



Reactive Oxygen and Nitrogen Species (RONS) and Cytokines—Myokines Involved in Glucose Uptake and Insulin Resistance in Skeletal Muscle


Abstract
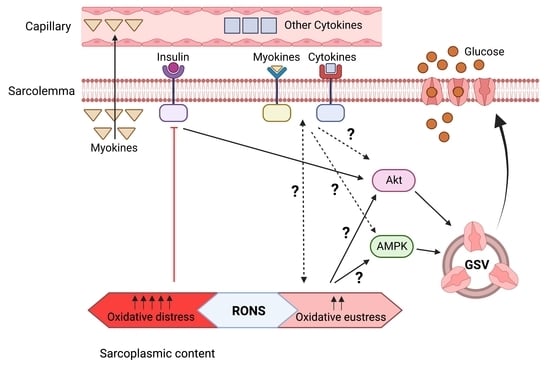
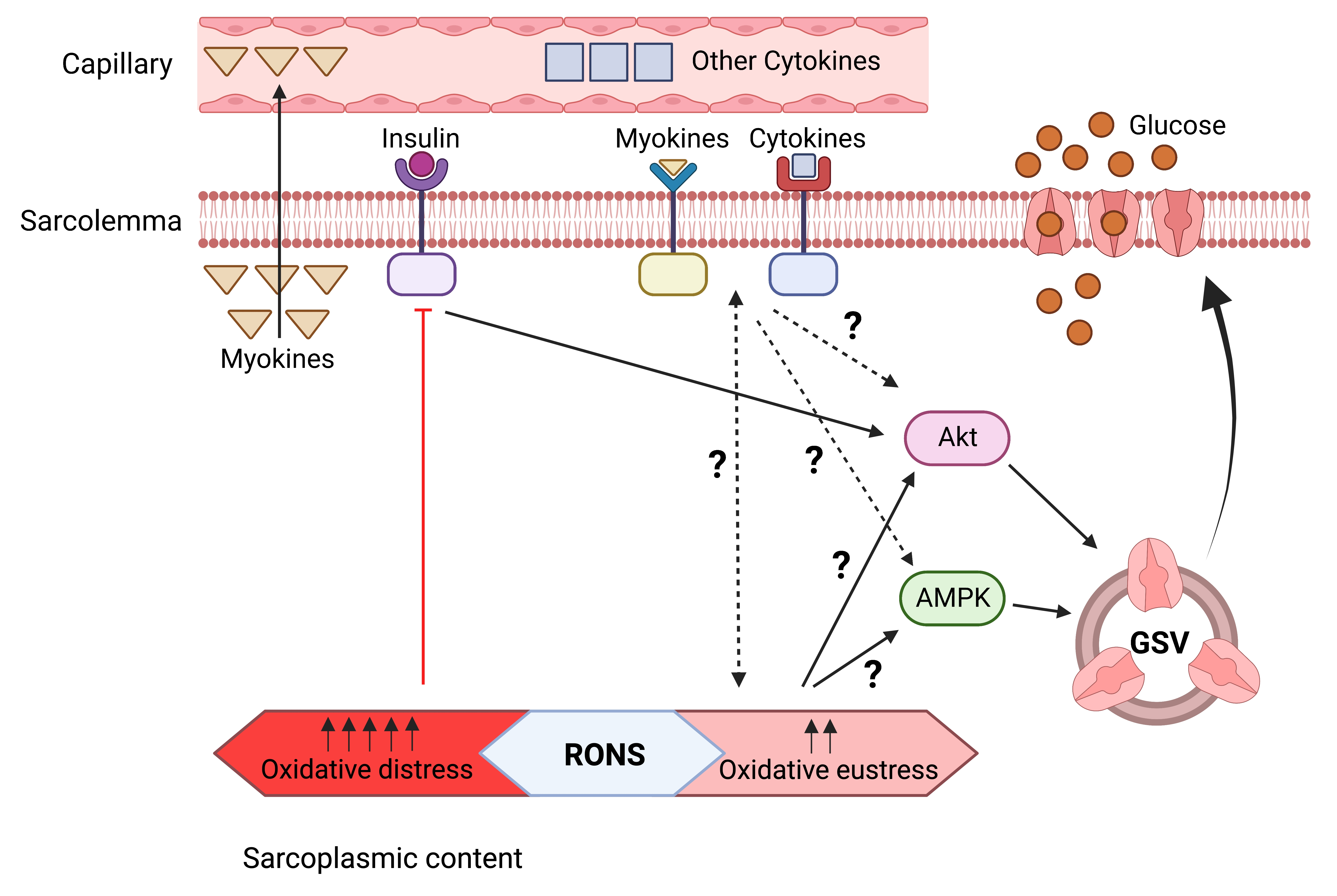
1. Introduction
2. Reactive Oxygen and Nitrogen Species in Skeletal Muscle
3. Glucose Uptake and Insulin Resistance in Skeletal Muscle
4. Effect of Cytokines on Skeletal Muscle
5. Myokines Involved in Glucose Uptake in Skeletal Muscle
6. Potential Crosstalk between RONS and Cytokines - Myokines in Glucose Uptake and Insulin Resistance in Skeletal Muscle
7. Conclusions
8. Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Frontera, W.R.; Ochala, J. Skeletal Muscle: A Brief Review of Structure and Function. Calcif. Tissue Int. 2015, 96, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Reid, K.F.; Fielding, R.A. Skeletal muscle power: A critical determinant of physical functioning in older adults. Exerc. Sport Sci. Rev. 2012, 40, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Richter, E.A.; Hargreaves, M. Exercise, GLUT4, and Skeletal Muscle Glucose Uptake. Physiol. Rev. 2013, 93, 993–1017. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, E.J.; LeRoith, D.; Karnieli, E. Insulin Resistance in Obesity as the Underlying Cause for the Metabolic Syndrome. Mt. Sinai J. Med. A J. Transl. Pers. Med. 2010, 77, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Rani, V.; Deep, G.; Singh, R.K.; Palle, K.; Yadav, U.C.S. Oxidative stress and metabolic disorders: Pathogenesis and therapeutic strategies. Life Sci. 2016, 148, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Boyle, J.P.; Thompson, T.J.; Gregg, E.W.; Barker, L.E.; Williamson, D.F. Projection of the Year 2050 Burden of Diabetes in the US Adult Population: Dynamic Modeling of Incidence, Mortality, and Prediabetes Prevalence. Popul. Health Metr. 2010, 8. [Google Scholar] [CrossRef]
- Dumont, N.A.; Bentzinger, C.F.; Sincennes, M.C.; Rudnicki, M.A. Satellite Cells and Skeletal Muscle Regeneration. Compr. Physiol. 2015, 5, 1027–1059. [Google Scholar] [CrossRef]
- Gillies, A.R.; Lieber, R.L. Structure and function of the skeletal muscle extracellular matrix. Muscle Nerve 2011, 44, 318–331. [Google Scholar] [CrossRef]
- Palomero, J.; Jackson, M. Redox regulation in skeletal muscle during contractile activity and aging 1. J. Anim. Sci. 2010, 88, 1307–1313. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Sylow, L.; Møller, L.L.V.; Kleinert, M.; Richter, E.A.; Jensen, T.E. Rac1-a novel regulator of contraction-stimulated glucose uptake in skeletal muscle. Exp. Physiol. 2014, 99, 1574–1580. [Google Scholar] [CrossRef] [PubMed]
- Henríquez-Olguin, C.; Knudsen, J.R.; Raun, S.H.; Li, Z.; Dalbram, E.; Treebak, J.T.; Sylow, L.; Holmdahl, R.; Richter, E.A.; Jaimovich, E.; et al. Cytosolic ROS production by NADPH oxidase 2 regulates muscle glucose uptake during exercise. Nat. Commun. 2019, 10, 4623. [Google Scholar] [CrossRef] [PubMed]
- Merry, T.L.; McConell, G.K. Skeletal muscle glucose uptake during exercise: A focus on reactive oxygen species and nitric oxide signaling. IUBMB Life 2009, 61, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K. Muscle as a Secretory Organ. Compr. Physiol. 2013, 3, 1337–1362. [Google Scholar] [CrossRef] [PubMed]
- Parkin, J.M.; Carey, M.F.; Zhao, S.; Febbraio, M.A. Effect of ambient temperature on human skeletal muscle metabolism during fatiguing submaximal exercise. J. Appl. Physiol. 1999, 86, 902–908. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, C.; Weigert, C. Skeletal Muscle as an Endocrine Organ: The Role of Myokines in Exercise Adaptations. Cold Spring Harb. Perspect. Med. 2017, 7, a029793. [Google Scholar] [CrossRef]
- Commoner, B.; Townsend, J.; Pake, G.E. Free Radicals in Biological Materials. Nature 1954, 174, 689–691. [Google Scholar] [CrossRef]
- Dillard, C.J.; Litov, R.E.; Savin, W.M.; Dumelin, E.E.; Tappel, A.L. Effects of exercise, vitamin E, and ozone on pulmonary function and lipid peroxidation. J. Appl. Physiol. 1978, 45, 927–932. [Google Scholar] [CrossRef]
- Brady, P.S.; Brady, L.J.; Ullrey, D.E. Selenium, Vitamin E and the Response to Swimming Stress in the Rat. J. Nutr. 1979, 109, 1103–1109. [Google Scholar] [CrossRef]
- Davies, K.J.; Quintanilha, A.T.; Brooks, G.A.; Packer, L. Free radicals and tissue damage produced by exercise. Biochem. Biophys. Res. Commun. 1982, 107, 1198–1205. [Google Scholar] [CrossRef]
- Reid, M.B.; Shoji, T.; Moody, M.R.; Entman, M.L. Reactive oxygen in skeletal muscle. II. Extracellular release of free radicals. J. Appl. Physiol. 1992, 73, 1805–1809. [Google Scholar] [CrossRef] [PubMed]
- Balon, T.W.; Nadler, J.L. Nitric oxide release is present from incubated skeletal muscle preparations. J. Appl. Physiol. 1994, 77, 2519–2521. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, C.A.; Stebbins, C.L.; Bonigut, S.; Halliwell, B.; Longhurst, J.C. Production of hydroxyl radicals in contracting skeletal muscle of cats. J. Appl. Physiol. 1996, 81, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.J.; Pye, D.; Palomero, J. The production of reactive oxygen and nitrogen species by skeletal muscle. J. Appl. Physiol. 2007, 102, 1664–1670. [Google Scholar] [CrossRef]
- Powers, S.K.; Jackson, M.J. Exercise-Induced Oxidative Stress: Cellular Mechanisms and Impact on Muscle Force Production. Physiol. Rev. 2008, 88, 1243–1276. [Google Scholar] [CrossRef]
- Jackson, M.J. Control of Reactive Oxygen Species Production in Contracting Skeletal Muscle. Antioxid. Redox Signal. 2011, 15, 2477–2486. [Google Scholar] [CrossRef]
- Silveira, L.R.; Pereira-Da-Silva, L.; Juel, C.; Hellsten, Y. Formation of hydrogen peroxide and nitric oxide in rat skeletal muscle cells during contractions. Free Radic. Biol. Med. 2003, 35, 455–464. [Google Scholar] [CrossRef]
- Palomero, J.; Pye, D.; Kabayo, T.; Spiller, D.G.; Jackson, M.J. In Situ Detection and Measurement of Intracellular Reactive Oxygen Species in Single Isolated Mature Skeletal Muscle Fibers by Real Time Fluorescence Microscopy. Antioxid. Redox Signal. 2008, 10, 1463–1474. [Google Scholar] [CrossRef]
- Pye, D.; Palomero, J.; Kabayo, T.; Jackson, M.J. Real-time measurement of nitric oxide in single mature mouse skeletal muscle fibres during contractions. J. Physiol. 2007, 581, 309–318. [Google Scholar] [CrossRef]
- Sakellariou, G.K.; Pye, D.; Vasilaki, A.; Zibrik, L.; Palomero, J.; Kabayo, T.; McArdle, F.; Van Remmen, H.; Richardson, A.; Tidball, J.G.; et al. Role of superoxide-nitric oxide interactions in the accelerated age-related loss of muscle mass in mice lacking Cu, Zn superoxide dismutase. Aging Cell 2011, 10, 749–760. [Google Scholar] [CrossRef]
- McArdle, A.; Pattwell, D.; Vasilaki, A.; Griffiths, R.D.; Jackson, M.J. Contractile activity-induced oxidative stress: Cellular origin and adaptive responses. Am. J. Physiol. Physiol. 2001, 280, C621–C627. [Google Scholar] [CrossRef] [PubMed]
- Patwell, D.M.; McArdle, A.; Morgan, J.E.; Patridge, T.A.; Jackson, M. Release of reactive oxygen and nitrogen species from contracting skeletal muscle cells. Free Radic. Biol. Med. 2004, 37, 1064–1072. [Google Scholar] [CrossRef] [PubMed]
- Javesghani, D.; Magder, S.A.; Barreiro, E.; Quinn, M.T.; Hussain, S.N.A. Molecular Characterization of a Superoxide-Generating NAD(P)H Oxidase in the Ventilatory Muscles. Am. J. Respir. Crit. Care Med. 2002, 165, 412–418. [Google Scholar] [CrossRef]
- Xia, R.; Webb, J.A.; Gnall, L.L.M.; Cutler, K.; Abramson, J.J. Skeletal muscle sarcoplasmic reticulum contains a NADH-dependent oxidase that generates superoxide. Am. J. Physiol. Physiol. 2003, 285, C215–C221. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, A.; Leiva, A.; Peña, M.; Müller, M.; Debandi, A.; Hidalgo, C.; Carrasco, M.A.; Jaimovich, E. Myotube depolarization generates reactive oxygen species through NAD(P)H oxidase; ROS-elicited Ca2+ stimulates ERK, CREB, early genes. J. Cell. Physiol. 2006, 209, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Sakellariou, G.K.; Vasilaki, A.; Palomero, J.; Kayani, A.; Zibrik, L.; McArdle, A.; Jackson, M. Studies of Mitochondrial and Nonmitochondrial Sources Implicate Nicotinamide Adenine Dinucleotide Phosphate Oxidase(s) in the Increased Skeletal Muscle Superoxide Generation That Occurs During Contractile Activity. Antioxid. Redox Signal. 2013, 18, 603–621. [Google Scholar] [CrossRef]
- Gong, M.C.; Arbogast, S.; Guo, Z.; Mathenia, J.; Su, W.; Reid, M.B. Calcium-independent phospholipase A2modulates cytosolic oxidant activity and contractile function in murine skeletal muscle cells. J. Appl. Physiol. 2006, 100, 399–405. [Google Scholar] [CrossRef]
- Zuo, L.; Christofi, F.L.; Wright, V.P.; Bao, S.; Clanton, T.L. Lipoxygenase-dependent superoxide release in skeletal muscle. J. Appl. Physiol. 2004, 97, 661–668. [Google Scholar] [CrossRef]
- Gomez-Cabrera, M.C.; Close, G.L.; Kayani, A.; McArdle, A.; Viña, J.; Jackson, M.J. Effect of xanthine oxidase-generated extracellular superoxide on skeletal muscle force generation. Am. J. Physiol. Integr. Comp. Physiol. 2010, 298, R2–R8. [Google Scholar] [CrossRef]
- Hirschfield, W.; Moody, M.R.; O’Brien, W.E.; Gregg, A.R.; Bryan, R.M.; Reid, M.B. Nitric oxide release and contractile properties of skeletal muscles from mice deficient in type III NOS. Am. J. Physiol. Integr. Comp. Physiol. 2000, 278, R95–R100. [Google Scholar] [CrossRef]
- Kobzik, L.; Reid, M.B.; Bredt, D.S.; Stamler, J.S. Nitric oxide in skeletal muscle. Nature 1994, 372, 546–548. [Google Scholar] [CrossRef] [PubMed]
- Palomero, J.; Pye, D.; Kabayo, T.; Jackson, M.J. Effect of passive stretch on intracellular nitric oxide and superoxide activities in single skeletal muscle fibres: Influence of ageing. Free. Radic. Res. 2012, 46, 30–40. [Google Scholar] [CrossRef]
- Sies, H.; Belousov, V.V.; Chandel, N.S.; Davies, M.J.; Jones, D.P.; Mann, G.E.; Murphy, M.P.; Yamamoto, M.; Winterbourn, C. Defining roles of specific reactive oxygen species (ROS) in cell biology and physiology. Nat. Rev. Mol. Cell Biol. 2022, 23, 499–515. [Google Scholar] [CrossRef]
- Szabó, C.; Ischiropoulos, H.; Radi, R. Peroxynitrite: Biochemistry, pathophysiology and development of therapeutics. Nat. Rev. Drug Discov. 2007, 6, 662–680. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.L.; Fu, R.; Mitchell, E.W. Glutathione and antioxidant enzymes in skeletal muscle: Effects of fiber type and exercise intensity. J. Appl. Physiol. 1992, 73, 1854–1859. [Google Scholar] [CrossRef]
- Leeuwenburgh, C.; Hollander, J.; Leichtweis, S.; Griffiths, M.; Gore, M.; Ji, L.L. Adaptations of glutathione antioxidant system to endurance training are tissue and muscle fiber specific. Am. J. Physiol. Integr. Comp. Physiol. 1997, 272, R363–R369. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Ji, L.L.; Leeuwenburgh, C. Exercise training-induced alterations in skeletal muscle antioxidant capacity: A brief review. Med. Sci. Sport. Exerc. 1999, 31, 987–997. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Ji, L.L.; Kavazis, A.N.; Jackson, M.J. Reactive Oxygen Species: Impact on Skeletal Muscle. Compr. Physiol. 2011, 1, 941–969. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Tripathy, D. Skeletal Muscle Insulin Resistance Is the Primary Defect in Type 2 Diabetes. Diabetes Care 2009, 32 (Suppl. 2), S157–S163. [Google Scholar] [CrossRef]
- Ejensen, J.; Rustad, P.I.; Kolnes, A.J.; Lai, Y.-C. The Role of Skeletal Muscle Glycogen Breakdown for Regulation of Insulin Sensitivity by Exercise. Front. Physiol. 2011, 2, 112. [Google Scholar] [CrossRef]
- Knuiman, P.; Hopman, M.T.E.; Mensink, M. Glycogen availability and skeletal muscle adaptations with endurance and resistance exercise. Nutr. Metab. 2015, 12, 59. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Lauritzen, H.P.; Galbo, H.; Brandauer, J.; Goodyear, L.J.; Ploug, T. Large GLUT4 Vesicles Are Stationary While Locally and Reversibly Depleted During Transient Insulin Stimulation of Skeletal Muscle of Living Mice. Diabetes 2008, 57, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Etgen, G.J.; Wilson, C.M.; Jensen, J.; Cushman, S.W.; Ivy, J.L. Glucose transport and cell surface GLUT-4 protein in skeletal muscle of the obese Zucker rat. Am. J. Physiol. Metab. 1996, 271, E294–E301. [Google Scholar] [CrossRef]
- Wang, Q.; Somwar, R.; Bilan, P.J.; Liu, Z.; Jin, J.; Woodgett, J.R.; Klip, A. Protein Kinase B/Akt Participates in GLUT4 Translocation by Insulin in L6 Myoblasts. Mol. Cell. Biol. 1999, 19, 4008–4018. [Google Scholar] [CrossRef]
- Bruss, M.D.; Arias, E.B.; Lienhard, G.E.; Cartee, G.D. Increased Phosphorylation of Akt Substrate of 160 kDa (AS160) in Rat Skeletal Muscle in Response to Insulin or Contractile Activity. Diabetes 2005, 54, 41–50. [Google Scholar] [CrossRef]
- Marette, A.; Richardson, J.M.; Ramlal, T.; Balon, T.W.; Vranic, M.; Pessin, J.E.; Klip, A. Abundance, localization, and insulin-induced translocation of glucose transporters in red and white muscle. Am. J. Physiol. Physiol. 1992, 263, C443–C452. [Google Scholar] [CrossRef]
- Tong, P.; Khayat, Z.A.; Huang, C.; Patel, N.; Ueyama, A.; Klip, A. Insulin-induced cortical actin remodeling promotes GLUT4 insertion at muscle cell membrane ruffles. J. Clin. Investig. 2001, 108, 371–381. [Google Scholar] [CrossRef]
- Merz, K.E.; Thurmond, D.C. Role of Skeletal Muscle in Insulin Resistance and Glucose Uptake. Compr. Physiol. 2020, 10, 785–809. [Google Scholar] [CrossRef]
- Mueckler, M. Insulin resistance and the disruption of Glut4 trafficking in skeletal muscle. J. Clin. Investig. 2001, 107, 1211–1213. [Google Scholar] [CrossRef]
- Katz, A.; Broberg, S.; Sahlin, K.; Wahren, J. Leg glucose uptake during maximal dynamic exercise in humans. Am. J. Physiol. Metab. 1986, 251, E65–E70. [Google Scholar] [CrossRef] [PubMed]
- Katz, A. Modulation of glucose transport in skeletal muscle by reactive oxygen species. J. Appl. Physiol. 2007, 102, 1671–1676. [Google Scholar] [CrossRef] [PubMed]
- Higaki, Y.; Mikami, T.; Fujii, N.; Hirshman, M.F.; Koyama, K.; Seino, T.; Tanaka, K.; Goodyear, L.J. Oxidative stress stimulates skeletal muscle glucose uptake through a phosphatidylinositol 3-kinase-dependent pathway. Am. J. Physiol. Metab. 2008, 294, E889–E897. [Google Scholar] [CrossRef] [PubMed]
- Higaki, Y.; Hirshman, M.F.; Fujii, N.; Goodyear, L.J. Nitric Oxide Increases Glucose Uptake Through a Mechanism That Is Distinct from the Insulin and Contraction Pathways in Rat Skeletal Muscle. Diabetes 2001, 50, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, B.J.; Mahadev, K.; Wu, X.; Zhu, L.; Motoshima, H. Role of Insulin-Induced Reactive Oxygen Species in the Insulin Signaling Pathway. Antioxid. Redox Signal. 2005, 7, 1021–1031. [Google Scholar] [CrossRef]
- Zorzano, A.; Palacin, M.; Gumà, A. Mechanisms regulating GLUT4 glucose transporter expression and glucose transport in skeletal muscle. Acta Physiol. Scand. 2005, 183, 43–58. [Google Scholar] [CrossRef]
- Américo-Da-Silva, L.; Aguilera, J.; Quinteros-Waltemath, O.; Sánchez-Aguilera, P.; Russell, J.; Cadagan, C.; Meneses-Valdés, R.; Sánchez, G.; Estrada, M.; Jorquera, G.; et al. Activation of the NLRP3 Inflammasome Increases the IL-1β Level and Decreases GLUT4 Translocation in Skeletal Muscle during Insulin Resistance. Int. J. Mol. Sci. 2021, 22, 10212. [Google Scholar] [CrossRef]
- Loh, K.; Deng, H.; Fukushima, A.; Cai, X.; Boivin, B.; Galic, S.; Bruce, C.; Shields, B.J.; Skiba, B.; Ooms, L.M.; et al. Reactive Oxygen Species Enhance Insulin Sensitivity. Cell Metab. 2009, 10, 260–272. [Google Scholar] [CrossRef]
- Dinarello, C.A. Historical insights into cytokines. Eur. J. Immunol. 2007, 37 (Suppl. 1), S34–S45. [Google Scholar] [CrossRef]
- Shi, J.; Fan, J.; Su, Q.; Yang, Z. Cytokines and Abnormal Glucose and Lipid Metabolism. Front. Endocrinol. 2019, 10, 703. [Google Scholar] [CrossRef]
- Tomas, E.; Tsao, T.-S.; Saha, A.K.; Murrey, H.E.; Zhang, C.C.; Itani, S.I.; Lodish, H.F.; Ruderman, N.B. Enhanced muscle fat oxidation and glucose transport by ACRP30 globular domain: Acetyl–CoA carboxylase inhibition and AMP-activated protein kinase activation. Proc. Natl. Acad. Sci. USA 2002, 99, 16309–16313. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 2002, 8, 1288–1295. [Google Scholar] [CrossRef]
- Ceddia, R.B.; Somwar, R.; Maida, A.; Fang, X.; Bikopoulos, G.; Sweeney, G. Globular adiponectin increases GLUT4 translocation and glucose uptake but reduces glycogen synthesis in rat skeletal muscle cells. Diabetologia 2005, 48, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Shiuchi, T.; Toda, C.; Okamoto, S.; Coutinho, E.A.; Saito, K.; Miura, S.; Ezaki, O.; Minokoshi, Y. Induction of glucose uptake in skeletal muscle by central leptin is mediated by muscle β2-adrenergic receptor but not by AMPK. Sci. Rep. 2017, 7, 15141. [Google Scholar] [CrossRef] [PubMed]
- Sáinz, N.; Rodríguez, A.; Catalán, V.; Becerril, S.; Ramírez, B.; Lancha, A.; Burgos-Ramos, E.; Gómez-Ambrosi, J.; Frühbeck, G. Leptin Reduces the Expression and Increases the Phosphorylation of the Negative Regulators of GLUT4 Traffic TBC1D1 and TBC1D4 in Muscle of ob/ob Mice. PLoS ONE 2012, 7, e29389. [Google Scholar] [CrossRef]
- Patsouris, D.; Cao, J.-J.; Vial, G.; Bravard, A.; Lefai, E.; Durand, A.; Durand, C.; Chauvin, M.-A.; Laugerette, F.; Debard, C.; et al. Insulin Resistance is Associated with MCP1-Mediated Macrophage Accumulation in Skeletal Muscle in Mice and Humans. PLoS ONE 2014, 9, e110653. [Google Scholar] [CrossRef]
- Sell, H.; Dietze-Schroeder, D.; Kaiser, U.; Eckel, J. Monocyte Chemotactic Protein-1 Is a Potential Player in the Negative Cross-Talk between Adipose Tissue and Skeletal Muscle. Endocrinology 2006, 147, 2458–2467. [Google Scholar] [CrossRef]
- Cho, K.-A.; Kang, P.B. PLIN2 inhibits insulin-induced glucose uptake in myoblasts through the activation of the NLRP3 inflammasome. Int. J. Mol. Med. 2015, 36, 839–844. [Google Scholar] [CrossRef]
- Plomgaard, P.; Bouzakri, K.; Krogh-Madsen, R.; Mittendorfer, B.; Zierath, J.R.; Pedersen, B.K. Tumor Necrosis Factor-α Induces Skeletal Muscle Insulin Resistance in Healthy Human Subjects via Inhibition of Akt Substrate 160 Phosphorylation. Diabetes 2005, 54, 2939–2945. [Google Scholar] [CrossRef]
- De Alvaro, C.; Teruel, T.; Hernandez, R.; Lorenzo, M. Tumor Necrosis Factor α Produces Insulin Resistance in Skeletal Muscle by Activation of Inhibitor κB Kinase in a p38 MAPK-dependent Manner. J. Biol. Chem. 2004, 279, 17070–17078. [Google Scholar] [CrossRef]
- Rosales-Soto, G.; Diaz-Vegas, A.; Casas, M.; Contreras-Ferrat, A.; Jaimovich, E. Fibroblast growth factor-21 potentiates glucose transport in skeletal muscle fibers. J. Mol. Endocrinol. 2020, 65, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Lee, J.O.; Kim, N.; Kim, J.K.; Kim, H.I.; Lee, Y.W.; Kim, S.J.; Choi, J.-I.; Oh, Y.; Kim, J.H.; et al. Irisin, a novel myokine, regulates glucose uptake in skeletal muscle cells via AMPK. Mol. Endocrinol. 2015, 29, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Yano, N.; Zhang, L.; Wei, D.; Dubielecka, P.M.; Wei, L.; Zhuang, S.; Zhu, P.; Qin, G.; Liu, P.Y.; Chin, Y.E.; et al. Irisin counteracts high glucose and fatty acid-induced cytotoxicity by preserving the AMPK-insulin receptor signaling axis in C2C12 myoblasts. Am. J. Physiol. Metab. 2020, 318, E791–E805. [Google Scholar] [CrossRef]
- Ye, X.; Shen, Y.; Ni, C.; Ye, J.; Xin, Y.; Zhang, W.; Ren, Y. Irisin reverses insulin resistance in C2C12 cells via the p38-MAPK-PGC-1α pathway. Peptides 2019, 119, 170120. [Google Scholar] [CrossRef] [PubMed]
- Xin, C.; Liu, J.; Zhang, J.D.; Zhu, D.; Wang, H.; Xiong, L.; Lee, Y.; Ye, J.; Lian, K.; Xu, C.; et al. Irisin improves fatty acid oxidation and glucose utilization in type 2 diabetes by regulating the AMPK signaling pathway. Int. J. Obes. 2016, 40, 443–451. [Google Scholar] [CrossRef]
- Carey, A.L.; Steinberg, G.R.; Macaulay, S.L.; Thomas, W.G.; Holmes, A.G.; Ramm, G.; Prelovsek, O.; Hohnen-Behrens, C.; Watt, M.J.; James, D.E.; et al. Interleukin-6 Increases Insulin-Stimulated Glucose Disposal in Humans and Glucose Uptake and Fatty Acid Oxidation In Vitro via AMP-Activated Protein Kinase. Diabetes 2006, 55, 2688–2697. [Google Scholar] [CrossRef]
- Glund, S.; Deshmukh, A.; Long, Y.C.; Moller, T.; Koistinen, H.A.; Caidahl, K.; Zierath, J.R.; Krook, A. Interleukin-6 Directly Increases Glucose Metabolism in Resting Human Skeletal Muscle. Diabetes 2007, 56, 1630–1637. [Google Scholar] [CrossRef]
- Yue, P.; Jin, H.; Aillaud, M.; Deng, A.C.; Azuma, J.; Asagami, T.; Kundu, R.K.; Reaven, G.M.; Quertermous, T.; Tsao, P.S. Apelin is necessary for the maintenance of insulin sensitivity. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E59–E67. [Google Scholar] [CrossRef]
- Dray, C.; Knauf, C.; Daviaud, D.; Waget, A.; Boucher, J.; Buléon, M.; Cani, P.D.; Attané, C.; Guigné, C.; Carpéné, C.; et al. Apelin Stimulates Glucose Utilization in Normal and Obese Insulin-Resistant Mice. Cell Metab. 2008, 8, 437–445. [Google Scholar] [CrossRef]
- Attané, C.; Foussal, C.; Le Gonidec, S.; Benani, A.; Daviaud, D.; Wanecq, E.; Guzmán-Ruiz, R.; Dray, C.; Bezaire, V.; Rancoule, C.; et al. Apelin Treatment Increases Complete Fatty Acid Oxidation, Mitochondrial Oxidative Capacity, and Biogenesis in Muscle of Insulin-Resistant Mice. Diabetes 2012, 61, 310–320. [Google Scholar] [CrossRef]
- Liu, X.-H.; Bauman, W.A.; Cardozo, C.P. Myostatin inhibits glucose uptake via suppression of insulin-dependent and -independent signaling pathways in myoblasts. Physiol. Rep. 2018, 6, e13837. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ye, J.; Cao, L.; Zhang, Y.; Xia, W.; Zhu, D. Myostatin regulates glucose metabolism via the AMP-activated protein kinase pathway in skeletal muscle cells. Int. J. Biochem. Cell Biol. 2010, 42, 2072–2081. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Huo, X.; Pang, X.; Zong, Z.; Meng, X.; Liu, G. Musclin Inhibits Insulin Activation of Akt/Protein Kinase B in Rat Skeletal Muscle. J. Int. Med. Res. 2008, 36, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-J.; Liu, Y.; Sui, Y.-B.; Yang, H.-T.; Chang, J.-R.; Tang, C.-S.; Qi, Y.-F.; Zhang, J.; Yin, X.-H. Positive association between musclin and insulin resistance in obesity: Evidence of a human study and an animal experiment. Nutr. Metab. 2017, 14, 46. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zheng, J.; Liu, X.; Feng, Z.; Zhang, X.; Cao, L.; Zhou, Z. Exercise improved lipid metabolism and insulin sensitivity in rats fed a high-fat diet by regulating glucose transporter 4 (GLUT4) and musclin expression. Braz. J. Med. Biol. Res. 2016, 49, e5129. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, M.; Tsuchida, A.; Nakagawa, T.; Nonomura, T.; Ono-Kishino, M.; Sugaru, E.; Noguchi, H.; Taiji, M. Brain-derived neurotrophic factor enhances glucose utilization in peripheral tissues of diabetic mice. Diabetes Obes. Metab. 2007, 9, 59–64. [Google Scholar] [CrossRef]
- Suwa, M.; Yamamoto, K.-I.; Nakano, H.; Sasaki, H.; Radak, Z.; Kumagai, S. Brain-Derived Neurotrophic Factor Treatment Increases the Skeletal Muscle Glucose Transporter 4 Protein Expression in Mice. Physiol. Res. 2010, 59, 619–623. [Google Scholar] [CrossRef]
- Scherer, P.E.; Williams, S.; Fogliano, M.; Baldini, G.; Lodish, H.F. A Novel Serum Protein Similar to C1q, Produced Exclusively in Adipocytes. J. Biol. Chem. 1995, 270, 26746–26749. [Google Scholar] [CrossRef]
- Spranger, J.; Kroke, A.; Möhlig, M.; Bergmann, M.M.; Ristow, M.; Boeing, H.; Pfeiffer, A.F. Adiponectin and protection against type 2 diabetes mellitus. Lancet 2003, 361, 226–228. [Google Scholar] [CrossRef]
- Krause, M.P.; Milne, K.J.; Hawke, T.J. Adiponectin—Consideration for its Role in Skeletal Muscle Health. Int. J. Mol. Sci. 2019, 20, 1528. [Google Scholar] [CrossRef]
- Yamauchi, T.; Kamon, J.; Ito, Y.; Tsuchida, A.; Yokomizo, T.; Kita, S.; Sugiyama, T.; Miyagishi, M.; Hara, K.; Tsunoda, M.; et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 2003, 423, 762–769. [Google Scholar] [CrossRef] [PubMed]
- Bruce, C.R.; Mertz, V.A.; Heigenhauser, G.J.; Dyck, D.J. The Stimulatory Effect of Globular Adiponectin on Insulin-Stimulated Glucose Uptake and Fatty Acid Oxidation Is Impaired in Skeletal Muscle from Obese Subjects. Diabetes 2005, 54, 3154–3160. [Google Scholar] [CrossRef] [PubMed]
- Myers, M.G.; Cowley, M.A.; Münzberg, H. Mechanisms of Leptin Action and Leptin Resistance. Annu. Rev. Physiol. 2008, 70, 537–556. [Google Scholar] [CrossRef] [PubMed]
- Cummings, B.P.; Bettaieb, A.; Graham, J.L.; Stanhope, K.L.; Dill, R.; Morton, G.J.; Haj, F.G.; Havel, P.J. Subcutaneous administration of leptin normalizes fasting plasma glucose in obese type 2 diabetic UCD-T2DM rats. Proc. Natl. Acad. Sci. USA 2011, 108, 14670–14675. [Google Scholar] [CrossRef] [PubMed]
- Morton, G.J.; Gelling, R.W.; Niswender, K.D.; Morrison, C.; Rhodes, C.J.; Schwartz, M.W. Leptin regulates insulin sensitivity via phosphatidylinositol-3-OH kinase signaling in mediobasal hypothalamic neurons. Cell Metab. 2005, 2, 411–420. [Google Scholar] [CrossRef]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte Chemoattractant Protein-1 (MCP-1): An Overview. J. Interf. Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef]
- Sartipy, P.; Loskutoff, D.J. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2003, 100, 7265–7270. [Google Scholar] [CrossRef]
- Estrov, Z.; Kurzrock, R.; Talpaz, M. Interleukin-1 and its inhibitors: Implications for disease biology and therapy. Cytokines Interleukins Recept. 1995, 80, 51–82. [Google Scholar] [CrossRef]
- Cho, K.-A.; Park, M.; Kim, Y.-H.; Woo, S.-Y.; Ryu, K.-H. Conditioned media from human palatine tonsil mesenchymal stem cells regulates the interaction between myotubes and fibroblasts by IL-1Ra activity. J. Cell. Mol. Med. 2016, 21, 130–141. [Google Scholar] [CrossRef]
- Jorquera, G.; Russell, J.; Monsalves-Álvarez, M.; Cruz, G.; Valladares-Ide, D.; Basualto-Alarcón, C.; Barrientos, G.; Estrada, M.; Llanos, P. NLRP3 Inflammasome: Potential Role in Obesity Related Low-Grade Inflammation and Insulin Resistance in Skeletal Muscle. Int. J. Mol. Sci. 2021, 22, 3254. [Google Scholar] [CrossRef]
- Jorquera, G.; Meneses-Valdés, R.; Rosales-Soto, G.; Valladares-Ide, D.; Campos, C.; Silva-Monasterio, M.; Llanos, P.; Cruz, G.; Jaimovich, E.; Casas, M. High extracellular ATP levels released through pannexin-1 channels mediate inflammation and insulin resistance in skeletal muscle fibres of diet-induced obese mice. Diabetologia 2021, 64, 1389–1401. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.G.; Fielding, R.A.; Fiatarone, M.A.; Orencole, S.F.; Dinarello, C.A.; Evans, W.J. Increased interleukin 1 beta in human skeletal muscle after exercise. Am. J. Physiol. Integr. Comp. Physiol. 1989, 257, R451–R455. [Google Scholar] [CrossRef] [PubMed]
- Terink, R.; Bongers, C.C.W.G.; Witkamp, R.F.; Mensink, M.; Eijsvogels, T.M.; Klein Gunnewiek, J.M.T.; Hopman, M.T.E. Changes in cytokine levels after prolonged and repeated moderate intensity exercise in middle-aged men and women. Transl. Sport. Med. 2018, 1, 110–119. [Google Scholar] [CrossRef]
- Idriss, H.T.; Naismith, J.H. TNFα and the TNF Receptor Superfamily: Structure-Function Relationship(s). Microsc. Res. Tech. 2000, 50, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-P.; Reid, M.B. NF-κB mediates the protein loss induced by TNF-α in differentiated skeletal muscle myotubes. Am. J. Physiol. Integr. Comp. Physiol. 2000, 279, R1165–R1170. [Google Scholar] [CrossRef] [PubMed]
- Stasko, S.A.; Hardin, B.J.; Smith, J.; Moylan, J.S.; Reid, M.B. TNF signals via neuronal-type nitric oxide synthase and reactive oxygen species to depress specific force of skeletal muscle. J. Appl. Physiol. 2013, 114, 1629–1636. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Olson, N.C.; Callas, P.W.; Hanley, A.J.G.; Festa, A.; Haffner, S.M.; Wagenknecht, L.E.; Tracy, R.P. Circulating Levels of TNF-α Are Associated with Impaired Glucose Tolerance, Increased Insulin Resistance, and Ethnicity: The Insulin Resistance Atherosclerosis Study. J. Clin. Endocrinol. Metab. 2012, 97, 1032–1040. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K.; Febbraio, M.A. Muscles, exercise and obesity: Skeletal muscle as a secretory organ. Nat. Rev. Endocrinol. 2012, 8, 457–465. [Google Scholar] [CrossRef]
- Lightfoot, A.; Cooper, R.G. The role of myokines in muscle health and disease. Curr. Opin. Rheumatol. 2016, 28, 661–666. [Google Scholar] [CrossRef]
- Eckel, J. Myokines in metabolic homeostasis and diabetes. Diabetologia 2019, 62, 1523–1528. [Google Scholar] [CrossRef]
- Giudice, J.; Taylor, J.M. Muscle as a paracrine and endocrine organ. Curr. Opin. Pharmacol. 2017, 34, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Aguer, C.; Loro, E.; Di Raimondo, D. Editorial: The Role of the Muscle Secretome in Health and Disease. Front. Physiol. 2020, 11, 1101. [Google Scholar] [CrossRef] [PubMed]
- Goörgens, S.W.; Eckardt, K.; Jensen, J.; Drevon, C.A.; Eckel, J. Exercise and Regulation of Adipokine and Myokine Production. Prog. Mol. Biol. Transl. Sci. 2015, 135, 313–336. [Google Scholar] [CrossRef]
- Balakrishnan, R.; Thurmond, D.C. Mechanisms by Which Skeletal Muscle Myokines Ameliorate Insulin Resistance. Int. J. Mol. Sci. 2022, 23, 4636. [Google Scholar] [CrossRef]
- Kharitonenkov, A.; Shiyanova, T.L.; Koester, A.; Ford, A.M.; Micanovic, R.; Galbreath, E.J.; Sandusky, G.E.; Hammond, L.J.; Moyers, J.S.; Owens, R.A.; et al. FGF-21 as a novel metabolic regulator. J. Clin. Investig. 2005, 115, 1627–1635. [Google Scholar] [CrossRef] [PubMed]
- Izumiya, Y.; Bina, H.A.; Ouchi, N.; Akasaki, Y.; Kharitonenkov, A.; Walsh, K. FGF21 is an Akt-regulated myokine. FEBS Lett. 2008, 582, 3805–3810. [Google Scholar] [CrossRef]
- Von Holstein-Rathlou, S.; BonDurant, L.D.; Peltekian, L.; Naber, M.C.; Yin, T.C.; Claflin, K.E.; Urizar, A.I.; Madsen, A.N.; Ratner, C.; Holst, B.; et al. FGF21 Mediates Endocrine Control of Simple Sugar Intake and Sweet Taste Preference by the Liver. Cell Metab. 2016, 23, 335–343. [Google Scholar] [CrossRef]
- Potthoff, M.J.; Kliewer, S.A.; Mangelsdorf, D.J. Endocrine fibroblast growth factors 15/19 and 21: From feast to famine. Genes Dev. 2012, 26, 312–324. [Google Scholar] [CrossRef]
- Jimenez, V.; Jambrina, C.; Casana, E.; Sacristan, V.; Muñoz, S.; Darriba, S.; Rodó, J.; Mallol, C.; Garcia, M.; León, X.; et al. FGF21 gene therapy as treatment for obesity and insulin resistance. EMBO Mol. Med. 2018, 10, e8791. [Google Scholar] [CrossRef]
- Mahajan, R.D.; Patra, S.K. Irisin, a Novel Myokine Responsible for Exercise Induced Browning of White Adipose Tissue. Indian J. Clin. Biochem. 2013, 28, 102–103. [Google Scholar] [CrossRef]
- Yang, Z.; Chen, X.; Chen, Y.; Zhao, Q. Decreased irisin secretion contributes to muscle insulin resistance in high-fat diet mice. Int. J. Clin. Exp. Pathol. 2015, 8, 6490–6497. [Google Scholar] [PubMed]
- Kistner, T.M.; Pedersen, B.K.; Lieberman, D.E. Interleukin 6 as an energy allocator in muscle tissue. Nat. Metab. 2022, 4, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K.; Åkerström, T.C.; Nielsen, A.R.; Fischer, C.P. Role of myokines in exercise and metabolism. J. Appl. Physiol. 2007, 103, 1093–1098. [Google Scholar] [CrossRef] [PubMed]
- Steensberg, A.; Keller, C.; Starkie, R.L.; Osada, T.; Febbraio, M.A.; Pedersen, B.K. IL-6 and TNF-α expression in, and release from, contracting human skeletal muscle. Am. J. Physiol. Metab. 2002, 283, E1272–E1278. [Google Scholar] [CrossRef]
- Febbraio, M.A.; Hiscock, N.; Sacchetti, M.; Fischer, C.P.; Pedersen, B.K. Interleukin-6 Is a Novel Factor Mediating Glucose Homeostasis During Skeletal Muscle Contraction. Diabetes 2004, 53, 1643–1648. [Google Scholar] [CrossRef]
- Knudsen, J.G.; Gudiksen, A.; Bertholdt, L.; Overby, P.; Villesen, I.; Schwartz, C.L.; Pilegaard, H. Skeletal muscle IL-6 regulates muscle substrate utilization and adipose tissue metabolism during recovery from an acute bout of exercise. PLoS ONE 2017, 12, e0189301. [Google Scholar] [CrossRef]
- Shoelson, S.E.; Lee, J.; Goldfine, A.B. Inflammation and insulin resistance. J. Clin. Investig. 2006, 116, 1793–1801. [Google Scholar] [CrossRef]
- Nelke, C.; Dziewas, R.; Minnerup, J.; Meuth, S.G.; Ruck, T. Skeletal muscle as potential central link between sarcopenia and immune senescence. eBioMedicine 2019, 49, 381–388. [Google Scholar] [CrossRef]
- Chaves-Almagro, C.; Castan-Laurell, I.; Dray, C.; Knauf, C.; Valet, P.; Masri, B. Apelin receptors: From signaling to antidiabetic strategy. Eur. J. Pharmacol. 2015, 763, 149–159. [Google Scholar] [CrossRef]
- Castan-Laurell, I.; Dray, C.; Knauf, C.; Kunduzova, O.; Valet, P. Apelin, a promising target for type 2 diabetes treatment? Trends Endocrinol. Metab. 2012, 23, 234–241. [Google Scholar] [CrossRef]
- Besse-Patin, A.; Montastier, E.; Vinel, C.; Castan-Laurell, I.; Louche, K.; Dray, C.; Daviaud, D.; Mir, L.; Marques, M.-A.; Thalamas, C.; et al. Effect of endurance training on skeletal muscle myokine expression in obese men: Identification of apelin as a novel myokine. Int. J. Obes. 2013, 38, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Waller, J.D.; McNeill, E.H.; Zhong, F.; Vervaecke, L.S.; Goldfarb, A.H. Plasma Apelin Unchanged with Acute Exercise Insulin Sensitization. J. Sport. Sci. Med. 2019, 18, 537–543. [Google Scholar]
- Rodgers, B.D.; Ward, C.W. Myostatin/Activin Receptor Ligands in Muscle and the Development Status of Attenuating Drugs. Endocr. Rev. 2021, 43, 329–365. [Google Scholar] [CrossRef] [PubMed]
- Amor, M.; Itariu, B.K.; Moreno-Viedma, V.; Keindl, M.; Jürets, A.; Prager, G.; Langer, F.; Grablowitz, V.; Zeyda, M.; Stulnig, T.M. Serum Myostatin is Upregulated in Obesity and Correlates with Insulin Resistance in Humans. Exp. Clin. Endocrinol. Diabetes 2019, 127, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.L.; Hittel, D.S.; McPherron, A. Expression and Function of Myostatin in Obesity, Diabetes, and Exercise Adaptation. Med. Sci. Sport. Exerc. 2011, 43, 1828–1835. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; McFarlane, C.; Lokireddy, S.; Masuda, S.; Ge, X.; Gluckman, P.D.; Sharma, M.; Kambadur, R. Inhibition of myostatin protects against diet-induced obesity by enhancing fatty acid oxidation and promoting a brown adipose phenotype in mice. Diabetologia 2011, 55, 183–193. [Google Scholar] [CrossRef]
- Dong, J.; Dong, Y.; Chen, F.; Mitch, W.E.; Zhang, L. Inhibition of myostatin in mice improves insulin sensitivity via irisin-mediated cross talk between muscle and adipose tissues. Int. J. Obes. 2016, 40, 434–442. [Google Scholar] [CrossRef]
- Nishizawa, H.; Matsuda, M.; Yamada, Y.; Kawai, K.; Suzuki, E.; Makishima, M.; Kitamura, T.; Shimomura, I. Musclin, a Novel Skeletal Muscle-derived Secretory Factor. J. Biol. Chem. 2004, 279, 19391–19395. [Google Scholar] [CrossRef]
- Chen, W.-J.; Liu, Y.; Sui, Y.-B.; Zhang, B.; Zhang, X.-H.; Yin, X.-H. Increased circulating levels of musclin in newly diagnosed type 2 diabetic patients. Diabetes Vasc. Dis. Res. 2017, 14, 116–121. [Google Scholar] [CrossRef]
- Sánchez, Y.L.; Yepes-Calderón, M.; Valbuena, L.; Milán, A.F.; Trillos-Almanza, M.C.; Granados, S.; Peña, M.; Estrada-Castrillón, M.; Aristizábal, J.C.; Narvez-Sanchez, R.; et al. Musclin Is Related to Insulin Resistance and Body Composition, but Not to Body Mass Index or Cardiorespiratory Capacity in Adults. Endocrinol. Metab. 2021, 36, 1055–1068. [Google Scholar] [CrossRef]
- Shimomura, M.; Horii, N.; Fujie, S.; Inoue, K.; Hasegawa, N.; Iemitsu, K.; Uchida, M.; Iemitsu, M. Decreased muscle-derived musclin by chronic resistance exercise is associated with improved insulin resistance in rats with type 2 diabetes. Physiol. Rep. 2021, 9, e14823. [Google Scholar] [CrossRef] [PubMed]
- Miranda, M.; Morici, J.F.; Zanoni, M.B.; Bekinschtein, P. Brain-Derived Neurotrophic Factor: A Key Molecule for Memory in the Healthy and the Pathological Brain. Front. Cell. Neurosci. 2019, 13, 363. [Google Scholar] [CrossRef] [PubMed]
- Matthews, V.B.; Åström, M.-B.; Chan, S.; Bruce, C.; Krabbe, K.S.; Prelovsek, O.; Åkerström, T.; Yfanti, C.; Broholm, C.; Mortensen, O.H.; et al. Brain-derived neurotrophic factor is produced by skeletal muscle cells in response to contraction and enhances fat oxidation via activation of AMP-activated protein kinase. Diabetologia 2009, 52, 1409–1418. [Google Scholar] [CrossRef] [PubMed]
- Vega, S.R.; Strüder, H.K.; Wahrmann, B.V.; Schmidt, A.; Bloch, W.; Hollmann, W. Acute BDNF and cortisol response to low intensity exercise and following ramp incremental exercise to exhaustion in humans. Brain Res. 2006, 1121, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Krabbe, K.S.; Nielsen, A.R.; Krogh-Madsen, R.; Plomgaard, P.; Rasmussen, P.; Erikstrup, C.; Fischer, C.; Lindegaard, B.; Petersen, A.M.W.; Taudorf, S.; et al. Brain-derived neurotrophic factor (BDNF) and type 2 diabetes. Diabetologia 2007, 50, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Lightfoot, A.P.; Sakellariou, G.K.; Nye, G.A.; McArdle, F.; Jackson, M.J.; Griffiths, R.D.; McArdle, A. SS-31 attenuates TNF-α induced cytokine release from C2C12 myotubes. Redox Biol. 2015, 6, 253–259. [Google Scholar] [CrossRef]
- Contreras-Ferrat, A.; Llanos, P.; Vásquez, C.; Espinosa, A.; Osorio-Fuentealba, C.; Arias-Calderon, M.; Lavandero, S.; Klip, A.; Hidalgo, C.; Jaimovich, E. Insulin elicits a ROS-activated and an IP3-dependent Ca2+ release; both impinge on GLUT4 translocation. J. Cell Sci. 2014, 127, 1911–1923. [Google Scholar] [CrossRef]
- Sánchez-Aguilera, P.; Vegas, A.D.; Campos, C.; Quinteros-Waltemath, O.; Cerda-Kohler, H.; Barrientos, G.; Contreras-Ferrat, A.; Llanos, P. Role of ABCA1 on membrane cholesterol content, insulin-dependent Akt phosphorylation and glucose uptake in adult skeletal muscle fibers from mice. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2018, 1863, 1469–1477. [Google Scholar] [CrossRef]
- Llanos, P.; Contreras-Ferrat, A.; Georgiev, T.; Osorio-Fuentealba, C.; Espinosa, A.; Hidalgo, J.; Hidalgo, C.; Jaimovich, E. The cholesterol-lowering agent methyl-β-cyclodextrin promotes glucose uptake via GLUT4 in adult muscle fibers and reduces insulin resistance in obese mice. Am. J. Physiol. Metab. 2015, 308, E294–E305. [Google Scholar] [CrossRef]
- Barrientos, G.; Llanos, P.; Hidalgo, J.; Bolaños, P.; Caputo, C.; Riquelme, A.; Sánchez, G.; Quest, A.F.G.; Hidalgo, C. Cholesterol removal from adult skeletal muscle impairs excitation–contraction coupling and aging reduces caveolin-3 and alters the expression of other triadic proteins. Front. Physiol. 2015, 6, 105. [Google Scholar] [CrossRef]
- Palomero, J.; Vasilaki, A.; Pye, D.; McArdle, A.; Jackson, M.J. Aging increases the oxidation of dichlorohydrofluorescein in single isolated skeletal muscle fibers at rest, but not during contractions. Am. J. Physiol. Integr. Comp. Physiol. 2013, 305, R351–R358. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Puente, E.; Palomero, J. Genetically Encoded Biosensors to Monitor Intracellular Reactive Oxygen and Nitrogen Species and Glutathione Redox Potential in Skeletal Muscle Cells. Int. J. Mol. Sci. 2021, 22, 10876. [Google Scholar] [CrossRef] [PubMed]
- Puente, E.F.; Sánchez-Martín, M.A.; De Andrés, J.; Rodríguez-Izquierdo, L.; Méndez, L.; Palomero, J. Expression and functional analysis of the hydrogen peroxide biosensors HyPer and HyPer2 in C2C12 myoblasts/myotubes and single skeletal muscle fibres. Sci. Rep. 2020, 10, 871. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Peptide | Type | Main Effects | References |
|---|---|---|---|
| Adiponectin | Adipokine | ↑ Glucose uptake. ↑ GLUT4 translocation via AMPK. | [71,72] [73] |
| Leptin | Adipokine | ↑ Glucose uptake. ↑ GLUT4 translocation independent of AMPK. ↓ Expression and activity of the negative regulators of AS160. | [74] [75] [75] |
| Monocyte chemotactic protein 1 | Inflammatory cytokines | ↑ Recruitment of macrophages and altered local insulin sensitivity. ↓ Insulin signaling and glucose uptake. | [76] [77] |
| Interleukin-1β | Inflammatory cytokines and myokine | ↓ GLUT4 translocation, glucose uptake in response to insulin and the expression of IRS-1. | [67,78] |
| Tumor necrosis factor-α | Inflammatory cytokines | ¬ AS160 phosphorylation. ↑ Serine phosphorylation at insulin receptor and IRS-1. | [79] [80] |
| Fibroblast growth factor 21 | Hepatokine and myokine | ↑ Glucose uptake and GLUT4 translocation dependent on PKC-ζ. | [81] |
| Irisin | Myokine | ↑ Glucose uptake via ROS-mediated AMPK pathway. Reverses insulin resistance via p38-MAPK-PGC-1α. ↑ Fatty acid oxidation and glucose utilization by AMPK. | [82,83] [84] [85] |
| Interleukin-6 | Myokine | ↑ Insulin sensitivity via AMP-activated protein kinase. ↑ Glucose metabolism while resting, without changing insulin-stimulated glucose transport and insulin signaling. | [86] [87] |
| Apelin | Adipokine and myokine | ↑ Glucose uptake via endothelial NO synthase, AMPK and Akt. | [88,89,90] |
| Myostatin | Myokine | ¬ Glucose uptake via suppression of insulin-dependent and -independent signaling pathways in skeletal muscle. Acceleration of glucose utilization via AMPK. Up-regulation of several glucose metabolism-related genes. | [91] [92] |
| Musclin | Myokine | ↓ Insulin-stimulated glucose uptake via Akt inhibition and PPARγ/LXRα. ↓ Glucose metabolism and, at least in part, through endoplasmic reticulum stress. Exercise training improves ↑ insulin sensitivity by upregulating GLUT4 and downregulating musclin in skeletal muscle. | [93] [94] [95] |
| Brain-derived neurotrophic factor | Myokine | ↑ Glucose uptake. ↑ GLUT4 protein expression. | [96] [97] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Llanos, P.; Palomero, J. Reactive Oxygen and Nitrogen Species (RONS) and Cytokines—Myokines Involved in Glucose Uptake and Insulin Resistance in Skeletal Muscle. Cells 2022, 11, 4008. https://doi.org/10.3390/cells11244008
Llanos P, Palomero J. Reactive Oxygen and Nitrogen Species (RONS) and Cytokines—Myokines Involved in Glucose Uptake and Insulin Resistance in Skeletal Muscle. Cells. 2022; 11(24):4008. https://doi.org/10.3390/cells11244008
Chicago/Turabian StyleLlanos, Paola, and Jesus Palomero. 2022. "Reactive Oxygen and Nitrogen Species (RONS) and Cytokines—Myokines Involved in Glucose Uptake and Insulin Resistance in Skeletal Muscle" Cells 11, no. 24: 4008. https://doi.org/10.3390/cells11244008
APA StyleLlanos, P., & Palomero, J. (2022). Reactive Oxygen and Nitrogen Species (RONS) and Cytokines—Myokines Involved in Glucose Uptake and Insulin Resistance in Skeletal Muscle. Cells, 11(24), 4008. https://doi.org/10.3390/cells11244008