

The Role of Oxytocin in Abnormal Brain Development: Effect on Glial Cells and Neuroinflammation

 , , ,
, , ,
Abstract
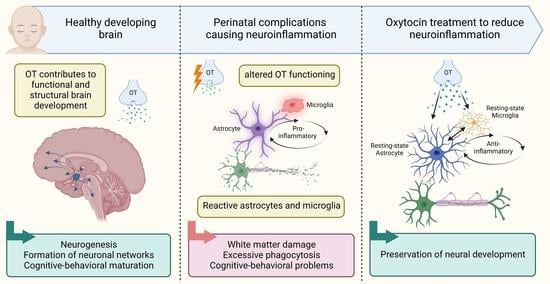
1. Introduction
2. The Oxytocin System in the Brain
2.1. Oxytocin Synthesis and Release to the Oxytocin Receptor
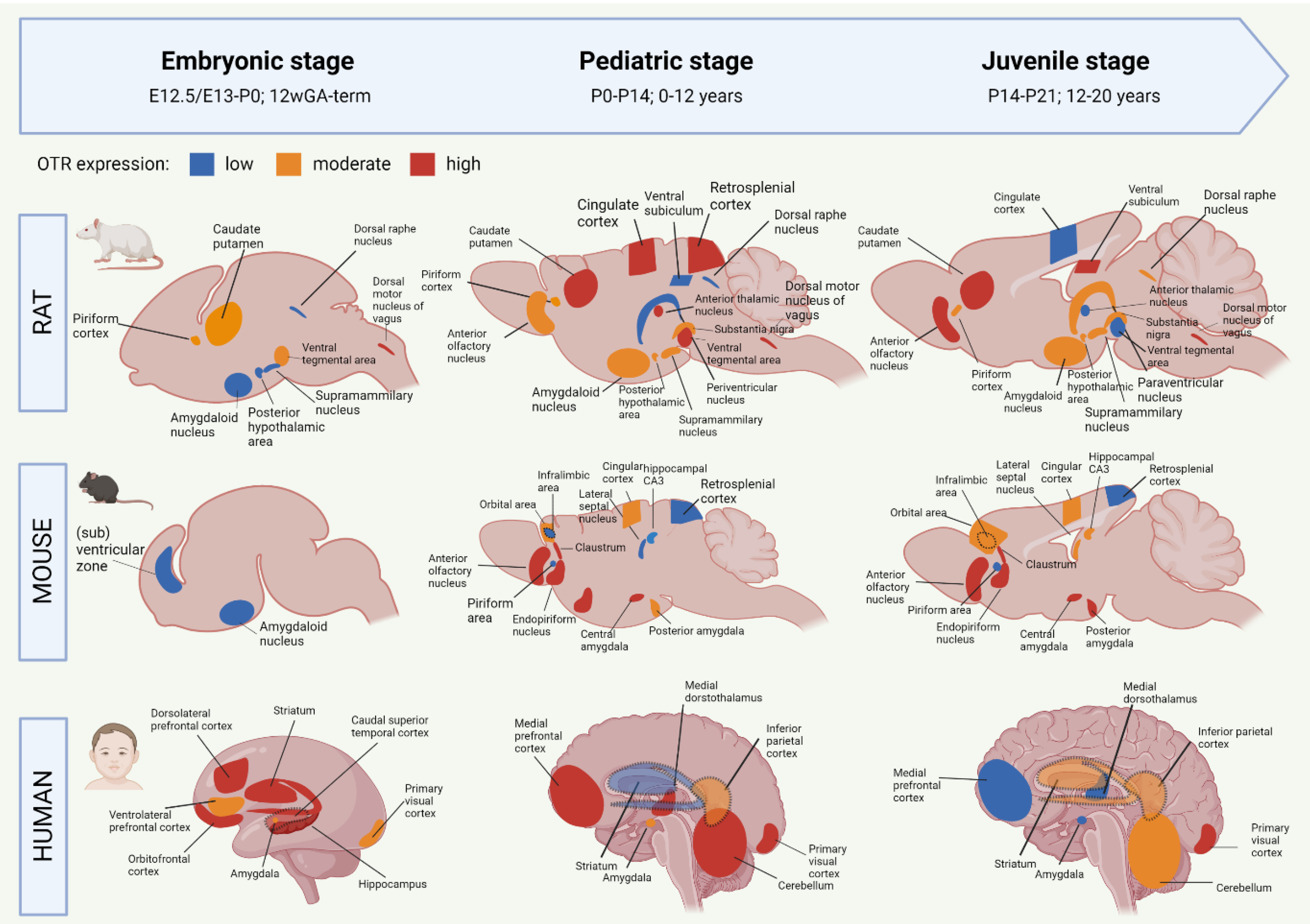
2.2. Development of the Oxytocin System from the Embryonic to Juvenile Age
2.2.1. Oxytocin Receptor System of the Developing Rat Brain
2.2.2. Oxytocin Receptor System of the Developing Mouse Brain
2.2.3. Oxytocin Receptor System of the Developing Human Brain
2.3. The Functions of Oxytocin in the Brain
Major Functional Part of the Oxytocin Network: The Amygdala
3. Oxytocin Functioning in Abnormal Brain Development
3.1. Neuroinflammation as a Common Factor of Perinatal Complications
3.2. Altered Oxytocin Functioning in Neuroinflammation and Other Neurodevelopmental Disorders
4. Experimental Studies on Neonatal Oxytocin Treatment
Neonatal Oxytocin Treatment to Alleviate Inflammation-Induced Brain Injury
5. The Underlying Mechanisms for the Neuroprotective Effect of Oxytocin in Neonatal Neuroinflammation: Astrocytes and Microglia
5.1. Oxytocin and Astrocytes
5.1.1. Shift to Neuroprotective Astrocytes
5.1.2. Astrocyte Process Retraction to Reduce Neurotoxic Effects
5.2. Oxytocin and Microglia
5.2.1. Reducing Phagocytic Activity via NADPH/ROS Signaling
5.2.2. Regulating Cytokine Secretion via the NF-кB/MAPK Pathway
5.2.3. ER Stress-Related eIF-2a-ATF4 Pathway
5.3. Glial Crosstalk in the Effect of Oxytocin: The Astrocyte Versus Microglia Battle
5.3.1. Oxytocin Acts to Beneficially Influence the Control of Astrocytes on Microglia
5.3.2. How Does Oxytocin Reach Microglia: The Microglial Oxytocin Receptor Debate

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study; Journal | Species; Strain; Sex | Age of Assessment | Assessment Method | In Vivo or In Vitro? | OTR Expression Found? | Type of Inflammation | Effect of OT Treatment |
|---|---|---|---|---|---|---|---|
| [142] Nat Commun. | Primary microglia from C57BL/6 mice; sex unspecified | P5 | Bulk RNA Seq: Illumina HiSeq 4000 | In vitro; cultured for 7 days | yes (Mean = 1.139 SD = 0.904 FPKM) | LPS stimulation (50 ng/mL) for 24 h | LPS decreased OTR expression in C57Bl/6 mice (Mean = 0.486 SD = 0.486 FPKM) |
| [155] Am J Physiol Endocrinol Metab | (1) Primary macrophages from male and female humans (2) Peritoneal macrophages from male C57BL/6 mice | Human: Not disclosed Mouse: 12 weeks | qPCR and Western blot | In vitro; cultured for 7 days | Human: yes Mouse: yes | LPS stimulation (100 ng/mL) for 6 h | Human: LPS increased OTR protein expression of both 67 kDa and 46 kDa forms. Mouse: no effect |
| [153] Cells | Microglial cell line MG6 from C57BL/6 mice; sex unspecified | Not applicable (cell line) | RT-qPCR; Western blot; ELISA | In vitro; cultured for 2–3 days; including 30 min OT pretreatment before inflammation | Not assessed directly, but OTR antagonist L-371,257 pre-treatment reversed the anti-inflammatory effects of OT | LPS stimulation (100 ng/mL) for 24 h | OT treatment suppressed proinflammatory cytokine production in LPS-stimulated MG6 microglia |
| [131] J Neuro-inflammation | BV-2 cells and primary microglia; sex unspecified | P1-P2 | RT-qPCR; immuno-histochemistry | In vitro; cultured for 21 days (14 days mixed-glia culture, 7 days microglia culture); including 2 h OT pretreatment before inflammation | BV-2 cells: yes Primary microglia: yes | LPS stimulation (500 ng/mL) for 24 h | LPS increased OTR mRNA expression in BV-2 cells and primary microglia. OT pre-treatment suppressed LPS-stimulated expression of TNF-α, IL-1β, COX-2 and iNOS at the protein and transcriptome level. |
| [174] Stroke | CD11b-positive microglia from male C57BL/6 mice | Adult | RT-PCR and flow cytometry | In vivo and in vitro In vitro: primary microglia, 2 h pre-treatment with OT or OT antagonist | In vivo: 16% of CD11b-positive cells expressed OTR In vitro: Not assessed directly, but OTR antagonist application blocked the anti-inflammatory effect of OT | LPS stimulation (1 μg/mL) for 22 h | In vivo: not assessed. In vitro: OT incubation before LPS stimulation attenuated major histocompatibility complex class II expression. |
| [169] Proc Natl Acad Sci U S A | FACS-sorted microglia from C57BL/6 mice, male and female | E17, P7, P14, P21 and P60 | Bulk RNA Seq. Database: http://www.brainrnaseq.org/, accessed on 1 October 2022 | In vivo | E17: no P7: no P14: no P21: no P60: yes (Mean = 0.120 SD = 0.120 FPKM) | LPS stimulation (5 mg/kg, single intraperitoneal injection at P60) | LPS treatment depleted OTR expression at P60 (Mean = 0, SD = 0 FPKM) |
| [173] J Neurosc | Microglia and macrophages from Tie2–EGFP transgenic mice; sex unspecified | P7 | Bulk RNA Seq: Illumina HiSeq 2000. Database: http://www.brainrnaseq.org/, accessed on 1 October 2022 | In vivo | Yes: (Mean = 0.1 SD = 0 FPKM) | None | Not applicable |
| [170] Immunity | FACS-sorted microglia from C57BL/6 mice, male and female | E14.5, P4/P5, P30, P100, P450 | Single cell RNA Seq: Illumina NextSeq500 Database: http://www.microgliasinglecell.com/, accessed on 1 October 2022 | In vivo | Not detected at any time-point. | None | Not applicable |
| [171] Neuron | FACS-sorted microglia from male C57BL/6 mice | E14.5 (whole brain), P7 and P60 | Single cell RNA Seq: Smart-seq2. Database: https://myeloidsc.appspot.com/, accessed on 1 October 2022 | In vivo | P60 clusters: 0–0.1 CPM P7 clusters: 0–0.1 CPM Embryo cluster: 0–0.1 CPM (Did not reach cut-off threshold of >2CPM) | None | Not applicable |
| [172] Nat. Neurosci. | FACS-sorted microglia from male C57BL/6 mice | P90 | Single cell RNA Seq: Illumina NextSeq500 | In vivo | No | None | Not applicable |
6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Blencowe, H.; Cousens, S.; Chou, D.; Oestergaard, M.; Say, L.; Moller, A.-B.; Kinney, M.; Lawn, J.; the Born Too Soon Preterm Birth Action Group. Born Too Soon: The global epidemiology of 15 million preterm births. Reprod. Health 2013, 10 (Suppl. 1), S2. [Google Scholar] [CrossRef] [PubMed]
- Vogel, J.P.; Chawanpaiboon, S.; Moller, A.-B.; Watananirun, K.; Bonet, M.; Lumbiganon, P. The global epidemiology of preterm birth. Best Pr. Res. Clin. Obstet. Gynaecol. 2018, 52, 3–12. [Google Scholar] [CrossRef]
- Liu, L.; Oza, S.; Hogan, D.; Chu, Y.; Perin, J.; Zhu, J.; Lawn, J.E.; Cousens, S.; Mathers, C.; Black, R.E. Global, regional, and national causes of under-5 mortality in 2000–15: An updated systematic analysis with implications for the Sustainable Development Goals. Lancet 2016, 388, 3027–3035. [Google Scholar] [CrossRef] [PubMed]
- Meldrum, S.J.; Strunk, T.; Currie, A.; Prescott, S.L.; Simmer, K.; Whitehouse, A.J. Autism spectrum disorder in children born preterm—Role of exposure to perinatal inflammation. Front. Neurosci. 2013, 7, 123. [Google Scholar] [CrossRef]
- Sanches, E.; Carvalho, A.; van de Looij, Y.; Toulotte, A.; Wyse, A.; Netto, C.; Sizonenko, S. Experimental cerebral palsy causes microstructural brain damage in areas associated to motor deficits but no spatial memory impairments in the developing rat. Brain Res. 2021, 1761, 147389. [Google Scholar] [CrossRef]
- Trønnes, H.; Wilcox, A.J.; Lie, R.T.; Markestad, T.; Moster, D. Risk of cerebral palsy in relation to pregnancy disorders and preterm birth: A national cohort study. Dev. Med. Child Neurol. 2014, 56, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, H.; Gressens, P.; Mallard, C. Inflammation during fetal and neonatal life: Implications for neurologic and neuropsychiatric disease in children and adults. Ann. Neurol. 2011, 71, 444–457. [Google Scholar] [CrossRef]
- Volpe, J.J. Brain injury in premature infants: A complex amalgam of destructive and developmental disturbances. Lancet Neurol. 2009, 8, 110–124. [Google Scholar] [CrossRef]
- Van Steenwinckel, J.; Schang, A.L.; Sigaut, S.; Chhor, V.; Degos, V.; Hagberg, H.; Baud, O.; Fleiss, B.; Gressens, P. Brain damage of the preterm infant: New insights into the role of inflammation. Biochem. Soc. Trans. 2014, 42, 557–563. [Google Scholar] [CrossRef]
- Gui, L.; Loukas, S.; Lazeyras, F.; Hüppi, P.; Meskaldji, D.; Tolsa, C.B. Longitudinal study of neonatal brain tissue volumes in preterm infants and their ability to predict neurodevelopmental outcome. NeuroImage 2019, 185, 728–741. [Google Scholar] [CrossRef]
- Cismaru, A.L.; Gui, L.; Vasung, L.; Lejeune, F.; Barisnikov, K.; Truttmann, A.; Tolsa, C.B.; Hüppi, P.S. Altered Amygdala Development and Fear Processing in Prematurely Born Infants. Front. Neuroanat. 2016, 10, 55. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Choi, Y.-H.; Cha, J.H.; Lee, J.Y.; Lee, Y.-J.; Kim, B.-H.; Kim, I.-K.; Lee, J.-M.; Lee, H.J. Altered asymmetries of the structural networks comprising the fronto-limbic brain circuitry of preterm infants. Sci. Rep. 2021, 11, 1–12. [Google Scholar] [CrossRef]
- Kalpakidou, A.K.; Allin, M.P.G.; Walshe, M.; Giampietro, V.; McGuire, P.; Rifkin, L.; Murray, R.; Nosarti, C. Functional Neuroanatomy of Executive Function after Neonatal Brain Injury in Adults Who Were Born Very Preterm. PLoS ONE 2014, 9, e113975. [Google Scholar] [CrossRef] [PubMed]
- Lejeune, F.; Réveillon, M.; Monnier, M.; Hüppi, P.S.; Tolsa, C.B.; Barisnikov, K. Social reasoning abilities in preterm and full-term children aged 5–7 years. Early Hum. Dev. 2016, 103, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Moreno, E.; Fischi-Gomez, E.; Batalle, D.; Borradori-Tolsa, C.; Eixarch, E.; Thiran, J.-P.; Gratacós, E.; Hüppi, P.S. Structural Brain Network Reorganization and Social Cognition Related to Adverse Perinatal Condition from Infancy to Early Adolescence. Front. Neurosci. 2016, 10, 560. [Google Scholar] [CrossRef]
- Réveillon, M.; Tolsa, C.B.; Monnier, M.; Hüppi, P.; Barisnikov, K. Response inhibition difficulties in preterm children aged 9–12 years: Relations with emotion and behavior. Child Neuropsychol. 2015, 22, 420–442. [Google Scholar] [CrossRef]
- Urben, S.; Jonge, L.V.H.D.; Barisnikov, K.; Pizzo, R.; Monnier, M.; Lazeyras, F.; Tolsa, C.B.; Hüppi, P.S. Gestational age and gender influence on executive control and its related neural structures in preterm-born children at 6 years of age. Child Neuropsychol. 2015, 23, 188–207. [Google Scholar] [CrossRef]
- Witt, A.; Theurel, A.; Tolsa, C.B.; Lejeune, F.; Fernandes, L.; van Hanswijck de Jonge, L.; Monnier, M.; Graz, M.B.; Barisnikov, K.; Gentaz, E.; et al. Emotional and effortful control abilities in 42-month-old very preterm and full-term children. Early Hum. Dev. 2014, 90, 565–569. [Google Scholar] [CrossRef]
- Kommers, D.; Oei, G.; Chen, W.; Feijs, L.; Bambang Oetomo, S. Suboptimal bonding impairs hormonal, epigenetic and neuronal development in preterm infants, but these impairments can be reversed. Acta Paediatr. 2016, 105, 738–751. [Google Scholar] [CrossRef]
- Filippa, M.; Poisbeau, P.; Mairesse, J.; Monaci, M.G.; Baud, O.; Hüppi, P.; Grandjean, D.; Kuhn, P. Pain, Parental Involvement, and Oxytocin in the Neonatal Intensive Care Unit. Front. Psychol. 2019, 10. [Google Scholar] [CrossRef]
- Lammertink, F.; van den Heuvel, M.P.; Hermans, E.J.; Dudink, J.; Tataranno, M.L.; Benders, M.J.N.L.; Vinkers, C.H. Early-life stress exposure and large-scale covariance brain networks in extremely preterm-born infants. Transl. Psychiatry 2022, 12, 1–9. [Google Scholar]
- Palazzi, A.; Meschini, R.; Piccinini, C.A. NICU music therapy effects on maternal mental health and preterm infant’s emotional arousal. Infant Ment. Heal. J. 2021, 42, 672–689. [Google Scholar] [CrossRef] [PubMed]
- Standley, J. Music Therapy Research in the NICU: An Updated Meta-Analysis. Neonatal Netw. 2012, 31, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Vittner, D.; McGrath, J.; Robinson, J.; Lawhon, G.; Cusson, R.; Eisenfeld, L.; Walsh, S.; Young, E.; Cong, X. Increase in Oxytocin From Skin-to-Skin Contact Enhances Development of Parent–Infant Relationship. Biol. Res. Nurs. 2017, 20, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Vittner, D.; Butler, S.; Smith, K.; Makris, N.; Brownell, E.; Samra, H.; McGrath, J. Parent Engagement Correlates With Parent and Preterm Infant Oxytocin Release During Skin-to-Skin Contact. Adv. Neonatal Care 2019, 19, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Filippa, M.; Monaci, M.G.; Spagnuolo, C.; Serravalle, P.; Daniele, R.; Grandjean, D. Maternal speech decreases pain scores and increases oxytocin levels in preterm infants during painful procedures. Sci. Rep. 2021, 11, 1–10. [Google Scholar] [CrossRef]
- Udry-Jørgensen, L.; Pierrehumbert, B.; Borghini, A.; Habersaat, S.; Forcada-Guex, M.; Ansermet, F.; Muller-Nix, C. Quality of attachment, perinatal risk, and mother–infant interaction in a high-risk premature sample. Infant Ment. Health J. 2011, 32, 305–318. [Google Scholar] [CrossRef]
- Gimpl, G.; Fahrenholz, F. The Oxytocin Receptor System: Structure, Function, and Regulation. Physiol. Rev. 2001, 81, 629–683. [Google Scholar] [CrossRef]
- Hasan, M.T.; Althammer, F.; da Gouveia, M.S.; Goyon, S.; Eliava, M.; Lefevre, A.; Kerspern, D.; Schimmer, J.; Raftogianni, A.; Wahis, J.; et al. A Fear Memory Engram and Its Plasticity in the Hypothalamic Oxytocin System. Neuron 2019, 103, 133–146.e8. [Google Scholar] [CrossRef]
- Knobloch, H.S.; Charlet, A.; Hoffmann, L.C.; Eliava, M.; Khrulev, S.; Cetin, A.H.; Osten, P.; Schwarz, M.K.; Seeburg, P.H.; Stoop, R.; et al. Evoked Axonal Oxytocin Release in the Central Amygdala Attenuates Fear Response. Neuron 2012, 73, 553–566. [Google Scholar] [CrossRef]
- Viviani, D.; Charlet, A.; Burg, E.V.D.; Robinet, C.; Hurni, N.; Abatis, M.; Magara, F.; Stoop, R. Oxytocin Selectively Gates Fear Responses Through Distinct Outputs from the Central Amygdala. Science 2011, 333, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Wahis, J.; Baudon, A.; Althammer, F.; Kerspern, D.; Goyon, S.; Hagiwara, D.; Lefevre, A.; Barteczko, L.; Boury-Jamot, B.; Bellanger, B.; et al. Astrocytes mediate the effect of oxytocin in the central amygdala on neuronal activity and affective states in rodents. Nat. Neurosci. 2021, 24, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Schmitz-Koep, B.; Zimmermann, J.; Menegaux, A.; Nuttall, R.; Bäuml, J.G.; Schneider, S.C.; Daamen, M.; Boecker, H.; Zimmer, C.; Wolke, D.; et al. Decreased amygdala volume in adults after premature birth. Sci. Rep. 2021, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kanel, D.; Vanes, L.D.; Ball, G.; Hadaya, L.; Falconer, S.; Counsell, S.J.; Edwards, A.D.; Nosarti, C. Neonatal amygdala resting-state functional connectivity and socio-emotional development in very preterm children. Brain Commun. 2022, 4. [Google Scholar] [CrossRef] [PubMed]
- Alaerts, K.; Bernaerts, S.; Prinsen, J.; Dillen, C.; Steyaert, J.; Wenderoth, N. Oxytocin induces long-lasting adaptations within amygdala circuitry in autism: A treatment-mechanism study with randomized placebo-controlled design. Neuropsychopharmacology 2020, 45, 1141–1149. [Google Scholar] [CrossRef]
- Scharrer, E. Die Lichtempfindlichkeit Blinder Elritzen. (Untersuchungen Über das Zwischenhirn der Fische I.). Z. Für Vgl. Physiol. 1928, 7, 1–38. [Google Scholar] [CrossRef]
- du Vigneaud, V.; Ressler, C.; Swan, J.M.; Roberts, C.W.; Katsoyannis, P.G. The Synthesis of Oxytocin. J. Am. Chem. Soc. 1954, 76, 3115–3121. [Google Scholar] [CrossRef]
- Grinevich, V.; Desarmã©Nien, M.G.; Chini, B.; Tauber, M.; Muscatelli, F. Ontogenesis of oxytocin pathways in the mammalian brain: Late maturation and psychosocial disorders. Front. Neuroanat. 2015, 8, 164. [Google Scholar] [CrossRef]
- Muscatelli, F.; Desarménien, M.G.; Matarazzo, V.; Grinevich, V. Oxytocin Signaling in the Early Life of Mammals: Link to Neurodevelopmental Disorders Associated with ASD. Curr. Top Behav. Neurosci. 2018, 35, 239–268. [Google Scholar]
- Matsunaga, W.; Miyata, S.; Takamata, A.; Bun, H.; Nakashima, T.; Kiyohara, T. LPS-induced Fos expression in oxytocin and vasopressin neurons of the rat hypothalamus. Brain Res. 2000, 858, 9–18. [Google Scholar] [CrossRef]
- Grinevich, V.; Ludwig, M. The multiple faces of the oxytocin and vasopressin systems in the brain. J. Neuroendocr. 2021, 33, e13004. [Google Scholar] [CrossRef] [PubMed]
- Bakos, J.; Srancikova, A.; Havranek, T.; Bacova, Z. Molecular Mechanisms of Oxytocin Signaling at the Synaptic Connection. Neural Plast. 2018, 2018, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chini, B.; Verhage, M.; Grinevich, V. The Action Radius of Oxytocin Release in the Mammalian CNS: From Single Vesicles to Behavior. Trends Pharmacol. Sci. 2017, 38, 982–991. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, M.; Apps, D.; Menzies, J.; Patel, J.C.; Rice, M.E. Dendritic Release of Neurotransmitters. Compr. Physiol. 2016, 7, 235–252. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, M.; Leng, G. Dendritic peptide release and peptide-dependent behaviours. Nat. Rev. Neurosci. 2006, 7, 126–136. [Google Scholar] [CrossRef]
- Ludwig, M.; Stern, J. Multiple signalling modalities mediated by dendritic exocytosis of oxytocin and vasopressin. Philos Trans R Soc Lond B Biol Sci. 2015, 370, 20140182. [Google Scholar] [CrossRef]
- Zheng, J.-J.; Li, S.-J.; Zhang, X.-D.; Miao, W.-Y.; Zhang, D.; Yao, H.; Yu, X. Oxytocin mediates early experience–dependent cross-modal plasticity in the sensory cortices. Nat. Neurosci. 2014, 17, 391–399. [Google Scholar] [CrossRef]
- Goaillard, J.-M.; Moubarak, E.; Tapia, M.; Tell, F. Diversity of Axonal and Dendritic Contributions to Neuronal Output. Front. Cell. Neurosci. 2020, 13. [Google Scholar] [CrossRef]
- Grinevich, V.; Hurlemann, R. (Eds.) Behavioral Pharmacology of Neuropeptides: Oxytocin, 1st ed.; Current Topics in Behavioral Neurosciences; Springer International Publishing: Cham, Switzerland, 2018; p. 1. [Google Scholar]
- Neumann, I. Stimuli and consequences of dendritic release of oxytocin within the brain. Biochem. Soc. Trans. 2007, 35, 1252–1257. [Google Scholar] [CrossRef]
- Carter, C.S. Sex differences in oxytocin and vasopressin: Implications for autism spectrum disorders? Behav Brain Res. 2007, 176, 170–186. [Google Scholar] [CrossRef]
- Busnelli, M.; Chini, B. Molecular Basis of Oxytocin Receptor Signalling in the Brain: What We Know and What We Need to Know. Curr. Top Behav. Neurosci. 2017, 35, 3–29. [Google Scholar] [CrossRef]
- Manning, M.; Misicka, A.; Olma, A.; Bankowski, K.; Stoev, S.; Chini, B.; Durroux, T.; Mouillac, B.; Corbani, M.; Guillon, G. Oxytocin and Vasopressin Agonists and Antagonists as Research Tools and Potential Therapeutics. J. Neuroendocr. 2012, 24, 609–628. [Google Scholar] [CrossRef] [PubMed]
- Hammock, E.A.D. Developmental Perspectives on Oxytocin and Vasopressin. Neuropsychopharmacology 2014, 40, 24–42. [Google Scholar] [CrossRef] [PubMed]
- Newmaster, K.T.; Nolan, Z.T.; Chon, U.; Vanselow, D.J.; Weit, A.R.; Tabbaa, M.; Hidema, S.; Nishimori, K.; Hammock, E.A.D.; Kim, Y. Quantitative cellular-resolution map of the oxytocin receptor in postnatally developing mouse brains. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Kang, H.J.; Kawasawa, Y.I.; Cheng, F.; Zhu, Y.; Xu, X.; Li, M.; Sousa, A.M.M.; Pletikos, M.; Meyer, K.A.; Sedmak, G.; et al. Spatio-temporal transcriptome of the human brain. Nature 2011, 478, 483–489. [Google Scholar] [CrossRef]
- Rokicki, J.; Kaufmann, T.; de Lange, A.-M.G.; van der Meer, D.; Bahrami, S.; Sartorius, A.M.; Haukvik, U.K.; Steen, N.E.; Schwarz, E.; Stein, D.J.; et al. Oxytocin receptor expression patterns in the human brain across development. Neuropsychopharmacology 2022, 47, 1550–1560. [Google Scholar] [CrossRef]
- Yoshimura, R.; Kimura, T.; Watanabe, D.; Kiyama, H. Differential expression of oxytocin receptor mRNA in the developing rat brain. Neurosci. Res. 1996, 24, 291–304. [Google Scholar] [CrossRef]
- Hammock, E.A.D.; Levitt, P. Oxytocin receptor ligand binding in embryonic tissue and postnatal brain development of the C57BL/6J mouse. Front. Behav. Neurosci. 2013, 7. [Google Scholar] [CrossRef]
- Grinevich, V.; Knobloch-Bollmann, H.S.; Eliava, M.; Busnelli, M.; Chini, B. Assembling the Puzzle: Pathways of Oxytocin Signaling in the Brain. Biol. Psychiatry 2015, 79, 155–164. [Google Scholar] [CrossRef]
- Chatterjee, O.; Patil, K.; Sahu, A.; Gopalakrishnan, L.; Mol, P.; Advani, J.; Mukherjee, S.; Christopher, R.; Prasad, T.S.K. An overview of the oxytocin-oxytocin receptor signaling network. J. Cell Commun. Signal. 2016, 10, 355–360. [Google Scholar] [CrossRef]
- Mitre, M.; Minder, J.; Morina, E.X.; Chao, M.V.; Froemke, R.C. Oxytocin Modulation of Neural Circuits. Curr. Top Behav. Neurosci. 2017, 35, 31–53. [Google Scholar] [CrossRef]
- Kimura, T. Differential expression of oxytocin receptor mRNA in the developing rat brain. Neuroscience Research [Internet]. 1 January 1996. Available online: https://www.academia.edu/27604227/Differential_expression_of_oxytocin_receptor_mRNA_in_the_developing_rat_brain (accessed on 3 November 2022).
- Tribollet, E.; Dubois-Dauphin, M.; Dreifuss, J.J.; Barberis, C.; Jard, S. Oxytocin Receptors in the Central Nervous System. Ann. New York Acad. Sci. 1992, 652, 29–38. [Google Scholar] [CrossRef] [PubMed]
- McCutcheon, J.E.; Conrad, K.L.; Carr, S.B.; Ford, K.A.; McGehee, D.S.; Marinelli, M. Dopamine neurons in the ventral tegmental area fire faster in adolescent rats than in adults. J. Neurophysiol. 2012, 108, 1620–1630. [Google Scholar] [CrossRef] [PubMed]
- Madrigal, M.P.; Jurado, S. Specification of oxytocinergic and vasopressinergic circuits in the developing mouse brain. Commun Biol. 2021, 4, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Schaller, F.; Watrin, F.; Sturny, R.; Massacrier, A.; Szepetowski, P.; Muscatelli, F. A single postnatal injection of oxytocin rescues the lethal feeding behaviour in mouse newborns deficient for the imprinted Magel2 gene. Hum. Mol. Genet. 2010, 19, 4895–4905. [Google Scholar] [CrossRef]
- Tamborski, S.; Mintz, E.M.; Caldwell, H.K. Sex Differences in the Embryonic Development of the Central Oxytocin System in Mice. J. Neuroendocr. 2016, 28. [Google Scholar] [CrossRef]
- Olazábal, D.E.; Alsina-Llanes, M. Are age and sex differences in brain oxytocin receptors related to maternal and infanticidal behavior in naïve mice? Horm. Behav. 2016, 77, 132–140. [Google Scholar] [CrossRef]
- Onaka, T.; Takayanagi, Y. The oxytocin system and early-life experience-dependent plastic changes. J. Neuroendocrinol. 2021, 33, e13049. [Google Scholar] [CrossRef]
- Netser, S.; Meyer, A.; Magalnik, H.; Zylbertal, A.; De La Zerda, S.H.; Briller, M.; Bizer, A.; Grinevich, V.; Wagner, S. Distinct dynamics of social motivation drive differential social behavior in laboratory rat and mouse strains. Nat. Commun. 2020, 11, 5908. [Google Scholar] [CrossRef]
- Sachuriga; Nishimaru, H.; Takamura, Y.; Matsumoto, J.; Ferreira Pereira de Araújo, M.; Ono, T.; Nishijo, H. Neuronal Representation of Locomotion During Motivated Behavior in the Mouse Anterior Cingulate Cortex. Front. Syst. Neurosci. 2021, 15, 655110. [Google Scholar] [CrossRef]
- Craig, A.; Luo, N.L.; Beardsley, D.J.; Wingate-Pearse, N.; Walker, D.W.; Hohimer, A.; A Back, S. Quantitative analysis of perinatal rodent oligodendrocyte lineage progression and its correlation with human. Exp. Neurol. 2003, 181, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Dobbing, J.; Sands, J. Comparative aspects of the brain growth spurt. Early Hum. Dev. 1979, 3, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Semple, B.D.; Blomgren, K.; Gimlin, K.; Ferriero, D.M.; Noble-Haeusslein, L.J. Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol. 2013, 106-107, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Workman, A.D.; Charvet, C.J.; Clancy, B.; Darlington, R.B.; Finlay, B.L. Modeling Transformations of Neurodevelopmental Sequences across Mammalian Species. J. Neurosci. 2013, 33, 7368–7383. [Google Scholar] [CrossRef]
- Swaab, D. Development of the human hypothalamus. Neurochem. Res. 1995, 20, 509–519. [Google Scholar] [CrossRef]
- Cratty, B.J. Perceptual and Motor Development in Infants and Children, 2nd ed.; 1979. [Google Scholar]
- Peters, S.; Crone, E.A. Increased striatal activity in adolescence benefits learning. Nat. Commun. 2017, 8, 1–9. [Google Scholar] [CrossRef]
- Bosch, O.; Meddle, S.L.; Beiderbeck, D.I.; Douglas, A.J.; Neumann, I.D. Brain Oxytocin Correlates with Maternal Aggression: Link to Anxiety. J. Neurosci. 2005, 25, 6807–6815. [Google Scholar] [CrossRef]
- Lukas, M.; Toth, I.; Veenema, A.H.; Neumann, I.D. Oxytocin mediates rodent social memory within the lateral septum and the medial amygdala depending on the relevance of the social stimulus: Male juvenile versus female adult conspecifics. Psychoneuroendocrinology 2012, 38, 916–926. [Google Scholar] [CrossRef]
- Pedersen, C.A.; Prange, A.J. Induction of maternal behavior in virgin rats after intracerebroventricular administration of oxytocin. Proc. Natl. Acad. Sci. USA 1979, 76, 6661–6665. [Google Scholar] [CrossRef]
- Tapp, D.N.; Singstock, M.D.; Gottliebson, M.S.; McMurray, M.S. Central but not peripheral oxytocin administration reduces risk-based decision-making in male rats. Horm. Behav. 2020, 125, 104840. [Google Scholar] [CrossRef]
- Eliava, M.; Melchior, M.; Knobloch-Bollmann, H.S.; Wahis, J.; Gouveia, M.D.S.; Tang, Y.; Ciobanu, A.C.; del Rio, R.T.; Roth, L.C.; Althammer, F.; et al. A New Population of Parvocellular Oxytocin Neurons Controlling Magnocellular Neuron Activity and Inflammatory Pain Processing. Neuron 2016, 89, 1291–1304. [Google Scholar] [CrossRef] [PubMed]
- Manjila, S.B.; Betty, R.; Kim, Y. Missing pieces in decoding the brain oxytocin puzzle: Functional insights from mouse brain wiring diagrams. Front. Neurosci. 2020, 16, 1044736. [Google Scholar] [CrossRef]
- Son, S.; Manjila, S.B.; Newmaster, K.T.; Wu, Y.-T.; Vanselow, D.J.; Ciarletta, M.; Anthony, T.E.; Cheng, K.C.; Kim, Y. Whole-Brain Wiring Diagram of Oxytocin System in Adult Mice. J. Neurosci. 2022, 42, 5021–5033. [Google Scholar] [CrossRef] [PubMed]
- Raam, T.; McAvoy, K.M.; Besnard, A.; Veenema, A.H.; Sahay, A. Hippocampal oxytocin receptors are necessary for discrimination of social stimuli. Nat. Commun. 2017, 8, 2001. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.T.; Hsieh, T.Y.; Tsai, T.C.; Chen, C.C.; Huang, C.C.; Hsu, K.S. Conditional Deletion of Hippocampal CA2/CA3a Oxytocin Receptors Impairs the Persistence of Long-Term Social Recognition Memory in Mice. J. Neurosci. 2018, 38, 1218–1231. [Google Scholar] [CrossRef]
- Tsai, T.-C.; Fang, Y.-S.; Hung, Y.-C.; Hung, L.-C.; Hsu, K.-S. A dorsal CA2 to ventral CA1 circuit contributes to oxytocinergic modulation of long-term social recognition memory. J. Biomed. Sci. 2022, 29, 1–20. [Google Scholar] [CrossRef]
- Quintana, D.S.; Rokicki, J.; van der Meer, D.; Alnæs, D.; Kaufmann, T.; Córdova-Palomera, A.; Dieset, I.; Andreassen, O.A.; Westlye, L.T. Oxytocin pathway gene networks in the human brain. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Bakos, J.; Zatkova, M.; Bacova, Z.; Ostatnikova, D. The Role of Hypothalamic Neuropeptides in Neurogenesis and Neuritogenesis. Neural Plast. 2016, 2016, 1–10. [Google Scholar] [CrossRef]
- Lin, Y.-T.; Chen, C.-C.; Huang, C.-C.; Nishimori, K.; Hsu, K.-S. Oxytocin stimulates hippocampal neurogenesis via oxytocin receptor expressed in CA3 pyramidal neurons. Nat. Commun. 2017, 8, 1–16. [Google Scholar] [CrossRef]
- Han, B.; Bellemer, A.; Koelle, M.R. An Evolutionarily Conserved Switch in Response to GABA Affects Development and Behavior of the Locomotor Circuit of Caenorhabditis elegans. Genetics 2015, 199, 1159–1172. [Google Scholar] [CrossRef]
- Leonzino, M.; Busnelli, M.; Antonucci, F.; Verderio, C.; Mazzanti, M.; Chini, B. The Timing of the Excitatory-to-Inhibitory GABA Switch Is Regulated by the Oxytocin Receptor via KCC2. Cell Rep. 2016, 15, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Tyzio, R.; Cossart, R.; Khalilov, I.; Minlebaev, M.; Hübner, C.A.; Represa, A.; Ben-Ari, Y.; Khazipov, R. Maternal Oxytocin Triggers a Transient Inhibitory Switch in GABA Signaling in the Fetal Brain During Delivery. Science 2006, 314, 1788–1792. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ari, Y. Oxytocin and Vasopressin, and the GABA Developmental Shift During Labor and Birth: Friends or Foes? Front. Cell Neurosci. 2018, 12, 254. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, M.; Tsukahara, T.; Tomita, K.; Iwai, H.; Sonomura, T.; Miyawaki, S.; Sato, T. Neonatal maternal separation delays the GABA excitatory-to-inhibitory functional switch by inhibiting KCC2 expression. Biochem. Biophys. Res. Commun. 2017, 493, 1243–1249. [Google Scholar] [CrossRef]
- Chang, S.W.C.; Fagan, N.A.; Toda, K.; Utevsky, A.V.; Pearson, J.M.; Platt, M.L. Neural mechanisms of social decision-making in the primate amygdala. Proc. Natl. Acad. Sci. USA 2015, 112, 16012–16017. [Google Scholar] [CrossRef]
- Petrovic, P.; Kalisch, R.; Singer, T.; Dolan, R.J. Oxytocin Attenuates Affective Evaluations of Conditioned Faces and Amygdala Activity. J Neurosci. 2008, 28, 6607–6615. [Google Scholar] [CrossRef]
- Geng, Y.; Zhao, W.; Zhou, F.; Ma, X.; Yao, S.; Hurlemann, R.; Becker, B.; Kendrick, K.M. Oxytocin Enhancement of Emotional Empathy: Generalization Across Cultures and Effects on Amygdala Activity. Front. Neurosci. 2018, 12, 512. [Google Scholar] [CrossRef] [PubMed]
- Hurlemann, R.; Patin, A.; Onur, O.A.; Cohen, M.X.; Baumgartner, T.; Metzler, S.; Dziobek, I.; Gallinat, J.; Wagner, M.; Maier, W.; et al. Oxytocin Enhances Amygdala-Dependent, Socially Reinforced Learning and Emotional Empathy in Humans. J. Neurosci. 2010, 30, 4999–5007. [Google Scholar] [CrossRef]
- Haubensak, W.; Kunwar, P.S.; Cai, H.; Ciocchi, S.; Wall, N.R.; Ponnusamy, R.; Biag, J.; Dong, H.-W.; Deisseroth, K.; Callaway, E.M.; et al. Genetic dissection of an amygdala microcircuit that gates conditioned fear. Nature 2010, 468, 270–276. [Google Scholar] [CrossRef]
- Huber, D.; Veinante, P.; Stoop, R. Vasopressin and Oxytocin Excite Distinct Neuronal Populations in the Central Amygdala. Science 2005, 308, 245–248. [Google Scholar] [CrossRef]
- Arpino, C.; Compagnone, E.; Montanaro, M.L.; Cacciatore, D.; De Luca, A.; Cerulli, A.; Di Girolamo, S.; Curatolo, P. Preterm birth and neurodevelopmental outcome: A review. Child’s Nerv. Syst. 2010, 26, 1139–1149. [Google Scholar] [CrossRef] [PubMed]
- Johnston, M.V.; Hoon, A.H. Cerebral palsy. Neuromolecular Med. 2006, 8, 435–450. [Google Scholar] [CrossRef] [PubMed]
- Ream, M.A.; Lehwald, L. Neurologic Consequences of Preterm Birth. Curr. Neurol. Neurosci. Rep. 2018, 18, 48. [Google Scholar] [CrossRef]
- Zinni, M.; Pansiot, J.; Colella, M.; Faivre, V.; Delahaye-Duriez, A.; Guillonneau, F.; Bruce, J.; Salnot, V.; Mairesse, J.; Knoop, M.; et al. Impact of Fetal Growth Restriction on the Neonatal Microglial Proteome in the Rat. Nutrients 2021, 13, 3719. [Google Scholar] [CrossRef]
- Block, M.L.; Zecca, L.; Hong, J.-S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef]
- von Bartheld, C.S.; Bahney, J.; Herculano-Houzel, S. The Search for True Numbers of Neurons and Glial Cells in the Human Brain: A Review of 150 Years of Cell Counting. J. Comp. Neurol. 2016, 524, 3865–3895. [Google Scholar] [CrossRef]
- Jiang, X.; He, H.; Mo, L.; Liu, Q.; Yang, F.; Zhou, Y.; Li, L.; Su, D.; Yi, S.; Zhang, J. Mapping the Plasticity of Morphology, Molecular Properties and Function in Mouse Primary Microglia. Front. Cell. Neurosci. 2022, 15. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Sierra, A.; Stevens, B.; Tremblay, M.-E.; Aguzzi, A.; Ajami, B.; Amit, I.; Audinat, E.; Bechmann, I.; Bennett, M.; et al. Microglia states and nomenclature: A field at its crossroads. Neuron 2022, 110, 3458–3483. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T. Dynamic Aspects of Glial Reactions in Altered Brains. Pathol. Res. Pr. 1980, 168, 301–343. [Google Scholar] [CrossRef]
- Woodburn, S.C.; Bollinger, J.L.; Wohleb, E.S. The semantics of microglia activation: Neuroinflammation, homeostasis, and stress. J. Neuroinflammation 2021, 18, 1–16. [Google Scholar] [CrossRef]
- Matejuk, A.; Ransohoff, R.M. Crosstalk Between Astrocytes and Microglia: An Overview. Front. Immunol. 2020, 11, 1416. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Tang, Y.; Feng, J. Cross talk between activation of microglia and astrocytes in pathological conditions in the central nervous system. Life Sci. 2011, 89, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.-R.; Liu, J.-C.; Bao, J.-S.; Bai, Q.-Q.; Wang, G. Interaction of Microglia and Astrocytes in the Neurovascular Unit. Front. Immunol. 2020, 11, 1024. [Google Scholar] [CrossRef] [PubMed]
- Panaro, M.A.; Benameur, T.; Porro, C. Hypothalamic Neuropeptide Brain Protection: Focus on Oxytocin. J. Clin. Med. 2020, 9, 1534. [Google Scholar] [CrossRef] [PubMed]
- Kuban, K.C.K.; O’Shea, T.M.; Allred, E.N.; Fichorova, R.N.; Heeren, T.; Paneth, N.; Hirtz, D.; Dammann, O.; Leviton, A.; ELGAN Study Investigators. The breadth and type of systemic inflammation and the risk of adverse neurological outcomes in extremely low gestational age newborns. Pediatr Neurol. 2015, 52, 42–48. [Google Scholar] [CrossRef]
- Leviton, A.; Joseph, R.M.; Allred, E.N.; Fichorova, R.N.; O’Shea, T.M.; Kuban, K.K.; Dammann, O. The risk of neurodevelopmental disorders at age 10 years associated with blood concentrations of interleukins 4 and 10 during the first postnatal month of children born extremely preterm. Cytokine 2018, 110, 181–188. [Google Scholar] [CrossRef] [PubMed]
- O’Loughlin, E.; Pakan, J.M.P.; Yilmazer-Hanke, D.; McDermott, K.W. Acute in utero exposure to lipopolysaccharide induces inflammation in the pre- and postnatal brain and alters the glial cytoarchitecture in the developing amygdala. J. Neuroinflammation 2017, 14, 1–12. [Google Scholar] [CrossRef]
- Moerkerke, M.; Peeters, M.; de Vries, L.; Daniels, N.; Steyaert, J.; Alaerts, K.; Boets, B. Endogenous Oxytocin Levels in Autism—A Meta-Analysis. Brain Sci. 2021, 11, 1545. [Google Scholar] [CrossRef]
- Meziane, H.; Schaller, F.; Bauer, S.; Villard, C.; Matarazzo, V.; Riet, F.; Guillon, G.; Lafitte, D.; Desarmenien, M.G.; Tauber, M.; et al. An Early Postnatal Oxytocin Treatment Prevents Social and Learning Deficits in Adult Mice Deficient for Magel2, a Gene Involved in Prader-Willi Syndrome and Autism. Biol. Psychiatry 2014, 78, 85–94. [Google Scholar] [CrossRef]
- Dai, Y.-C.; Zhang, H.-F.; Schön, M.; Böckers, T.M.; Han, S.-P.; Han, J.-S.; Zhang, R. Neonatal Oxytocin Treatment Ameliorates Autistic-Like Behaviors and Oxytocin Deficiency in Valproic Acid-Induced Rat Model of Autism. Front. Cell. Neurosci. 2018, 12, 355. [Google Scholar] [CrossRef]
- Bertoni, A.; Schaller, F.; Tyzio, R.; Gaillard, S.; Santini, F.; Xolin, M.; Diabira, D.; Vaidyanathan, R.; Matarazzo, V.; Medina, I.; et al. Oxytocin administration in neonates shapes hippocampal circuitry and restores social behavior in a mouse model of autism. Mol. Psychiatry 2021, 26, 7582–7595. [Google Scholar] [CrossRef] [PubMed]
- Maejima, Y.; Yokota, S.; Ono, T.; Yu, Z.; Yamachi, M.; Hidema, S.; Nollet, K.E.; Nishimori, K.; Tomita, H.; Yaginuma, H.; et al. Identification of oxytocin expression in human and murine microglia. Prog. Neuro-Psychopharmacology Biol. Psychiatry 2022, 119. [Google Scholar] [CrossRef] [PubMed]
- Mairesse, J.; Zinni, M.; Pansiot, J.; Hassan-Abdi, R.; Demene, C.; Colella, M.; Charriaut-Marlangue, C.; Novais, A.R.B.; Tanter, M.; Maccari, S.; et al. Oxytocin receptor agonist reduces perinatal brain damage by targeting microglia. Glia 2018, 67, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Favrais, G.; van de Looij, Y.; Fleiss, B.; Ramanantsoa, N.; Bonnin, P.; Stoltenburg-Didinger, G.; Lacaud, A.; Saliba, E.; Dammann, O.; Gallego, J.; et al. Systemic inflammation disrupts the developmental program of white matter. Ann. Neurol. 2011, 70, 550–565. [Google Scholar] [CrossRef] [PubMed]
- Van Steenwinckel, J.; Schang, A.L.; Krishnan, M.L.; Degos, V.; Delahaye-Duriez, A.; Bokobza, C.; Csaba, Z.; Verdonk, F.; Montané, A.; Sigaut, S. Decreased microglial Wnt/β-catenin signalling drives microglial pro-inflammatory activation in the developing brain. Brain 2019, 142, 3806–3833. [Google Scholar] [CrossRef]
- van Tilborg, E.; Achterberg, E.J.M.; van Kammen, C.M.; van der Toorn, A.; Groenendaal, F.; Dijkhuizen, R.M.; Heijnen, C.J.; Vanderschuren, L.J.M.J.; Benders, M.N.J.L.; Nijboer, C.H.A. Combined fetal inflammation and postnatal hypoxia causes myelin deficits and autism-like behavior in a rat model of diffuse white matter injury. Glia 2018, 66, 78–93. [Google Scholar] [CrossRef]
- Sünnetçi, E.; Solmaz, V.; Erbaş, O. Chronic Oxytocin treatment has long lasting therapeutic potential in a rat model of neonatal hypercapnic-hypoxia injury, through enhanced GABAergic signaling and by reducing hippocampal gliosis with its anti-inflammatory feature. Peptides 2020, 135, 170398. [Google Scholar] [CrossRef]
- Yuan, L.; Liu, S.; Bai, X.; Gao, Y.; Liu, G.; Wang, X.; Liu, D.; Li, T.; Hao, A.; Wang, Z. Oxytocin inhibits lipopolysaccharide-induced inflammation in microglial cells and attenuates microglial activation in lipopolysaccharide-treated mice. J. Neuroinflammation 2016, 13, 1–17. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, S.; Liu, X.; Zheng, Y.; Li, L.; Meng, S. Oxytocin improves animal behaviors and ameliorates oxidative stress and inflammation in autistic mice. Biomed. Pharmacother. 2018, 107, 262–269. [Google Scholar] [CrossRef]
- Shigemori, T.; Sakai, A.; Takumi, T.; Itoh, Y.; Suzuki, H. Altered Microglia in the Amygdala Are Involved in Anxiety-related Behaviors of a Copy Number Variation Mouse Model of Autism. J. Nippon Med Sch. 2015, 82, 92–99. [Google Scholar] [CrossRef]
- Heuer, L.S.; Croen, L.A.; Jones, K.L.; Yoshida, C.K.; Hansen, R.L.; Yolken, R.; Zerbo, O.; DeLorenze, G.; Kharrazi, M.; Ashwood, P.; et al. An Exploratory Examination of Neonatal Cytokines and Chemokines as Predictors of Autism Risk: The Early Markers for Autism Study. Biol. Psychiatry 2019, 86, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Kordulewska, N.K.; Kostyra, E.; Piskorz-Ogórek, K.; Moszyńska, M.; Cieślińska, A.; Fiedorowicz, E.; Jarmołowska, B. Serum cytokine levels in children with spectrum autism disorder: Differences in pro- and anti-inflammatory balance. J. Neuroimmunol. 2019, 337, 577066. [Google Scholar] [CrossRef] [PubMed]
- Saghazadeh, A.; Ataeinia, B.; Keynejad, K.; Abdolalizadeh, A.; Hirbod-Mobarakeh, A.; Rezaei, N. A meta-analysis of pro-inflammatory cytokines in autism spectrum disorders: Effects of age, gender, and latitude. J. Psychiatr. Res. 2019, 115, 90–102. [Google Scholar] [CrossRef]
- Akhondzadeh, S.; Fallah, J.; Mohammadi, M.-R.; Imani, R.; Salehi, B.; Ghanizadeh, A.; Raznahan, M.; Mohebbi-Rasa, S.; Rezazadeh, S.-A.; Forghani, S. Double-blind placebo-controlled trial of pentoxifylline added to risperidone: Effects on aberrant behavior in children with autism. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2010, 34, 32–36. [Google Scholar] [CrossRef]
- Asadabadi, M.; Mohammadi, M.-R.; Ghanizadeh, A.; Modabbernia, A.; Ashrafi, M.; Hassanzadeh, E.; Forghani, S.; Akhondzadeh, S. Celecoxib as adjunctive treatment to risperidone in children with autistic disorder: A randomized, double-blind, placebo-controlled trial. Psychopharmacology 2012, 225, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Ghaleiha, A.; Rasa, S.M.; Nikoo, M.; Farokhnia, M.; Mohammadi, M.-R.; Akhondzadeh, S. A pilot double-blind placebo-controlled trial of pioglitazone as adjunctive treatment to risperidone: Effects on aberrant behavior in children with autism. Psychiatry Res. 2015, 229, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Masi, A.; Glozier, N.; Dale, R.; Guastella, A.J. The Immune System, Cytokines, and Biomarkers in Autism Spectrum Disorder. Neurosci. Bull. 2017, 33, 194–204. [Google Scholar] [CrossRef]
- Barbar, L.; Jain, T.; Zimmer, M.; Kruglikov, I.; Sadick, J.S.; Wang, M.; Kalpana, K.; Rose, I.V.L.; Burstein, S.R.; Rusielewicz, T.; et al. CD49f Is a Novel Marker of Functional and Reactive Human iPSC-Derived Astrocytes. Neuron 2020, 107, 436–453.e12. [Google Scholar] [CrossRef]
- Guttenplan, K.A.; Weigel, M.K.; Adler, D.I.; Couthouis, J.; Liddelow, S.A.; Gitler, A.D.; Barres, B.A. Knockout of reactive astrocyte activating factors slows disease progression in an ALS mouse model. Nat. Commun. 2020, 11, 1–9. [Google Scholar] [CrossRef]
- Baudon, A.; Creusot, E.C.; Althammer, F.; Schaaf, C.P.; Charlet, A. Emerging role of astrocytes in oxytocin-mediated control of neural circuits and brain functions. Prog. Neurobiol. 2022, 217. [Google Scholar] [CrossRef]
- Kuo, J.; Hariri, O.R.; Micevych, P. An Interaction of Oxytocin Receptors with Metabotropic Glutamate Receptors in Hypothalamic Astrocytes. J. Neuroendocr. 2009, 21, 1001–1006. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Qin, D.; Wang, Y.-F. Oxytocin Rapidly Changes Astrocytic GFAP Plasticity by Differentially Modulating the Expressions of pERK 1/2 and Protein Kinase A. Front. Mol. Neurosci. 2017, 10, 262. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Hasel, P.; Rose, I.V.L.; Sadick, J.S.; Kim, R.D.; Liddelow, S.A. Neuroinflammatory astrocyte subtypes in the mouse brain. Nat. Neurosci. 2021, 24, 1475–1487. [Google Scholar] [CrossRef] [PubMed]
- Mittaud, P.; Labourdette, G.; Zingg, H.; Guenot-Di Scala, D. Neurons modulate oxytocin receptor expression in rat cultured astrocytes: Involvement of TGF-β and membrane components. Glia 2002, 37, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Zicari, A.; Ticconi, C.; Realacci, M.; Cela, O.; Santangelo, C.; Pietropolli, A.; Russo, M.; Piccione, E. Hormonal regulation of cytokine release by human fetal membranes at term gestation: Effects of oxytocin, hydrocortisone and progesterone on tumour necrosis factor-α and transforming growth factor-β 1 output. J. Reprod. Immunol. 2002, 56, 123–136. [Google Scholar] [CrossRef]
- Wang, Y.; Pan, Q.; Tian, R.; Wen, Q.; Qin, G.; Zhang, D.; Chen, L.; Zhang, Y.; Zhou, J. Repeated oxytocin prevents central sensitization by regulating synaptic plasticity via oxytocin receptor in a chronic migraine mouse model. J. Headache Pain 2021, 22, 1–16. [Google Scholar] [CrossRef]
- Wang, Y.F.; Hatton, G.I. Interaction of Extracellular Signal-Regulated Protein Kinase 1/2 with Actin Cytoskeleton in Supraoptic Oxytocin Neurons and Astrocytes: Role in Burst Firing. J. Neurosci. 2007, 27, 13822–13834. [Google Scholar] [CrossRef]
- Meinung, C.P. Oxytocin Receptor-Mediated Signaling in Astrocytes; Universität Regensburg: Regensburg, Germany, 2020. [Google Scholar]
- Inoue, T.; Yamakage, H.; Tanaka, M.; Kusakabe, T.; Shimatsu, A.; Satoh-Asahara, N. Oxytocin Suppresses Inflammatory Responses Induced by Lipopolysaccharide through Inhibition of the eIF-2α–ATF4 Pathway in Mouse Microglia. Cells 2019, 8, 527. [Google Scholar] [CrossRef]
- Szeto, A.; Nation, D.A.; Mendez, A.J.; Dominguez-Bendala, J.; Brooks, L.G.; Schneiderman, N.; McCabe, P.M. Oxytocin attenuates NADPH-dependent superoxide activity and IL-6 secretion in macrophages and vascular cells. Am. J. Physiol. Metab. 2008, 295, E1495–E1501. [Google Scholar] [CrossRef]
- Szeto, A.; Sun-Suslow, N.; Mendez, A.J.; Hernandez, R.I.; Wagner, K.V.; McCabe, P.M. Regulation of the macrophage oxytocin receptor in response to inflammation. Am. J. Physiol. Metab. 2017, 312, E183–E189. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, S.; Hiraoka, Y.; Hidema, S.; Nishimori, K. Prenatal minocycline treatment alters synaptic protein expression, and rescues reduced mother call rate in oxytocin receptor-knockout mice. Biochem. Biophys. Res. Commun. 2016, 472, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Smith, C.J.; Van Eldik, L.J. Importance of MAPK pathways for microglial pro-inflammatory cytokine IL-1 beta production. Neurobiol. Aging 2004, 25, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Nijboer, C.H.; Heijnen, C.J.; Groenendaal, F.; May, M.J.; van Bel, F.; Kavelaars, A. A dual role of the NF-kappaB pathway in neonatal hypoxic-ischemic brain damage. Stroke 2008, 39, 2578–2586. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Qin, B.; Shi, A.; Zhao, J.; Guo, X.; Dong, L. Oxytocin inhibited stress induced visceral hypersensitivity, enteric glial cells activation, and release of proinflammatory cytokines in maternal separated rats. Eur. J. Pharmacol. 2018, 818, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Cheng, M.; Ma, L.; Zhang, T.; Sun, Z.; Yu, C.; Wang, J.; Dou, Y. Oxytocin Nanogels Inhibit Innate Inflammatory Response for Early Intervention in Alzheimer’s Disease. ACS Appl. Mater. Interfaces 2022, 14, 21822–21835. [Google Scholar] [CrossRef]
- Bordt, E.A.; Smith, C.J.; Demarest, T.G.; Bilbo, S.D.; Kingsbury, M.A. Mitochondria, Oxytocin, and Vasopressin: Unfolding the Inflammatory Protein Response. Neurotox. Res. 2018, 36, 239–256. [Google Scholar] [CrossRef] [PubMed]
- Fernández, D.; Geisse, A.; Bernales, J.I.; Lira, A.; Osorio, F. The Unfolded Protein Response in Immune Cells as an Emerging Regulator of Neuroinflammation. Front. Aging Neurosci. 2021, 13. [Google Scholar] [CrossRef]
- Havranek, T.; Lešťanová, Z.; Mravec, B.; Štrbák, V.; Bakos, J.; Bacova, Z. Oxytocin Modulates Expression of Neuron and Glial Markers in the Rat Hippocampus. Folia Biol. 2017, 63, 91–97. [Google Scholar]
- Zöller, T.; Schneider, A.; Kleimeyer, C.; Masuda, T.; Potru, P.S.; Pfeifer, D.; Blank, T.; Prinz, M.; Spittau, B. Silencing of TGFβ signalling in microglia results in impaired homeostasis. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Baxter, P.S.; Dando, O.; Emelianova, K.; He, X.; McKay, S.; Hardingham, G.E.; Qiu, J. Microglial identity and inflammatory responses are controlled by the combined effects of neurons and astrocytes. Cell Rep. 2021, 34, 108882. [Google Scholar] [CrossRef] [PubMed]
- Sperlágh, B.; Illes, P. Purinergic modulation of microglial cell activation. Purinergic Signal 2006, 3, 117–127. [Google Scholar] [CrossRef]
- Du, D.; Jiang, M.; Liu, M.; Wang, J.; Xia, C.; Guan, R.; Shen, L.; Ji, Y.; Zhu, D. Microglial P2X7 receptor in the hypothalamic paraventricular nuclei contributes to sympathoexcitatory responses in acute myocardial infarction rat. Neurosci. Lett. 2015, 587, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Zhang, Y.; Zhang, J.; Zhu, Z.; Lv, Q.; Su, J. Astrocyte-derived CCL7 promotes microglia-mediated inflammation following traumatic brain injury. Int. Immunopharmacol. 2021, 99, 107975. [Google Scholar] [CrossRef]
- Bennett, M.L.; Bennett, F.C.; Liddelow, S.A.; Ajami, B.; Zamanian, J.L.; Fernhoff, N.B.; Mulinyawe, S.B.; Bohlen, C.J.; Adil, A.; Tucker, A.; et al. New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. USA 2016, 113, E1738–E1746. [Google Scholar] [CrossRef]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 2019, 50, 253–271.E6. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Cheng, Z.; Zhou, L.; Darmanis, S.; Neff, N.F.; Okamoto, J.; Gulati, G.; Bennett, M.L.; Sun, L.O.; Clarke, L.E.; et al. Developmental Heterogeneity of Microglia and Brain Myeloid Cells Revealed by Deep Single-Cell RNA Sequencing. Neuron 2018, 101, 207–223.e10. [Google Scholar] [CrossRef]
- Marsh, S.E.; Walker, A.J.; Kamath, T.; Dissing-Olesen, L.; Hammond, T.R.; de Soysa, T.Y.; Young, A.M.H.; Murphy, S.; Abdulraouf, A.; Nadaf, N.; et al. Dissection of artifactual and confounding glial signatures by single-cell sequencing of mouse and human brain. Nat. Neurosci. 2022, 25, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef]
- Karelina, K.; Stuller, K.A.; Jarrett, B.; Zhang, N.; Wells, J.; Norman, G.J.; Devries, A.C. Oxytocin Mediates Social Neuroprotection After Cerebral Ischemia. Stroke 2011, 42, 3606–3611. [Google Scholar] [CrossRef]
- Béchade, C.; D’Andrea, I.; Etienne, F.; Verdonk, F.; Moutkine, I.; Banas, S.M.; Kolodziejczak, M.; Diaz, S.L.; Parkhurst, C.N.; Gan, W.B.; et al. The serotonin 2B receptor is required in neonatal microglia to limit neuroinflammation and sickness behavior in adulthood. Glia 2020, 69, 638–654. [Google Scholar] [CrossRef]
- Dölen, G.; Darvishzadeh, A.; Huang, K.W.; Malenka, R.C. Social reward requires coordinated activity of nucleus accumbens oxytocin and serotonin. Nature 2013, 501, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Yoshimura, M.; Shimizu, M.; Sanada, K.; Sonoda, S.; Nishimura, K.; Baba, K.; Ikeda, N.; Motojima, Y.; Maruyama, T.; et al. Endogenous oxytocin exerts anti-nociceptive and anti-inflammatory effects in rats. Commun. Biol. 2022, 5, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Lefevre, A.; Richard, N.; Jazayeri, M.; Beuriat, P.-A.; Fieux, S.; Zimmer, L.; Duhamel, J.-R.; Sirigu, A. Oxytocin and Serotonin Brain Mechanisms in the Nonhuman Primate. J. Neurosci. 2017, 37, 6741–6750. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Takayanagi, Y.; Inoue, K.; Kimura, T.; Young, L.J.; Onaka, T.; Nishimori, K. Evidence That Oxytocin Exerts Anxiolytic Effects via Oxytocin Receptor Expressed in Serotonergic Neurons in Mice. J. Neurosci. 2009, 29, 2259–2271. [Google Scholar] [CrossRef]
- Chruścicka, B.; Fitzsimons, S.E.W.; Borroto-Escuela, D.O.; Druelle, C.; Stamou, P.; Nally, K.; Dinan, T.G.; Cryan, J.F.; Fuxe, K.; Schellekens, H. Attenuation of Oxytocin and Serotonin 2A Receptor Signaling through Novel Heteroreceptor Formation. ACS Chem. Neurosci. 2019, 10, 3225–3240. [Google Scholar] [CrossRef]



| Study; Journal | Species; Strain; Sex | Disorder Studied/Simulated + Method of Induction | Age at OT(RA) Administration | OT(RA) Administration + Concentration | Age at Readout of Effect | Readouts Assessed | Effect OT on Neuro-inflammation |
Effect OT on Neuro-structural/ Functional Outcomes |
|---|---|---|---|---|---|---|---|---|
| [126] Glia | Rat; Sprague Dawley; both sexes | Intrauterine-growth-restriction; Low-protein diet and postnatal IL-1b injection | Twice daily administration at P1 and P2 | I.p. injection of OTR agonist carbetocin (1 mg/kg) | P2; P4; P10; behavior: one to two-months old | Microarray analysis; qRT-PCR; immunohistochemistry of Iba1/MBP/APC/Olig2; functional ultrasound imaging; behavioral tests (Open field; Y-maze) | Decrease in P4 microglial activation (pro-inflammatory cytokines + gene pathways involved in inflammation). | Rescue of P10 decrease in MBP density, number of APC oligodendrocytes. No effect on total oligo number. Rescue of anxious behavior and Y-maze learning deficiencies |
| [122] Biol. Psychiatry | Mouse; C57BL/6J background; males only | ASD; Genetic; Magel2+m/−p deficiency | Daily administration from P0-P6 First injection 3–5 h after birth | S.c. Injection 2 μg OT dissolved in 20 μL of isotonic saline | Adulthood (4 months old) | Behavioral tests: social recognition and social interaction tests, Morris water maze and Open field, Y-maze, object recognition task | Not reported | Restoration of normal quantity OT immunoreactivity in the medial amygdala. Restoration of deficits in social behavior and Morris water maze performance. |
| [124] Molec. Psychiatry | Mouse; C57BL/6J background; males only | ASD; Genetic; Magel2tm1.1Mus deficiency | P0,P2,P4,P6 | s.c. administration of 2 μg OT (1 mg/kg) | Adulthood | RNAscope (ISH) in hippocampus (aDG, CA2/CA3) Behavior: social behavior, locomotor and vertical activity, anxiety-like behavior and nonsocial memory | Not reported | Rescue of social memory deficit. No effect on sociability and social discrimination. Females: rescue Normalization of increased OT-binding sites in aDG, and increased SST+ neurons in aCA2/CA3d and aDG regions. Normalization of increased GABAergic activity in aCA2/CA3d neurons. Rescue of delayed excitatory-to-inhibitory GABA shift in hippocampal neurons. |
| [132] Biomed Pharmacother. | Mouse; C57BL/6J background; males only | ASD; Valproate acid treatment of pregnant dam | P30 (juvenile age) | Intranasal OT administration (200 μg/kg) 30 min before behavioral testing | P30 | RT-qPCR; immunohistochemistry; behavioral assays. | Normalization of increased TNF-α, IL-1β and IL-6 expression, and attenuation of increased oxidative stress response in amygdala and hippocampus. | Rescue of autism-like behavior in the open field, tail suspension test, social interaction test and marble burying test. |
| [130] Peptides | Rat; Sprague Dawley; both sexes | neonatal hypercapnic-hypoxia injury; P0 exposure to 100% CO2 (5 or 10 min) At 6 months: pentylenetetrazol-induced seizures | P1–P28 (1 month) | I.P., 100 IU/kg/day oxytocin | 6 months | GFAP Immunohistochemistry in CA1, ELISA (TNF-α + GAD-67) | Rescue of increased TNF-α levels, and of decreased GAD-67 levels. Reduction in astrogliosis (number of GFAP+ cells in CA1) | Not reported |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Knoop, M.; Possovre, M.-L.; Jacquens, A.; Charlet, A.; Baud, O.; Darbon, P. The Role of Oxytocin in Abnormal Brain Development: Effect on Glial Cells and Neuroinflammation. Cells 2022, 11, 3899. https://doi.org/10.3390/cells11233899
Knoop M, Possovre M-L, Jacquens A, Charlet A, Baud O, Darbon P. The Role of Oxytocin in Abnormal Brain Development: Effect on Glial Cells and Neuroinflammation. Cells. 2022; 11(23):3899. https://doi.org/10.3390/cells11233899
Chicago/Turabian StyleKnoop, Marit, Marie-Laure Possovre, Alice Jacquens, Alexandre Charlet, Olivier Baud, and Pascal Darbon. 2022. "The Role of Oxytocin in Abnormal Brain Development: Effect on Glial Cells and Neuroinflammation" Cells 11, no. 23: 3899. https://doi.org/10.3390/cells11233899
APA StyleKnoop, M., Possovre, M.-L., Jacquens, A., Charlet, A., Baud, O., & Darbon, P. (2022). The Role of Oxytocin in Abnormal Brain Development: Effect on Glial Cells and Neuroinflammation. Cells, 11(23), 3899. https://doi.org/10.3390/cells11233899