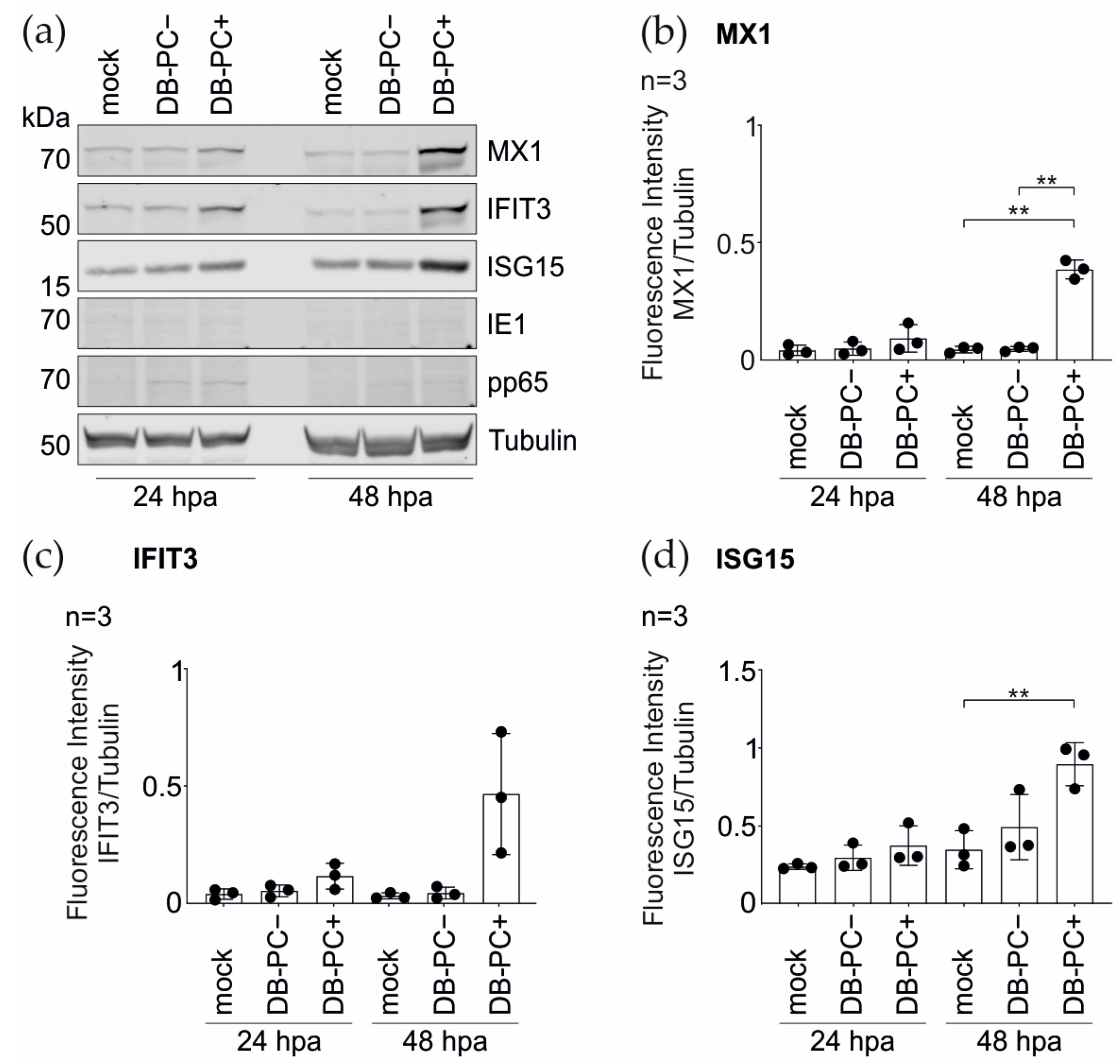
3.1.2. Confirmation of the Mass Spectrometry Results by the Immunoblot Analyses
3.1.3. IRG Induction in the HFFs Is Not Dependent on the PC
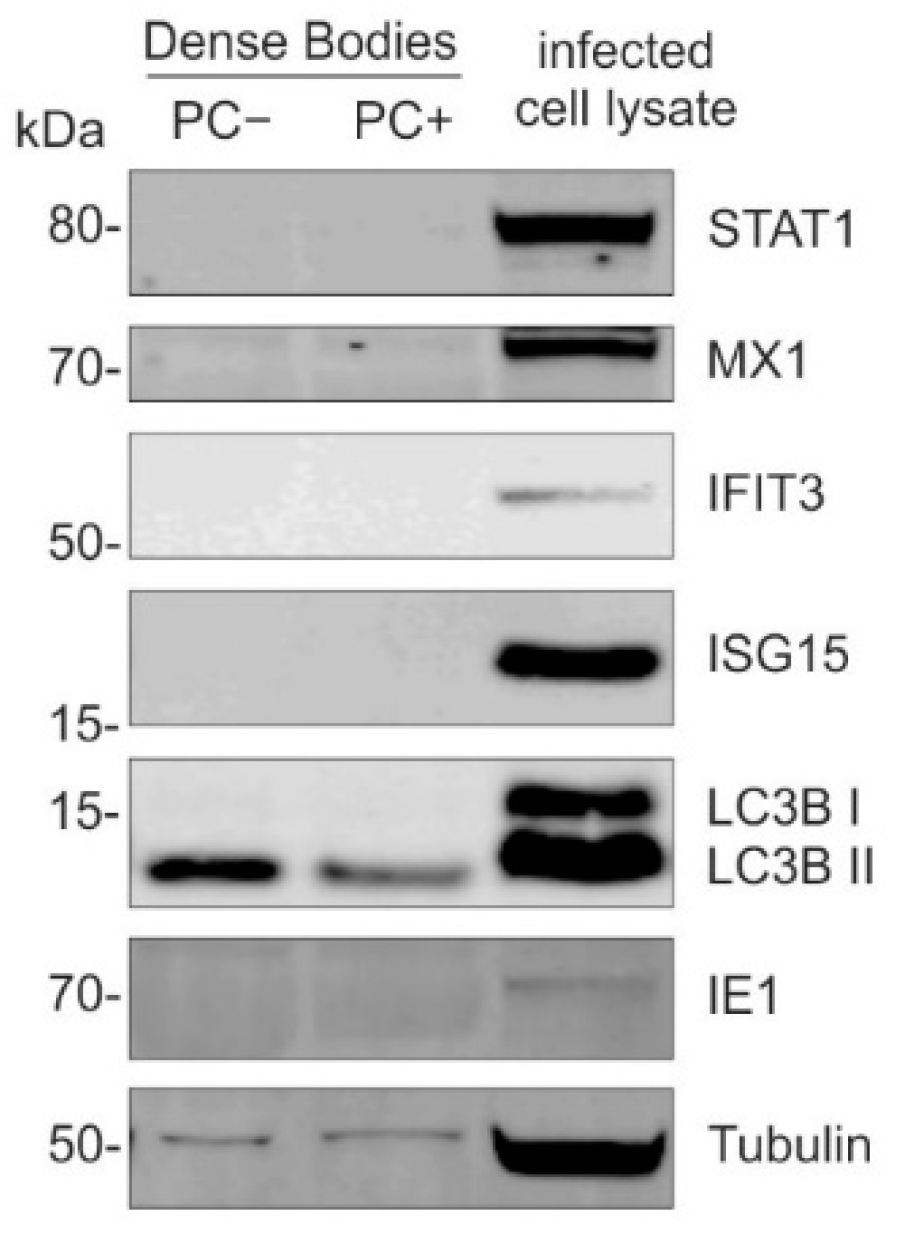
3.1.4. DBs Do Not Package STAT1, MX1, IFIT3, or ISG15
3.2. The Pentameric Complex Is Crucial for the DB-Induced IRG Expression in the Endothelial Cells
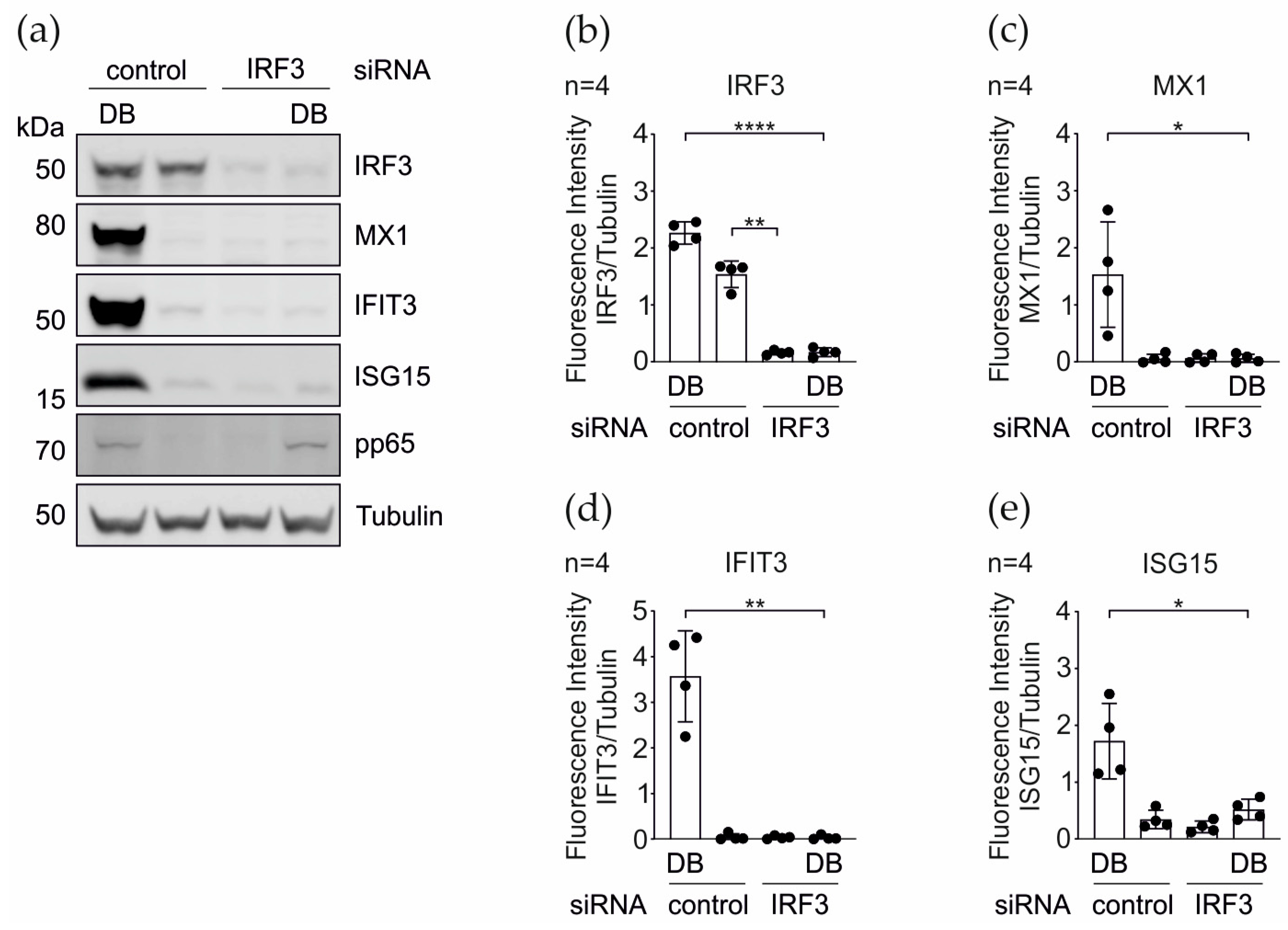
3.3. IRG Induction upon the DB-Application Requires IRF3
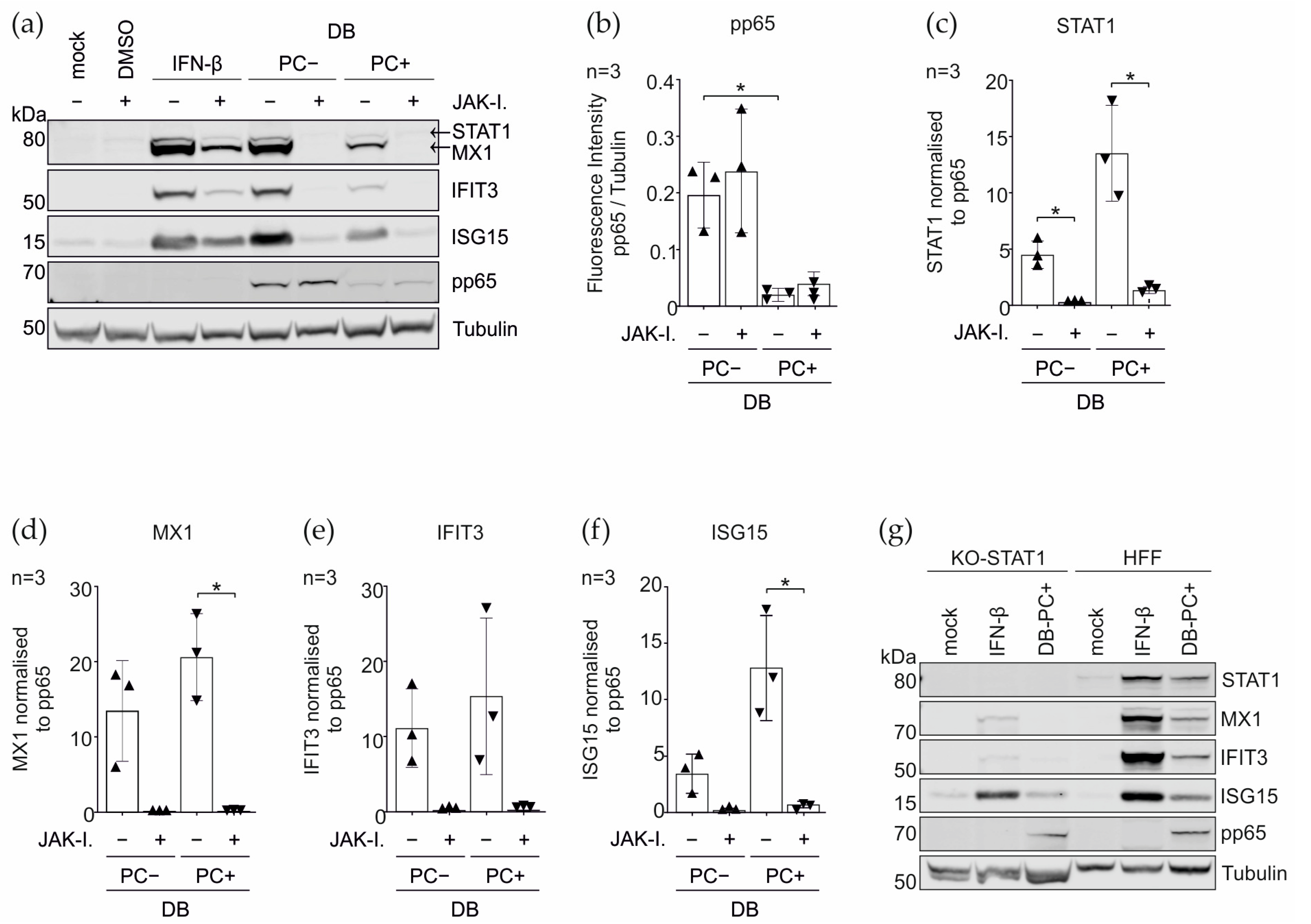
3.4. Induction of the IRGs by the DBs Depends on the Functionality of the IFN Signaling Pathway
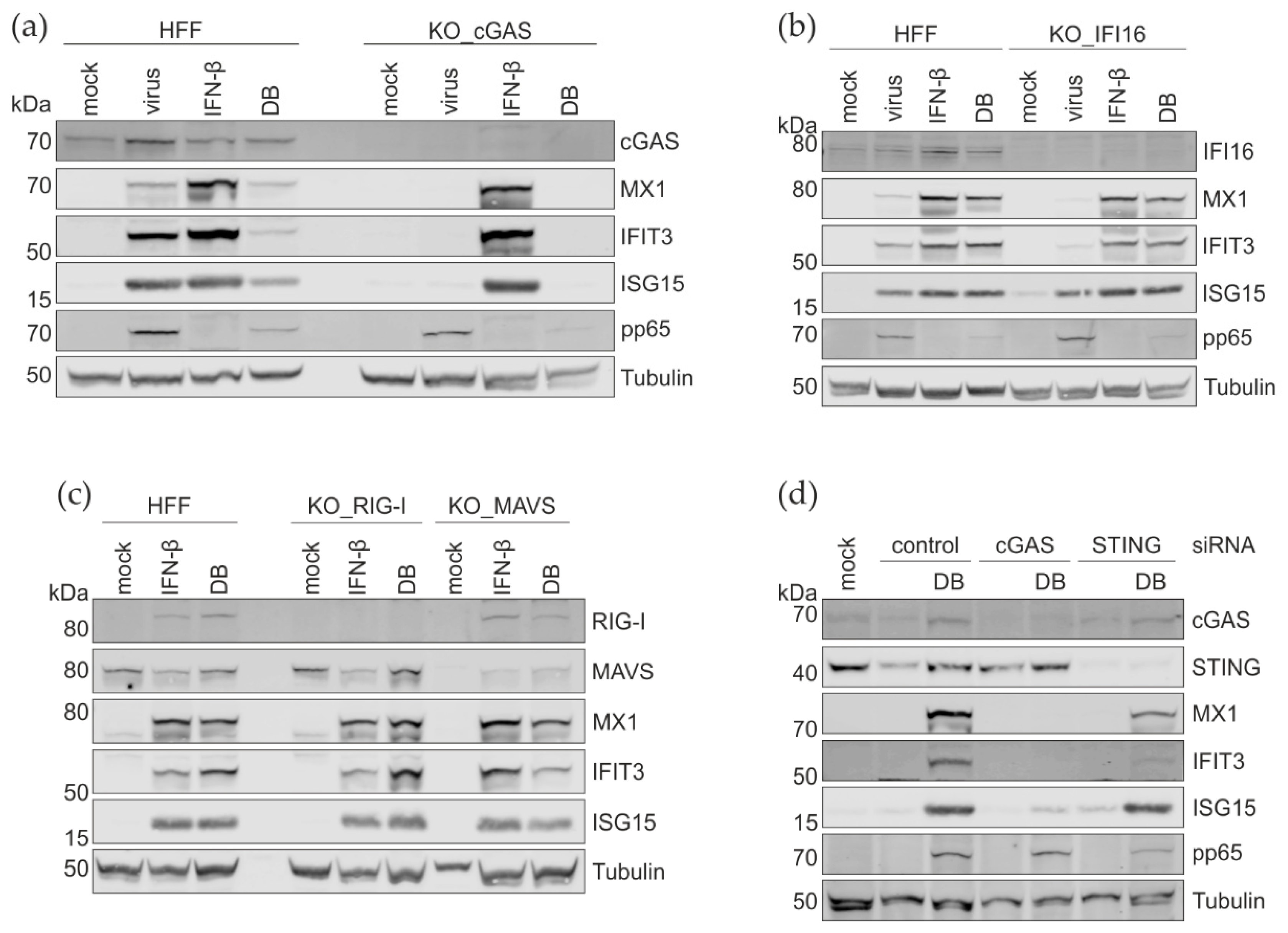
3.5. CRISPR Cas9 Knockout and the siRNA Knockdown of PRRs and Their Adaptor Proteins
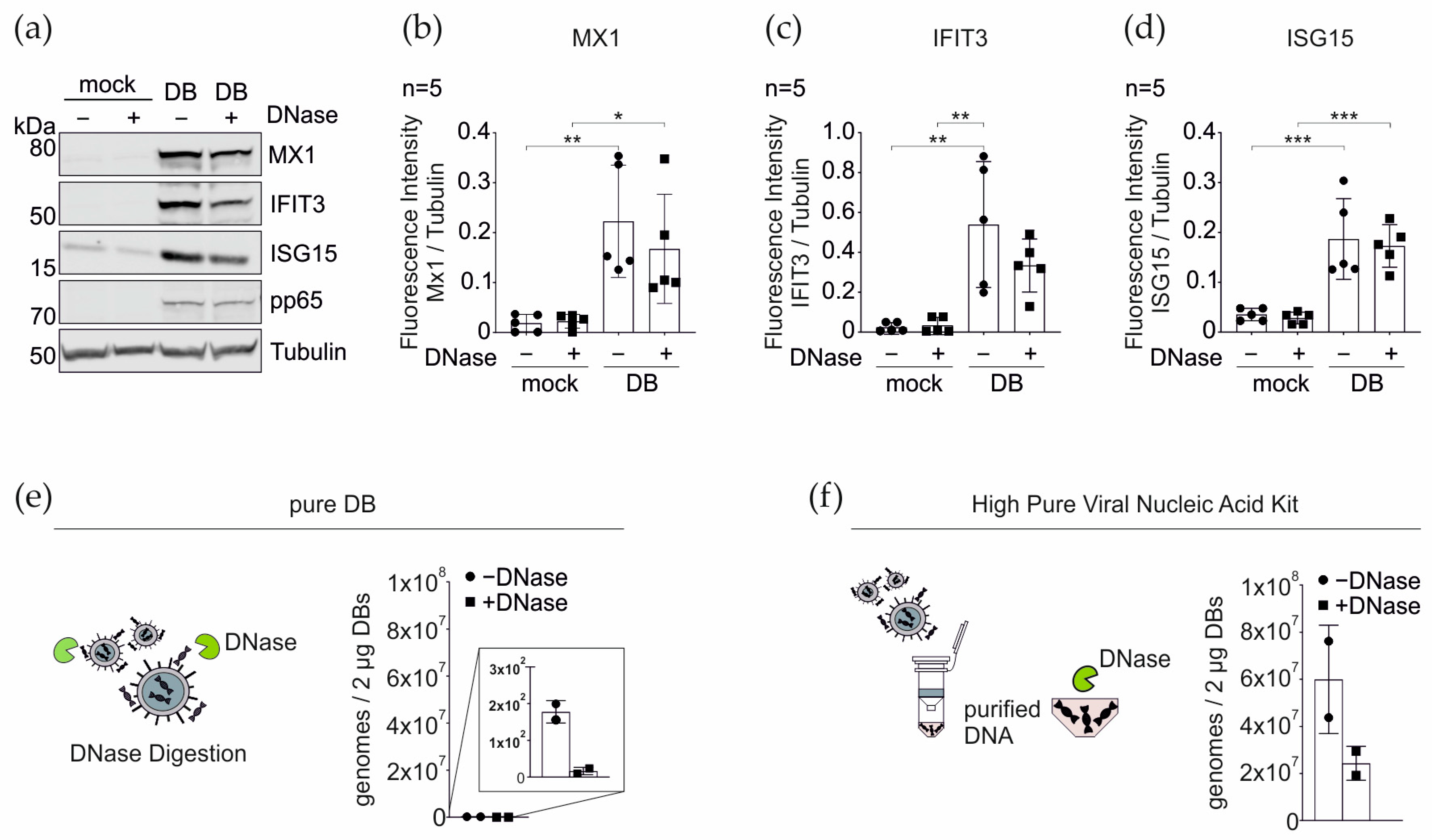
3.6. DNase Treatment of DBs Does Not Alter the IRG Expression
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stempel, M.; Chan, B.; Brinkmann, M.M. Coevolution pays off: Herpesviruses have the license to escape the DNA sensing pathway. Med. Microbiol. Immunol. 2019, 208, 495–512. [Google Scholar] [CrossRef] [PubMed]
- Biolatti, M.; Gugliesi, F.; Dell’Oste, V.; Landolfo, S. Modulation of the innate immune response by human cytomegalovirus. Infect. Genet. Evol. 2018, 64, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Kaiser, W.J.; Mocarski, E.S. Manipulation of apoptosis and necroptosis signaling by herpesviruses. Med. Microbiol. Immunol. 2015, 204, 439–448. [Google Scholar] [CrossRef]
- Fliss, P.M.; Brune, W. Prevention of cellular suicide by cytomegaloviruses. Viruses 2012, 4, 1928–1949. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, C.; Krämer, N.; Krauter, S.; Strand, D.; Sehn, E.; Wolfrum, U.; Freiwald, A.; Butter, F.; Plachter, B. Autophagy interferes with human cytomegalovirus genome replication, morphogenesis, and progeny release. Autophagy 2020, 17, 779–795. [Google Scholar] [CrossRef] [PubMed]
- Taisne, C.; Lussignol, M.; Hernandez, E.; Moris, A.; Mouna, L.; Esclatine, A. Human cytomegalovirus hijacks the autophagic machinery and LC3 homologs in order to optimize cytoplasmic envelopment of mature infectious particles. Sci. Rep. 2019, 9, 4560. [Google Scholar] [CrossRef] [PubMed]
- Craighead, J.E.; Kanich, R.E.; Almeida, J.D. Nonviral microbodies with viral antigenicity produced in cytomegalovirus-infected cells. J. Virol. 1972, 10, 766–775. [Google Scholar] [CrossRef]
- Irmiere, A.; Gibson, W. Isolation and characterization of a noninfectious virion-like particle released from cells infected with human strains of cytomegalovirus. Virology 1983, 130, 118–133. [Google Scholar] [CrossRef]
- Grefte, A.; Blom, N.; van der Giessen, M.; van Son, W.; The, T.H. Ultrastructural Analysis of Circulating Cytomegalic Cells in Patients with Active Cytomegalovirus Infection: Evidence for Virus Production and Endothelial Origin. J. Infect. Dis. 1993, 168, 1110–1118. [Google Scholar] [CrossRef]
- Schneider-Ohrum, K.; Cayatte, C.; Liu, Y.; Wang, Z.; Irrinki, A.; Cataniag, F.; Nguyen, N.; Lambert, S.; Liu, H.; Aslam, S.; et al. Production of Cytomegalovirus Dense Bodies by Scalable Bioprocess Methods Maintains Immunogenicity and Improves Neutralizing Antibody Titers. J. Virol. 2016, 90, 10133–10144. [Google Scholar] [CrossRef][Green Version]
- Cayatte, C.; Schneider-Ohrum, K.; Wang, Z.; Irrinki, A.; Nguyen, N.; Lu, J.; Nelson, C.; Servat, E.; Gemmell, L.; Citkowicz, A.; et al. Cytomegalovirus vaccine strain towne-derived dense bodies induce broad cellular immune responses and neutralizing antibodies that prevent infection of fibroblasts and epithelial cells. J. Virol. 2013, 87, 11107–11120. [Google Scholar] [CrossRef] [PubMed]
- Pepperl-Klindworth, S.; Besold, K.; Frankenberg, N.; Farkas, M.; Kuball, J.; Theobald, M.; Plachter, B. Cytomegalovirus interleukin-10 expression in infected cells does not impair MHC class I restricted peptide presentation on bystanding antigen-presenting cells. Viral Immunol. 2006, 19, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, C.; Falk, J.J.; Büscher, N.; Penner, I.; Zimmermann, C.; Gogesch, P.; Sinzger, C.; Plachter, B. Dense bodies of a gH/gL/UL128-131 pentamer repaired Towne strain of human cytomegalovirus induce an enhanced neutralizing antibody response. J. Virol. 2019, 93, e00931-19. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, S.A.; Plachter, B. Cytomegalovirus Vaccine: On the Way to the Future? In Cytomegaloviruses: From Molecular Pathogenesis to Intervention, 2nd ed.; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; pp. 424–449. [Google Scholar]
- Sauer, C.; Klobuch, S.; Herr, W.; Thomas, S.; Plachter, B. Subviral dense bodies of human cytomegalovirus stimulate maturation and activation of monocyte-derived immature dendritic cells. J. Virol. 2013, 87, 11287–11291. [Google Scholar] [CrossRef]
- Pepperl, S.; Münster, J.; Mach, M.; Harris, J.R.; Plachter, B. Dense bodies of human cytomegalovirus induce both humoral and cellular immune responses in the absence of viral gene expression. J. Virol. 2000, 74, 6132–6146. [Google Scholar] [CrossRef]
- Sinzger, C.; Plachter, B.; Grefte, A.; The, T.H.; Jahn, G. Tissue macrophages are infected by human cytomegalovirus in vivo. J. Infect. Dis. 1996, 173, 240–245. [Google Scholar] [CrossRef]
- Plachter, B.; Sinzger, C.; Jahn, G. Cell types involved in replication and distribution of human cytomegalovirus. Adv. Virus Res. 1996, 46, 195–261. [Google Scholar]
- Pepperl-Klindworth, S.; Frankenberg, N.; Riegler, S.; Plachter, B. Protein delivery by subviral particles of human cytomegalovirus. Gene Ther. 2003, 10, 278–284. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gerna, G.; Kabanova, A.; Lilleri, D. Human Cytomegalovirus Cell Tropism and Host Cell Receptors. Vaccines 2019, 7, 70. [Google Scholar] [CrossRef]
- Büscher, N.; Paulus, C.; Nevels, M.; Tenzer, S.; Plachter, B. The proteome of human cytomegalovirus virions and dense bodies is conserved across different strains. Med. Microbiol. Immunol. 2015, 204, 285–293. [Google Scholar] [CrossRef]
- Varnum, S.M.; Streblow, D.N.; Monroe, M.E.; Smith, P.; Auberry, K.J.; Pasa-Tolic, L.; Wang, D.; Camp, D.G.; Rodland, K.; Wiley, S.; et al. Identification of proteins in human cytomegalovirus (HCMV) particles: The HCMV proteome. J. Virol. 2004, 78, 10960–10966. [Google Scholar] [CrossRef] [PubMed]
- Vanarsdall, A.L.; Johnson, D.C. Human cytomegalovirus entry into cells. Curr. Opin. Virol. 2012, 2, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Shenk, T. Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc. Natl. Acad. Sci. USA 2005, 102, 18153–18158. [Google Scholar] [CrossRef]
- Zhou, M.; Lanchy, J.M.; Ryckman, B.J. Human Cytomegalovirus gH/gL/gO Promotes the Fusion Step of Entry into All Cell Types, whereas gH/gL/UL128-131 Broadens Virus Tropism through a Distinct Mechanism. J. Virol. 2015, 89, 8999–9009. [Google Scholar] [CrossRef] [PubMed]
- Kabanova, A.; Marcandalli, J.; Zhou, T.; Bianchi, S.; Baxa, U.; Tsybovsky, Y.; Lilleri, D.; Silacci-Fregni, C.; Foglierini, M.; Fernandez-Rodriguez, B.M.; et al. Platelet-derived growth factor-alpha receptor is the cellular receptor for human cytomegalovirus gHgLgO trimer. Nat. Microbiol. 2016, 1, 16082. [Google Scholar] [CrossRef]
- Wu, Y.; Prager, A.; Boos, S.; Resch, M.; Brizic, I.; Mach, M.; Wildner, S.; Scrivano, L.; Adler, B. Human cytomegalovirus glycoprotein complex gH/gL/gO uses PDGFR-alpha as a key for entry. PLoS Pathog. 2017, 13, e1006281. [Google Scholar] [CrossRef]
- Martinez-Martin, N.; Marcandalli, J.; Huang, C.S.; Arthur, C.P.; Perotti, M.; Foglierini, M.; Ho, H.; Dosey, A.M.; Shriver, S.; Payandeh, J.; et al. An Unbiased Screen for Human Cytomegalovirus Identifies Neuropilin-2 as a Central Viral Receptor. Cell 2018, 174, 1158–1171.e1119. [Google Scholar] [CrossRef]
- Nguyen, C.C.; Kamil, J.P. Pathogen at the Gates: Human Cytomegalovirus Entry and Cell Tropism. Viruses 2018, 10, 704. [Google Scholar] [CrossRef]
- Liu, J.; Jardetzky, T.S.; Chin, A.L.; Johnson, D.C.; Vanarsdall, A.L. The Human Cytomegalovirus Trimer and Pentamer Promote Sequential Steps in Entry into Epithelial and Endothelial Cells at Cell Surfaces and Endosomes. J. Virol. 2018, 92, e01336-18. [Google Scholar] [CrossRef]
- Vanarsdall, A.L.; Pritchard, S.R.; Wisner, T.W.; Liu, J.; Jardetzky, T.S.; Johnson, D.C. CD147 Promotes Entry of Pentamer-Expressing Human Cytomegalovirus into Epithelial and Endothelial Cells. mBio 2018, 9, e00781-18. [Google Scholar] [CrossRef]
- Stein, K.R.; Gardner, T.J.; Hernandez, R.E.; Kraus, T.A.; Duty, J.A.; Ubarretxena-Belandia, I.; Moran, T.M.; Tortorella, D. CD46 facilitates entry and dissemination of human cytomegalovirus. Nat. Commun. 2019, 10, 2699. [Google Scholar] [CrossRef]
- Xiaofei, E.; Meraner, P.; Lu, P.; Perreira, J.M.; Aker, A.M.; McDougall, W.M.; Zhuge, R.; Chan, G.C.; Gerstein, R.M.; Caposio, P.; et al. OR14I1 is a receptor for the human cytomegalovirus pentameric complex and defines viral epithelial cell tropism. Proc. Natl. Acad. Sci. USA 2019, 116, 7043–7052. [Google Scholar] [CrossRef]
- Topilko, A.; Michelson, S. Hyperimmediate entry of human cytomegalovirus virions and dense bodies into human fibroblasts. Res. Virol. 1994, 145, 75–82. [Google Scholar] [CrossRef]
- Abe, T.; Shapira, S.D. Negative Regulation of Cytosolic Sensing of DNA. Int. Rev. Cell Mol. Biol. 2019, 344, 91–115. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef]
- Motwani, M.; Pesiridis, S.; Fitzgerald, K.A. DNA sensing by the cGAS-STING pathway in health and disease. Nat. Rev. Genet. 2019, 20, 657–674. [Google Scholar] [CrossRef]
- Marques, M.; Ferreira, A.R.; Ribeiro, D. The Interplay between Human Cytomegalovirus and Pathogen Recognition Receptor Signaling. Viruses 2018, 10, 514. [Google Scholar] [CrossRef]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef] [PubMed]
- Aaronson, D.S.; Horvath, C.M. A road map for those who don’t know JAK-STAT. Science 2002, 296, 1653–1655. [Google Scholar] [CrossRef] [PubMed]
- Compton, T.; Kurt-Jones, E.A.; Boehme, K.W.; Belko, J.; Latz, E.; Golenbock, D.T.; Finberg, R.W. Human cytomegalovirus activates inflammatory cytokine responses via CD14 and Toll-like receptor 2. J. Virol. 2003, 77, 4588–4596. [Google Scholar] [CrossRef]
- Besold, K.; Wills, M.; Plachter, B. Immune evasion proteins gpUS2 and gpUS11 of human cytomegalovirus incompletely protect infected cells from CD8 T cell recognition. Virology 2009, 391, 5–19. [Google Scholar] [CrossRef]
- May, T.; Butueva, M.; Bantner, S.; Markusic, D.; Seppen, J.; MacLeod, R.A.; Weich, H.; Hauser, H.; Wirth, D. Synthetic gene regulation circuits for control of cell expansion. Tissue Eng. Part A 2010, 16, 441–452. [Google Scholar] [CrossRef]
- Lieber, D.; Hochdorfer, D.; Stoehr, D.; Schubert, A.; Lotfi, R.; May, T.; Wirth, D.; Sinzger, C. A permanently growing human endothelial cell line supports productive infection with human cytomegalovirus under conditional cell growth arrest. Biotechniques 2015, 59, 127–136. [Google Scholar] [CrossRef]
- Marchini, A.; Liu, H.; Zhu, H. Human cytomegalovirus with IE-2 (UL122) deleted fails to express early lytic genes. J. Virol. 2001, 75, 1870–1878. [Google Scholar] [CrossRef]
- Zimmermann, C.; Büscher, N.; Krauter, S.; Kramer, N.; Wolfrum, U.; Sehn, E.; Tenzer, S.; Plachter, B. The Abundant Tegument Protein pUL25 of Human Cytomegalovirus Prevents Proteasomal Degradation of pUL26 and Supports Its Suppression of ISGylation. J. Virol. 2018, 92, e01180-18. [Google Scholar] [CrossRef]
- Roby, C.; Gibson, W. Characterization of phosphoproteins and protein kinase activity of virions, noninfectious enveloped particles, and dense bodies of human cytomegalovirus. J. Virol. 1986, 59, 714–727. [Google Scholar] [CrossRef]
- Gonzalez-Perez, A.C.; Stempel, M.; Wyler, E.; Urban, C.; Piras, A.; Hennig, T.; Ganskih, S.; Wei, Y.; Heim, A.; Landthaler, M.; et al. The Zinc Finger Antiviral Protein ZAP Restricts Human Cytomegalovirus and Selectively Binds and Destabilizes Viral UL4/UL5 Transcripts. mBio 2021, 12, e02683-20. [Google Scholar] [CrossRef]
- Heckl, D.; Kowalczyk, M.S.; Yudovich, D.; Belizaire, R.; Puram, R.V.; McConkey, M.E.; Thielke, A.; Aster, J.C.; Regev, A.; Ebert, B.L. Generation of mouse models of myeloid malignancy with combinatorial genetic lesions using CRISPR-Cas9 genome editing. Nat. Biotechnol. 2014, 32, 941–946. [Google Scholar] [CrossRef]
- Sassenscheidt, J.; Rohayem, J.; Illmer, T.; Bandt, D. Detection of beta-herpesviruses in allogenic stem cell recipients by quantitative real-time PCR. J. Virol. Methods 2006, 138, 40–48. [Google Scholar] [CrossRef]
- Yun, Z.; Lewensohn-Fuchs, I.; Ljungman, P.; Vahlne, A. Real-time monitoring of cytomegalovirus infections after stem cell transplantation using the TaqMan polymerase chain reaction assays. Transplantation 2000, 69, 1733–1736. [Google Scholar] [CrossRef]
- Fleckenstein, B.; Müller, I.; Collins, J. Cloning of the complete human cytomegalovirus genome in cosmids. Gene 1982, 18, 39–46. [Google Scholar] [CrossRef]
- Krauter, S.; Büscher, N.; Brauchle, E.; Ortega Iannazzo, S.; Penner, I.; Kramer, N.; Gogesch, P.; Thomas, S.; Kreutz, M.; Dejung, M.; et al. An Attenuated Strain of Human Cytomegalovirus for the Establishment of a Subviral Particle Vaccine. Vaccines 2022, 10, 1326. [Google Scholar] [CrossRef]
- Schoggins, J.W.; Rice, C.M. Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 2011, 1, 519–525. [Google Scholar] [CrossRef]
- Baldick, C.J., Jr.; Shenk, T. Proteins associated with purified human cytomegalovirus particles. J. Virol. 1996, 70, 6097–6105. [Google Scholar] [CrossRef]
- Sinzger, C.; Grefte, A.; Plachter, B.; Gouw, A.S.; The, T.H.; Jahn, G. Fibroblasts, epithelial cells, endothelial cells and smooth muscle cells are major targets of human cytomegalovirus infection in lung and gastrointestinal tissues. J. Gen. Virol. 1995, 76 Pt 4, 741–750. [Google Scholar] [CrossRef]
- Hahn, G.; Revello, M.G.; Patrone, M.; Percivalle, E.; Campanini, G.; Sarasini, A.; Wagner, M.; Gallina, A.; Milanesi, G.; Koszinowski, U.; et al. Human cytomegalovirus UL131-128 genes are indispensable for virus growth in endothelial cells and virus transfer to leukocytes. J. Virol. 2004, 78, 10023–10033. [Google Scholar] [CrossRef]
- Ho, D.D.; Rota, T.R.; Andrews, C.A.; Hirsch, M.S. Replication of human cytomegalovirus in endothelial cells. J. Infect. Dis. 1984, 150, 956–957. [Google Scholar] [CrossRef] [PubMed]
- Weekes, M.P.; Tomasec, P.; Huttlin, E.L.; Fielding, C.A.; Nusinow, D.; Stanton, R.J.; Wang, E.C.; Aicheler, R.; Murrell, I.; Wilkinson, G.W.; et al. Quantitative temporal viromics: An approach to investigate host-pathogen interaction. Cell 2014, 157, 1460–1472. [Google Scholar] [CrossRef]
- Ashley, C.L.; Abendroth, A.; McSharry, B.P.; Slobedman, B. Interferon-Independent Upregulation of Interferon-Stimulated Genes during Human Cytomegalovirus Infection is Dependent on IRF3 Expression. Viruses 2019, 11, 246. [Google Scholar] [CrossRef]
- Simmen, K.A.; Singh, J.; Luukkonen, B.G.; Lopper, M.; Bittner, A.; Miller, N.E.; Jackson, M.R.; Compton, T.; Früh, K. Global modulation of cellular transcription by human cytomegalovirus is initiated by viral glycoprotein B. Proc. Natl. Acad. Sci. USA 2001, 98, 7140–7145. [Google Scholar] [CrossRef] [PubMed]
- Gogesch, P.; Penner, I.; Krauter, S.; Büscher, N.; Grode, L.; Aydin, I.; Plachter, B. Production Strategies for Pentamer-Positive Subviral Dense Bodies as a Safe Human Cytomegalovirus Vaccine. Vaccines 2019, 7, 104. [Google Scholar] [CrossRef] [PubMed]
- Kiu, H.; Nicholson, S.E. Biology and significance of the JAK/STAT signalling pathways. Growth Factors 2012, 30, 88–106. [Google Scholar] [CrossRef] [PubMed]
- Shuai, K.; Liu, B. Regulation of JAK-STAT signalling in the immune system. Nat. Rev. Immunol. 2003, 3, 900–911. [Google Scholar] [CrossRef] [PubMed]
- Luse, S.A.; Smith, M.G. Electron microscopy of salivary gland viruses. J. Exp. Med. 1958, 107, 623–632. [Google Scholar] [CrossRef] [PubMed]
- McGavran, M.H.; Smith, M.G. Ultrastructural, Cytochemical, and Microchemical Observations on Cytomegalovirus (Salivary Gland Virus) Infection of Human Cells in Tissue Culture. Exp. Mol. Pathol. 1965, 4, 1–10. [Google Scholar] [CrossRef]
- Sarov, I.; Abady, I. The morphogenesis of human cytomegalovirus. Isolation and polypeptide characterization of cytomegalovirions and dense bodies. Virology 1975, 66, 464–473. [Google Scholar] [CrossRef]
- Grefte, A.; van-der-Giessen, M.; van-Son, W.; The, T.H. Circulating cytomegalovirus (CMV)-infected endothelial cells in patients with an active CMV infection. J. Infect. Dis. 1993, 167, 270–277. [Google Scholar] [CrossRef]
- Schmolke, S.; Drescher, P.; Jahn, G.; Plachter, B. Nuclear targeting of the tegument protein pp65 (UL83) of human cytomegalovirus: An unusual bipartite nuclear localization signal functions with other portions of the protein to mediate its efficient nuclear transport. J. Virol. 1995, 69, 1071–1078. [Google Scholar] [CrossRef]
- Liu, B.; Stinski, M.F. Human cytomegalovirus contains a tegument protein that enhances transcription from promoters with upstream ATF and AP-1 cis- acting elements. J. Virol. 1992, 66, 4434–4444. [Google Scholar] [CrossRef]
- Kalejta, R.F. Pre-immediate early tegument protein functions. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention, 2nd ed.; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, VA, USA, 2013; pp. 141–151. [Google Scholar]
- Boyle, K.A.; Pietropaolo, R.L.; Compton, T. Engagement of the cellular receptor for glycoprotein B of human cytomegalovirus activates the interferon-responsive pathway. Mol. Cell. Biol. 1999, 19, 3607–3613. [Google Scholar] [CrossRef]
- Netterwald, J.R.; Jones, T.R.; Britt, W.J.; Yang, S.J.; McCrone, I.P.; Zhu, H. Postattachment events associated with viral entry are necessary for induction of interferon-stimulated genes by human cytomegalovirus. J. Virol. 2004, 78, 6688–6691. [Google Scholar] [CrossRef]
- Marshall, E.E.; Bierle, C.J.; Brune, W.; Geballe, A.P. Essential role for either TRS1 or IRS1 in human cytomegalovirus replication. J. Virol. 2009, 83, 4112–4120. [Google Scholar] [CrossRef] [PubMed]
- Scott, I. Degradation of RIG-I following cytomegalovirus infection is independent of apoptosis. Microbes Infect. 2009, 11, 973–979. [Google Scholar] [CrossRef]
- Choi, H.J.; Park, A.; Kang, S.; Lee, E.; Lee, T.A.; Ra, E.A.; Lee, J.; Lee, S.; Park, B. Human cytomegalovirus-encoded US9 targets MAVS and STING signaling to evade type I interferon immune responses. Nat. Commun. 2018, 9, 125. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, A.A.; Costa, H.; Landazuri, N.; Lui, W.O.; Hultenby, K.; Rahbar, A.; Yaiw, K.C.; Soderberg-Naucler, C. Human cytomegalovirus microRNAs are carried by virions and dense bodies and are delivered to target cells. J. Gen. Virol. 2017, 98, 1058–1072. [Google Scholar] [CrossRef]
- Terhune, S.S.; Schroer, J.; Shenk, T. RNAs are packaged into human cytomegalovirus virions in proportion to their intracellular concentration. J. Virol. 2004, 78, 10390–10398. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chen, J.; Cristea, I.M. Human cytomegalovirus tegument protein pUL83 inhibits IFI16-mediated DNA sensing for immune evasion. Cell Host Microbe 2013, 14, 591–599. [Google Scholar] [CrossRef]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Latz, E.; Xiao, T.S.; et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010, 11, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef]
- Dell’Oste, V.; Gatti, D.; Gugliesi, F.; De Andrea, M.; Bawadekar, M.; Lo Cigno, I.; Biolatti, M.; Vallino, M.; Marschall, M.; Gariglio, M.; et al. Innate nuclear sensor IFI16 translocates into the cytoplasm during the early stage of in vitro human cytomegalovirus infection and is entrapped in the egressing virions during the late stage. J. Virol. 2014, 88, 6970–6982. [Google Scholar] [CrossRef]
- Biolatti, M.; Dell’Oste, V.; Pautasso, S.; von Einem, J.; Marschall, M.; Plachter, B.; Gariglio, M.; De Andrea, M.; Landolfo, S. Regulatory Interaction between the Cellular Restriction Factor IFI16 and Viral pp65 (pUL83) Modulates Viral Gene Expression and IFI16 Protein Stability. J. Virol. 2016, 90, 8238–8250. [Google Scholar] [CrossRef] [PubMed]
- Abate, D.A.; Watanabe, S.; Mocarski, E.S. Major human cytomegalovirus structural protein pp65 (ppUL83) prevents interferon response factor 3 activation in the interferon response. J. Virol. 2004, 78, 10995–11006. [Google Scholar] [CrossRef] [PubMed]
- Biolatti, M.; Dell’Oste, V.; Pautasso, S.; Gugliesi, F.; von Einem, J.; Krapp, C.; Jakobsen, M.R.; Borgogna, C.; Gariglio, M.; De Andrea, M.; et al. Human Cytomegalovirus Tegument Protein pp65 (pUL83) Dampens Type I Interferon Production by Inactivating the DNA Sensor cGAS without Affecting STING. J. Virol. 2018, 92, e01774-17. [Google Scholar] [CrossRef]
- Fu, Y.Z.; Su, S.; Gao, Y.Q.; Wang, P.P.; Huang, Z.F.; Hu, M.M.; Luo, W.W.; Li, S.; Luo, M.H.; Wang, Y.Y.; et al. Human Cytomegalovirus Tegument Protein UL82 Inhibits STING-Mediated Signaling to Evade Antiviral Immunity. Cell Host Microbe 2017, 21, 231–243. [Google Scholar] [CrossRef]
- Chan, Y.K.; Gack, M.U. Viral evasion of intracellular DNA and RNA sensing. Nat. Rev. Microbiol. 2016, 14, 360–373. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Yin, Y.; Chen, Y.; Mao, L. The Role of Viral Proteins in the Regulation of Exosomes Biogenesis. Front. Cell. Infect. Microbiol. 2021, 11, 671625. [Google Scholar] [CrossRef]
- Alenquer, M.; Amorim, M.J. Exosome Biogenesis, Regulation, and Function in Viral Infection. Viruses 2015, 7, 5066–5083. [Google Scholar] [CrossRef]
- Sadeghipour, S.; Mathias, R.A. Herpesviruses hijack host exosomes for viral pathogenesis. Semin. Cell Dev. Biol. 2017, 67, 91–100. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Penner, I.; Büscher, N.; Dejung, M.; Freiwald, A.; Butter, F.; Plachter, B. Subviral Dense Bodies of Human Cytomegalovirus Induce an Antiviral Type I Interferon Response. Cells 2022, 11, 4028. https://doi.org/10.3390/cells11244028
Penner I, Büscher N, Dejung M, Freiwald A, Butter F, Plachter B. Subviral Dense Bodies of Human Cytomegalovirus Induce an Antiviral Type I Interferon Response. Cells. 2022; 11(24):4028. https://doi.org/10.3390/cells11244028
Chicago/Turabian StylePenner, Inessa, Nicole Büscher, Mario Dejung, Anja Freiwald, Falk Butter, and Bodo Plachter. 2022. "Subviral Dense Bodies of Human Cytomegalovirus Induce an Antiviral Type I Interferon Response" Cells 11, no. 24: 4028. https://doi.org/10.3390/cells11244028
APA StylePenner, I., Büscher, N., Dejung, M., Freiwald, A., Butter, F., & Plachter, B. (2022). Subviral Dense Bodies of Human Cytomegalovirus Induce an Antiviral Type I Interferon Response. Cells, 11(24), 4028. https://doi.org/10.3390/cells11244028