
TGF-β2 Regulates Transcription of the K+/Cl− Cotransporter 2 (KCC2) in Immature Neurons and Its Phosphorylation at T1007 in Differentiated Neurons


Abstract
1. Introduction
2. Materials and Methods
2.1. Antibodies and Reagents/Chemicals
2.2. Animals
2.3. Primary Cultures of Mouse E17.5-18.5 Hippocampal Neurons
2.4. Quantitative RT-PCR
2.5. Chromatin Immunoprecipitation Assay (ChIP)
2.6. Immunoblotting
2.7. Immunocytochemistry
2.8. Image Acquisition and Analysis
2.9. Acute Brainstem Slices
2.10. Immunofluorescence on Tissue Sections
2.11. Surface Biotinylation of Acute Brainstem Slices
2.12. Statistical Analysis
3. Results
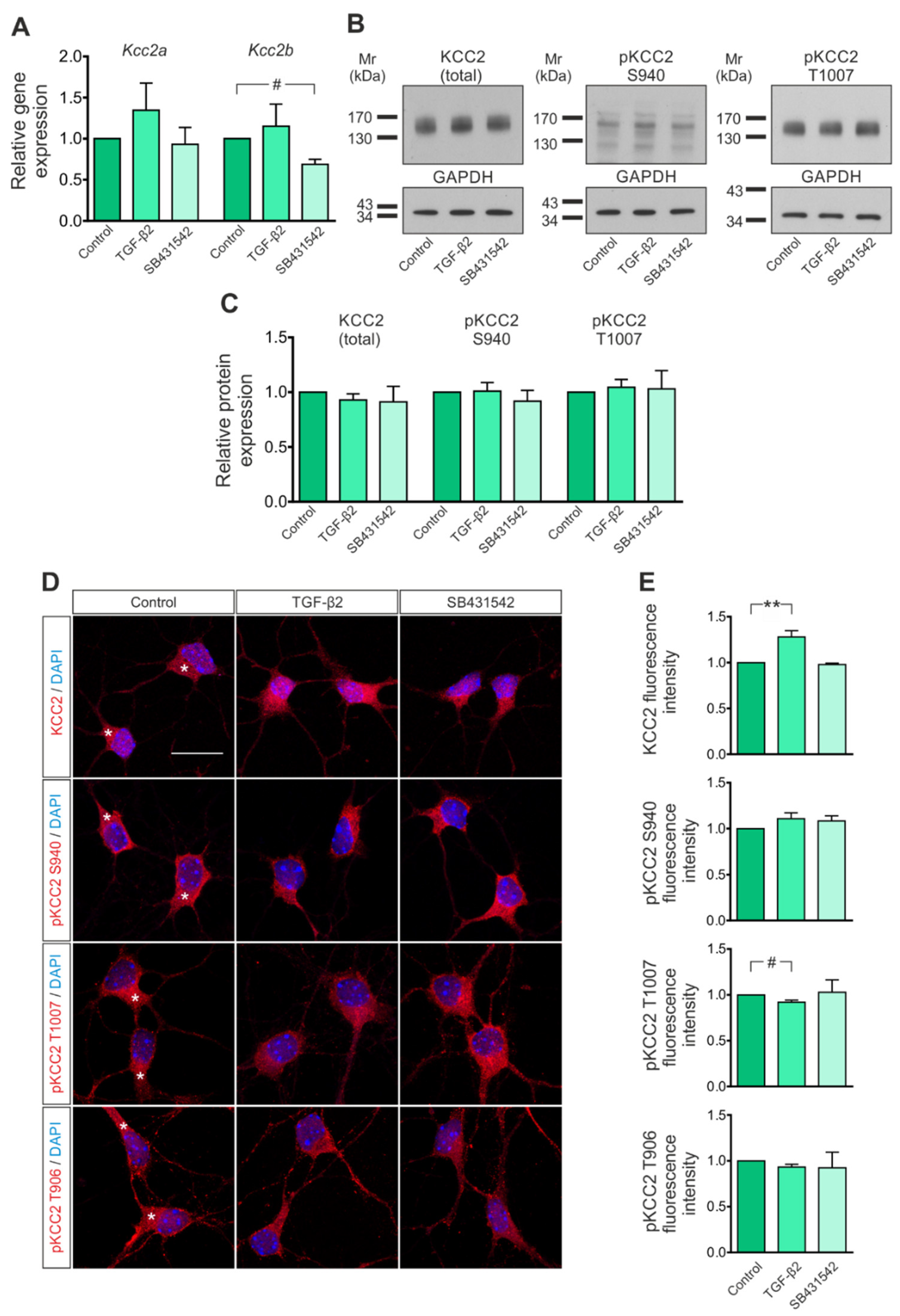
3.1. KCC2 Expression in Immature Neurons Is Regulated by TGF-β/Activin Signaling
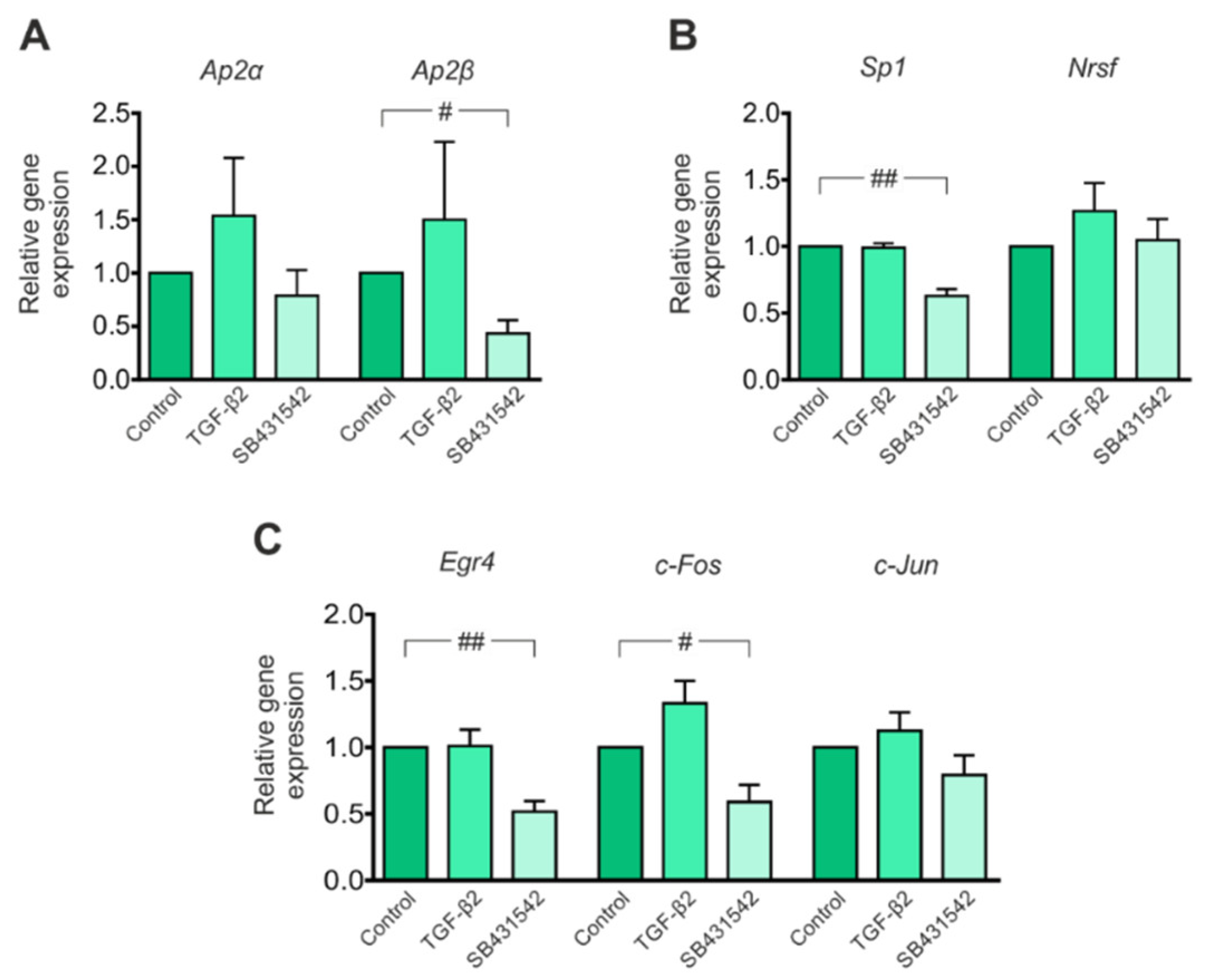
3.2. Expression of Transcription Factors That Potentially Modulate Kcc2 Promoter Activity Is Regulated by TGF-β/Activin Signaling
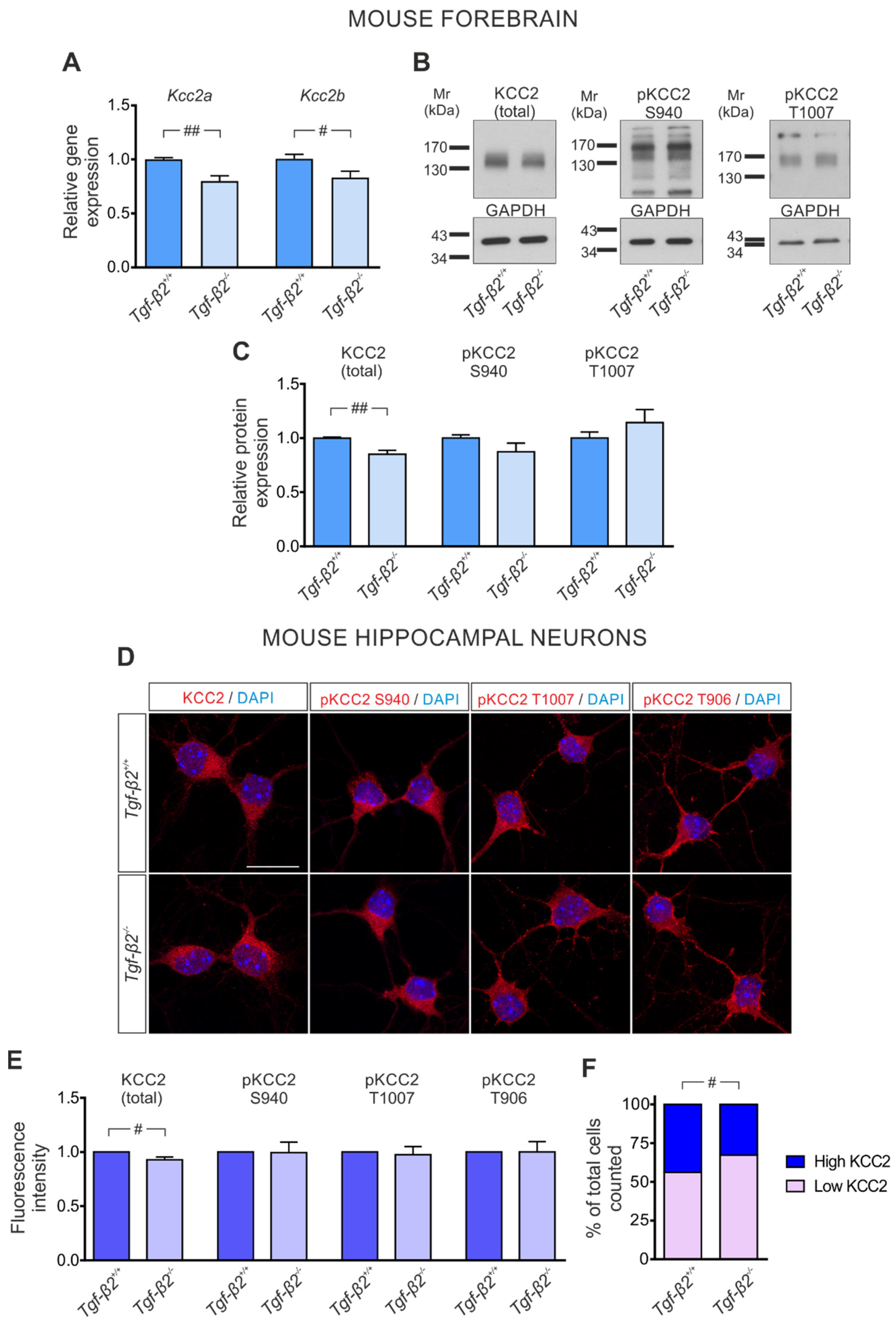
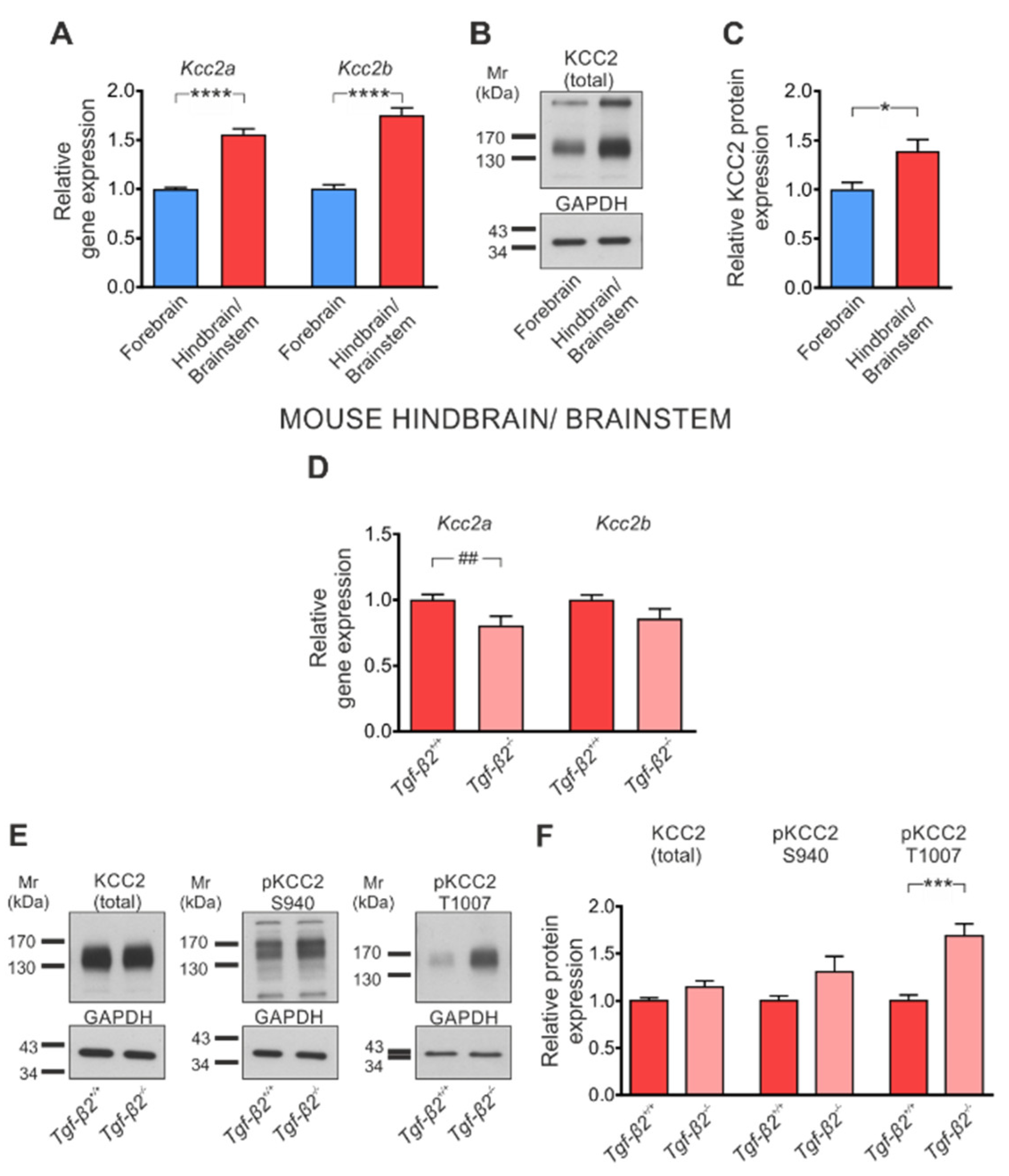
3.3. TGF-β2-Dependent Regulation of Kcc2 Gene Expression and KCC2 Phosphorylation State in the Developing Mouse Forebrain
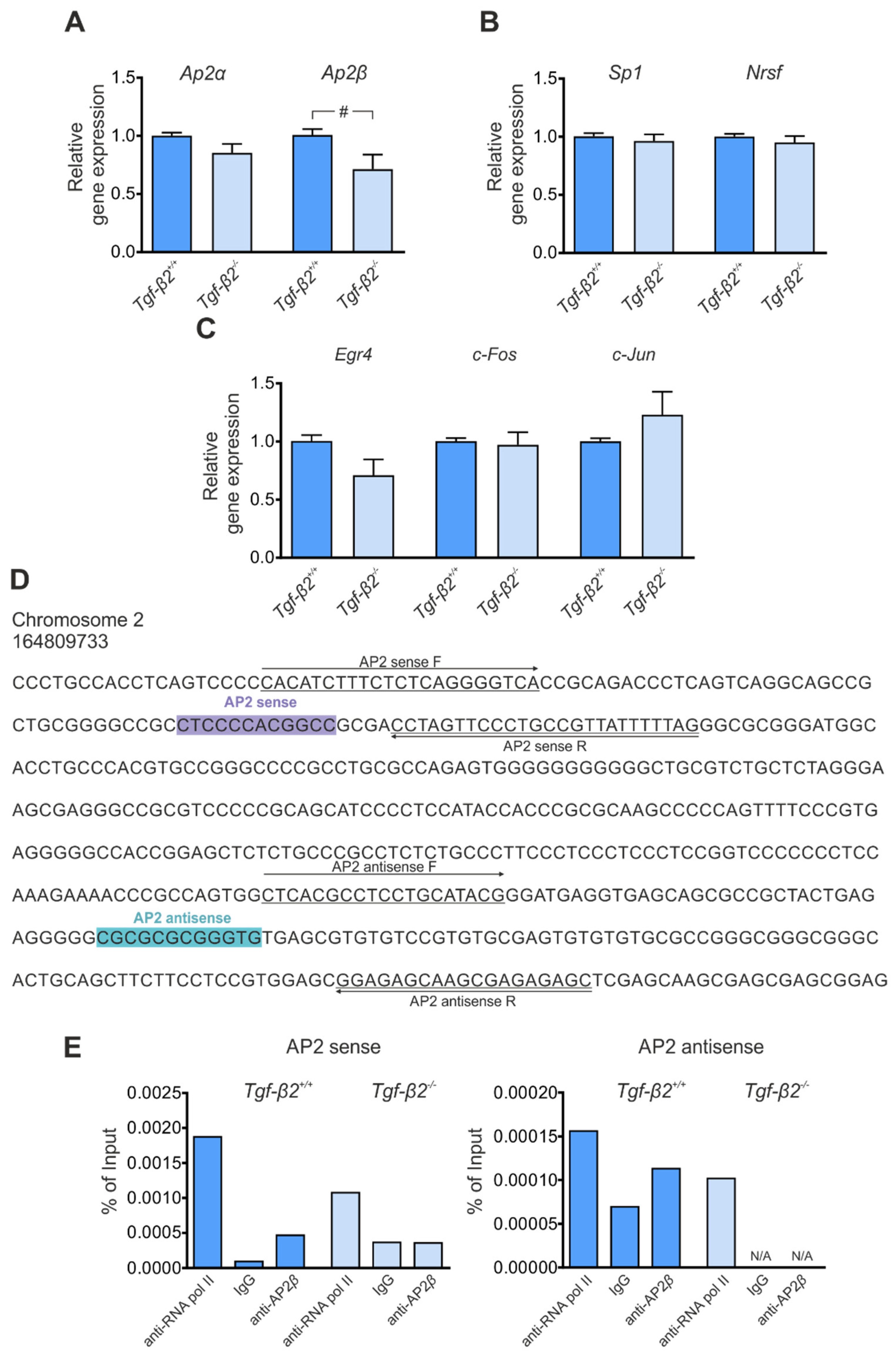
3.4. Transcriptional Regulation of Kcc2 via TGF-β2
3.5. Loss of TGF-β2 Prevents Developmental Dephosphorylation of KCC2 at T1007
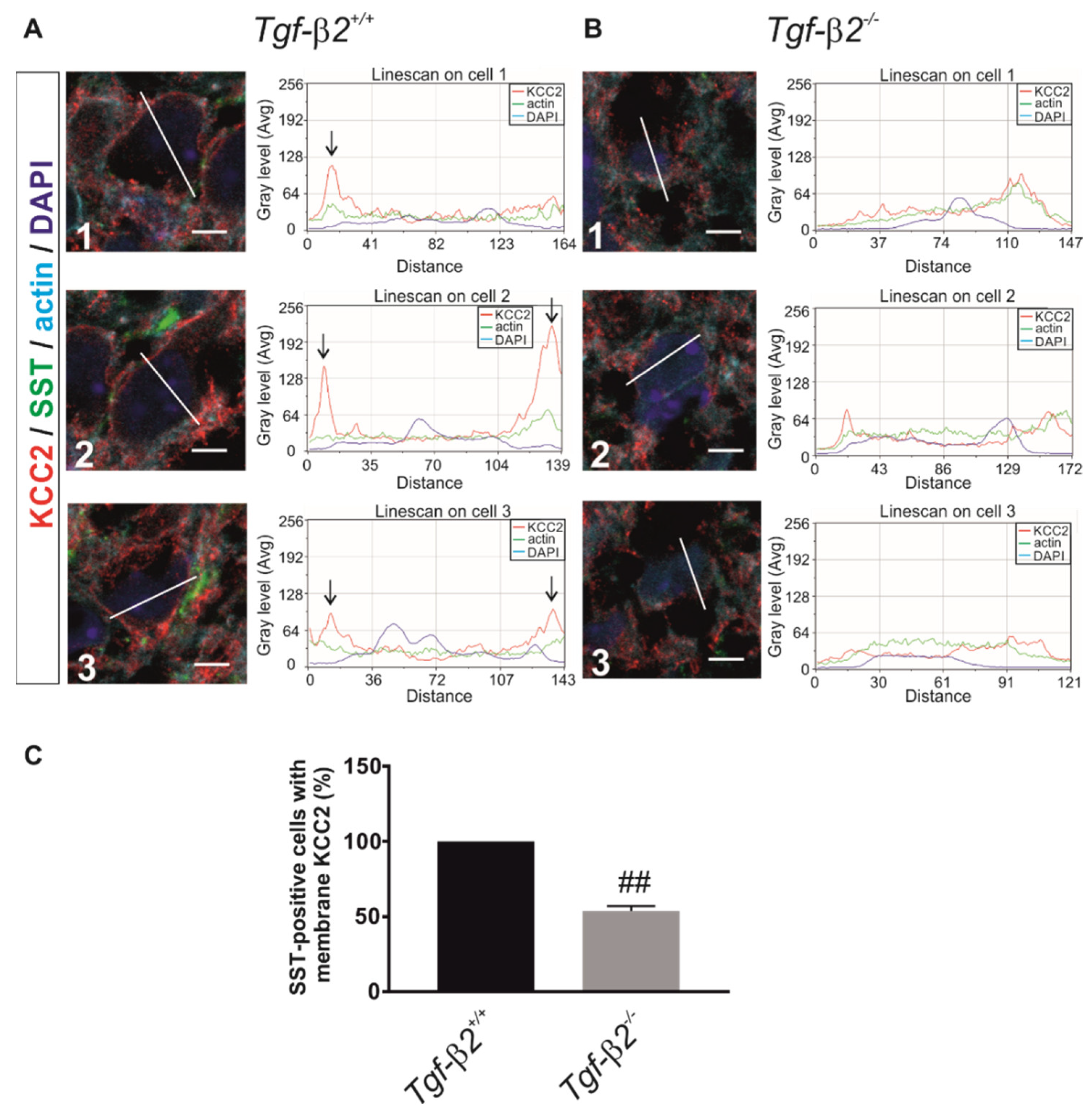
3.6. Impaired KCC2 Membrane Expression in Pre-Bötzinger Complex of Tgf-β2−/− Mice
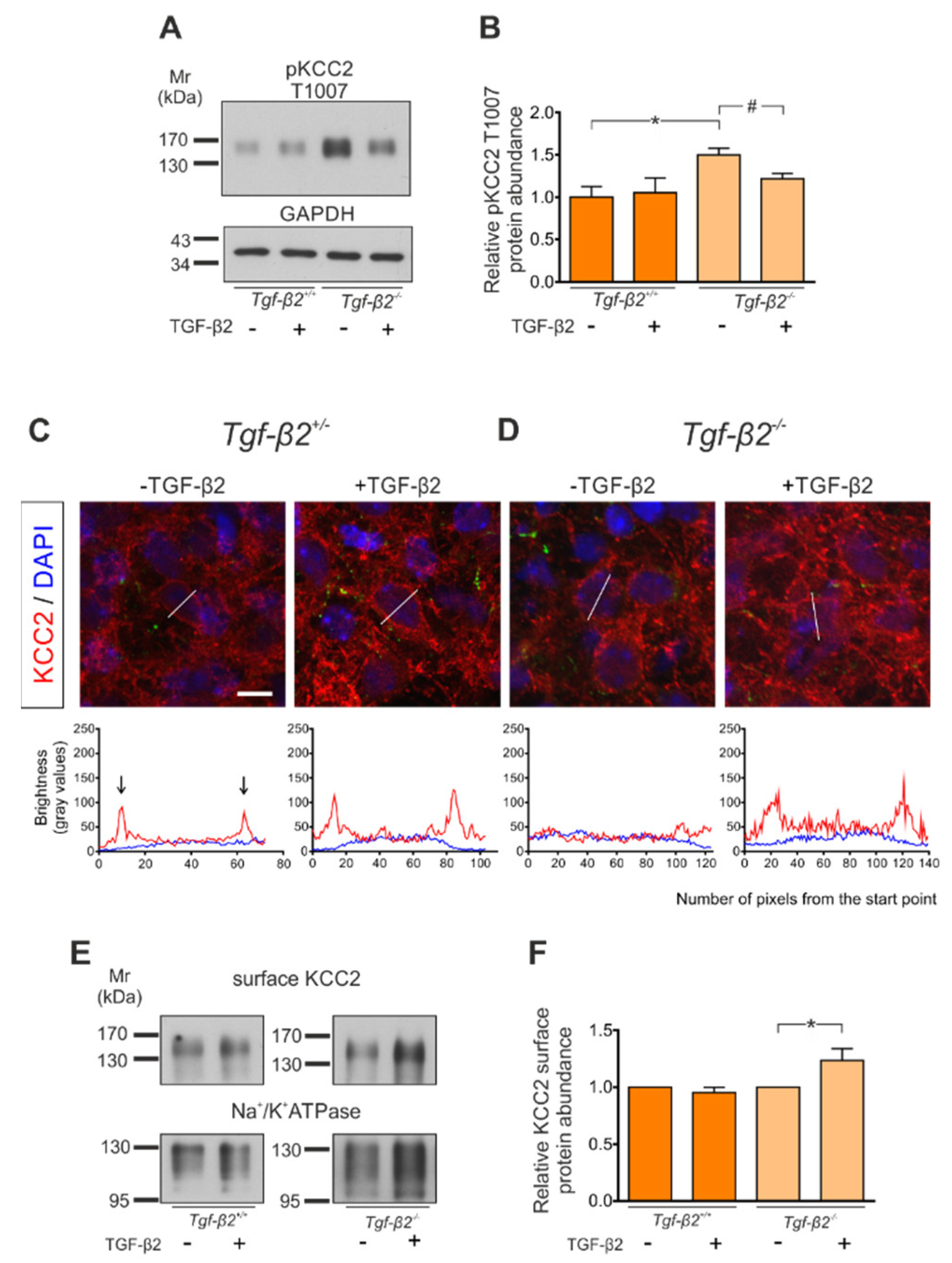
3.7. Rescue of KCC2 Phenotype in Tgf-β2−/− Neurons by Exogenous TGF-β2
4. Discussion
4.1. TGF-β2-Dependent Regulation of KCC2 Transcription in Immature Neurons
4.2. TGF-β2-Dependent Regulation of KCC2 Phosphorylation at T1007
4.3. TGF-β2-Dependent Regulation of Membrane KCC2 in the PreBötC
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Rivera, C.; Voipio, J.; Payne, J.; Ruusuvuori, E.; Lahtinen, H.; Lamsa, K.; Pirvola, U.; Saarma, M.; Kaila, K. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 1999, 397, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Khirug, S.; Cai, C.; Ludwig, A.; Blaesse, P.; Kolikova, J.; Afzalov, R.; Coleman, S.K.; Lauri, S.; Airaksinen, M.S.; et al. KCC2 interacts with the dendritic cytoskeleton to promore spine development. Neuron 2007, 56, 1019–1033. [Google Scholar] [CrossRef] [PubMed]
- Fiumelli, H.; Briner, A.; Puskarjov, M.; Blaesse, P.; Belem, B.J.; Dayer, A.G.; Kaila, K.; Martin, J.L.; Vutskits, L. An ion transport-independent role for the cation-chloride cotransporter KCC2 in dendritic spinogenesis in vivo. Cereb. Cortex 2013, 23, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Llano, O.; Smirnov, S.; Soni, S.; Golubtsov, A.; Guillemin, I.; Hotulainen, P.; Medina, I.; Nothwang, H.G.; Rivera, C.; Ludwig, A. KCC2 regulates actin dynamics in dendritic spines via interaction with β-PIX. J. Cell Biol. 2015, 609, 671–686. [Google Scholar] [CrossRef]
- Mavrovic, M.; Uvarov, P.; Delpire, E.; Vutskits, L.; Kaila, K.; Puskarjov, M. Loss of non-canonical KCC2 functions promotes developmental apoptosis of cortical projection neurons. EMBO Rep. 2020, 21, e48880. [Google Scholar] [CrossRef]
- Kaila, K.; Price, T.J.; Payne, J.A.; Puskarjov, M.; Voipio, J. Cation-chloride transporters in neuronal development, plasticity and disease. Nat. Rev. Neurosci. 2014, 15, 637–654. [Google Scholar] [CrossRef]
- Moore, Y.E.; Kelley, M.R.; Brandon, N.J.; Deeb, T.Z.; Moss, S.J. Seizing control of KCC2: A new therapeutic target for epilepsy. Trends Neurosci. 2017, 40, 555–571. [Google Scholar] [CrossRef]
- Rinehart, J.; Maksimova, Y.D.; Tanis, J.E.; Stone, K.L.; Hodson, C.A.; Zhang, J.; Risinger, M.; Pan, W.; Wu, D.; Colangela, C.M.; et al. Sites of regulated phosphorylation that control K-Cl cotransporter activity. Cell 2009, 138, 525–536. [Google Scholar] [CrossRef]
- Kahle, K.T.; Deeb, T.Z.; Puskarjov, M.; Silayeva, L.; Liang, B.; Kaila, K.; Moss, S.J. Modulation of neuronal activity by phosphorylation of the K-Cl cotransporter KCC2. Trends Neurosci. 2013, 36, 726–737. [Google Scholar] [CrossRef]
- Lee, H.H.; Walker, J.A.; Williams, J.R.; Goodier, R.J.; Payne, J.A.; Moss, S.J. Direct protein kinase C-dependent phosphorylation regulates the cell surface stability and activity of the potassium chloride cotransporter KCC2. J. Biol. Chem. 2007, 282, 29777–29784. [Google Scholar] [CrossRef]
- Friedel, P.; Kahle, K.T.; Zhang, J.; Hertz, N.; Pisella, L.I.; Buhler, E.; Schaller, F.; Duan, J.; Khanna, A.R.; Bishop, P.N.; et al. WNK1-regulated inhibitory phosphorylation of the KCC2 cotransporter maintains the depolarizing action of GABA in immature neurons. Sci. Signal. 2015, 8, ra65. [Google Scholar] [CrossRef] [PubMed]
- Cordshagen, A.; Busch, W.; Winklhofer, M.; Nothwang, H.G.; Hartmann, A.M. Phosphoregulation of the intracellular termini of K+-Cl− cotransporter 2 (KCC2) enables flexible control of its activity. J. Biol. Chem. 2018, 293, 16984–16993. [Google Scholar] [CrossRef]
- Zhang, J.; Cordshagen, A.; Medina, I.; Nothwang, H.G.; Wisniewski, J.R.; Winklhofer, M.; Hartmann, A.M. Staurosporine and NEM mainly impair WNK-SPAK/OSR1 mediated phosphorylation of KCC2 and NKCC1. PLoS ONE 2020, 15, e0232967. [Google Scholar] [CrossRef]
- Watanabe, M.; Zhang, J.; Mansuri, M.S.; Duan, J.; Karimy, J.K.; Delpire, E.; Alper, S.L.; Lifton, R.P.; Fukuda, A.; Kahle, K.T. Developmentally regulated KCC2 phosphorylation is essential for dynamic GABA-mediated inhibition and survival. Sci. Signal. 2019, 12, eaaw9315. [Google Scholar] [CrossRef]
- Pisella, L.I.; Gaiarsa, J.L.; Diabira, D.; Zhang, J.; Khalilov, I.; Duan, J.; Kahle, K.T.; Medina, I. Impaired regulation of KCC2 phosphorylation leads to neuronal network dysfunction and neurodevelopmental pathology. Sci. Signal. 2019, 12, eaay0300. [Google Scholar] [CrossRef] [PubMed]
- Moore, Y.E.; Deeb, T.Z.; Chadchankar, H.; Brandon, N.J.; Moss, S.J. Potentiating KCC2 activity is sufficient to limit the onset and severity of seizures. Proc. Natl. Acad. Sci. USA 2018, 115, 10166–10171. [Google Scholar] [CrossRef]
- Silayeva, L.; Deeb, T.Z.; Hines, R.M.; Kelley, M.R.; Munoz, M.B.; Lee, H.H.C.; Brandon, N.J.; Dunlop, J.; Maguire, J.; Davies, P.A.; et al. KCC2 activity is critical in limiting the onset and severity of status epilepticus. Proc. Natl. Acad. Sci. USA 2015, 112, 3523–3528. [Google Scholar] [CrossRef] [PubMed]
- Uvarov, P.; Ludwig, A.; Markkanen, M.; Pruunsild, P.; Kaila, K.; Delpire, E.; Timmusk, T.; Rivery, C.; Airaksinen, M.S. A novel N-terminal isoform of the neuron-specific K-Cl cotransporter KCC2. J. Biol. Chem. 2007, 282, 30570–30576. [Google Scholar] [CrossRef]
- Markkanen, M.; Uvarov, P.; Airaksinen, M.S. Role of upstream stimulating factors in the transcriptional regulation of the neuron-specific K-Cl cotransporter KCC2. Brain Res. 2008, 1236, 8–15. [Google Scholar] [CrossRef]
- Uvarov, P.; Ludwig, A.; Markkanen, M.; Rivera, C.; Airaksinen, M.S. Upregulation of the neuron-specific K+/Cl− contransporter expression by transcription factor early growth response 4. J. Neurosci. 2006, 26, 13463–13473. [Google Scholar] [CrossRef]
- Uvarov, P.; Pruunsild, P.; Timmusk, T.; Airaksinen, M.S. Neuronal K+/Cl− co-transporter (KCC2) transgenes lacking neurone restrictive silencer element recapitulate CNS neurone-specific expression and developmental up-regulation of endogenous KCC2 gene. J. Neurochem. 2005, 95, 1144–1155. [Google Scholar] [CrossRef] [PubMed]
- Yeo, M.; Berglund, K.B.; Augustine, G.; Liedtke, W. Novel repression of Kcc2 transcription by REST-RE-1 controls developmental switch in neuronal chloride. J. Neurosci. 2009, 29, 14652–14662. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, A.; Uvarov, P.; Soni, S.; Thomas-Crusells, J.; Airaksinen, M.S.; Rivera, C. Early growth response 4 mediates BDNF induction of potassium chloride cotransporter 2 transcription. J. Neurosci. 2011, 31, 644–649. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, A.; Uvarov, P.; Pellegrino, C.; Thomas-Crisells, J.; Schuchmann, S.; Saarma, M.; Airaksinen, M.S.; Rivera, C. Neurtrurin evokes MAPK-dependent upregulation of Egr4 and KCC2 in developing neurons. Neural Plast. 2011, 2011, 641248. [Google Scholar] [CrossRef]
- Rivera, C.; Li, H.; Thomas-Crusells, J.; Lahtinen, H.; Viitanen, T.; Nanobashvili, A.; Kokaia, Z.; Airaksinen, M.S.; Voipio, J.; Kaila, K.; et al. BDNF-induced TrkB activation down-regulates the K+-Cl− cotransporter KCC2 and impairs neuronal Cl− extrusion. J. Cell Biol. 2002, 159, 747–752. [Google Scholar] [CrossRef]
- Rivera, C.; Voipio, J.; Thomas-Crusells, J.; Li, H.; Emri, Z.; Sipilä, S.; Payne, J.A.; Minichiello, L.; Saarma, M.; Kaila, K. Mechanism of activity-dependent downregulation of the neuron-specific K-Cl cotransporter KCC2. J. Neurosci. 2004, 24, 4683–4691. [Google Scholar] [CrossRef]
- Krieglstein, K.; Zheng, F.; Unsicker, K.; Alzheimer, C. More than being protective: Functional roles for TGF-β/activin signaling pathways at central synapses. Trends Neurosci. 2011, 34, 421–429. [Google Scholar] [CrossRef]
- Roussa, E.; Wiehle, M.; Dünker, N.; Becker-Katins, S.; Oehlke, O.; Krieglstein, K. Transforming growth factor beta is required for differentiation of mouse mesencephalic progenitors into dopaminergic neurons in vitro and in vivo: Ectopic induction in dorsal mesencephalon. Stem Cells 2006, 9, 2120–2129. [Google Scholar] [CrossRef]
- Roux, J.; Carles, M.; Koh, H.; Goolaerts, A.; Ganter, M.T.; Chesebro, B.B.; Howard, M.; Houseman, B.T.; Finkbeiner, W.; Shokat, K.M.; et al. Transforming growth factor beta1 inhibits cystic fibrosis transmembrane conductance regulator-dependent cAMP-stimulated alveolar epithelial fluid transport via a phosphatidylinositol 3-kinase-dependent mechanism. J. Biol. Chem. 2010, 285, 4278–4290. [Google Scholar] [CrossRef]
- Yi, S.; Pierucci-Alves, F.; Schultz, B.D. Transforming growth factor-b1 impairs CFTR-mediated anion secretion across cultures porcine vas deferens epithelial monolayer via the p38 MAPK pathway. Am. J. Physiol. Cell Physiol. 2013, 305, C867–C876. [Google Scholar] [CrossRef][Green Version]
- Kluge, M.; Namkoong, E.; Khakipoor, S.; Park, K.; Roussa, E. Differential regulation of vacuolar H+-ATPase subunits by transforming growth factor-β1 in salivary ducts. J. Cell. Physiol. 2019, 234, 15061–15079. [Google Scholar] [CrossRef]
- Khakipoor, S.; Ophoven, C.; Schrödl-Häußel, M.; Feuerstein, M.; Heimrich, B.; Deitmer, J.W.; Roussa, E. TGF-β signaling directly regulates transcription and functional expression of the electrogenic sodium bicarbonate cotransporter 1, NBCe1 (SLC4A4), via Smad4 in mouse astrocytes. Glia 2017, 65, 1361–1375. [Google Scholar] [CrossRef] [PubMed]
- Roussa, E.; Speer, J.M.; Chudotvorova, I.; Khakipoor, S.; Smirnov, S.; Rivera, C.; Krieglstein, K. The membrane trafficking and functionality of the K+-Cl− co-transporter KCC2 is regulated by TGF-β2. J. Cell Sci. 2016, 129, 3485–3498. [Google Scholar] [PubMed]
- Sanford, L.P.; Ormsby, I.; Gittenberger-de Groot, A.C.; Sariola, H.; Friedman, R.; Boivin, G.P.; Cardell, E.L.; Doetschman, T. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development 1997, 124, 2659–2670. [Google Scholar] [CrossRef]
- Inman, G.J.; Nicolás, F.J.; Callahan, J.F.; Harling, J.D.; Gaster, L.M.; Reith, A.D.; Laping, N.J.; Hill, C.S. SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol. Pharmacol. 2002, 62, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Xu, H.; Sandell, L. Negative Regulation of Chondrocyte Differentiation by Transcription Factor AP-2α. J. Bone Miner. Res. 2004, 19, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Schulien, I.; Hockenjos, B.; Schmitt-Graeff, A.; Perdekamp, M.G.; Follo, M.; Thimme, R.; Hasselblatt, P. The transcription factor c-Jun/AP-1 promotes liver fibrosis during non-alcoholic steatohepatitis by regulating Osteopontin expression. Cell Death Differ. 2019, 26, 1688–1699. [Google Scholar] [CrossRef] [PubMed]
- Markkanen, M.; Ludwig, A.; Khirug, S.; Pryazhnikov, E.; Soni, S.; Khiroug, L.; Delpire, E.; Rivera, C.; Airaksinen, M.S.; Uvarov, P. Implications of the N-terminal heterogeneity for the neuronal K-Cl cotransporter KCC2 function. Brain Res. 2017, 1675, 87–101. [Google Scholar] [CrossRef]
- Li, H.; Liu, Z.; Wu, Y.; Chen, Y.; Wang, J.; Wang, Z.; Huang, D.; Wang, M.; Yu, M.; Fei, J.; et al. The deficiency of NRSF/REST enhances the pro-inflammatory function of astrocytes in a model of Parkinson’s disease. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2020, 1866, 165590. [Google Scholar] [CrossRef] [PubMed]
- García-Morales, V.; Rodríguez-Bey, G.; Gómez-Pérez, L.; Domínguez-Vías, G.; González-Forero, D.; Portillo, F.; Campos-Caro, A.; Gento-Caro, Á.; Issaoui, N.; Soler, R.M.; et al. Sp1-regulated expression of p11 contributes to motor neuron degeneration by membrane insertion of TASK1. Nat. Commun. 2019, 10, 3784. [Google Scholar] [CrossRef]
- Chleilat, E.; Pethe, A.; Pfeifer, D.; Krieglstein, K.; Roussa, E. TGF-β signaling regulates SLC8A3 expression and prevents oxidative stress in developing midbrain dopaminergic and dorsal raphe serotonergic neurons. Int. J. Mol. Sci. 2020, 21, 2735. [Google Scholar] [CrossRef]
- Ludwig, A.; Li, H.; Saarma, M.; Kaila, K.; Rivera, C. Developmental up-regulation of KCC2 in the absence of GABAergic and glutamatergic transmission. Eur. J. Neurosci. 2003, 18, 3199–3206. [Google Scholar] [CrossRef]
- Feng, X.H.; Lin, X.; Derynck, R. Smad2, Smad3 and Smad4 cooparate with Sp1 to induce p15(Ink4B) transcription in response to TGF-beta. EMBO J. 2000, 19, 5178–5193. [Google Scholar] [CrossRef] [PubMed]
- Koinuma, D.; Tsutsumi, S.; Kamimura, N.; Taniguchi, H.; Miyazawa, K.; Sunamura, M.; Imamura, T.; Miyazono, K.; Aburatani, H. Chromatin immunoprecipitation on Microarray analysis of Smad2/3 binding sites reveals roles of ETS1 and TFAP2A in transforming growth factor b signaling. Mol. Cell Biol. 2009, 29, 172–186. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.D.; Haensel, D.; Gaddam, S.; Patel, T.; Atwood, S.X.; Sarin, K.Y.; Whitson, R.J.; McKellar, S.; Shankar, G.; Aasi, S.; et al. AP-1 and TGF-b cooperativity drives non-canonical Hedgehog signaling in resistant basal cell carcinoma. Nat. Commun. 2020, 11, 5079. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.C.; Ellenberger, H.H.; Ballanyi, K.; Richter, D.W.; Feldman, J.L. Pre-Bötzinger complex: A brainstem region that may generate respiratory rhythm in mammals. Science 1991, 254, 726–729. [Google Scholar] [CrossRef] [PubMed]
- Hübner, C.A.; Stein, V.; Hermans-Borgmeyer, I.; Meyer, T.; Ballanyi, K.; Jentsch, T.J. Disruption of KCC2 reveals an essential role of K-Cl cotransport already in early synaptic inhibition. Neuron 2001, 30, 515–524. [Google Scholar] [CrossRef]
- Koizumi, H.; Koshiya, N.; Chia, X.J.; Cao, F.; Nugent, J.; Zhang, R.; Smith, J.C. Structural-functional properties of identified excitatory and inhibitory interneurons within Pre-Bötzinger complex respiratory microcircuits. J. Neurosci. 2013, 33, 2994–3009. [Google Scholar] [CrossRef] [PubMed]
- Heupel, K.; Sargsyan, V.; Plomp, J.J.; Rickmann, M.; Varoqueaux, F.; Zhang, W.; Krieglstein, K. Loss of transforming growth factor-beta 2 leads to impairment of central synapse function. Neural Dev. 2008, 3, 25. [Google Scholar] [CrossRef] [PubMed]
- Stornetta, R.L.; Rosin, D.L.; Wang, H.; Sevigny, C.P.; Weston, M.C.; Guyenet, P.G. A group of glutamatergic interneurons expressing high levels of both neurokinin-1 receptors and somatostatin identifies the region of the pre-Bötzinger complex. J. Comp. Neurol. 2003, 455, 499–512. [Google Scholar] [CrossRef] [PubMed]
- Thoby-Brisson, M.; Karlen, M.; Wu, N.; Charnay, P.; Champagnat, J.; Fortin, G. Genetic identification of an embryonic parafacial oscillator coupling to the preBotzinger complex. Nat. Neurosci. 2009, 12, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, V.; Becker, M.; Löhrke, S.; Nothwang, H.G.; Güresir, E.; Friauf, E. Expression and function of chloride transporters during development of inhibitory neurotransmission in the auditory brainstem. J. Neurosci. 2003, 23, 4134–4145. [Google Scholar] [CrossRef] [PubMed]
- Khiroug, L.; Huttu, K.; Ludwig, A.; Smirnov, S.; Voipio, J.; Rivera, C.; Kaila, K.; Khiroug, L. Dinstinct properties of functional KCC2 expression in immature mouse hippocampal neurons in culture and in acute slices. Eur. J. Neurosci. 2005, 21, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Aguado, F.; Carmona, M.A.; Pozas, E.; Aguilo, A.; Martinez-Guijarro, F.J.; Alcantara, S.; Borrell, V.; Yuste, R.; Ibañez, C.F.; Soriano, E. BDNF regulates spontaneous correlated activity at early developmental stages by increasing synaprogenesis and expression of the K+/Cl− co-transporter KCC2. Development 2003, 130, 1267–1280. [Google Scholar] [CrossRef] [PubMed]
- Carmona, M.A.; Pozas, E.; Martinaz, A.; Espinoza-Parrilla, J.F.; Soriano, E.; Aguado, F. Age-dependent spontaneous hyperexcitability and impairment of GABAergic function in the hippocampus of mice lacking trkB. Cereb. Cortex 2006, 16, 47–63. [Google Scholar] [CrossRef] [PubMed]
- Puskarjov, M.; Ahmad, F.; Khirug, S.; Sivakumaran, S.; Kaila, K.; Blaesse, P. BDNF is required for seizure-induced but not developmental up-regulation of KCC2 in the neonatal hippocampus. Neuropharmacology 2015, 88, 103–109. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers Used for QRT-PCR | |||
| Primer Name | Sequence | GenBank Accession No. | Source |
| Ap2α F | GCGGCCCAATCCTATCCT | BC007471 | [36] |
| Ap2α R | CCATGGGAGATGAGGTTGAAG | ||
| Ap2β F | AAAGCTGTCTCACGCACTTCAGT | NM_009334 | |
| Ap2β R | AGCGCAGCGCAAATGG | ||
| c-Fos F | CCAGTCCTCACCTCTTCCAG | NM_010234.3 | [37] |
| c-Fos R | TCCAGCACCAGGTTAATTCC | ||
| c-Jun F | AAAACCTTGAAAGCGCAAAA | NM_010591.2 | |
| c-Jun R | CGCAACCAGTCAAGTTCTCA | ||
| Egr4 F | TCTCTCCAAGCCCACCGAAG | NM_020596.3 | [20,23] |
| Egr4 R | AACCGCCTGGATGAAGAAGC | ||
| Gapdh F | TGCACCACCAACTGCTTAGC | NM_001289726.1 | [23] |
| Gapdh R | TGGCATGGACTGTGGTCATG | ||
| Kcc2a F | GCCGGCTCCCGAGGGAAG | NM_001355480.1/NM_001355481.1 | [38] |
| Kcc2a R | CTTCTCCGTGTCCGTGCTGTTGATG | ||
| Kcc2b F | GCCACCATGCTCAACAACCT | NM_020333.2 | |
| Kcc2b R | ACTGAGCAGGGAGGATACCA | ||
| Nrsf F | TGAGAAACCATTTCCCCAGG | NM_011263.2 | [39] |
| Nrsf R | TGAGAGCTTGAGTAAGGACAAAG | ||
| Sp1 F | TCTCAGGCAGGCACTATCAGCA | NM_013672.2 | [40] |
| Sp1 R | TTGTGATGACACCAAGCTGGC | ||
| Primers Used for ChIP-QPCR | |||
| Primer Name | Sequence | GenBank Accession No. | Source |
| AP2 sense F | CACATCTTTCTCTCAGGGGTCA | NC_000068.8 | |
| AP2 sense R | CTAAAAATAACGGCAGGGAACTAGG | ||
| AP2 antisense F | CTCACGCCTCCTGCATACG | ||
| AP2 antisense R | GCTCTCTCGCTTGCTCTCC | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rigkou, A.; Magyar, A.; Speer, J.M.; Roussa, E. TGF-β2 Regulates Transcription of the K+/Cl− Cotransporter 2 (KCC2) in Immature Neurons and Its Phosphorylation at T1007 in Differentiated Neurons. Cells 2022, 11, 3861. https://doi.org/10.3390/cells11233861
Rigkou A, Magyar A, Speer JM, Roussa E. TGF-β2 Regulates Transcription of the K+/Cl− Cotransporter 2 (KCC2) in Immature Neurons and Its Phosphorylation at T1007 in Differentiated Neurons. Cells. 2022; 11(23):3861. https://doi.org/10.3390/cells11233861
Chicago/Turabian StyleRigkou, Anastasia, Attila Magyar, Jan Manuel Speer, and Eleni Roussa. 2022. "TGF-β2 Regulates Transcription of the K+/Cl− Cotransporter 2 (KCC2) in Immature Neurons and Its Phosphorylation at T1007 in Differentiated Neurons" Cells 11, no. 23: 3861. https://doi.org/10.3390/cells11233861
APA StyleRigkou, A., Magyar, A., Speer, J. M., & Roussa, E. (2022). TGF-β2 Regulates Transcription of the K+/Cl− Cotransporter 2 (KCC2) in Immature Neurons and Its Phosphorylation at T1007 in Differentiated Neurons. Cells, 11(23), 3861. https://doi.org/10.3390/cells11233861