Abstract
Numerous studies recently showed that the inhibitory neurotransmitter, γ-aminobutyric acid (GABA), can stimulate cerebral angiogenesis and promote neurovascular coupling by activating the ionotropic GABAA receptors on cerebrovascular endothelial cells, whereas the endothelial role of the metabotropic GABAB receptors is still unknown. Preliminary evidence showed that GABAA receptor stimulation can induce an increase in endothelial Ca2+ levels, but the underlying signaling pathway remains to be fully unraveled. In the present investigation, we found that GABA evoked a biphasic elevation in [Ca2+]i that was initiated by inositol-1,4,5-trisphosphate- and nicotinic acid adenine dinucleotide phosphate-dependent Ca2+ release from neutral and acidic Ca2+ stores, respectively, and sustained by store-operated Ca2+ entry. GABAA and GABAB receptors were both required to trigger the endothelial Ca2+ response. Unexpectedly, we found that the GABAA receptors signal in a flux-independent manner via the metabotropic GABAB receptors. Likewise, the full Ca2+ response to GABAB receptors requires functional GABAA receptors. This study, therefore, sheds novel light on the molecular mechanisms by which GABA controls endothelial signaling at the neurovascular unit.
1. Introduction
The neurotransmitter γ-aminobutyric acid (GABA) is not only crucial to maintain a proper excitatory:inhibitory balance in neuronal networks by inhibiting glutamatergic pyramidal neurons [1,2,3], but is also instrumental to generate synchronized network oscillations that underpin optimal cognitive functions [4,5]. GABA dampens neuronal excitability by gating Cl−-permeable GABAA receptors, thereby enabling Cl− influx down the electrochemical gradient and hyperpolarizing the membrane potential [4]. GABAA receptors typically consist of pentameric channels formed by the combination of three distinct subunits, according to the following stoichiometry: 2α:2β:1γ [3,6]. GABA-dependent inhibition of neurotransmitter release and neuronal activity may also be mediated by metabotropic Gi/o protein–coupled GABAB receptors, which are widely distributed in the central nervous system [7,8]. GABAB receptors are composed of an obligatory heterodimer of the GABAB1 and GABAB2 subunits and induce slow postsynaptic inhibitory potentials by stimulating Gi/o protein-coupled inward-rectifying K+ channels (GIRK) [7,8]. In addition, it has been documented that GABAB receptors can induce an increase in intracellular Ca2+ concentrations ([Ca2+]i) in rat cortical neurons [9] and mouse cerebellar granule neurons [10]. GABAB receptors trigger intracellular Ca2+ signals via a signaling pathway that utilizes heterotrimeric Gi/o proteins to stimulate phospholipase Cβ (PLCβ) and cleave phosphatidylinositol 4,5-bisphosphate (PIP2) into the two intracellular second messengers inositol-1,4,5-trisphosphate (InsP3) and diacylglycerol (DAG). InsP3, in turn, binds to InsP3 receptors (InsP3Rs), which are located on the endoplasmic reticulum (ER), and promotes Ca2+ release into the cytoplasm. The following reduction in ER Ca2+ levels activates a Ca2+ entry pathway that resides on the plasma membrane, known as store-operated Ca2+ entry (SOCE), which prolongs over time the Ca2+ response to GABAB receptor stimulation [9,10,11].
GABAergic signaling within the neurovascular unit (NVU) has also been described in non-neuronal cells, including astrocytes [12,13] and cerebrovascular endothelial cells [14]. Cortical microvessels, as well as perivascular astrocytes, receive an extensive GABAergic input from local GABA interneurons [15]. Moreover, it has been clearly demonstrated that, during brain development, the pre-formed vascular network guides the tangential journey of GABAergic neurons from the dorsal to the basal telencephalon [16]. Thus, GABAergic neurons can establish a bidirectional communication with cerebrovascular endothelial cells to effectively coordinate these neurovascular interactions [16,17]. Mouse brain microvascular endothelial cells are endowed with the α1, α2, α6, β1, β2, β3, γ1, γ2 and γ3 subunits of GABAA receptors [18], while only functional evidence has been documented in favor of GABAB receptor expression [19]. The role played by endothelial GABAA receptors at the NVU has gathered growing interest upon the discovery that they control cerebral angiogenesis [17] and guide the radial migration of GABA interneurons [16] during embryonic development in mice, and regulate cerebral blood flow (CBF) and prevent neurological deficits in the adult [20]. The signaling pathways that are activated downstream of GABAA receptors in cerebrovascular endothelial cells are yet to be fully unraveled [21]. Preliminary evidence showed that muscimol, a selective GABAA receptor agonist, induced an inward Cl− current and a transient increase in [Ca2+]i in mice cerebrovascular endothelial cells [17]. Nevertheless, it is unclear how the inward (i.e., hyperpolarizing) current carried by the ionotropic GABAA receptors could elevate the [Ca2+]i in GABA-stimulated cells. Of note, recent studies have revealed that GABAA receptors may trigger a metabotropic (i.e., flux-independent) signaling pathway that leads to InsP3-induced Ca2+ release from the ER [22,23,24]. Since an increase in endothelial [Ca2+]i regulates both angiogenesis [25,26] and CBF [27,28], deciphering the mechanisms whereby GABA induces intracellular Ca2+ signals is mandatory to understand the molecular interactions between inhibitory interneurons and adjacent microvessels.
The hCMEC/D3 cell line represents the most suitable in vitro model of human cerebral microvascular endothelial cells [29,30] and has been largely exploited to unveil how cerebrovascular endothelium perceives and transduces neural activity with an increase in [Ca2+]i [27,31]. For instance, acetylcholine [32], adenosine trisphosphate (ATP) [33,34], glutamate [35], and histamine [36] bind to their specific Gq-protein coupled receptors (GqPCRs) and stimulate PLCβ to trigger InsP3-dependent Ca2+ release from the ER. This initial Ca2+ peak can be supported by lysosomal Ca2+ mobilization through two-pore channels 1 and 2 (respectively, TPC1 and TPC2) and is maintained over time by SOCE activation [27,31]. Intriguingly, the ionotropic N-methyl-D-aspartate (NMDA) receptors (NMDARs) were recently shown to signal an increase in [Ca2+]i in hCMEC/D3 cells in a flux-independent mode by interacting with metabotropic glutamate receptors [37]. Herein, we adopted an array of approaches, ranging from real-time quantitative reverse transcription PCR (qRT-PCR) to single-cell Fura-2 imaging to investigate the mechanisms whereby GABA elicits intracellular Ca2+ signals in hCMEC/D3 cells. We found that both GABAA and GABAB receptors are expressed and that GABA triggers a biphasic increase in [Ca2+]i. Pharmacological manipulation revealed that GABA-induced intracellular Ca2+ release was mediated by ER Ca2+ mobilization through InsP3Rs and lysosomal Ca2+ discharge via TPC1-2, whereas Ca2+ entry was mediated by SOCE. However, GABAA receptors did not mediate any detectable inward current, but they rather signaled in a flux-independent manner to cause intracellular Ca2+ release. Conversely, the selective stimulation of GABAB receptors induced the expected metabotropic Ca2+ signal that has been described in other cell types. Intriguingly, we provide evidence that GABAA and GABAB receptors interact to elicit a full Ca2+ response to GABA in hCMEC/D3 cells. These data provide the first elucidation of the signaling pathways whereby GABA can increase the [Ca2+]i in cerebrovascular endothelial cells.
2. Materials and Methods
2.1. Cell Culture
Human cerebral microvascular endothelial cells (hCMEC/D3) were obtained from the Institut National de la Santé et de la Recherche Médicale (INSERM, Paris, France). hCMEC/D3 cells cultured between passage 25 and 35 were used. As described in [38], the cells were seeded at a concentration of 27.000 cells/cm2 and grown in tissue culture flasks coated with 0.1 mg/mL rat tail collagen type 1, in the following medium: EBM-2 medium (Lonza, Basel, Switzerland) supplemented with 5% fetal bovine serum, 1% penicillin–streptomycin, 1.4 μM hydrocortisone, 5 μg/mL ascorbic acid, 1/100 chemically defined lipid concentrate (Life Technologies, Milan, Italy), 10 mM HEPES and 1 ng/mL basic fibroblast growth factor. The cells were cultured at 37 °C, 5% CO2 saturated humidity.
2.2. Solutions
Physiological salt solution (PSS) had the following composition (in mM): 150 NaCl, 6 KCl, 1.5 CaCl2, 1 MgCl2, 10 glucose, 10 HEPES. In Ca2+-free solution (0Ca2+), Ca2+ was substituted with 2 mM NaCl, and 0.5 mM EGTA was added. Solutions were titrated to pH 7.4 with NaOH. The osmolality of PSS as measured with an osmometer (Wescor 5500, Logan, UT, USA) was 300–310 mOsm/L.
2.3. [Ca2+]i Imaging
We utilized the Ca2+ imaging set-up that we have described elsewhere [32]. hCMEC/D3 cells were loaded with 4 µM Fura-2 acetoxymethyl ester (Fura-2/AM; 1 mM stock in dimethyl sulfoxide) in PSS for 30 min at 37 °C and 5% CO2 saturated humidity. After washing in PSS, the coverslip was fixed to the bottom of a Petri dish and the cells were observed by an upright epifluorescence Axiolab microscope (Carl Zeiss, Oberkochen, Germany), usually equipped with a Zeiss ×40 Achroplan objective (water-immersion, 2.0 mm working distance, 0.9 numerical aperture). The cells were excited alternately at 340 and 380 nm, and the emitted light was detected at 510 nm. A neutral density filter (1 or 0.3 optical density) reduced the overall intensity of the excitation light, and a second neutral density filter (optical density = 0.3) was coupled to the 380 nm filter to approach the intensity of the 340 nm light. A round diaphragm was used to increase the contrast. The excitation filters were mounted on a filter wheel (Lambda 10, Sutter Instrument, Novato, CA, USA). Custom software, working in the LINUX environment, was used to drive the camera (Extended-ISIS Camera, Photonic Science, Millham, UK) and the filter wheel, and to measure and plot on-line the fluorescence from 15–25 rectangular “regions of interest” (ROI) enclosing a corresponding number of single cells. Each ROI was identified by a number. Adjacent ROIs never superimposed. [Ca2+]i was monitored by measuring, for each ROI, the ratio of the mean fluorescence emitted at 510 nm when exciting alternatively at 340 and 380 nm [Ratio (F340/F380)]. An increase in [Ca2+]i causes an increase in the ratio [39,40]. Ratio measurements were performed and plotted on-line every 3 s. The experiments were performed at room temperature (22 °C).
2.4. Real-Time Reverse Transcription Quantitative PCR (qRT-PCR)
Total RNA was isolated from hCMEC/D3 cells (passage 33) using Trizol reagent (Thermo Fisher Scientific, Milan, Italy) according to the manufacturer’s instructions. After DNAse treatment (Turbo DNA-free™ kit, Thermo Fisher Scientific, Milan, Italy), RNA was quantified using a BioPhotometer D30 (Eppendorf, Hamburg, Germany). For cDNA synthesis, 100 ng RNA was reverse transcribed into 20 µL total volume using the iScriptTM cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA). Real-time reverse transcription quantitative PCRs with specific primers, designed on an exon–intron junction using the NCBI Primer tool (https://www.ncbi.nlm.nih.gov/tools/primer-blast/ (accessed on 3 March 2022)) [41] (Table S1) were performed using SsoFast™ EvaGreen® Supermix (Bio-Rad) on a CFX Connect Real-Time System (Bio-Rad) programmed as following: an initial step of 95 °C for 30 s, 40 cycles of 5 s at 95 °C, 5 s at 58 °C. A fluorescence reading was made at the end of each extension step. The PCR mixture consisted of 10 µL SsoFast™ EvaGreen® Supermix (Bio-Rad), 7 µL nuclease-free water, 1 µL cDNA and 1 µL of each forward and reverse primer. All primers were validated by melting curve analyses after each qRT-PCR run and determination of their efficiencies with at least four different cDNA concentrations. Gene expression was evaluated using the ΔΔCt method [42,43]. The genes actin beta (NM_001101) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (M17851) were used as endogenous reference for normalizing target mRNA [44]. For each sample, the 2−ΔΔCt value was calculated and represents the gene expression fold-change normalized to the reference gene and relative to the internal calibrator. Data are represented as mean ± SEM of fold-change values. Statistical analysis was performed using log-transformed values of the raw 2−ΔΔCt data. The statistical comparison of fold-changes in gene expression was analyzed with a one-way analysis of variance (ANOVA) followed by Bonferroni’s post-hoc test.
2.5. SDS-PAGE and Immunoblotting
hCMEC/D3 cells were lysed with Lysis Buffer (50 mM Tris–HCl, 150 mM NaCl, 1% Nonidet P-40, 1 mM EDTA, 0.25% deoxycholic acid, 0.1% SDS, pH 7.4, 1 mM PMSF, 5 μg/mL leupeptin, and 5 μg/mL aprotinin). Then, 10 μg of total cell proteins was separated by SDS-PAGE and transferred to a PVDF membrane. Membrane probing was performed using the different antibodies diluted 1:1000 in TBS (20 mM Tris, 500 mM NaCl, pH 7.5) containing 5% BSA and 0.1% Tween-20 in combination with the appropriate HRP-conjugated secondary antibodies (1:2000 in PBS plus 0.1% Tween-20). The following antibodies were used: anti-GABA A Receptor α1, clone 3H10 (ZRB1626) from Sigma-Aldrich (Saint Louis, MO, USA), anti-GABA B Receptor 1, clone 2D7 (ab55051) from Abcam (Cambridge, UK). The chemiluminescence reaction was performed using Immobilon Western (Millipore) and images were acquired by the Chemidoc XRS (Bio-Rad, Segrate, MI, Italy).
2.6. Electrophysiological Recordings
The presence of GABAA-receptor-mediated Cl− currents was assessed by using a port-a-patch planar patch-clamp system (Nanion Technologies, Munich, Germany) in the whole-cell, voltage-clamp configuration, at room temperature (22 °C), as described in [45]. Cultured cells (2–3 days after plating) were detached with Detachin and suspended at a cell density of 1–5 × 106 cells/mL in an external recording solution containing (in mM): 145 NaCl, 2.8 KCl, 2 MgCl2, 10 CaCl2, 10 HEPES, 10 D-glucose (pH = 7.4). Suspended cells were placed on the NPC© chip surface, and the whole cell configuration was achieved. Internal recording solution, containing (in mM) 10 CsCl, 110 CsF, 10 NaCl, 10 HEPES, 10 EGTA (pH = 7.2, adjusted with CsOH), was deposited in recording chips, having resistances of 3–5 MΩ. To test the ability of GABAA receptors to conduct inward Cl− currents, 100 µM GABA or 30 µM muscimol was added to the external solution. The bioelectrical response to agonist stimulation was recorded in the voltage-clamp mode at a holding potential of −70 mV, as described in [17], by using an EPC-10 patch-clamp amplifier (HEKA, Munich, Germany). Immediately after the whole-cell configuration was established, the cell capacitance and the series resistances (<10 MΩ) were measured. During the recordings, these two parameters were measured, and if exceeding ≥10% with respect to the initial value, the experiment was discontinued [45]. Liquid junction potential and capacitive currents were cancelled using the automatic compensation of the EPC-10 [46]. Data were filtered at 10 kHz and sampled at 5 kHz.
2.7. Statistical Analysis of Ca2+ Signals
All the data have been obtained from hCMEC/D3 cells from at least three independent experiments. The amplitude of agonist-evoked Ca2+ signals was measured as the difference between the ratio at the Ca2+ peak and the mean ratio of 30 s baseline before the peak. Pooled data are given as mean ± SEM, while the number of cells analyzed is indicated above the corresponding histogram bars (number of responding cells/total number of analyzed cells). Comparisons between the two groups were done using the Student’s t-test, whereas multiple comparisons were performed using ANOVA with the Bonferroni and Dunnett’s post-hoc test, as appropriate. The Bonferroni post-hoc test was used to evaluate multiple comparisons between different means, while Dunnett’s post-hoc test was used to compare each mean to a control mean. p-values less than 0.05 were considered statistically significant.
2.8. Chemicals
Fura-2/AM was purchased from Molecular Probes (Molecular Probes Europe BV, Leiden, The Netherlands). Nigericin, NED-19, NED-K, and GABA were obtained from Tocris (Bristol, UK). BTP-2 was purchased from Merck Millipore (Darmstadt, Germany). All the other chemicals were of analytical grade and obtained from Sigma Chemical Co. (St. Louis, MO, USA).
3. Results
3.1. GABAA and GABAB Receptors Are Expressed in hCMEC/D3 Cells
A thorough qRT-PCR analysis was carried out to assess whether and which GABA receptor subunits are expressed in hCMEC/D3 cells, as previously shown in mouse brain cerebrovascular endothelial cells [17,18], by using the specific primers reported in Table S1. The transcripts encoding for the following GABAA receptor subunits were expressed: α1, α5, β1, β2, and γ1 (Figure 1A). Comparison of the mean fold-change values revealed the following mRNA expression profile: α1 > β1 > α5 = β2 = γ1 (Figure 1A). GABAB1 and GABAB2 subunit mRNAs were also expressed (Figure 1B), although the transcripts encoding for the GABAB1 isoform were significantly up-regulated as compared to GABAB2 (Figure 1B). Immunoblotting confirmed that both GABAA α1 and GABAB1 subunits were expressed at the protein level (Figure 1C). These data, therefore, demonstrate that GABA receptors are expressed also in the human cerebrovascular endothelial cell line, hCMEC/D3.
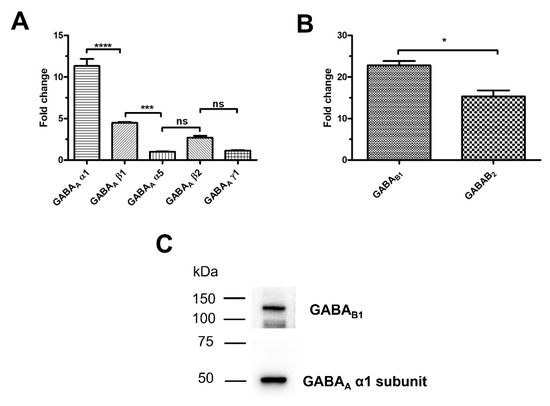
Figure 1.
GABAA and GABAB receptors are expressed in hCMEC/D3 cells. (A), a panel of GABAA receptor subunit transcripts are expressed in hCMEC/D3 cells based on qRT-PCR data. **** indicate p < 0.0001 *** indicate p < 0.001 (One-way ANOVA followed by the post-hoc Bonferroni test). NS indicates not significant. (B), both GABAB1 and GABAB2 receptor subunit transcripts are expressed in hCMEC/D3 cells. * indicates p < 0.05 (Student’s t-test). (C), representative immunoblots showing that both GABAA α1 and GABAB1 subunits are expressed at protein level. qRT-PCR and immunoblotting analyses were carried out on three distinct biological replicates.
3.2. GABA Induces a Dose-Dependent Increase in [Ca2+]i in hCMEC/D3 Cells
In order to assess whether GABA was able to increase the [Ca2+]i, hCMEC/D3 cells were loaded with the Ca2+-sensitive fluorophore, Fura-2/AM, as described in [35,37]. GABA was found to induce an elevation in [Ca2+]i already at a dose as low as 1 pM (Figure 2A). The Ca2+ response was consistently observed by challenging hCMEC/D3 cells with increasing concentrations of GABA, ranging from 1 pM to 100 µM (Figure 2A,B). At each dose tested, the Ca2+ signal comprised an initial Ca2+ peak which decayed to a plateau level that was maintained as long as the agonist was presented to the cells, as shown for the Ca2+ response to 100 µM GABA (Figure 2C). Figure 2C shows that, after a short washout, hCMEC/D3 cells were able to promptly respond to a second application of 100 µM GABA. The dose–response relationship did not show the typical S-shaped curve that is mediated by membrane receptors coupled to PLC; accordingly, the amplitude of the initial Ca2+ transient was relatively constant between 1 pM and 100 nM (Figure 2B). The highest Ca2+ response was elicited by 1 µM GABA, although a robust increase in [Ca2+]i could also be observed at higher agonist concentrations, i.e., 10 and 100 µM (Figure 2A,B). Nevertheless, 100 µM is the concentration range that has been exploited by most of the studies investigating how GABA induced intracellular Ca2+ signals in neurons and astrocytes [9,10,11,12,13]. Furthermore, GABA concentrations at the synaptic cleft can increase up to 80 µM during neuronal activity [47], although they can reach over-saturating levels (≈3 mM) in response to tetanic stimulation [48]. Therefore, 100 µM GABA, which reliably induces robust elevations in [Ca2+]i in hCMEC/D3 cells, was chosen to characterize the underlying signaling pathways.
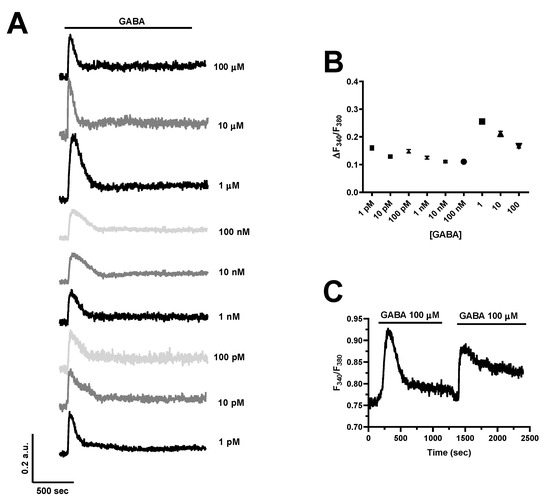
Figure 2.
GABA evokes a dose–response increase in [Ca2+]i in hCMEC/D3 cells. (A), the inhibitory neurotransmitter, GABA, elicits a dose-dependent elevation in [Ca2+]i, which remains relatively stable between 1 pM and 100 nM and achieves its peak at 1 µM. In this and the following figures, the black bar above the Ca2+ tracings indicates the time of agonist addition. (B), mean ± SE of the amplitude of the initial Ca2+ peak evoked by GABA at each agonist concentration ([GABA]). The number of analyzed cells ranges between 103 and 117 from three independent experiments for each dose. (C), GABA (100 μM) elicits an additional increase in [Ca2+]i upon 3 min washout, which indicates that GABA receptors do not desensitize at this agonist concentration.
3.3. GABA-Induced Intracellular Ca2+ Signals Are Sustained by Extracellular Ca2+ Entry through the SOCE Pathway in hCMEC/D3 Cells
The endothelial Ca2+ response to extracellular autacoids can be shaped by extracellular Ca2+ entry through the plasma membrane and intracellular Ca2+ mobilization from endogenous organelles [26,49,50], as also demonstrated in hCMEC/D3 cells [32,33,34,35,36,51]. Therefore, in order to disentangle the contribution of intra- vs. extracellular Ca2+ sources to GABA-induced intracellular Ca2+ signals, we stimulated the cells in the absence of extracellular Ca2+ (0Ca2+). Figure 3A–C show that the removal of external Ca2+ significantly (p < 0.0001) reduced the amplitude of the initial Ca2+ peak and abolished the plateau phase, thereby turning the biphasic elevation in [Ca2+]i into a transient Ca2+ signal. The subsequent restitution of Ca2+ to the perfusate induced a second elevation in [Ca2+]i, which was due to extracellular Ca2+ entry (Figure 3B). GABA was removed from the bath 100 s before reintroducing Ca2+ into the external saline to prevent the activation of second messenger-operated channels (SMOCs). As discussed elsewhere [32,35,38], the main physiological stimulus coupling GABA receptor activation to extracellular Ca2+ influx is, therefore, represented by the initial depletion of the endogenous Ca2+ reservoir. In order to confirm that SOCE sustains GABA-induced Ca2+ entry, hCMEC/D3 cells were pre-treated with either Pyr6 (10 µM), BTP-2 (20 µM) or Gd3+ (10 µM), three distinct inhibitors of Orai1 [35,52,53,54], i.e., the Ca2+-selective channel that is activated upon ER Ca2+ depletion in these cells [32]. The pharmacological blockade of SOCE with Pyr6 or BTP-2 suppressed GABA-evoked extracellular Ca2+ influx in most cells and significantly reduced the amplitude of the residual Ca2+ entry in the remaining ones (Figure 3D,E). In accord, 10 µM Gd3+, which plugs the pore of Orai1 channels [55,56], fully abolished GABA-evoked extracellular Ca2+ entry (Figure 3C,D). To rule out the involvement of SMOCs in the Ca2+ response to GABA, we inhibited Transient Receptor Potential Vanilloid 4 (TRPV) channels, which are expressed in hCMEC/D3 cells and are sensitive to RN-1734 [57]. Supplementary Figure S1 shows that pre-treatment with RN-1734 (20 µM) did not affect GABA-evoked extracellular Ca2+ influx. Conversely, hCMEC/D3 cells express only very low levels of TRP Canonical 7 (TRPC7) channels [32], while they lack other TRPC channel isoforms that can support agonist-dependent Ca2+ influx in vascular endothelial cells, such as TRPC3 and TRPC6 [26,58]. Altogether, these findings indicate that SOCE supports GABA-induced intracellular Ca2+ signals in hCMEC/D3 cells.
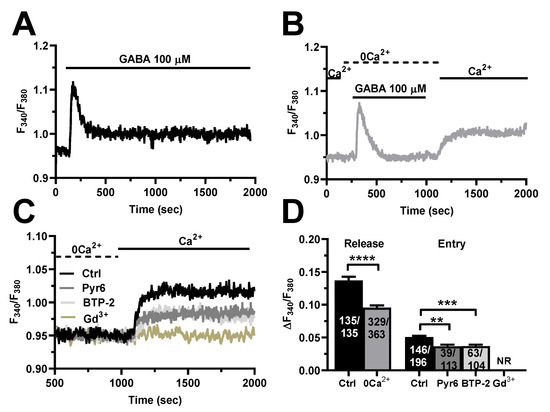
Figure 3.
GABA evokes intracellular Ca2+ release and SOCE activation in hCMEC/D3 cells. (A), representative tracing of the biphasic Ca2+ response induced by GABA (100 µM) in the presence of extracellular Ca2+ (Ctrl). (B), representative tracing of the two distinct components of GABA-evoked intracellular Ca2+ signals in hCMEC/D3 cells. GABA induced a transient increase in [Ca2+]i in the absence of extracellular Ca2+ (0Ca2+), which is due to endogenous Ca2+ mobilization. Restoration of extracellular Ca2+ (1.5 mM) after removal of the agonist induced a second increase in [Ca2+]i, which is indicative of SOCE activation. (C), GABA-evoked extracellular Ca2+ entry was significantly reduced in the presence of Pyr6 (10 µM, 10 min), BTP-2 (20 µM, 10 min), or Gd3+ (20 µM, 10 min), three specific SOCE inhibitors. Intracellular Ca2+ release is not shown. (D), mean ± SE of the amplitude of GABA-evoked Ca2+ peaks in the presence (Ctrl) and absence (0Ca2+) of extracellular Ca2+. **** indicate p < 0.0001 (Student’s t-test). Mean ± SE of the amplitude of GABA-evoked extracellular Ca2+ entry in the absence (Ctrl) and presence of Pyr6, BTP-2 or Gd3+. *** indicate p < 0.001 and ** indicate p < 0.01 (one-way ANOVA followed by the post-hoc Dunnett’s test). NR indicates “No Response” as evaluated in 89 cells from three different experiments.
3.4. InsP3 and Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP) Trigger GABA-Induced Intracellular Ca2+ Release in hCMEC/D3 Cells
Growing evidence indicates that neurotransmitters and neuromodulators elicit intracellular Ca2+ release in the hCMEC/D3 cell line that is triggered by InsP3-induced ER Ca2+ mobilization through InsP3Rs and supported by NAADP-dependent lysosomal Ca2+ release via TPCs [35,36,37]. In accord, GABA-evoked intracellular Ca2+ mobilization was suppressed by blocking PLCβ activity with the aminosteroid U73122 (10 µM) [32,35,36,37] (Figure 4A) and by inhibiting InsP3Rs with the non-competitive antagonist 2-aminoethoxydiphenyl borate (2-APB; 50 µM) [32,35,36,37] (Figure 4A). Furthermore, the endogenous Ca2+ response to GABA was repressed by depleting the ER Ca2+ store with cyclopiazonic acid (CPA; 30 µM) (Figure 4B), which selectively affects the sarco-endoplasmic reticulum Ca2+ ATPase (SERCA) activity [32,35,36,37]. The statistical analysis of these experiments has been reported in Figure 4C. Lysosomal Ca2+ release via TPCs can recruit juxtaposed InsP3Rs through the mechanism of Ca2+-induced Ca2+ release, thereby triggering the Ca2+ response to extracellular stimuli in hCMEC/D3 cells [35,36,37], as well as in other endothelial cell types [59]. Figure 4D shows that GABA-induced intracellular Ca2+ release was abrogated by depleting the lysosomal Ca2+ pool with nigericin (50 µM), which acts as a H+/K+ antiporter and thereby dissipates the H+ gradient that maintains lysosomal Ca2+ refilling [60,61,62]. Moreover, the endogenous Ca2+ response to GABA (100 µM) was strongly inhibited by two specific NAADP antagonists, NED-19 (100 µM) (Figure 4E) and its chemically modified analogue, NED-K (100 µM) (Figure 4E) [60,63]. The analysis of these results has been illustrated in Figure 4F. Taken together, these findings demonstrate that InsP3-induced ER Ca2+ release and NAADP-evoked lysosomal Ca2+ discharge shape GABA-evoked intracellular Ca2+ mobilization in hCMEC/D3 cells.
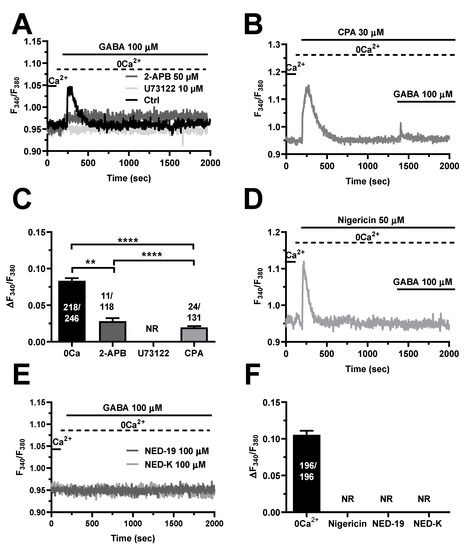
Figure 4.
InsP3 and NAADP mediate GABA-evoked intracellular Ca2+ release in hCMEC/D3 cells. (A), representative tracings of GABA-evoked intracellular Ca2+ release in the absence (Ctrl) and presence of U73122 (10 µM, 10 min) or 2-APB (50 µM, 30 min), which, respectively, inhibit PLC and InsP3Rs. GABA was administered at 100 µM. (B), CPA (30 µM), a selective blocker of SERCA activity, induced a transient elevation in [Ca2+]i under 0Ca2+ conditions due to passive ER Ca2+ efflux. The subsequent addition of GABA (100 µM) induced only a small Ca2+ transient, which was due to the lower ER Ca2+ content. (C), mean ± SE of the peak amplitude of GABA-evoked intracellular Ca2+ release under the designated treatments. **** indicate p < 0.0001, ** indicate p < 0.001 (One-way ANOVA followed by the post-hoc Bonferroni test). NR indicates “No Response” as evaluated in 102 cells from three different experiments. (D), depleting the lysosomal Ca2+ pool with the selective H+/K+ antiporter, nigericin (50 µM), caused a transient elevation in [Ca2+]i. The following addition of GABA (100 µM) failed to elicit a detectable Ca2+ signal. (E), inhibiting NAADP signaling with NED-19 (100 µM) or NED-K (100 µM) abolished the intracellular Ca2+ mobilization induced by GABA (100 µM). (F), mean ± SE of the peak amplitude of GABA-evoked intracellular Ca2+ release under the designated treatments. NR indicates “No Response” as evaluated in 104 cells treated with nigericin, with 101 cells treated with NED-19 and 99 cells treated with NED-K.
3.5. GABAA and GABAB Receptors Mediate GABA-Induced Intracellular Ca2+ Signals in hCMEC/D3 Cells
Recent work showed that GABAA receptors initiate the Ca2+ response to GABA in mouse cerebrovascular endothelial cells [17]. However, GABAB receptors, which trigger an increase in [Ca2+]i in both neurons [9,10] and astrocytes [12,13], were recently found to mediate intracellular Ca2+ signals also in human aortic endothelial cells (HAECs) [64]. To disentangle the GABA receptors involved in the endothelial Ca2+ signals (Figure 5A), we first exploited a battery of selective inhibitors of GABAA and GABAB receptors. Blocking GABAA receptors with SR95531 (gabazine; 10 µM) strongly reduced the percentage of responding cells and significantly reduced the peak Ca2+ response in the minority of hCMEC/D3 cells displaying a detectable Ca2+ signal [2,22] (Figure 5B,D,E). The same inhibitory effects were achieved by selectively inhibiting GABAB receptors with either saclofen (200 µM) or CGP35348 (100 µM) (Figure 5C–E). These data, therefore, demonstrate that both GABAA and GABAB receptors mediate the Ca2+ response to GABA in hCMEC/D3 cells.
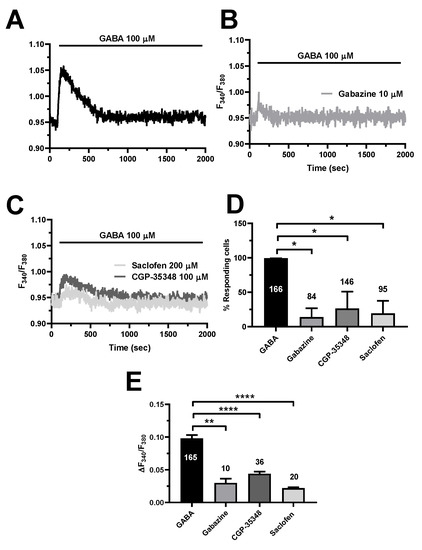
Figure 5.
The pharmacological blockade of GABAA and GABAB receptors inhibit GABA-evoked intracellular Ca2+ signals in hCMEC/D3 cells. (A), representative tracing of the Ca2+ response induced by GABA (100 µM) under control (Ctrl) conditions. (B), blocking GABAA receptors with gabazine (10 µM, 5 min) significantly reduced GABA-evoked intracellular Ca2+ signals. (C), blocking GABAB receptors with either saclofen (200 µM, 5 min) or CGP-35348 (100 µM, 5 min) significantly reduced GABA-evoked intracellular Ca2+ signals. (D), mean ± SE of the percentage of hCMEC/D3 cells responding to GABA under the designated treatments. * indicates p < 0.05 (one-way ANOVA followed by the post-hoc Dunnett’s test). (E), mean ± SE of the amplitude of the peak Ca2+ response to GABA under the designated treatments. **** indicates p < 0.0001, ** indicates p < 0.01 (one-way ANOVA followed by the post-hoc Dunnett’s test).
To further corroborate these findings, we took advantage of two different agonists of GABAA and GABAB receptors, i.e., respectively, muscimol [17,22] and baclofen [12,13,22]. Figure 6A shows that both muscimol (15 µM) and baclofen (100 µM) induced a biphasic increase in [Ca2+]i that closely resembled the Ca2+ response to GABA (100 µM). There was no significant difference in the percentage of responding cells (Figure 6B), while the amplitude of the peak Ca2+ response to GABA was significantly (p < 0.0001) higher as compared to both muscimol and baclofen (Figure 6C). The Ca2+ response to the combined application of muscimol (15 µM) and baclofen (100 µM) was also significantly (p < 0.0001) lower as compared to GABA (Figure 6). Nevertheless, these results confirm that both GABAA and GABAB contribute to triggering GABA-induced intracellular Ca2+ signals in the human cerebrovascular endothelial cell line hCMEC/D3.
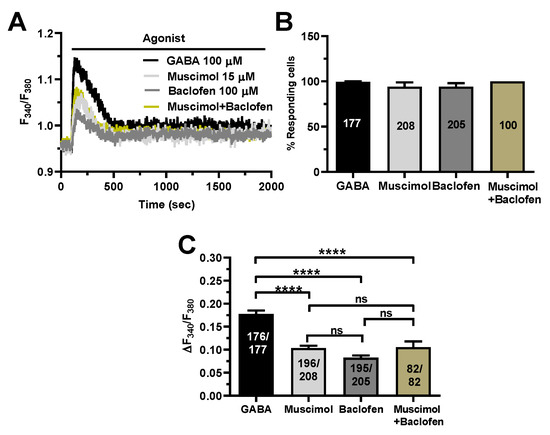
Figure 6.
Selective stimulation of GABAA and GABAB receptors induce intracellular Ca2+ signals in hCMEC/D3 cells. (A), representative tracings of the biphasic Ca2+ signals induced by GABA (100 µM), the GABAA receptor agonist, muscimol (15 µM), the GABAB receptor agonist, baclofen (100 µM), and muscimol (15 µM) + baclofen (100 µM), in hCMEC/D3 cells. (B), mean ± SE of the percentage of hCMEC/D3 cells responding to GABA, muscimol and baclofen. (C), mean ± SE of the amplitude of the peak Ca2+ response to GABA, muscimol and baclofen. **** indicate p < 0.0001 (one-way ANOVA followed by the post-hoc Bonferroni test). NS indicates not significant.
3.6. Evidence That GABAA and GABAB Receptors Must Interact to Increase the [Ca2+]i
The mechanistic analysis described in Section 3.3 and Section 3.4 revealed that the Ca2+ response to GABA is triggered by endogenous Ca2+ release from non-acidic (ER) and acidic (lysosomes) Ca2+ stores and maintained over time by SOCE. It has long been known that GABAB receptors are coupled to InsP3-induced ER Ca2+ mobilization and SOCE activation [7,8,9,10,12,13]. Interestingly, emerging evidence indicates that GABAA receptors could signal an increase in [Ca2+]i in a flux-independent manner [22,23,24], i.e., without the need for Cl− fluxes, possibly via the metabotropic GABAB receptors [22]. In agreement with these observations, planar patch-clamp recordings revealed that neither GABA (100 µM) (Figure 7A) nor muscimol (15 µM) (Figure 7B) were able to elicit detectable transmembrane currents in hCMEC/D3 cells. Furthermore, muscimol (15 µM) evoked robust intracellular Ca2+ mobilization (Figure 7C), which demonstrates that GABAA receptor stimulation is able to mobilize the intracellular Ca2+ pool. In agreement with this observation, restoration of extracellular Ca2+ led to SOCE activation in 101 out of 147 hCMEC/D3 cells (Figure 7C). These findings suggest that the GABAA subunits present in hCMEC/D3 cells assemble into a pentameric complex that is able to trigger intracellular Ca2+ signals by promoting intracellular Ca2+ release, but not to conduct sizeable Cl− currents, as recently shown in mouse ciliated oviductal cells [22]. In accord, the Ca2+ response to muscimol (15 µM) was significantly inhibited by blocking GABAA receptors with gabazine (10 µM) (Figure 7D,E). Notably, muscimol-evoked intracellular Ca2+ signals in hCMEC/D3 cells were also strongly reduced by inhibiting GABAB receptors with either saclofen (200 µM) or CGP-35348 (100 µM) (Figure 7D,E). Therefore, GABAA receptors require functional GABAB receptors to signal the increase in [Ca2+]i in a flux-independent manner. Similarly, the Ca2+ response to baclofen (100 µM) was sensitive to the pharmacological blockade of either GABAA receptors with gabazine (10 µM) (Figure 7F,G), or GABAB receptors with saclofen (200 µM) and CGP-35348 (100 µM) (Figure 7F,G). Therefore, the sequence of signaling events leading to GABA-induced intracellular Ca2+ signals in hCMEC/D3 cells requires the functional interaction between both GABAA and GABAB receptors, with GABAA receptors operating in a non-canonical (i.e., flux-independent) manner via GABAB receptors.
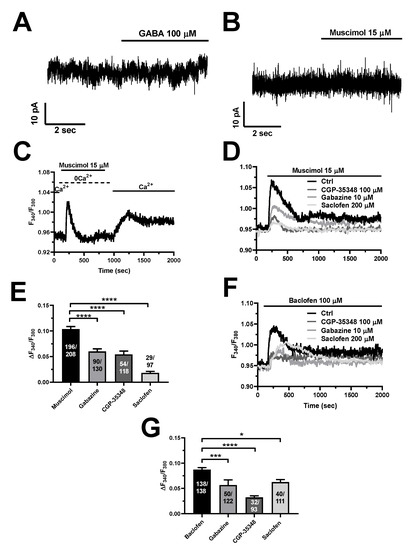
Figure 7.
GABA-evoked intracellular Ca2+ signals require the functional interaction between GABAA and GABAB receptors in hCMEC/D3 cells. Planar whole-cell patch-clamp recordings revealed that neither GABA (100 µM) (A) nor muscimol (15 µM) (B) induced any inward current at a Vh = −70 mV. (C), the Ca2+ add-back protocol showed that muscimol (15 µM) was able to mobilize the intracellular Ca2+ pool under 0Ca2+ conditions and to activate SOCE on restoration of extracellular Ca2+ levels (1.5 mM) in the absence of the agonist. (D), the Ca2+ response to muscimol (15 µM) was reduced by blocking either GABAA receptors with gabazine (10 µM, 5 min), or GABAB receptors with saclofen (200 µM, 5 min) or CGP-35348 (100 µM, 5 min). (E), mean ± SE of the amplitude of the peak Ca2+ response to muscimol in the absence (Ctrl) or in the presence of gabazine, CGP-35348 or saclofen. **** indicate p < 0.0001 (one-way ANOVA followed by the post-hoc Dunnett’s test). (F), the Ca2+ response to baclofen (100 µM) was reduced by blocking either GABAA receptors with gabazine (10 µM, 5 min), or GABAB receptors with saclofen (200 µM, 5 min) or CGP-35348 (100 µM, 5 min). (G), mean ± SE of the amplitude of the peak Ca2+ response to baclofen in the absence (Ctrl) or in the presence of gabazine, CGP-35348 or saclofen. **** indicate p < 0.0001, *** indicate p < 0.001, * indicates p < 0.05 (one-way ANOVA followed by the post-hoc Dunnett’s test).
To further confirm this hypothesis, we pretreated hCMEC/D3 cells with pertussis toxin (PTx), which blocks the heterotrimeric Gi/o proteins that recruit PLCβ upon GABAB receptor activation [10,12,22]. Figure 8 shows that PTx (1 ng/mL) significantly reduced the Ca2+ response to GABA (100 µM), muscimol (15 µM), or baclofen (100 µM). These findings confirm that GABAB receptors signal the increase in [Ca2+]i via Gi/o proteins and lend further support to the evidence that GABAB receptors are also crucial to the onset of GABAA-receptor-mediated Ca2+ signals.
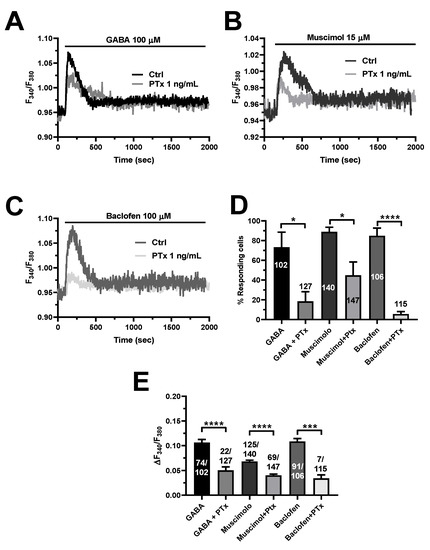
Figure 8.
Selective inhibition of heterotrimeric Gi/o proteins with pertussis toxin (PTX) reduces GABAA- and GABAB-induced intracellular Ca2+ signals in hCMEC/D3 cells. (A), representative tracings of the biphasic Ca2+ signals induced by GABA (100 µM), in the presence and in the absence of PTx (1 ng/mL). (B), Representative traces of the Ca2+ response induced by the GABAA receptor agonist, muscimol (15 µM), in the presence and in the absence of PTx (1 ng/mL). (C), Representative Ca2+ traces evoked by the GABAB receptor agonist, baclofen (100 µM), in the presence and in the absence of PTx (1 ng/mL). (D), mean ± SE of the percentage of hCMEC/D3 cells responding under the designated treatments. **** indicate p < 0.0001, * indicates p < 0.05 (Student’s t-test). (E), mean ± SE of the amplitude of the peak Ca2+ response under the designated treatments. **** indicate p < 0.0001, *** indicates p < 0.001 (Student’s t-test).
4. Discussion
Herein, we provided the first evidence that both GABAA and GABAB receptors mediate the Ca2+ response to the inhibitory neurotransmitter GABA in the widely employed human cerebrovascular endothelial cell line, hCMEC/D3. We further show that GABAA receptors operate in a metabotropic, i.e., flux-independent, mode, to signal the downstream increase in [Ca2+]i via the metabotropic GABAB receptors. In line with this evidence, GABAB receptors require functional GABAA receptors to elevate the [Ca2+]i in hCMEC/D3 cells. Unravelling the molecular mechanisms involved in endothelial GABA signaling will contribute to gather further insights into the mechanisms whereby endothelial GABAergic signaling regulates the NVU.
4.1. The Expression Profile of GABAA and GABAB Receptors in hCMEC/D3 Cells
The GABAergic innervation of intracortical microvessels by local GABA interneurons, rather than basal forebrain GABAergic terminals, has long been identified [15,65,66]. Subsequently, autoradiography with use of the GABAA receptor agonist, muscimol, demonstrated specific binding sites in cerebral arterioles [67]. More recently, a thorough RT-PCR characterization showed that mouse brain cerebrovascular endothelial cells express the α1, α2, α6, β1, β2, β3, γ1, γ2 and γ3 subunits of GABAA receptors [18]. A similar pattern of expression has been also detected in mouse embryonic forebrain endothelial cells [17]. In the present investigation, we found that hCMEC/D3 cells express the following GABAA receptor subunits: α1 (also confirmed by immunoblotting), α5, β1, β2, and γ1. Therefore, human cerebrovascular endothelial cells possess all the three distinct GABAA receptor subunits that can arrange into a pentameric ion channel with the likely stoichiometry 2α:2β:1γ [3,6]. Based upon their expression levels, GABAA receptors in hCMEC/D3 cells are predicted to incorporate the α1, β1, and γ1 subunits. Surprisingly, human cerebrovascular endothelial cells do not present the GABAA receptor β3 subunit, which is a crucial component of mouse GABAA receptors [17,20,21]. The expression of the GABAB receptor has hitherto been suggested only by an early study reporting on baclofen-induced nitric oxide (NO) release and collagen constriction in mouse cortical microvascular endothelial cells [19]. Herein, we provided the first evidence that both GABAB1 and GABAB2 subunits are expressed in hCMEC/D3 cells with GABAB1 showing enriched expression. Thus, a functional GABAB receptor heterodimer can be assembled in human cerebrovascular endothelium.
4.2. GABA Induces Intracellular Ca2+ Signals in hCMEC/D3 Cells
GABA is emerging as a crucial mediator of neuro-to-vascular communication at the NVU [68]; mouse cerebrovascular endothelial cells express ionotropic GABAA receptors that perceive GABA released during neuronal activity from inhibitory interneurons and trigger a signaling pathway that finely controls cerebral angiogenesis [17] and CBF [20]. It has been suggested that GABA activates GABAA receptors to evoke intracellular Ca2+ signals [17,20], which could, in turn, drive endothelial cell proliferation and angiogenesis [25,26,69] and recruit endothelial nitric oxide synthase (eNOS) to produce NO, i.e., the most important vaso-relaxing mediator in the brain [27,28,31,68]. Nevertheless, GABAA receptors are ionotropic receptors that are permeable to Cl− and, therefore, are not expected to directly raise the [Ca2+]i during GABAergic signaling [6]. The metabotropic GABAB receptors have recently been shown to cause a transient increase in [Ca2+]i in HAECs, but it is still unknown whether they are able to regulate the [Ca2+]i also in cerebrovascular endothelial cells. We found that GABA evoked a biphasic Ca2+ response over a wide concentration range in hCMEC/D3 cells. The Ca2+ response to GABA was already detectable as concentrations as low as 1 pM and achieved the peak at 1 µM. The amplitude of the initial Ca2+ peak progressively decreased with further increases in agonist concentration. This dose–response relationship is quite different from the sigmoidal curve that has been described for GABA-evoked intracellular Ca2+ signals in neurons and astrocytes [70], in which the Ca2+ response is exclusively mediated by the metabotropic GABAB receptors [9,10,11,12,13,70]. The evidence, which is further illustrated below, that the endothelial Ca2+ response to GABA is triggered by both GABAA and GABAB receptors could explain the peculiar profile of the dose–response relationship obtained in hCMEC/D3 cells. These preliminary observations confirmed that intracellular Ca2+ signaling could be instrumental for GABA to control endothelial cell functions [17,20]. A novel GABA biosensor based upon a dual-enzyme immobilization approach recently showed that, during neuronal activity, GABA concentration may transiently raise up to ≈80 µM in the cortex [47]. Ca2+ imaging recordings showed that 100 µM GABA elicits a robust biphasic elevation in [Ca2+]i in hCMEC/D3 cells, and this concentration was used to unveil the underlying signaling pathways. Notably, GABA-induced intracellular Ca2+ waves have been observed in many cell types that do not belong to the NVU, including HAECs [64], human aortic smooth muscle cells [71], mouse embryonic stem cells [72], and the human breast cancer cell line, MCF7 [24].
4.3. The Complex Mechanisms of GABA-Induced Intracellular Ca2+ Signals in hCMEC/D3 Cells: InsP3Rs, TPCs and SOCE
Previous studies showed that, in neurons and astrocytes, the Ca2+ response to GABA was initiated by GABAB receptors and comprised a rapid Ca2+ transient that is shaped by ER Ca2+ release through InsP3Rs followed by SOCE [9,10,11,12,13]. The PLCβ signaling pathway has been invoked also as molecular driver of GABA-induced intracellular Ca2+ signals outside the NVU [24,64,71,72]. A recent series of studies demonstrated that, in hCMEC/D3 cells, the Ca2+ response to neurotransmitters and neuromodulators, such as acetylcholine [32], ATP [33,34], glutamate [35,37], histamine [36] and arachidonic acid [57], is triggered by InsP3-evoked Ca2+ release from the ER and maintained over time by SOCE. In addition, NAADP-induced lysosomal Ca2+ discharge via TPCs can support InsP3-dependent ER Ca2+ release [59,61], thereby adding a further layer of complexity to the molecular mechanisms that pattern endothelial Ca2+ signals. Conversely, hCMEC/D3 cells lack ryanodine receptors [32], which may amplify InsP3-induced ER Ca2+ release through the Ca2+-induced Ca2+-release (CICR) mechanism in other endothelial cell types [73].
4.3.1. InsP3Rs and TPCs
Herein, pharmacological manipulation of extracellular Ca2+ concentration confirmed that also the Ca2+ response to GABA was dependent upon intra- and extracellular Ca2+ sources. In the absence of external Ca2+, GABA evoked a smaller and transient elevation in [Ca2+]i, as previously reported in cortical neurons [9] and astrocytes [13], as well as in HAECs [64] and human aortic smooth muscle cells [71]. The GABA-evoked intracellular Ca2+ release in hCMEC/D3 cells was strongly impaired by blocking PLCβ activity with U73122, by inhibiting InsP3Rs with 2-APB, and by depleting ER Ca2+ content with CPA. As previously shown for acetylcholine [32], glutamate [35], ATP [33], and histamine [36], these findings convincingly indicate that InsP3-induced ER Ca2+ mobilization initiates the Ca2+ response to GABA in hCMEC/D3 cells. NAADP-evoked lysosomal Ca2+ discharge via TPCs is emerging as a crucial mechanism shaping endothelial Ca2+ signaling in peripheral vasculature [59,60,61,74,75,76,77]. Likewise, GABA-induced intracellular Ca2+ release was disrupted by pharmacologically emptying the lysosomal Ca2+ store with nigericin and by inhibiting TPCs with either NED-19 or NED-K. These results lend further support to the emerging notion that TPCs finely tune the Ca2+ response to extracellular stimulation not only in cerebrovascular endothelial cells [32,35,36,37,57], but also in neurons and astrocytes [78,79,80]. It has been suggested that lysosomal Ca2+ release through TPCs could trigger cytosolic CICR responses from the ER through InsP3Rs at juxtaposed ER–lysosome contact sites [61,62,63,81]. An alternative, but not mutually exclusive, mechanism whereby the lysosomal Ca2+ store could contribute to InsP3-driven Ca2+ signals is by refilling the ER with Ca2+ [82]. On the other hand, InsP3-induced Ca2+ release could induce the Ca2+-dependent production of NAADP [83] or favor lysosomal Ca2+ loading [84], which could activate TPC2 even in the absence of its ligand [85]. Although elucidating the Ca2+-dependent cross-talk between the ER and lysosomal Ca2+ stores in hCMEC/D3 cells is far beyond the scope of the present investigation, there is no doubt that both InsP3Rs and TPCs contribute to GABA-evoked intracellular Ca2+ release.
4.3.2. SOCE
SOCE represents the Ca2+ entry pathway that mediates extracellular Ca2+ entry evoked by chemical cues in endothelial cells across the whole peripheral vasculature [25,86,87]. Likewise, also in hCMEC/D3 cells, SOCE sustains extracellular Ca2+ influx in response to acetylcholine [32], glutamate [35], and histamine [36]. In addition, SOCE can also be activated downstream of NMDARs, which signal in a flux-independent manner by recruiting the PLCβ signaling pathway [37]. Herein, we found that the re-addition of external Ca2+ after GABA-dependent depletion of the ER Ca2+ stores, and in the absence of the agonist, evoked robust extracellular Ca2+ influx. Under these conditions, GABA is no longer bound to its membrane receptors and, therefore, it is unlikely to stimulate the production of intracellular second messengers, such as arachidonic acid [57], which are able to gate Ca2+-permeable channels on the plasma membrane [32,35,38]. In agreement with these observations, blocking SOCE with either Pyr6 or BTP-2 significantly reduced or abolished GABA-evoked extracellular Ca2+ influx, whereas low micromolar doses of Gd3+ suppressed it. The different extents of SOCE inhibition between the pyrazole derivatives, Pyr6 and BTP-2, and the trivalent cation, Gd3+, could be due to their distinct mechanisms of action. In accord, while Gd3+ directly plugs the channel pore of Orai1 protein, Pyr6 and BTP-2 are likely to interfere with Orai1 recruitment by STIM1 [55,88,89]. Of note, the pharmacological blockade of TRPV4 channels, which are also expressed in hCMEC/D3 cells [57], with RN-1734 did not affect GABA-evoked Ca2+ entry. This finding, therefore, extends the repertoire of neurotransmitters that impinge on SOCE to generate long-lasting Ca2+ signals within the NVU and raises the question as to whether SOCE is recruited by GABA also in HAECs [64], human aortic smooth muscle cells [71], mouse embryonic stem cells [72], and MCF-7 breast cancer cells [24].
4.4. GABAA and GABAB Receptors Mediate GABA-Induced Intracellular Ca2+ Signals in hCMEC/D3 Cells
Although the metabotropic GABAB receptors are known to induce intracellular Ca2+ signals both within [9,10,11] and outside [24,64,71,72] the NVU, the increase in the [Ca2+]i whereby GABA regulates multiple endothelium-dependent functions in brain microvessels is mediated by the ionotropic GABAA receptors [17,20]. GABAA receptor activation by the selective agonist muscimol elicits an inward Cl− current in mouse cerebrovascular endothelial cells, thereby elevating the [Ca2+]i [17]. A potential explanation for this finding is that GABA-induced hyperpolarization enhances the driving-force sustaining the constitutive influx of Ca2+ occurring in these cells [90]. If this hypothesis holds true, muscimol should not increase the [Ca2+]i in the absence of extracellular Ca2+.
Preliminary experiments revealed that both GABAA and GABAB receptors mediate the Ca2+ response to GABA. In accord, GABA-evoked intracellular Ca2+ signals were significantly reduced by inhibiting both the ionotropic GABAA receptors with gabazine and the metabotropic GABAB receptors with saclofen or CGP35348. Moreover, muscimol and baclofen, which, respectively, activate GABAA and GABAB receptors, also elicited a biphasic Ca2+ response in hCMEC/D3 cells. The following pieces of evidence support the notion that the ionotropic GABAA receptor may signal the increase in [Ca2+]i in a flux-independent (i.e., metabotropic) mode. First, whole-cell patch-clamp recordings showed that neither GABA nor muscimol evoked a sizeable membrane current in hCMEC/D3 cells. Second, the Ca2+ response to muscimol was sensitive to gabazine. Third, muscimol induced a robust Ca2+ signal also under 0Ca2+ conditions, which demonstrates that GABAA receptors are able to mobilize the endogenous Ca2+ pool and thereby can operate in a metabotropic manner [91,92]. Intriguingly, a recent investigation revealed that, in hCMEC/D3 cells, the ionotropic NMDARs do not mediate detectable non-selective cation currents, but signal increases in [Ca2+]i in a flux-independent manner by interacting with mGluR1 and mGluR5 [37]. In agreement with our observations, a body of studies has recently showed that also GABAA receptors present metabotropic activity and can induce InsP3-dependent ER Ca2+ release in MCF-7 breast cancer cells [24] and rat cortical neurons [23]. Furthermore, it has been demonstrated that GABAA receptors may transactivate the Gi/o protein coupled GABAB receptors in a PTX-dependent manner and promote InsP3-dependent ER Ca2+ release in mice ciliated oviductal cells [22]. Intriguingly, also in these cells, GABAA receptor stimulation with muscimol did not activate any inward Cl- current [22]. Likewise, we found that the Ca2+ response to direct GABAA receptor stimulation with muscimol was hampered by blocking GABAB receptor signaling with either saclofen, CGP35348 or PTx. Unlike the oviduct [22], however, the Ca2+ response to baclofen was in turn inhibited by blocking GABAA receptors with gabazine, as well as by preventing Gi/o protein activation with PTx.
These findings shed light upon an unusual mode of GABAergic signaling in the cerebrovascular endothelium. The Ca2+ response to GABA, which underpins GABA-induced cerebral angiogenesis [17] and the GABA-induced local increase in CBF [20], requires the functional interaction between GABAA and GABAB receptors. GABAA receptors can engage metabolic signaling via GABAB receptors, and GABAB receptors fail to generate a full Ca2+ signal if GABAA receptors are inhibited. In line with these findings, GABAA and GABAB1 receptors have been shown to physically interact in mice ciliated oviductal cells [22] and in rat brain lysates [93], and to co-localize at multiple synaptic and extra-synaptic sites in the brain [94]. Future work will seek to understand the molecular underpinnings of the functional interaction between GABAA and GABAB receptors in hCMEC/D3 cells. The preliminary evidence that muscimol and baclofen do not exert an additive effect (described in Figure 6C), however, strongly suggests that the PLCβ signaling pathway is engaged by only one GABA receptor isoform, which is likely to be GABAB [7,8]. This model is supported by the evidence that PTx also attenuates muscimol-evoked intracellular Ca2+ signals. Since the Ca2+ response to the mixture of muscimol and baclofen is lower as compared to GABA alone, we hypothesize that the two GABA receptor isoforms must somehow be “synchronized” to interact and elicit the full Ca2+ response and that this requires the presence of the physiological agonist.
5. Conclusions
Herein, we unveiled the complex signaling pathway whereby the inhibitory neurotransmitter GABA can induce an increase in [Ca2+]i in human cerebrovascular endothelial cells. The Ca2+ response to GABA is triggered by InsP3-induced ER Ca2+ release and NAADP-dependent lysosomal Ca2+ mobilization, whereas it is mainly maintained by SOCE (Figure 9). Both GABAA and GABAB receptors support GABA-evoked intracellular Ca2+ signals (Figure 9). The ionotropic GABAA receptors signal in a flux-independent manner via the metabotropic GABAB receptors. Likewise, the full Ca2+ response to GABAB receptor stimulation requires functional GABAA receptors. Endothelial Ca2+ signals finely tune a myriad of vascular functions, including those regulated by GABA, such as angiogenesis and CBF control via NO release. Therefore, this study sheds novel light on the molecular mechanisms by which GABA controls endothelial signaling at the NVU.
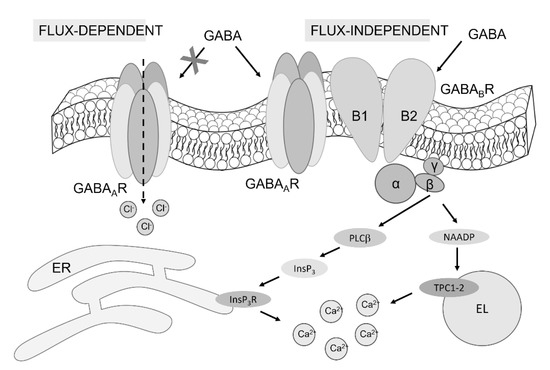
Figure 9.
The mechanism of GABA-evoked intracellular Ca2+ signals in hCMEC/D3 cells. The evidence presented in this investigation demonstrates that both GABAA and GABAB receptors support the Ca2+ response to GABA in hCMEC/D3 cells. GABAA receptors are unlikely to signal the increase in [Ca2+]i in a flux-dependent manner, whereas they can operate in a metabotropic manner by interacting with GABAB receptors. This interaction leads to InsP3-induced Ca2+ mobilization from the ER and NAADP-induced EL Ca2+ release through TPCs. The following reduction in ER Ca2+ concentrations, in turn, activates SOCE (not illustrated here).
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cells11233860/s1, Figure S1: TRPV4 inhibition does not affect GABA-evoked extracellular Ca2+ entry in hCMEC/D3 cells; Table S1: Primers used for real time qPCRs.
Author Contributions
Conceptualization, F.M.; methodology, S.N., F.S., M.V., V.B., P.F. and G.T.; formal analysis, S.N. and F.S.; investigation, S.N., F.S., M.V. and P.F.; resources, F.M.; data curation, S.N. and F.S.; writing—original draft preparation, F.M.; writing—review and editing, G.S., R.B.-R. and F.M.; supervision, F.M.; project administration, F.M.; funding acquisition, F.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by: Regione Lombardia, regional law n° 9/2020, resolution n° 3776/2020 (F.M.); Italian Ministry of Education, University and Research (MIUR): Dipartimenti di Eccellenza Program (2018–2022)-Dept. of Biology and Biotechnology “L. Spallanzani”, University of Pavia (F.M.), Fondo Ricerca Giovani from the University of Pavia (F.M.), and the EU Horizon 2020 FETOPEN-2018-2020 Program under Grant Agreement N. 828984, LION-HEARTED (F.M.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data supporting reported results can be obtained by the corresponding author upon reasonable request.
Acknowledgments
The authors gratefully acknowledge the Laboratory of Cellular Electrophysiology, Centro Grandi Strumenti of the University of Pavia, for the use of the Port-a-Patch automated system.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mapelli, L.; Soda, T.; D’Angelo, E.; Prestori, F. The Cerebellar Involvement in Autism Spectrum Disorders: From the Social Brain to Mouse Models. Int. J. Mol. Sci. 2022, 23, 3894. [Google Scholar] [CrossRef] [PubMed]
- Soda, T.; Mapelli, L.; Locatelli, F.; Botta, L.; Goldfarb, M.; Prestori, F.; D’Angelo, E. Hyperexcitability and Hyperplasticity Disrupt Cerebellar Signal Transfer in the IB2 KO Mouse Model of Autism. J. Neurosci. 2019, 39, 2383–2397. [Google Scholar] [CrossRef] [PubMed]
- Sakimoto, Y.; Oo, P.M.; Goshima, M.; Kanehisa, I.; Tsukada, Y.; Mitsushima, D. Significance of GABAA Receptor for Cognitive Function and Hippocampal Pathology. Int. J. Mol. Sci. 2021, 22, 12456. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, A. Chloride homeodynamics underlying modal shifts in cellular and network oscillations. Neurosci. Res. 2020, 156, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Cherubini, E.; Di Cristo, G.; Avoli, M. Dysregulation of GABAergic Signaling in Neurodevelomental Disorders: Targeting Cation-Chloride Co-transporters to Re-establish a Proper E/I Balance. Front. Cell. Neurosci. 2021, 15, 813441. [Google Scholar] [CrossRef]
- Alexander, S.P.; Mathie, A.; Peters, J.A.; Veale, E.L.; Striessnig, J.; Kelly, E.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; et al. The Concise Guide to Pharmacology 2021/22: Ion channels. Br. J. Pharmacol. 2021, 178 (Suppl. S1), S157–S245. [Google Scholar] [CrossRef]
- Bassetti, D. Keeping the Balance: GABAB Receptors in the Developing Brain and Beyond. Brain Sci. 2022, 12, 419. [Google Scholar] [CrossRef]
- Vlachou, S. A Brief History and the Significance of the GABAB Receptor. Curr. Top. Behav. Neurosci. 2022, 52, 1–17. [Google Scholar] [CrossRef]
- New, D.C.; An, H.; Ip, N.Y.; Wong, Y.H. GABAB heterodimeric receptors promote Ca2+ influx via store-operated channels in rat cortical neurons and transfected Chinese hamster ovary cells. Neuroscience 2006, 137, 1347–1358. [Google Scholar] [CrossRef]
- Kardos, J.; Elster, L.; Damgaard, I.; Krogsgaard-Larsen, P.; Schousboe, A. Role of GABAB receptors in intracellular Ca2+ homeostasis and possible interaction between GABAA and GABAB receptors in regulation of transmitter release in cerebellar granule neurons. J. Neurosci. Res. 1994, 39, 646–655. [Google Scholar] [CrossRef]
- Nieto, A.; Bailey, T.; Kaczanowska, K.; McDonald, P. GABAB Receptor Chemistry and Pharmacology: Agonists, Antagonists, and Allosteric Modulators. Curr. Top. Behav. Neurosci. 2022, 52, 81–118. [Google Scholar] [CrossRef] [PubMed]
- Mariotti, L.; Losi, G.; Sessolo, M.; Marcon, I.; Carmignoto, G. The inhibitory neurotransmitter GABA evokes long-lasting Ca2+ oscillations in cortical astrocytes. Glia 2016, 64, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Jiang, L.; Goldman, S.A.; Nedergaard, M. Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat. Neurosci. 1998, 1, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.K.; Vasudevan, A. Endothelial GABA signaling: A phoenix awakened. Aging 2018, 10, 859–860. [Google Scholar] [CrossRef] [PubMed]
- Vaucher, E.; Tong, X.K.; Cholet, N.; Lantin, S.; Hamel, E. GABA neurons provide a rich input to microvessels but not nitric oxide neurons in the rat cerebral cortex: A means for direct regulation of local cerebral blood flow. J. Comp. Neurol. 2000, 421, 161–171. [Google Scholar] [CrossRef]
- Won, C.; Lin, Z.; Kumar, T.P.; Li, S.; Ding, L.; Elkhal, A.; Szabo, G.; Vasudevan, A. Autonomous vascular networks synchronize GABA neuron migration in the embryonic forebrain. Nat. Commun. 2013, 4, 2149. [Google Scholar] [CrossRef]
- Li, S.; Kumar, T.P.; Joshee, S.; Kirschstein, T.; Subburaju, S.; Khalili, J.S.; Kloepper, J.; Du, C.; Elkhal, A.; Szabo, G.; et al. Endothelial cell-derived GABA signaling modulates neuronal migration and postnatal behavior. Cell Res. 2018, 28, 221–248. [Google Scholar] [CrossRef]
- Tyagi, N.; Lominadze, D.; Gillespie, W.; Moshal, K.S.; Sen, U.; Rosenberger, D.S.; Steed, M.; Tyagi, S.C. Differential expression of gamma-aminobutyric acid receptor A (GABA(A)) and effects of homocysteine. Clin. Chem. Lab. Med. 2007, 45, 1777–1784. [Google Scholar] [CrossRef]
- Shastry, S.; Moning, L.; Tyagi, N.; Steed, M.; Tyagi, S.C. GABA receptors and nitric oxide ameliorate constrictive collagen remodeling in hyperhomocysteinemia. J. Cell. Physiol. 2005, 205, 422–427. [Google Scholar] [CrossRef]
- Agrud, A.; Subburaju, S.; Goel, P.; Ren, J.; Kumar, A.S.; Caldarone, B.J.; Dai, W.; Chavez, J.; Fukumura, D.; Jain, R.K.; et al. Gabrb3 endothelial cell-specific knockout mice display abnormal blood flow, hypertension, and behavioral dysfunction. Sci. Rep. 2022, 12, 4922. [Google Scholar] [CrossRef]
- Choi, Y.K.; Vasudevan, A. Mechanistic insights into autocrine and paracrine roles of endothelial GABA signaling in the embryonic forebrain. Sci. Rep. 2019, 9, 16256. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.; Fernandez-Duenas, V.; Plata, C.; Garcia-Elias, A.; Ciruela, F.; Fernandez-Fernandez, J.M.; Valverde, M.A. Functional coupling of GABAA/B receptors and the channel TRPV4 mediates rapid progesterone signaling in the oviduct. Sci. Signal. 2018, 11, eaam6558. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, M.W.; Sweeney, A.; Pekle, E.; Alam, S.; Ali, A.B.; Duchen, M.; Jovanovic, J.N. Diazepam-induced loss of inhibitory synapses mediated by PLCdelta/Ca2+/calcineurin signalling downstream of GABAA receptors. Mol. Psychiatry 2018, 23, 1851–1867. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Cao, Q.; Liao, R.; Wu, X.; Xun, S.; Huang, J.; Dong, C. Loss of ABAT-Mediated GABAergic System Promotes Basal-Like Breast Cancer Progression by Activating Ca2+-NFAT1 Axis. Theranostics 2019, 9, 34–47. [Google Scholar] [CrossRef]
- Moccia, F.; Negri, S.; Shekha, M.; Faris, P.; Guerra, G. Endothelial Ca2+ Signaling, Angiogenesis and Vasculogenesis: Just What It Takes to Make a Blood Vessel. Int. J. Mol. Sci. 2019, 20, 3962. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Berra-Romani, R.; Guerra, G.; Moccia, F. Endothelial Transient Receptor Potential Channels and Vascular Remodeling: Extracellular Ca2+ Entry for Angiogenesis, Arteriogenesis and Vasculogenesis. Front. Physiol. 2019, 10, 1618. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Soda, T.; Moccia, F. Endothelial signaling at the core of neurovascular coupling: The emerging role of endothelial inward-rectifier K(+) (Kir2.1) channels and N-methyl-d-aspartate receptors in the regulation of cerebral blood flow. Int. J. Biochem. Cell Biol. 2021, 135, 105983. [Google Scholar] [CrossRef]
- Guerra, G.; Lucariello, A.; Perna, A.; Botta, L.; de Luca, A.; Moccia, F. The Role of Endothelial Ca2+ Signaling in Neurovascular Coupling: A View from the Lumen. Int. J. Mol. Sci. 2018, 19, 938. [Google Scholar] [CrossRef]
- Helms, H.C.; Abbott, N.J.; Burek, M.; Cecchelli, R.; Couraud, P.O.; Deli, M.A.; Forster, C.; Galla, H.J.; Romero, I.A.; Shusta, E.V.; et al. In vitro models of the blood-brain barrier: An overview of commonly used brain endothelial cell culture models and guidelines for their use. J. Cereb. Blood Flow Metab. 2016, 36, 862–890. [Google Scholar] [CrossRef]
- Weksler, B.; Romero, I.A.; Couraud, P.O. The hCMEC/D3 cell line as a model of the human blood brain barrier. Fluids Barriers CNS 2013, 10, 16. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Negri, S.; Faris, P.; Angelone, T. Targeting endothelial ion signalling to rescue cerebral blood flow in cerebral disorders. Vascul. Pharmacol. 2022, 145, 106997. [Google Scholar] [CrossRef] [PubMed]
- Zuccolo, E.; Laforenza, U.; Negri, S.; Botta, L.; Berra-Romani, R.; Faris, P.; Scarpellino, G.; Forcaia, G.; Pellavio, G.; Sancini, G.; et al. Muscarinic M5 receptors trigger acetylcholine-induced Ca2+ signals and nitric oxide release in human brain microvascular endothelial cells. J. Cell. Physiol. 2019, 234, 4540–4562. [Google Scholar] [CrossRef] [PubMed]
- Bintig, W.; Begandt, D.; Schlingmann, B.; Gerhard, L.; Pangalos, M.; Dreyer, L.; Hohnjec, N.; Couraud, P.O.; Romero, I.A.; Weksler, B.B.; et al. Purine receptors and Ca2+ signalling in the human blood-brain barrier endothelial cell line hCMEC/D3. Purinergic Signal. 2012, 8, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Forcaia, G.; Formicola, B.; Terribile, G.; Negri, S.; Lim, D.; Biella, G.; Re, F.; Moccia, F.; Sancini, G. Multifunctional Liposomes Modulate Purinergic Receptor-Induced Calcium Wave in Cerebral Microvascular Endothelial Cells and Astrocytes: New Insights for Alzheimer’s disease. Mol. Neurobiol. 2021, 58, 2824–2835. [Google Scholar] [CrossRef] [PubMed]
- Negri, S.; Faris, P.; Pellavio, G.; Botta, L.; Orgiu, M.; Forcaia, G.; Sancini, G.; Laforenza, U.; Moccia, F. Group 1 metabotropic glutamate receptors trigger glutamate-induced intracellular Ca2+ signals and nitric oxide release in human brain microvascular endothelial cells. Cell. Mol. Life Sci. 2020, 77, 2235–2253. [Google Scholar] [CrossRef]
- Berra-Romani, R.; Faris, P.; Pellavio, G.; Orgiu, M.; Negri, S.; Forcaia, G.; Var-Gaz-Guadarrama, V.; Garcia-Carrasco, M.; Botta, L.; Sancini, G.; et al. Histamine induces intracellular Ca2+ oscillations and nitric oxide release in endothelial cells from brain microvascular circulation. J. Cell. Physiol. 2020, 235, 1515–1530. [Google Scholar] [CrossRef] [PubMed]
- Negri, S.; Faris, P.; Maniezzi, C.; Pellavio, G.; Spaiardi, P.; Botta, L.; Laforenza, U.; Biella, G.; Moccia, D.F. NMDA receptors elicit flux-independent intracellular Ca2+ signals via metabotropic glutamate receptors and flux-dependent nitric oxide release in human brain microvascular endothelial cells. Cell Calcium 2021, 99, 102454. [Google Scholar] [CrossRef]
- Zuccolo, E.; Kheder, D.A.; Lim, D.; Perna, A.; Nezza, F.D.; Botta, L.; Scarpellino, G.; Negri, S.; Martinotti, S.; Soda, T.; et al. Glutamate triggers intracellular Ca2+ oscillations and nitric oxide release by inducing NAADP- and InsP3 -dependent Ca2+ release in mouse brain endothelial cells. J. Cell. Physiol. 2019, 234, 3538–3554. [Google Scholar] [CrossRef]
- Gerbino, A.; Bottillo, I.; Milano, S.; Lipari, M.; Zio, R.; Morlino, S.; Mola, M.G.; Procino, G.; Re, F.; Zachara, E.; et al. Functional Characterization of a Novel Truncating Mutation in Lamin A/C Gene in a Family with a Severe Cardiomyopathy with Conduction Defects. Cell. Physiol. Biochem. 2017, 44, 1559–1577. [Google Scholar] [CrossRef]
- Milano, S.; Gerbino, A.; Schena, G.; Carmosino, M.; Svelto, M.; Procino, G. Human beta3-Adrenoreceptor is Resistant to Agonist-Induced Desensitization in Renal Epithelial Cells. Cell. Physiol. Biochem. 2018, 48, 847–862. [Google Scholar] [CrossRef]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Ferrera, L.; Barbieri, R.; Picco, C.; Zuccolini, P.; Remigante, A.; Bertelli, S.; Fumagalli, M.R.; Zifarelli, G.; La Porta, C.A.M.; Gavazzo, P.; et al. TRPM2 Oxidation Activates Two Distinct Potassium Channels in Melanoma Cells through Intracellular Calcium Increase. Int. J. Mol. Sci. 2021, 22, 8359. [Google Scholar] [CrossRef] [PubMed]
- Sobradillo, D.; Hernandez-Morales, M.; Ubierna, D.; Moyer, M.P.; Nunez, L.; Villalobos, C. A reciprocal shift in transient receptor potential channel 1 (TRPC1) and stromal interaction molecule 2 (STIM2) contributes to Ca2+ remodeling and cancer hallmarks in colorectal carcinoma cells. J. Biol. Chem. 2014, 289, 28765–28782. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.; Remigante, A.; Civello, D.A.; Bernardinelli, E.; Szabo, Z.; Morabito, R.; Marino, A.; Sarikas, A.; Patsch, W.; Paulmichl, M.; et al. O-GlcNAcylation Suppresses the Ion Current IClswell by Preventing the Binding of the Protein ICln to alpha-Integrin. Front. Cell Dev. Biol. 2020, 8, 607080. [Google Scholar] [CrossRef] [PubMed]
- Burmeister, J.J.; Price, D.A.; Pomerleau, F.; Huettl, P.; Quintero, J.E.; Gerhardt, G.A. Challenges of simultaneous measurements of brain extracellular GABA and glutamate in vivo using enzyme-coated microelectrode arrays. J. Neurosci. Methods 2020, 329, 108435. [Google Scholar] [CrossRef]
- Grabauskas, G. Time course of GABA in the synaptic clefts of inhibitory synapses in the rostral nucleus of the solitary tract. Neurosci. Lett. 2005, 373, 10–15. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Moccia, F. Reactive Oxygen Species and Endothelial Ca2+ Signaling: Brothers in Arms or Partners in Crime? Int. J. Mol. Sci. 2021, 22, 9821. [Google Scholar] [CrossRef]
- McCarron, J.G.; Lee, M.D.; Wilson, C. The Endothelium Solves Problems That Endothelial Cells Do Not Know Exist. Trends Pharmacol. Sci. 2017, 38, 322–338. [Google Scholar] [CrossRef]
- Bader, A.; Bintig, W.; Begandt, D.; Klett, A.; Siller, I.G.; Gregor, C.; Schaarschmidt, F.; Weksler, B.; Romero, I.; Couraud, P.O.; et al. Adenosine receptors regulate gap junction coupling of the human cerebral microvascular endothelial cells hCMEC/D3 by Ca2+ influx through cyclic nucleotide-gated channels. J. Physiol. 2017, 595, 2497–2517. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xin, P.; Yoast, R.E.; Emrich, S.M.; Johnson, M.T.; Pathak, T.; Benson, J.C.; Azimi, I.; Gill, D.L.; Monteith, G.R.; et al. Distinct pharmacological profiles of ORAI1, ORAI2, and ORAI3 channels. Cell Calcium 2020, 91, 102281. [Google Scholar] [CrossRef] [PubMed]
- Abdullaev, I.F.; Bisaillon, J.M.; Potier, M.; Gonzalez, J.C.; Motiani, R.K.; Trebak, M. Stim1 and Orai1 mediate CRAC currents and store-operated calcium entry important for endothelial cell proliferation. Circ. Res. 2008, 103, 1289–1299. [Google Scholar] [CrossRef]
- Lodola, F.; Laforenza, U.; Bonetti, E.; Lim, D.; Dragoni, S.; Bottino, C.; Ong, H.L.; Guerra, G.; Ganini, C.; Massa, M.; et al. Store-operated Ca2+ entry is remodelled and controls in vitro angiogenesis in endothelial progenitor cells isolated from tumoral patients. PLoS ONE 2012, 7, e42541. [Google Scholar] [CrossRef]
- Prakriya, M.; Lewis, R.S. Store-Operated Calcium Channels. Physiol. Rev. 2015, 95, 1383–1436. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Zuccolo, E.; Poletto, V.; Turin, I.; Guerra, G.; Pedrazzoli, P.; Rosti, V.; Porta, C.; Montagna, D. Targeting Stim and Orai Proteins as an Alternative Approach in Anticancer Therapy. Curr. Med. Chem. 2016, 23, 3450–3480. [Google Scholar] [CrossRef]
- Berra-Romani, R.; Faris, P.; Negri, S.; Botta, L.; Genova, T.; Moccia, F. Arachidonic Acid Evokes an Increase in Intracellular Ca2+ Concentration and Nitric Oxide Production in Endothelial Cells from Human Brain Microcirculation. Cells 2019, 8, 689. [Google Scholar] [CrossRef]
- Thakore, P.; Earley, S. Transient Receptor Potential Channels and Endothelial Cell Calcium Signaling. Compr. Physiol. 2019, 9, 1249–1277. [Google Scholar] [CrossRef]
- Moccia, F.; Negri, S.; Faris, P.; Perna, A.; de Luca, A.; Soda, T.; Romani, R.B.; Guerra, G. Targeting Endolysosomal Two-Pore Channels to Treat Cardiovascular Disorders in the Novel COronaVIrus Disease 2019. Front. Physiol. 2021, 12, 629119. [Google Scholar] [CrossRef]
- Moccia, F.; Zuccolo, E.; Di Nezza, F.; Pellavio, G.; Faris, P.S.; Negri, S.; de Luca, A.; Laforenza, U.; Ambrosone, L.; Rosti, V.; et al. Nicotinic acid adenine dinucleotide phosphate activates two-pore channel TPC1 to mediate lysosomal Ca2+ release in endothelial colony-forming cells. J. Cell. Physiol. 2021, 236, 688–705. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Moccia, F. Endolysosomal Ca2+ signaling in cardiovascular health and disease. Int. Rev. Cell Mol. Biol. 2021, 363, 203–269. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.J.; Platt, F.M.; Lloyd-Evans, E.; Galione, A. Molecular mechanisms of endolysosomal Ca2+ signalling in health and disease. Biochem. J. 2011, 439, 349–374. [Google Scholar] [CrossRef] [PubMed]
- Faris, P.; Casali, C.; Negri, S.; Iengo, L.; Biggiogera, M.; Maione, A.S.; Moccia, F. Nicotinic Acid Adenine Dinucleotide Phosphate Induces Intracellular Ca2+ Signalling and Stimulates Proliferation in Human Cardiac Mesenchymal Stromal Cells. Front. Cell Dev. Biol. 2022, 10, 874043. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.P.; Cheng, Z.Y.; Schmid, K.L. GABAB receptors expressed in human aortic endothelial cells mediate intracellular calcium concentration regulation and endothelial nitric oxide synthase translocation. BioMed Res. Int. 2014, 2014, 871735. [Google Scholar] [CrossRef] [PubMed]
- Hamel, E. Perivascular nerves and the regulation of cerebrovascular tone. J. Appl. Physiol. 2006, 100, 1059–1064. [Google Scholar] [CrossRef] [PubMed]
- Cauli, B.; Tong, X.K.; Rancillac, A.; Serluca, N.; Lambolez, B.; Rossier, J.; Hamel, E. Cortical GABA interneurons in neurovascular coupling: Relays for subcortical vasoactive pathways. J. Neurosci. 2004, 24, 8940–8949. [Google Scholar] [CrossRef]
- Napoleone, P.; Erdo, S.; Amenta, F. Autoradiographic localization of the GABAA receptor agonist [3H] muscimol in rat cerebral vessels. Brain Res. 1987, 423, 109–115. [Google Scholar] [CrossRef]
- Kaplan, L.; Chow, B.W.; Gu, C. Neuronal regulation of the blood-brain barrier and neurovascular coupling. Nat. Rev. Neurosci. 2020, 21, 416–432. [Google Scholar] [CrossRef]
- Moccia, F. Endothelial Ca2+ Signaling and the Resistance to Anticancer Treatments: Partners in Crime. Int. J. Mol. Sci. 2018, 19, 217. [Google Scholar] [CrossRef]
- Doengi, M.; Hirnet, D.; Coulon, P.; Pape, H.C.; Deitmer, J.W.; Lohr, C. GABA uptake-dependent Ca2+ signaling in developing olfactory bulb astrocytes. Proc. Natl. Acad. Sci. USA 2009, 106, 17570–17575. [Google Scholar] [CrossRef]
- Wang, X.P.; Cheng, Z.Y.; Schmid, K.L. GABAB receptors are expressed in human aortic smooth muscle cells and regulate the intracellular Ca2+ concentration. Heart Vessel. 2015, 30, 249–257. [Google Scholar] [CrossRef]
- Schwirtlich, M.; Emri, Z.; Antal, K.; Mate, Z.; Katarova, Z.; Szabo, G. GABA(A) and GABA(B) receptors of distinct properties affect oppositely the proliferation of mouse embryonic stem cells through synergistic elevation of intracellular Ca2+. FASEB J. 2010, 24, 1218–1228. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, A.M.; Thompson, M.D.; Weisbrod, R.M.; Pimental, D.R.; Tong, X.; Bolotina, V.M.; Cohen, R.A. Redox regulation of SERCA2 is required for vascular endothelial growth factor-induced signaling and endothelial cell migration. Antioxid. Redox Signal. 2012, 17, 1099–1108. [Google Scholar] [CrossRef] [PubMed]
- Brailoiu, G.C.; Gurzu, B.; Gao, X.; Parkesh, R.; Aley, P.K.; Trifa, D.I.; Galione, A.; Dun, N.J.; Madesh, M.; Patel, S.; et al. Acidic NAADP-sensitive calcium stores in the endothelium: Agonist-specific recruitment and role in regulating blood pressure. J. Biol. Chem. 2010, 285, 37133–37137. [Google Scholar] [CrossRef] [PubMed]
- Favia, A.; Desideri, M.; Gambara, G.; D’Alessio, A.; Ruas, M.; Esposito, B.; Del Bufalo, D.; Parrington, J.; Ziparo, E.; Palombi, F.; et al. VEGF-induced neoangiogenesis is mediated by NAADP and two-pore channel-2-dependent Ca2+ signaling. Proc. Natl. Acad. Sci. USA 2014, 111, E4706–E4715. [Google Scholar] [CrossRef]
- Gambara, G.; Billington, R.A.; Debidda, M.; D’Alessio, A.; Palombi, F.; Ziparo, E.; Genazzani, A.A.; Filippini, A. NAADP-induced Ca2+ signaling in response to endothelin is via the receptor subtype B and requires the integrity of lipid rafts/caveolae. J. Cell. Physiol. 2008, 216, 396–404. [Google Scholar] [CrossRef]
- Balbi, C.; Lodder, K.; Costa, A.; Moimas, S.; Moccia, F.; van Herwaarden, T.; Rosti, V.; Campagnoli, F.; Palmeri, A.; de Biasio, P.; et al. Reactivating endogenous mechanisms of cardiac regeneration via paracrine boosting using the human amniotic fluid stem cell secretome. Int. J. Cardiol. 2019, 287, 87–95. [Google Scholar] [CrossRef]
- Martucci, L.L.; Cancela, J.M. Neurophysiological functions and pharmacological tools of acidic and non-acidic Ca2+ stores. Cell Calcium 2022, 104, 102582. [Google Scholar] [CrossRef]
- Foster, W.J.; Taylor, H.B.C.; Padamsey, Z.; Jeans, A.F.; Galione, A.; Emptage, N.J. Hippocampal mGluR1-dependent long-term potentiation requires NAADP-mediated acidic store Ca2+ signaling. Sci. Signal. 2018, 11, eaat9093. [Google Scholar] [CrossRef]
- Pereira, G.J.; Hirata, H.; Fimia, G.M.; do Carmo, L.G.; Bincoletto, C.; Han, S.W.; Stilhano, R.S.; Ureshino, R.P.; Bloor-Young, D.; Churchill, G.; et al. Nicotinic acid adenine dinucleotide phosphate (NAADP) regulates autophagy in cultured astrocytes. J. Biol. Chem. 2011, 286, 27875–27881. [Google Scholar] [CrossRef]
- Galione, A. NAADP Receptors. Cold Spring Harb. Perspect. Biol. 2019, 11, a035071. [Google Scholar] [CrossRef] [PubMed]
- Ronco, V.; Potenza, D.M.; Denti, F.; Vullo, S.; Gagliano, G.; Tognolina, M.; Guerra, G.; Pinton, P.; Genazzani, A.A.; Mapelli, L.; et al. A novel Ca2+-mediated cross-talk between endoplasmic reticulum and acidic organelles: Implications for NAADP-dependent Ca2+ signalling. Cell Calcium 2015, 57, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.J.; Davis, L.C.; Wagner, S.K.; Lewis, A.M.; Parrington, J.; Churchill, G.C.; Galione, A. Bidirectional Ca2+ signaling occurs between the endoplasmic reticulum and acidic organelles. J. Cell Biol. 2013, 200, 789–805. [Google Scholar] [CrossRef] [PubMed]
- Atakpa, P.; Thillaiappan, N.B.; Mataragka, S.; Prole, D.L.; Taylor, C.W. IP3 Receptors Preferentially Associate with ER-Lysosome Contact Sites and Selectively Deliver Ca2+ to Lysosomes. Cell Rep. 2018, 25, 3180–3193.e7. [Google Scholar] [CrossRef]
- Pitt, S.J.; Reilly-O’Donnell, B.; Sitsapesan, R. Exploring the biophysical evidence that mammalian two-pore channels are NAADP-activated calcium-permeable channels. J. Physiol. 2016, 594, 4171–4179. [Google Scholar] [CrossRef]
- Blatter, L.A. Tissue Specificity: SOCE: Implications for Ca2+ Handling in Endothelial Cells. Adv. Exp. Med. Biol. 2017, 993, 343–361. [Google Scholar] [CrossRef]
- Avila-Medina, J.; Mayoral-Gonzalez, I.; Dominguez-Rodriguez, A.; Gallardo-Castillo, I.; Ribas, J.; Ordonez, A.; Rosado, J.A.; Smani, T. The Complex Role of Store Operated Calcium Entry Pathways and Related Proteins in the Function of Cardiac, Skeletal and Vascular Smooth Muscle Cells. Front. Physiol. 2018, 9, 257. [Google Scholar] [CrossRef]
- Moccia, F.; Dragoni, S.; Poletto, V.; Rosti, V.; Tanzi, F.; Ganini, C.; Porta, C. Orai1 and Transient Receptor Potential Channels as novel molecular targets to impair tumor neovascularisation in renal cell carcinoma and other malignancies. Anticancer. Agents Med. Chem. 2014, 14, 296–312. [Google Scholar] [CrossRef]
- Schleifer, H.; Doleschal, B.; Lichtenegger, M.; Oppenrieder, R.; Derler, I.; Frischauf, I.; Glasnov, T.N.; Kappe, C.O.; Romanin, C.; Groschner, K. Novel pyrazole compounds for pharmacological discrimination between receptor-operated and store-operated Ca2+ entry pathways. Br. J. Pharmacol. 2012, 167, 1712–1722. [Google Scholar] [CrossRef]
- Zuccolo, E.; Lim, D.; Kheder, D.A.; Perna, A.; Catarsi, P.; Botta, L.; Rosti, V.; Riboni, L.; Sancini, G.; Tanzi, F.; et al. Acetylcholine induces intracellular Ca2+ oscillations and nitric oxide release in mouse brain endothelial cells. Cell Calcium 2017, 66, 33–47. [Google Scholar] [CrossRef]
- Montes de Oca Balderas, P. Flux-Independent NMDAR Signaling: Molecular Mediators, Cellular Functions, and Complexities. Int. J. Mol. Sci. 2018, 19, 3800. [Google Scholar] [CrossRef] [PubMed]
- Dore, K.; Stein, I.S.; Brock, J.A.; Castillo, P.E.; Zito, K.; Sjostrom, P.J. Unconventional NMDA Receptor Signaling. J. Neurosci. 2017, 37, 10800–10807. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Teissere, J.A.; Raju, D.V.; Hall, R.A. Hetero-oligomerization between GABAA and GABAB receptors regulates GABAB receptor trafficking. J. Biol. Chem. 2004, 279, 18840–18850. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, A.N.; Triller, A.; Sieghart, W. GABA(A) Receptors: Post-Synaptic Co-Localization and Cross-Talk with Other Receptors. Front. Cell. Neurosci. 2011, 5, 7. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).