

Dying of Stress: Chemotherapy, Radiotherapy, and Small-Molecule Inhibitors in Immunogenic Cell Death and Immunogenic Modulation



Abstract
1. Introduction
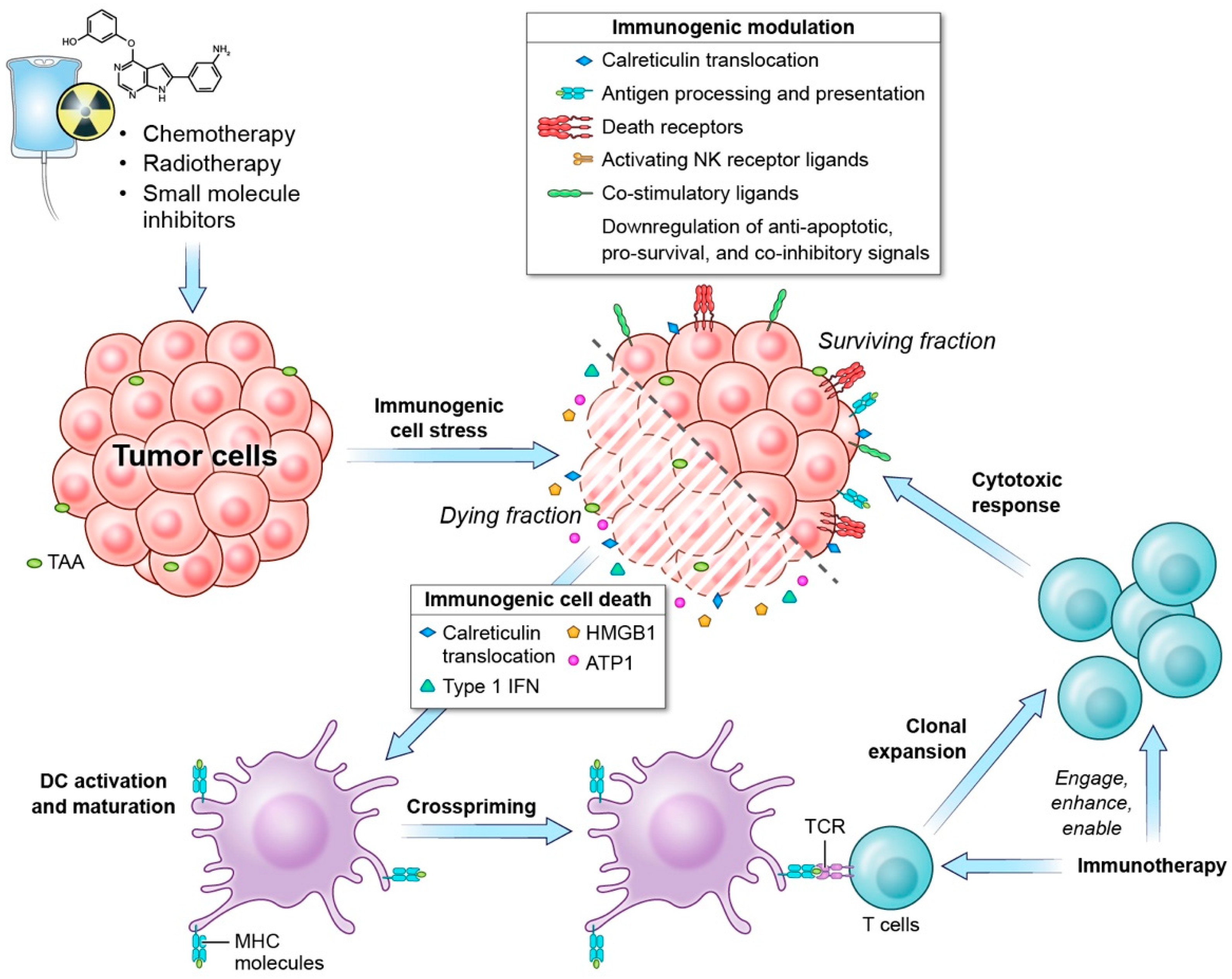
1.1. Immunogenic Cell Death
1.2. Immunogenic Modulation
1.3. Role of Immunogenic Cell-Stress Response in Anti-Tumor Response
2. Chemotherapy and Immunogenic Cell Stress
2.1. Mechanisms of Chemotherapy-Induced Immunogenic Cell Stress
2.2. Chemotherapy-Induced Immunogenic Cell-Stress Response in Preclinical Models
2.3. Chemotherapy-Induced Immunogenic Cell-Stress Response in the Clinic
3. Radiation and Immunogenic Cell Stress
3.1. Mechanisms of Radiation-Induced Immunogenic Cell Stress
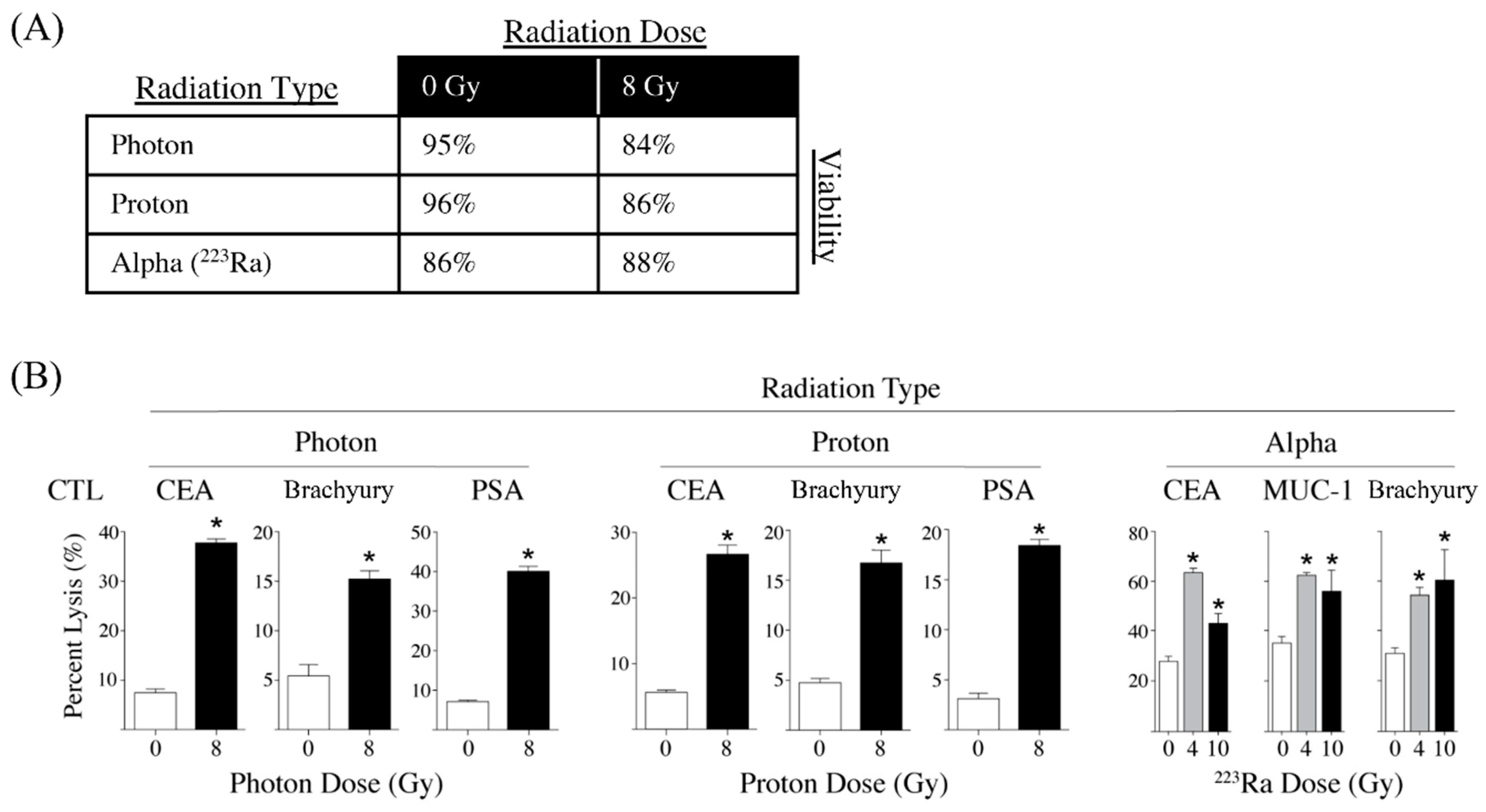
3.2. Different Modalities of Radiotherapy Induce Immunogenic Cell Stress
3.3. Radiotherapy-Induced Immunogenic Cell Stress Response in the Clinic
4. Small-Molecule Inhibitors and Immunogenic Cell Stress
Mechanisms of SMI-Induced Immunogenic Cell Stress
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hosoya, N.; Miyagawa, K. Targeting DNA damage response in cancer therapy. Cancer Sci. 2014, 105, 370–388. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Warren, S.; Adjemian, S.; Agostinis, P.; Martinez, A.B.; Chan, T.A.; Coukos, G.; Demaria, S.; Deutsch, E.; et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J. Immunother. Cancer 2020, 8, e000337. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galassi, C.; Zitvogel, L.; Galluzzi, L. Immunogenic cell stress and death. Nat. Immunol. 2022, 23, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Hodge, J.W.; Ardiani, A.; Farsaci, B.; Kwilas, A.R.; Gameiro, S.R. The tipping point for combination therapy: Cancer vaccines with radiation, chemotherapy, or targeted small molecule inhibitors. Semin. Oncol. 2012, 39, 323–339. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Kepp, O.; Menger, L.; Vacchelli, E.; Locher, C.; Adjemian, S.; Yamazaki, T.; Martins, I.; Sukkurwala, A.Q.; Michaud, M.; Senovilla, L.; et al. Crosstalk between ER stress and immunogenic cell death. Cytokine Growth Factor Rev. 2013, 24, 311–318. [Google Scholar] [CrossRef]
- Garg, A.D.; Dudek-Peric, A.M.; Romano, E.; Agostinis, P. Immunogenic cell death. Int. J. Dev. Biol. 2015, 59, 131–140. [Google Scholar] [CrossRef]
- Dudek, A.M.; Garg, A.D.; Krysko, D.V.; De Ruysscher, D.; Agostinis, P. Inducers of immunogenic cancer cell death. Cytokine Growth Factor Rev. 2013, 24, 319–333. [Google Scholar] [CrossRef]
- Workenhe, S.T.; Mossman, K.L. Oncolytic virotherapy and immunogenic cancer cell death: Sharpening the sword for improved cancer treatment strategies. Mol. Ther. 2014, 22, 251–256. [Google Scholar] [CrossRef]
- Pasquereau-Kotula, E.; Habault, J.; Kroemer, G.; Poyet, J.-L. The anticancer peptide RT53 induces immunogenic cell death. PLoS ONE 2018, 13, e0201220. [Google Scholar] [CrossRef]
- Zhou, H.; Forveille, S.; Sauvat, A.; Yamazaki, T.; Senovilla, L.; Ma, Y.; Liu, P.; Yang, H.; Bezu, L.; Müller, K.; et al. The oncolytic peptide LTX-315 triggers immunogenic cell death. Cell Death Dis. 2016, 7, e2134. [Google Scholar] [CrossRef]
- Pozzi, C.; Cuomo, A.; Spadoni, I.; Magni, E.; Silvola, A.; Conte, A.; Sigismund, S.; Ravenda, P.S.; Bonaldi, T.; Zampino, M.G.; et al. The EGFR-specific antibody cetuximab combined with chemotherapy triggers immunogenic cell death. Nat. Med. 2016, 22, 624–631. [Google Scholar] [CrossRef]
- Nuccitelli, R.; McDaniel, A.; Anand, S.; Cha, J.; Mallon, Z.; Berridge, J.C.; Uecker, D. Nano-pulse stimulation is a physical modality that can trigger immunogenic tumor cell death. J. Immunother. Cancer 2017, 5, 32. [Google Scholar] [CrossRef]
- Heemskerk, B.; Kvistborg, P.; Schumacher, T.N.M. The cancer antigenome. EMBO J. 2013, 32, 194–203. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Coulie, P.G.; Van den Eynde, B.J.; van der Bruggen, P.; Boon, T. Tumour antigens recognized by T lymphocytes: At the core of cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 135–146. [Google Scholar] [CrossRef]
- Smith, C.C.; Selitsky, S.R.; Chai, S.; Armistead, P.M.; Vincent, B.G.; Serody, J.S. Alternative tumour-specific antigens. Nat. Rev. Cancer 2019, 19, 465–478. [Google Scholar] [CrossRef]
- Garnett, C.T.; Palena, C.; Chakarborty, M.; Tsang, K.-Y.; Schlom, J.; Hodge, J.W. Sublethal irradiation of human tumor cells modulates phenotype resulting in enhanced killing by cytotoxic T lymphocytes. Cancer Res. 2004, 64, 7985–7994. [Google Scholar] [CrossRef]
- Lhuillier, C.; Rudqvist, N.-P.; Yamazaki, T.; Zhang, T.; Charpentier, M.; Galluzzi, L.; Dephoure, N.; Clement, C.C.; Santambrogio, L.; Zhou, X.K.; et al. Radiotherapy-exposed CD8+ and CD4+ neoantigens enhance tumor control. J. Clin. Investig. 2021, 131, e138740. [Google Scholar] [CrossRef]
- Hodge, J.W.; Garnett, C.T.; Farsaci, B.; Palena, C.; Tsang, K.Y.; Ferrone, S.; Gameiro, S.R. Chemotherapy-induced immunogenic modulation of tumor cells enhances killing by cytotoxic T lymphocytes and is distinct from immunogenic cell death. Int. J. Cancer 2013, 133, 624–636. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Fabian, K.P.; Wolfson, B.; Hodge, J.W. From immunogenic cell death to immunogenic modulation: Select chemotherapy regimens induce a spectrum of immune-enhancing activities in the tumor microenvironment. Front. Oncol. 2021, 11, 728018. [Google Scholar] [CrossRef] [PubMed]
- Fucikova, J.; Kepp, O.; Kasikova, L.; Petroni, G.; Yamazaki, T.; Liu, P.; Zhao, L.; Spisek, R.; Kroemer, G.; Galluzzi, L. Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis. 2020, 11, 1013. [Google Scholar] [CrossRef] [PubMed]
- Gardai, S.J.; McPhillips, K.A.; Frasch, S.C.; Janssen, W.J.; Starefeldt, A.; Murphy-Ullrich, J.E.; Bratton, D.L.; Oldenborg, P.A.; Michalak, M.; Henson, P.M. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 2005, 123, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.-L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef]
- Panaretakis, T.; Joza, N.; Modjtahedi, N.; Tesniere, A.; Vitale, I.; Durchschlag, M.; Fimia, G.M.; Kepp, O.; Piacentini, M.; Froehlich, K.U.; et al. The co-translocation of ERp57 and calreticulin determines the immunogenicity of cell death. Cell Death Differ. 2008, 15, 1499–1509. [Google Scholar] [CrossRef]
- Obeid, M. ERP57 membrane translocation dictates the immunogenicity of tumor cell death by controlling the membrane translocation of calreticulin. J. Immunol. 2008, 181, 2533. [Google Scholar] [CrossRef]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like receptor 4–dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef]
- Martins, I.; Wang, Y.; Michaud, M.; Ma, Y.; Sukkurwala, A.Q.; Shen, S.; Kepp, O.; Métivier, D.; Galluzzi, L.; Perfettini, J.L.; et al. Molecular mechanisms of ATP secretion during immunogenic cell death. Cell Death Differ. 2014, 21, 79–91. [Google Scholar] [CrossRef]
- Elliott, M.R.; Chekeni, F.B.; Trampont, P.C.; Lazarowski, E.R.; Kadl, A.; Walk, S.F.; Park, D.; Woodson, R.I.; Ostankovich, M.; Sharma, P.; et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 2009, 461, 282–286. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1β–dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef]
- Oh, J.H.; Kim, M.J.; Choi, S.J.; Ban, Y.H.; Lee, H.K.; Shin, E.-C.; Lee, K.-M.; Ha, S.-J. Sustained type I interferon reinforces NK cell–mediated cancer immunosurveillance during chronic virus infection. Cancer Immunol. Res. 2019, 7, 584–599. [Google Scholar] [CrossRef]
- Lu, C.; Klement, J.D.; Ibrahim, M.L.; Xiao, W.; Redd, P.S.; Nayak-Kapoor, A.; Zhou, G.; Liu, K. Type I interferon suppresses tumor growth through activating the STAT3-granzyme B pathway in tumor-infiltrating cytotoxic T lymphocytes. J. Immunother. Cancer 2019, 7, 157. [Google Scholar] [CrossRef]
- Bek, S.; Stritzke, F.; Wintges, A.; Nedelko, T.; Böhmer, D.F.R.; Fischer, J.C.; Haas, T.; Poeck, H.; Heidegger, S. Targeting intrinsic RIG-I signaling turns melanoma cells into type I interferon-releasing cellular antitumor vaccines. Oncoimmunology 2019, 8, e1570779. [Google Scholar] [CrossRef]
- Sistigu, A.; Yamazaki, T.; Vacchelli, E.; Chaba, K.; Enot, D.P.; Adam, J.; Vitale, I.; Goubar, A.; Baracco, E.E.; Remédios, C.; et al. Cancer cell–autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat. Med. 2014, 20, 1301–1309. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef]
- Hodge, J.W.; Kwilas, A.; Ardiani, A.; Gameiro, S.R. Attacking malignant cells that survive therapy: Exploiting immunogenic modulation. Oncoimmunology 2013, 2, e26937. [Google Scholar] [CrossRef][Green Version]
- Liu, W.M.; Fowler, D.W.; Smith, P.; Dalgleish, A.G. Pre-treatment with chemotherapy can enhance the antigenicity and immunogenicity of tumours by promoting adaptive immune responses. Br. J. Cancer 2010, 102, 115–123. [Google Scholar] [CrossRef]
- Kaneno, R.; Shurin, G.V.; Kaneno, F.M.; Naiditch, H.; Luo, J.; Shurin, M.R. Chemotherapeutic agents in low noncytotoxic concentrations increase immunogenicity of human colon cancer cells. Cell Oncol. 2011, 34, 97–106. [Google Scholar] [CrossRef]
- Jackaman, C.; Majewski, D.; Fox, S.A.; Nowak, A.K.; Nelson, D.J. Chemotherapy broadens the range of tumor antigens seen by cytotoxic CD8+ T cells in vivo. Cancer Immunol. Immunother. 2012, 61, 2343–2356. [Google Scholar] [CrossRef] [PubMed]
- Jaime-Sanchez, P.; Uranga-Murillo, I.; Aguilo, N.; Khouili, S.C.; Arias, M.A.; Sancho, D.; Pardo, J. Cell death induced by cytotoxic CD8(+) T cells is immunogenic and primes caspase-3-dependent spread immunity against endogenous tumor antigens. J. Immunother. Cancer 2020, 8, e000528. [Google Scholar] [CrossRef] [PubMed]
- Minute, L.; Teijeira, A.; Sanchez-Paulete, A.R.; Ochoa, M.C.; Alvarez, M.; Otano, I.; Etxeberrria, I.; Bolaños, E.; Azpilikueta, A.; Garasa, S.; et al. Cellular cytotoxicity is a form of immunogenic cell death. J. Immunother. Cancer 2020, 8, e000325. [Google Scholar] [CrossRef] [PubMed]
- Truxova, I.; Kasikova, L.; Salek, C.; Hensler, M.; Lysak, D.; Holicek, P.; Bilkova, P.; Holubova, M.; Chen, X.; Mikyskova, R.; et al. Calreticulin exposure on malignant blasts correlates with improved natural killer cell-mediated cytotoxicity in acute myeloid leukemia patients. Haematologica 2020, 105, 1868–1878. [Google Scholar] [CrossRef] [PubMed]
- Son, C.-H.; Keum, J.-H.; Yang, K.; Nam, J.; Kim, M.-J.; Kim, S.-H.; Kang, C.-D.; Oh, S.-O.; Kim, C.-D.; Park, Y.-S.; et al. Synergistic enhancement of NK cell-mediated cytotoxicity by combination of histone deacetylase inhibitor and ionizing radiation. Radiat. Oncol. 2014, 9, 49. [Google Scholar] [CrossRef]
- Cao, G.; Wang, J.; Zheng, X.; Wei, H.; Tian, Z.; Sun, R. Tumor therapeutics work as stress Inducers to enhance tumor sensitivity to natural killer (NK) cell cytolysis by up-regulating NKp30 ligand B7-H6. J. Biol. Chem. 2015, 290, 29964–29973. [Google Scholar] [CrossRef]
- Zingoni, A.; Fionda, C.; Borrelli, C.; Cippitelli, M.; Santoni, A.; Soriani, A. Natural killer cell response to chemotherapy-stressed cancer cells: Role in tumor immunosurveillance. Front. Immunol. 2017, 8, 1194. [Google Scholar] [CrossRef]
- Ramakrishnan, R.; Assudani, D.; Nagaraj, S.; Hunter, T.; Cho, H.I.; Antonia, S.; Altiok, S.; Celis, E.; Gabrilovich, D.I. Chemotherapy enhances tumor cell susceptibility to CTL-mediated killing during cancer immunotherapy in mice. J. Clin. Investig. 2010, 120, 1111–1124. [Google Scholar] [CrossRef]
- Kim, S.; Ramakrishnan, R.; Lavilla-Alonso, S.; Chinnaiyan, P.; Rao, N.; Fowler, E.; Heine, J.; Gabrilovich, D.I. Radiation-induced autophagy potentiates immunotherapy of cancer via up-regulation of mannose 6-phosphate receptor on tumor cells in mice. Cancer Immunol. Immunother. 2014, 63, 1009–1021. [Google Scholar] [CrossRef]
- Chakraborty, M.; Abrams, S.I.; Camphausen, K.; Liu, K.; Scott, T.; Coleman, C.N.; Hodge, J.W. Irradiation of tumor cells up-regulates Fas and enhances CTL lytic activity and CTL adoptive immunotherapy. J. Immunol. 2003, 170, 6338–6347. [Google Scholar] [CrossRef]
- Elrod, H.A.; Sun, S.-Y. Modulation of death receptors by cancer therapeutic agents. Cancer Biol. Ther. 2008, 7, 163–173. [Google Scholar] [CrossRef]
- Sojka, D.K.; Donepudi, M.; Bluestone, J.A.; Mokyr, M.B. Melphalan and other anticancer modalities up-regulate B7-1 gene expression in tumor cells. J. Immunol. 2000, 164, 6230. [Google Scholar] [CrossRef]
- Bernstein, M.B.; Garnett, C.T.; Zhang, H.; Velcich, A.; Wattenberg, M.M.; Gameiro, S.R.; Kalnicki, S.; Hodge, J.W.; Guha, C. Radiation-induced modulation of costimulatory and coinhibitory T-cell signaling molecules on human prostate carcinoma cells promotes productive antitumor immune interactions. Cancer Biother. Radiopharm. 2014, 29, 153–161. [Google Scholar] [CrossRef]
- Golden, E.B.; Apetoh, L. Radiotherapy and immunogenic cell death. Semin. Radiat. Oncol. 2015, 25, 11–17. [Google Scholar] [CrossRef]
- Fucikova, J.; Truxova, I.; Hensler, M.; Becht, E.; Kasikova, L.; Moserova, I.; Vosahlikova, S.; Klouckova, J.; Church, S.E.; Cremer, I.; et al. Calreticulin exposure by malignant blasts correlates with robust anticancer immunity and improved clinical outcome in AML patients. Blood 2016, 128, 3113–3124. [Google Scholar] [CrossRef]
- Kasikova, L.; Hensler, M.; Truxova, I.; Skapa, P.; Laco, J.; Belicova, L.; Praznovec, I.; Vosahlikova, S.; Halaska, M.J.; Brtnicky, T.; et al. Calreticulin exposure correlates with robust adaptive antitumor immunity and favorable prognosis in ovarian carcinoma patients. J. Immunother. Cancer 2019, 7, 312. [Google Scholar] [CrossRef]
- Suzuki, Y.; Mimura, K.; Yoshimoto, Y.; Watanabe, M.; Ohkubo, Y.; Izawa, S.; Murata, K.; Fujii, H.; Nakano, T.; Kono, K. Immunogenic tumor cell death induced by chemoradiotherapy in patients with esophageal squamous cell carcinoma. Cancer Res. 2012, 72, 3967–3976. [Google Scholar] [CrossRef]
- Tesniere, A.; Schlemmer, F.; Boige, V.; Kepp, O.; Martins, I.; Ghiringhelli, F.; Aymeric, L.; Michaud, M.; Apetoh, L.; Barault, L.; et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene 2010, 29, 482–491. [Google Scholar] [CrossRef]
- Bergmann, C.; Bachmann, H.S.; Bankfalvi, A.; Lotfi, R.; Pütter, C.; Wild, C.A.; Schuler, P.J.; Greve, J.; Hoffmann, T.K.; Lang, S.; et al. Toll-like receptor 4 single-nucleotide polymorphisms Asp299Gly and Thr399Ile in head and neck squamous cell carcinomas. J. Transl. Med. 2011, 9, 139. [Google Scholar] [CrossRef]
- DeVita, V.T., Jr.; Chu, E. A history of cancer chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef]
- Galmarini, D.; Galmarini, C.M.; Galmarini, F.C. Cancer chemotherapy: A critical analysis of its 60 years of history. Crit. Rev. Oncol./Hematol. 2012, 84, 181–199. [Google Scholar] [CrossRef] [PubMed]
- Fedele, P.; Ciccarese, M.; Surico, G.; Cinieri, S. An update on first line therapies for metastatic breast cancer. Expert Opin. Pharmacother. 2018, 19, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; Van Schil, P.E.; Hellmann, M.D.; et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv192–iv237. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhang, Z.; Wang, Q. Emerging therapies for small cell lung cancer. J. Hematol. Oncol. 2019, 12, 47. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, J.; Esposito, F.; Serrano, S.; Falco, E.; Escudero, P.; Ruiz-Casado, A.; Manzano, H.; Fernandez-Montes, A. Metastatic colorectal cancer. First line therapy for unresectable disease. J. Clin. Med. 2020, 9, 3889. [Google Scholar] [CrossRef]
- Springfeld, C.; Jäger, D.; Büchler, M.W.; Strobel, O.; Hackert, T.; Palmer, D.H.; Neoptolemos, J.P. Chemotherapy for pancreatic cancer. Presse Med. 2019, 48, e159–e174. [Google Scholar] [CrossRef]
- Kamat, A.M.; Hahn, N.M.; Efstathiou, J.A.; Lerner, S.P.; Malmström, P.-U.; Choi, W.; Guo, C.C.; Lotan, Y.; Kassouf, W. Bladder cancer. Lancet 2016, 388, 2796–2810. [Google Scholar] [CrossRef]
- Zitvogel, L.; Apetoh, L.; Ghiringhelli, F.; Kroemer, G. Immunological aspects of cancer chemotherapy. Nat. Rev. Immunol. 2008, 8, 59–73. [Google Scholar] [CrossRef]
- Sanchez-Perez, L.; Suryadevara, C.M.; Choi, B.D.; Reap, E.A.; Sampson, J.H. Leveraging chemotherapy-induced lymphopenia to potentiate cancer immunotherapy. OncoImmunology 2014, 3, e944054. [Google Scholar] [CrossRef][Green Version]
- Bracci, L.; Schiavoni, G.; Sistigu, A.; Belardelli, F. Immune-based mechanisms of cytotoxic chemotherapy: Implications for the design of novel and rationale-based combined treatments against cancer. Cell Death Differ. 2014, 21, 15–25. [Google Scholar] [CrossRef]
- McKnight, J.A. Principles of chemotherapy. Clin. Tech. Small Anim. Pract. 2003, 18, 67–72. [Google Scholar] [CrossRef]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Garcia-Bonilla, L.; Hu, J.; Harding, H.P.; Ron, D. Activation-dependent substrate recruitment by the eukaryotic translation initiation factor 2 kinase PERK. J. Cell Biol. 2006, 172, 201–209. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Panaretakis, T.; Kepp, O.; Brockmeier, U.; Tesniere, A.; Bjorklund, A.-C.; Chapman, D.C.; Durchschlag, M.; Joza, N.; Pierron, G.; van Endert, P.; et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 2009, 28, 578–590. [Google Scholar] [CrossRef]
- Rufo, N.; Garg, A.D.; Agostinis, P. The unfolded protein response in immunogenic cell death and cancer immunotherapy. Trends Cancer 2017, 3, 643–658. [Google Scholar] [CrossRef]
- Galluzzi, L.; Humeau, J.; Buqué, A.; Zitvogel, L.; Kroemer, G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2020, 17, 725–741. [Google Scholar] [CrossRef]
- Gameiro, S.R.; Caballero, J.A.; Hodge, J.W. Defining the molecular signature of chemotherapy-mediated lung tumor phenotype modulation and increased susceptibility to T-cell killing. Cancer Biother. Radiopharm. 2012, 27, 23–35. [Google Scholar] [CrossRef]
- Martins, I.; Kepp, O.; Schlemmer, F.; Adjemian, S.; Tailler, M.; Shen, S.; Michaud, M.; Menger, L.; Gdoura, A.; Tajeddine, N.; et al. Restoration of the immunogenicity of cisplatin-induced cancer cell death by endoplasmic reticulum stress. Oncogene 2011, 30, 1147–1158. [Google Scholar] [CrossRef]
- Terenzi, A.; Pirker, C.; Keppler, B.K.; Berger, W. Anticancer metal drugs and immunogenic cell death. J. Inorg. Biochem. 2016, 165, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, I.; Suzuki, H.; Kitamura, M.; Minamiya, Y.; Kawai, H.; Ogawa, J. Cisplatin induces fas expression in esophageal cancer cell lines and enhanced cytotoxicity in combination with LAK cells. Oncology 2000, 59, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Ohtsukasa, S.; Okabe, S.; Yamashita, H.; Iwai, T.; Sugihara, K. Increased expression of CEA and MHC class I in colorectal cancer cell lines exposed to chemotherapy drugs. J. Cancer Res. Clin. Oncol. 2003, 129, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Pestka, S.; Jubin, R.G.; Lyu, Y.L.; Tsai, Y.C.; Liu, L.F. Chemotherapeutics and radiation stimulate MHC class I expression through elevated interferon-beta signaling in breast cancer cells. PLoS ONE 2012, 7, e32542. [Google Scholar] [CrossRef] [PubMed]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Corrales, L.; Williams, J.; Horton, B.; Sivan, A.; Spranger, S. Cancer immunotherapy targets based on understanding the T cell-inflamed versus non-T cell-inflamed tumor microenvironment. In Tumor Immune Microenvironment in Cancer Progression and Cancer Therapy; Kalinski, P., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 19–31. [Google Scholar]
- Maleki Vareki, S. High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. J. Immunother. Cancer 2018, 6, 157. [Google Scholar] [CrossRef]
- Pfirschke, C.; Engblom, C.; Rickelt, S.; Cortez-Retamozo, V.; Garris, C.; Pucci, F.; Yamazaki, T.; Poirier-Colame, V.; Newton, A.; Redouane, Y.; et al. Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity 2016, 44, 343–354. [Google Scholar] [CrossRef]
- Rios-Doria, J.; Durham, N.; Wetzel, L.; Rothstein, R.; Chesebrough, J.; Holoweckyj, N.; Zhao, W.; Leow, C.C.; Hollingsworth, R. Doxil synergizes with cancer immunotherapies to enhance antitumor responses in syngeneic mouse models. Neoplasia 2015, 17, 661–670. [Google Scholar] [CrossRef]
- Yamazaki, T.; Buqué, A.; Ames, T.D.; Galluzzi, L. PT-112 induces immunogenic cell death and synergizes with immune checkpoint blockers in mouse tumor models. OncoImmunology 2020, 9, 1721810. [Google Scholar] [CrossRef]
- Sun, F.; Cui, L.; Li, T.; Chen, S.; Song, J.; Li, D. Oxaliplatin induces immunogenic cells death and enhances therapeutic efficacy of checkpoint inhibitor in a model of murine lung carcinoma. J. Recept. Signal. Transduct. Res. 2019, 39, 208–214. [Google Scholar] [CrossRef]
- Peng, J.; Hamanishi, J.; Matsumura, N.; Abiko, K.; Murat, K.; Baba, T.; Yamaguchi, K.; Horikawa, N.; Hosoe, Y.; Murphy, S.K.; et al. Chemotherapy induces programmed cell death-ligand 1 overexpression via the nuclear factor-κB to foster an immunosuppressive tumor microenvironment in ovarian cancer. Cancer Res. 2015, 75, 5034–5045. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Deng, Z.; Wang, H.; Ma, W.; Zhou, C.; Zhang, S. Repeated cycles of 5-fluorouracil chemotherapy impaired anti-tumor functions of cytotoxic T cells in a CT26 tumor-bearing mouse model. BMC Immunol. 2016, 17, 29. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Ye, W.; Xiao, R.; Silvin, C.; Padget, M.; Hodge, J.W.; Van Waes, C.; Schmitt, N.C. Cisplatin and oxaliplatin induce similar immunogenic changes in preclinical models of head and neck cancer. Oral Oncol. 2019, 95, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Grabosch, S.; Bulatovic, M.; Zeng, F.; Ma, T.; Zhang, L.; Ross, M.; Brozick, J.; Fang, Y.; Tseng, G.; Kim, E.; et al. Cisplatin-induced immune modulation in ovarian cancer mouse models with distinct inflammation profiles. Oncogene 2019, 38, 2380–2393. [Google Scholar] [CrossRef] [PubMed]
- Wolfson, B.; Hodge, J.W. Next Generation Therapeutic Strateg-Es: Evolving cancer immunotherapy through agents that Engage, Expand and Enable the anti-tumor immune response. ImmunoMedicine 2021, 1, e1020. [Google Scholar] [CrossRef]
- Garnett, C.T.; Schlom, J.; Hodge, J.W. Combination of docetaxel and recombinant vaccine enhances T-cell responses and antitumor activity: Effects of docetaxel on immune enhancement. Clin. Cancer Res. 2008, 14, 3536–3544. [Google Scholar] [CrossRef]
- Gameiro, S.R.; Caballero, J.A.; Higgins, J.P.; Apelian, D.; Hodge, J.W. Exploitation of differential homeostatic proliferation of T-cell subsets following chemotherapy to enhance the efficacy of vaccine-mediated antitumor responses. Cancer Immunol. Immunother. 2011, 60, 1227–1242. [Google Scholar] [CrossRef]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of indications of FDA-approved immune checkpoint inhibitors per NCCN guidelines with the level of evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef]
- Blick, S.K.; Scott, L.J. Cetuximab: A review of its use in squamous cell carcinoma of the head and neck and metastatic colorectal cancer. Drugs 2007, 67, 2585–2607. [Google Scholar] [CrossRef]
- Vacchelli, E.; Pol, J.; Bloy, N.; Eggermont, A.; Cremer, I.; Fridman, W.H.; Galon, J.; Marabelle, A.; Kohrt, H.; Zitvogel, L.; et al. Trial watch: Tumor-targeting monoclonal antibodies for oncological indications. OncoImmunology 2015, 4, e985940. [Google Scholar] [CrossRef]
- Martínez-Sáez, O.; Prat, A. Current and future management of HER2-positive metastatic breast cancer. JCO Oncol. Pract. 2021, 17, 594–604. [Google Scholar] [CrossRef]
- Rapoport, B.L.; Anderson, R. Realizing the clinical potential of immunogenic cell death in cancer chemotherapy and radiotherapy. Int. J. Mol. Sci 2019, 20, 959. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, L.; Arvin, A. Chemotherapy-induced immunosuppression. Environ. Health Perspect. 1982, 43, 21–25. [Google Scholar] [CrossRef]
- Wu, J.; Waxman, D.J. Immunogenic chemotherapy: Dose and schedule dependence and combination with immunotherapy. Cancer Lett. 2018, 419, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.L.; Chang, M.C.; Cheng, W.F. Metronomic chemotherapy and immunotherapy in cancer treatment. Cancer Lett. 2017, 400, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Emens, L.A.; Middleton, G. The interplay of immunotherapy and chemotherapy: Harnessing potential synergies. Cancer Immunol. Res. 2015, 3, 436. [Google Scholar] [CrossRef]
- Stoetzer, O.J.; Fersching, D.M.I.; Salat, C.; Steinkohl, O.; Gabka, C.J.; Hamann, U.; Braun, M.; Feller, A.-M.; Heinemann, V.; Siegele, B.; et al. Circulating immunogenic cell death biomarkers HMGB1 and RAGE in breast cancer patients during neoadjuvant chemotherapy. Tumor Biol. 2013, 34, 81–90. [Google Scholar] [CrossRef]
- Ladoire, S.; Enot, D.; Andre, F.; Zitvogel, L.; Kroemer, G. Immunogenic cell death-related biomarkers: Impact on the survival of breast cancer patients after adjuvant chemotherapy. Oncoimmunology 2015, 5, e1082706. [Google Scholar] [CrossRef]
- Østrup, O.; Dagenborg, V.J.; Rødland, E.A.; Skarpeteig, V.; Silwal-Pandit, L.; Grzyb, K.; Berstad, A.E.; Fretland, Å.A.; Mælandsmo, G.M.; Børresen-Dale, A.L.; et al. Molecular signatures reflecting microenvironmental metabolism and chemotherapy-induced immunogenic cell death in colorectal liver metastases. Oncotarget 2017, 8, 76290–76304. [Google Scholar] [CrossRef]
- Dagenborg, V.J.; Marshall, S.E.; Yaqub, S.; Grzyb, K.; Boye, K.; Lund-Iversen, M.; Høye, E.; Berstad, A.E.; Fretland, Å.A.; Edwin, B.; et al. Neoadjuvant chemotherapy is associated with a transient increase of intratumoral T-cell density in microsatellite stable colorectal liver metastases. Cancer Biol. Ther. 2020, 21, 432–440. [Google Scholar] [CrossRef]
- Garg, A.D.; More, S.; Rufo, N.; Mece, O.; Sassano, M.L.; Agostinis, P.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial watch: Immunogenic cell death induction by anticancer chemotherapeutics. Oncoimmunology 2017, 6, e1386829. [Google Scholar] [CrossRef] [PubMed]
- Vanmeerbeek, I.; Sprooten, J.; De Ruysscher, D.; Tejpar, S.; Vandenberghe, P.; Fucikova, J.; Spisek, R.; Zitvogel, L.; Kroemer, G.; Galluzzi, L.; et al. Trial watch: Chemotherapy-induced immunogenic cell death in immuno-oncology. Oncoimmunology 2020, 9, 1703449. [Google Scholar] [CrossRef] [PubMed]
- Voorwerk, L.; Slagter, M.; Horlings, H.M.; Sikorska, K.; van de Vijver, K.K.; de Maaker, M.; Nederlof, I.; Kluin, R.J.C.; Warren, S.; Ong, S.; et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: The TONIC trial. Nat. Med. 2019, 25, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Kyte, J.A.; Røssevold, A.; Falk, R.S.; Naume, B. ALICE: A randomized placebo-controlled phase II study evaluating atezolizumab combined with immunogenic chemotherapy in patients with metastatic triple-negative breast cancer. J. Transl. Med. 2020, 18, 252. [Google Scholar] [CrossRef] [PubMed]
- Kyte, J.A.; Andresen, N.K.; Russnes, H.G.; Fretland, S.Ø.; Falk, R.S.; Lingjærde, O.C.; Naume, B. ICON: A randomized phase IIb study evaluating immunogenic chemotherapy combined with ipilimumab and nivolumab in patients with metastatic hormone receptor positive breast cancer. J. Transl. Med. 2020, 18, 269. [Google Scholar] [CrossRef]
- Gatti-Mays, M.E.; Redman, J.M.; Collins, J.M.; Bilusic, M. Cancer vaccines: Enhanced immunogenic modulation through therapeutic combinations. Hum. Vacc. Immunother. 2017, 13, 2561–2574. [Google Scholar] [CrossRef]
- Heery, C.R.; Ibrahim, N.K.; Arlen, P.M.; Mohebtash, M.; Murray, J.L.; Koenig, K.; Madan, R.A.; McMahon, S.; Marté, J.L.; Steinberg, S.M.; et al. Docetaxel alone or in combination with a therapeutic cancer vaccine (PANVAC) in patients with metastatic breast cancer: A randomized clinical trial. JAMA Oncol 2015, 1, 1087–1095. [Google Scholar] [CrossRef]
- Chandran, E.B.A.; Atiq, M.O.; Donahue, R.N.; Karzai, F.; Bilusic, M.; Marte, J.L.; Arlen, P.M.; Cordes, L.M.; Owens, H.; Hankin, A.; et al. Evaluating the optimal sequence of immunotherapy and docetaxel in men with metastatic castration-sensitive prostate cancer. J. Clin. Oncol. 2022, 40, 130. [Google Scholar] [CrossRef]
- Friedman, E.J. Immune modulation by ionizing radiation and its implications for cancer immunotherapy. Curr. Pharm. Des. 2002, 8, 1765–1780. [Google Scholar] [CrossRef]
- Demaria, S.; Bhardwaj, N.; McBride, W.H.; Formenti, S.C. Combining radiotherapy and immunotherapy: A revived partnership. Int. J. Radiat. Oncol. Biol. Phys. 2005, 63, 655–666. [Google Scholar] [CrossRef]
- Matzinger, P. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 1994, 12, 991–1045. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Galluzzi, L.; Apetoh, L.; Baert, T.; Birge, R.B.; Bravo-San Pedro, J.M.; Breckpot, K.; Brough, D.; Chaurio, R.; Cirone, M.; et al. Molecular and translational classifications of DAMPs in immunogenic cell death. Front. Immunol. 2015, 6, 588. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Lee, S.; Seo, D.; Kim, D.; Kim, K.; Kim, E.; Kang, J.; Seong, K.M.; Youn, H.; Youn, B. Cellular stress responses in radiotherapy. Cells 2019, 8, 1105. [Google Scholar] [CrossRef] [PubMed]
- Obeid, M.; Panaretakis, T.; Joza, N.; Tufi, R.; Tesniere, A.; van Endert, P.; Zitvogel, L.; Kroemer, G. Calreticulin exposure is required for the immunogenicity of γ-irradiation and UVC light-induced apoptosis. Cell Death Diff. 2007, 14, 1848–1850. [Google Scholar] [CrossRef] [PubMed]
- Wattenberg, M.M.; Fahim, A.; Ahmed, M.M.; Hodge, J.W. Unlocking the combination: Potentiation of radiation-induced antitumor responses with immunotherapy. Radiat. Res. 2014, 182, 126–138. [Google Scholar] [CrossRef]
- Postow, M.A.; Callahan, M.K.; Barker, C.A.; Yamada, Y.; Yuan, J.; Kitano, S.; Mu, Z.; Rasalan, T.; Adamow, M.; Ritter, E.; et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N. Engl. J. Med. 2012, 366, 925–931. [Google Scholar] [CrossRef]
- Ngwa, W.; Irabor, O.C.; Schoenfeld, J.D.; Hesser, J.; Demaria, S.; Formenti, S.C. Using immunotherapy to boost the abscopal effect. Nat. Rev. Cancer 2018, 18, 313–322. [Google Scholar] [CrossRef]
- Demaria, S.; Ng, B.; Devitt, M.L.; Babb, J.S.; Kawashima, N.; Liebes, L.; Formenti, S.C. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int. J. Radiat. Oncol. Biol. Phys. 2004, 58, 862–870. [Google Scholar] [CrossRef]
- Kanagavelu, S.; Gupta, S.; Wu, X.; Philip, S.; Wattenberg, M.M.; Hodge, J.W.; Couto, M.D.; Chung, K.D.; Ahmed, M.M. In vivo effects of lattice radiation therapy on local and distant lung cancer: Potential role of immunomodulation. Radiat. Res. 2014, 182, 149–162. [Google Scholar] [CrossRef]
- Hodge, J.W.; Sharp, H.J.; Gameiro, S.R. Abscopal regression of antigen disparate tumors by antigen cascade after systemic tumor vaccination in combination with local tumor radiation. Cancer Biother. Radiopharm. 2012, 27, 12–22. [Google Scholar] [CrossRef]
- Gameiro, S.R.; Jammeh, M.L.; Wattenberg, M.M.; Tsang, K.Y.; Ferrone, S.; Hodge, J.W. Radiation-induced immunogenic modulation of tumor enhances antigen processing and calreticulin exposure, resulting in enhanced T-cell killing. Oncotarget 2014, 5, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, M.; Wansley, E.K.; Carrasquillo, J.A.; Yu, S.; Paik, C.H.; Camphausen, K.; Becker, M.D.; Goeckeler, W.F.; Schlom, J.; Hodge, J.W. The use of chelated radionuclide (samarium-153-ethylenediaminetetramethylenephosphonate) to modulate phenotype of tumor cells and enhance T cell-mediated killing. Clin. Cancer Res. 2008, 14, 4241–4249. [Google Scholar] [CrossRef] [PubMed]
- Reits, E.A.; Hodge, J.W.; Herberts, C.A.; Groothuis, T.A.; Chakraborty, M.; Wansley, E.K.; Camphausen, K.; Luiten, R.M.; de Ru, A.H.; Neijssen, J.; et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J. Exp. Med. 2006, 203, 1259–1271. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, M.; Abrams, S.I.; Coleman, C.N.; Camphausen, K.; Schlom, J.; Hodge, J.W. External beam radiation of tumors alters phenotype of tumor cells to render them susceptible to vaccine-mediated T-cell killing. Cancer Res. 2004, 64, 4328–4337. [Google Scholar] [CrossRef] [PubMed]
- Aryankalayil, M.J.; Makinde, A.Y.; Gameiro, S.R.; Hodge, J.W.; Rivera-Solis, P.P.; Palayoor, S.T.; Ahmed, M.M.; Coleman, C.N. Defining molecular signature of pro-immunogenic radiotherapy targets in human prostate cancer cells. Radiat. Res. 2014, 182, 139–148. [Google Scholar] [CrossRef]
- Baskar, R.; Lee, K.A.; Yeo, R.; Yeoh, K.W. Cancer and radiation therapy: Current advances and future directions. Int. J. Med. Sci. 2012, 9, 193–199. [Google Scholar] [CrossRef]
- Chakraborty, M.; Gelbard, A.; Carrasquillo, J.A.; Yu, S.; Mamede, M.; Paik, C.H.; Camphausen, K.; Schlom, J.; Hodge, J.W. Use of radiolabeled monoclonal antibody to enhance vaccine-mediated antitumor effects. Cancer Immunol. Immunother. 2008, 57, 1173–1183. [Google Scholar] [CrossRef]
- Grayson, J.M.; Harrington, L.E.; Lanier, J.G.; Wherry, E.J.; Ahmed, R. Differential sensitivity of naive and memory CD8+ T cells to apoptosis in vivo. J. Immunol. 2002, 169, 3760–3770. [Google Scholar] [CrossRef]
- Gameiro, S.R.; Malamas, A.S.; Bernstein, M.B.; Tsang, K.Y.; Vassantachart, A.; Sahoo, N.; Tailor, R.; Pidikiti, R.; Guha, C.P.; Hahn, S.M.; et al. Tumor cells surviving exposure to proton or photon radiation share a common immunogenic modulation signature, rendering them more sensitive to T cell-mediated killing. Int. J. Radiat. Oncol. Biol. Phys. 2016, 95, 120–130. [Google Scholar] [CrossRef]
- Malamas, A.S.; Gameiro, S.R.; Knudson, K.M.; Hodge, J.W. Sublethal exposure to alpha radiation (223Ra dichloride) enhances various carcinomas’ sensitivity to lysis by antigen-specific cytotoxic T lymphocytes through calreticulin-mediated immunogenic modulation. Oncotarget 2016, 7, 86937–86947. [Google Scholar] [CrossRef]
- Nelson, B.J.B.; Andersson, J.D.; Wuest, F. Targeted alpha therapy: Progress in radionuclide production, radiochemistry, and applications. Pharmaceutics 2021, 13, 49. [Google Scholar] [CrossRef] [PubMed]
- Tufano, R.P.; Pace-Asciak, P.; Russell, J.O.; Suárez, C.; Randolph, G.W.; López, F.; Shaha, A.R.; Mäkitie, A.; Rodrigo, J.P.; Kowalski, L.P.; et al. Update of radiofrequency ablation for treating benign and malignant thyroid nodules. The future is now. Front. Endocrinol. (Lausanne) 2021, 12, 698689. [Google Scholar] [CrossRef] [PubMed]
- Gameiro, S.R.; Higgins, J.P.; Dreher, M.R.; Woods, D.L.; Reddy, G.; Wood, B.J.; Guha, C.; Hodge, J.W. Combination therapy with local radiofrequency ablation and systemic vaccine enhances antitumor immunity and mediates local and distal tumor regression. PLoS ONE 2013, 8, e70417. [Google Scholar] [CrossRef] [PubMed]
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D.; et al. Photodynamic therapy of cancer: An update. CA Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, X.; Wang, X.; Guan, X.; Zhang, W.; Ma, J. Recent advances in selective photothermal therapy of tumor. J. Nanobiotechnol. 2021, 19, 335. [Google Scholar] [CrossRef]
- Sweeney, E.E.; Cano-Mejia, J.; Fernandes, R. Photothermal Therapy Generates a Thermal Window of Immunogenic Cell Death in Neuroblastoma. Small 2018, 14, e1800678. [Google Scholar] [CrossRef]
- Wang, M.; Rao, J.; Wang, M.; Li, X.; Liu, K.; Naylor, M.F.; Nordquist, R.E.; Chen, W.R.; Zhou, F. Cancer photo-immunotherapy: From bench to bedside. Theranostics 2021, 11, 2218–2231. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, X.; Liu, X.; Yu, J.; Bai, X.; Wu, X.; Guo, X.; Liu, Z.; Liu, X. Combination of phototherapy with immune checkpoint blockade: Theory and practice in cancer. Front. Immunol. 2022, 13, 955920. [Google Scholar] [CrossRef]
- Kobayashi, H.; Choyke, P.L. Near-Infrared Photoimmunotherapy of Cancer. Acc. Chem. Res. 2019, 52, 2332–2339. [Google Scholar] [CrossRef]
- Nagaya, T.; Nakamura, Y.; Sato, K.; Harada, T.; Choyke, P.L.; Hodge, J.W.; Schlom, J.; Kobayashi, H. Near infrared photoimmunotherapy with avelumab, an anti-programmed death-ligand 1 (PD-L1) antibody. Oncotarget 2017, 8, 8807–8817. [Google Scholar] [CrossRef]
- Bernstein, M.B.; Krishnan, S.; Hodge, J.W.; Chang, J.Y. Immunotherapy and stereotactic ablative radiotherapy (ISABR): A curative approach? Nat. Rev. Clin. Oncol. 2016, 13, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Demaria, S.; Guha, C.; Schoenfeld, J.; Morris, Z.; Monjazeb, A.; Sikora, A.; Crittenden, M.; Shiao, S.; Khleif, S.; Gupta, S.; et al. Radiation dose and fraction in immunotherapy: One-size regimen does not fit all settings, so how does one choose? J. Immunother. Cancer 2021, 9, e002038. [Google Scholar] [CrossRef] [PubMed]
- Nesslinger, N.J.; Sahota, R.A.; Stone, B.; Johnson, K.; Chima, N.; King, C.; Rasmussen, D.; Bishop, D.; Rennie, P.S.; Gleave, M.; et al. Standard treatments induce antigen-specific immune responses in prostate cancer. Clin. Cancer Res. 2007, 13, 1493–1502. [Google Scholar] [CrossRef] [PubMed]
- Schaue, D.; Comin-Anduix, B.; Ribas, A.; Zhang, L.; Goodglick, L.; Sayre, J.W.; Debucquoy, A.; Haustermans, K.; McBride, W.H. T-cell responses to survivin in cancer patients undergoing radiation therapy. Clin. Cancer Res. 2008, 14, 4883–4890. [Google Scholar] [CrossRef] [PubMed]
- Gulley, J.L.; Arlen, P.M.; Bastian, A.; Morin, S.; Marte, J.; Beetham, P.; Tsang, K.Y.; Yokokawa, J.; Hodge, J.W.; Menard, C.; et al. Combining a recombinant cancer vaccine with standard definitive radiotherapy in patients with localized prostate cancer. Clin. Cancer Res. 2005, 11, 3353–3362. [Google Scholar] [CrossRef]
- Heery, C.R.; Madan, R.A.; Stein, M.N.; Stadler, W.M.; Di Paola, R.S.; Rauckhorst, M.; Steinberg, S.M.; Marte, J.L.; Chen, C.C.; Grenga, I.; et al. Samarium-153-EDTMP (Quadramet(R)) with or without vaccine in metastatic castration-resistant prostate cancer: A randomized Phase 2 trial. Oncotarget 2016, 7, 69014–69023. [Google Scholar] [CrossRef]
- Rodriguez-Ruiz, M.E.; Perez-Gracia, J.L.; Rodriguez, I.; Alfaro, C.; Onate, C.; Perez, G.; Gil-Bazo, I.; Benito, A.; Inoges, S.; Lopez-Diaz de Cerio, A.; et al. Combined immunotherapy encompassing intratumoral poly-ICLC, dendritic-cell vaccination and radiotherapy in advanced cancer patients. Ann. Oncol. 2018, 29, 1312–1319. [Google Scholar] [CrossRef]
- Maity, A.; Mick, R.; Rengan, R.; Mitchell, T.C.; Amaravadi, R.K.; Schuchter, L.M.; Pryma, D.A.; Patsch, D.M.; Maity, A.P.; Minn, A.J.; et al. A stratified phase I dose escalation trial of hypofractionated radiotherapy followed by ipilimumab in metastatic melanoma: Long-term follow-up and final outcomes. Oncoimmunology 2021, 10, 1863631. [Google Scholar] [CrossRef]
- Twyman-Saint Victor, C.; Rech, A.J.; Maity, A.; Rengan, R.; Pauken, K.E.; Stelekati, E.; Benci, J.L.; Xu, B.; Dada, H.; Odorizzi, P.M.; et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015, 520, 373–377. [Google Scholar] [CrossRef]
- Imai, K.; Takaoka, A. Comparing antibody and small-molecule therapies for cancer. Nat. Rev. Cancer 2006, 6, 714–727. [Google Scholar] [CrossRef]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Signal Transduct. Target. Ther. 2021, 6, 201. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhao, R.; Jiang, X.; Li, Z.; Zhang, B. Progress on the application of bortezomib and bortezomib-based nanoformulations. Biomolecules 2022, 12, 51. [Google Scholar] [CrossRef] [PubMed]
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J., Jr.; Lee, K.P.; Boise, L.H. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006, 107, 4907–4916. [Google Scholar] [CrossRef] [PubMed]
- Spisek, R.; Charalambous, A.; Mazumder, A.; Vesole, D.H.; Jagannath, S.; Dhodapkar, M.V. Bortezomib enhances dendritic cell (DC)–mediated induction of immunity to human myeloma via exposure of cell surface heat shock protein 90 on dying tumor cells: Therapeutic implications. Blood 2007, 109, 4839–4845. [Google Scholar] [CrossRef] [PubMed]
- Gulla, A.; Morelli, E.; Samur, M.K.; Botta, C.; Hideshima, T.; Bianchi, G.; Fulciniti, M.; Malvestiti, S.; Prabhala, R.H.; Talluri, S.; et al. Bortezomib induces anti–multiple myeloma immune response mediated by cGAS/STING pathway activation. Blood Cancer Discov. 2021, 2, 468–483. [Google Scholar] [CrossRef]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP inhibitors: Clinical relevance, mechanisms of action and tumor resistance. Front. Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef]
- Meng, X.W.; Koh, B.D.; Zhang, J.S.; Flatten, K.S.; Schneider, P.A.; Billadeau, D.D.; Hess, A.D.; Smith, B.D.; Karp, J.E.; Kaufmann, S.H. Poly(ADP-ribose) polymerase inhibitors sensitize cancer cells to death receptor-mediated apoptosis by enhancing death receptor expression. J. Biol. Chem. 2014, 289, 20543–20558. [Google Scholar] [CrossRef]
- Fenerty, K.E.; Padget, M.; Wolfson, B.; Gameiro, S.R.; Su, Z.; Lee, J.H.; Rabizadeh, S.; Soon-Shiong, P.; Hodge, J.W. Immunotherapy utilizing the combination of natural killer- and antibody dependent cellular cytotoxicity (ADCC)-mediating agents with poly (ADP-ribose) polymerase (PARP) inhibition. J. Immunother. Cancer 2018, 6, 133. [Google Scholar] [CrossRef]
- Seyedin, S.N.; Hasibuzzaman, M.M.; Pham, V.; Petronek, M.S.; Callaghan, C.; Kalen, A.L.; Mapuskar, K.A.; Mott, S.L.; Spitz, D.R.; Allen, B.G.; et al. Combination therapy with radiation and PARP inhibition enhances responsiveness to anti-PD-1 therapy in colorectal tumor models. Int. J. Rad. Oncol. Biol. Phys. 2020, 108, 81–92. [Google Scholar] [CrossRef]
- Wu, Z.; Cui, P.; Tao, H.; Zhang, S.; Ma, J.; Liu, Z.; Wang, J.; Qian, Y.; Chen, S.; Huang, Z.; et al. The synergistic effect of PARP inhibitors and immune checkpoint inhibitors. Clin. Med. Insights Oncol. 2021, 15, 1179554921996288. [Google Scholar] [CrossRef]
- Rachner, T.D.; Coleman, R.; Hadji, P.; Hofbauer, L.C. Bone health during endocrine therapy for cancer. Lancet Diabetes Endocrinol. 2018, 6, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Ardiani, A.; Gameiro, S.R.; Kwilas, A.R.; Donahue, R.N.; Hodge, J.W. Androgen deprivation therapy sensitizes prostate cancer cells to T-cell killing through androgen receptor dependent modulation of the apoptotic pathway. Oncotarget 2014, 5, 9335–9348. [Google Scholar] [CrossRef] [PubMed]
- Ardiani, A.; Farsaci, B.; Rogers, C.J.; Protter, A.; Guo, Z.; King, T.H.; Apelian, D.; Hodge, J.W. Combination therapy with a second-generation androgen receptor antagonist and a metastasis vaccine improves survival in a spontaneous prostate cancer model. Clin. Cancer Res. 2013, 19, 6205–6218. [Google Scholar] [CrossRef] [PubMed]
- Kwilas, A.R.; Ardiani, A.; Gameiro, S.R.; Richards, J.; Hall, A.B.; Hodge, J.W. Androgen deprivation therapy sensitizes triple negative breast cancer cells to immune-mediated lysis through androgen receptor independent modulation of osteoprotegerin. Oncotarget 2016, 7, 23498–23511. [Google Scholar] [CrossRef]
- Wolfson, B.; Padget, M.R.; Schlom, J.; Hodge, J.W. Exploiting off-target effects of estrogen deprivation to sensitize estrogen receptor negative breast cancer to immune killing. J. Immunother. Cancer 2021, 9, e002258. [Google Scholar] [CrossRef]
- Kim, P.S.; Kwilas, A.R.; Xu, W.; Alter, S.; Jeng, E.K.; Wong, H.C.; Schlom, J.; Hodge, J.W. IL-15 superagonist/IL-15RalphaSushi-Fc fusion complex (IL-15SA/IL-15RalphaSu-Fc; ALT-803) markedly enhances specific subpopulations of NK and memory CD8+ T cells, and mediates potent anti-tumor activity against murine breast and colon carcinomas. Oncotarget 2016, 7, 16130–16145. [Google Scholar] [CrossRef]
- Williamson, C.W.; Sherer, M.V.; Zamarin, D.; Sharabi, A.B.; Dyer, B.A.; Mell, L.K.; Mayadev, J.S. Immunotherapy and radiation therapy sequencing: State of the data on timing, efficacy, and safety. Cancer 2021, 127, 1553–1567. [Google Scholar] [CrossRef]
- Weinberg, F.; Gadgeel, S. Combination pembrolizumab plus chemotherapy: A new standard of care for patients with advanced non-small-cell lung cancer. Lung Cancer 2019, 10, 47–56. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Clinical Trial | Trial Title | Conditions | Treatment | Phases |
|---|---|---|---|---|
| NCT03801304 | Trial to Evaluate Safety and Efficacy of Vinorelbine With Metronomic Administration in Combination With Atezolizumab as Second-line Treatment for Patients With Stage IV Non-small Cell Lung Cancer | Non-small-cell lung cancer | Atezolizumab Vinorelbine | Phase II |
| NCT04159818 | Immune Induction Strategies to Improve Response to Immune Checkpoint Blockade in Triple Negative Breast Cancer (TNBC) Patients (TONIC-2) | Metastatic breast cancer | Nivolumab Cisplatin Low-dose doxorubicin | Phase II |
| NCT04043195 | Nivolumab and Ipilimumab in Combination With Immunogenic Chemotherapy for Patients With Advanced NSCLC | Advanced non-small cell lung cancer (NSCLC) | Oxaliplatin Nivolumab Ipilimumab | Phase I Phase II |
| NCT04463368 | Isolated Hepatic Perfusion in Combination With Ipilimumab and Nivolumab in Patients With Uveal Melanoma Metastases (SCANDIUM II) | Uveal melanoma Liver metastases | Melphalan Ipilimumab Nivolumab | Phase I |
| NCT04072263 | Adoptive T Cell Therapy in Patients With Recurrent Ovarian Cancer (OVACURE) | Recurrent ovarian cancer | Tumor-infiltrating lymphocytes Interferon alfa 2A Carboplatin Paclitaxel | Phase I Phase II |
| NCT04262687 | Chemotherapy and Immunotherapy as Treatment for MSS Metastatic Colorectal Cancer with High Immune Infiltrate (POCHI) | Metastatic colorectal cancer High immune infiltrate Microsatellite stable (MSS) | Capecitabine Oxaliplatin Bevacizumab Pembrolizumab | Phase II |
| NCT05420584 | Neoadjuvant Arterial Embolization Chemotherapy Combined PD-1 Inhibitor for Locally Advanced Rectal Cancer (NECI) | Rectal neoplasms | Tislelizumab Capecitabine Oxaliplatin | Phase II |
| NCT04989218 | Durvalumab and Tremelimumab with Platinum-based Chemo- therapy in Intrahepatic Cholangiocarcinoma (ICC) | Cholangiocarcinoma | Gemcitabine Cisplatin Tremelimumab Durvalumab | Phase I Phase II |
| NCT05144698 | RAPA-201 Therapy of Solid Tumors | Breast cancer Small cell and non-small cell lung cancer Triple negative breast cancer Gastric cancer Esophageal adenocarcinoma Gastric junction adeno- carcinoma Esophageal squamous cell carcinoma Head and neck cancer Squamous cell carcinoma of oral cavity Squamous cell carcinoma of larynx Squamous cell carcinoma of nasopharynx Squamous cell carcinoma of other specified sites of skin Carcinoma of unknown primary Bladder cancer Malignant melanoma | RAPA-201 cells Carboplatin Paclitaxel | Phase II |
| NCT05307198 | Rectal Artery Infusion Chemotherapy Combined with Anti-PD1 Antibody for MSS LARC (RAIC) | Rectal neoplasms | Capecitabine Oxaliplatin Sintilimab | Phase II |
| NCT02499367 | Nivolumab After Induction Treatment in Triple-negative Breast Cancer (TNBC) Patients (TONIC) | Breast cancer | Nivolumab Radiation therapy Low dose doxorubicin Cyclophosphamide Cisplatin | Phase II |
| NCT03409198 | Phase IIb Study Evaluating Immunogenic Chemotherapy Combined with Ipilimumab and Nivolumab in Breast Cancer (ICON) | Breast cancer Hormone receptor positive tumor Metastatic breast cancer | Ipilimumab Nivolumab Pegylated liposomal doxorubicin Cyclophosphamide | Phase II |
| NCT03164993 | Atezolizumab Combined with Immunogenic Chemotherapy in Patients with Metastatic Triple-negative Breast Cancer (ALICE) | Breast cancer Triple-negative breast cancer | Atezolizumab Pegylated liposomal doxorubicin Cyclophosphamide | Phase II |
| NCT02649855 | Docetaxel and PROSTVAC for Metastatic Castration-Sensitive Prostate Cancer | Prostate cancer Prostate neoplasms | PROSTVAC-V PROSTVAC-F Docetaxel | Phase II |
| Clinical Trial | Trial Title | Conditions | Treatment | Phases |
|---|---|---|---|---|
| NCT04774133 | The Immunodynamic Effect of Radiotherapy in Prostate Cancer Patients | Prostate cancer | Radiation | N/A |
| NCT03942328 | Modified Immune Cells (Autologous Dendritic Cells) and a Vaccine (Prevnar) After High-Dose External Beam Radiation Therapy in Treating Patients With Unresectable Liver Cancer | Hepatocellular carcinoma Intrahepatic cholangiocarcinoma | External beam radiation, therapeutic autologous dendritic cells, Pneumococcal 13-valent conjugate vaccine | Phase I |
| NCT03789097 | Vaccination With Flt3L, Radiation, and Poly-ICLC | Non-Hodgkin’s lymphoma Metastatic breast cancer Head-and-neck squamous-cell carcinoma | Subtherapeutic radiation Fl3tL Poly-ICLC Pembrolizumab | Phase I Phase II |
| NCT03646617 | Ipilimumab and Nivolumab With or Without Hypofractionated Radiotherapy in Patients With Metastatic Melanoma (RadVax) | Metastatic melanoma | Hypofractionated radiation Ipilimumab Nivolumab | Phase II |
| NCT03313804 | Priming Immunotherapy in Advanced Disease With Radiation | Non-small-cell lung cancer Head-and-neck squamous-cell carcinoma | Stereotactic body radiation or fractionated radiation Nivolumab or pembrolizumab or atezolizumab | Phase II |
| NCT04454528 | BreastVAX: Radiation Boost to Enhance Immune Checkpoint Blockade Therapy (BreastVAX) | Breast cancer | Hypofractionated radiation Pembrolizumab | Phase I Phase II |
| Clinical Trial | Trial Title | Conditions | Treatment | Phases |
|---|---|---|---|---|
| NCT04265872 | Bortezomib Followed by Pembrolizumab and Cisplatin in metTNBC | Breast cancer | Bortezomib Pembrolizumab Cisplatin | Phase 1 |
| NCT04258683 | A Study of Pembrolizumab Added to the Standard First-Line Therapy of Cyclophosphamide, Bortezomib, and Dexamethasone (CyBorD) for NDMM NTE | Multiple myeloma | Cyclophosphamide Bortezomib Dexamethasone Pembrolizumab | Phase 2 |
| NCT04191096 NCT04934722 | Efficacy and Safety of Pembrolizumab (MK-3475) Plus Enzalutamide Plus Androgen Deprivation Therapy (ADT) Versus Placebo Plus Enzalutamide Plus ADT in Participants With Metastatic Hormone-Sensitive Prostate Cancer (mHSPC) (MK-3475-991/KEYNOTE-991) | Metastatic hormone-sensitive prostate cancer | Enzalutamide Pembrolizumab | Phase 3 |
| NCT04471974 | ZEN-3694, Enzalutamide, and Pembrolizumab for the Treatment of Metastatic Castration-Resistant Prostate Cancer | Castration-resistant prostate carcinoma Metastatic prostate adeno- carcinoma Metastatic prostate small cell carcinoma Stage IV/IVA/IVB prostate cancer AJCC v8 | ZEN-3694 Enzalutamide Pembrolizumab | Phase 2 |
| NCT04946370 | Maximizing Responses to Anti-PD1 Immunotherapy With PSMA-targeted Alpha Therapy in mCRPC | Prostate cancer | 225Ac-J591 Pembrolizumab Androgen receptor pathway inhibitor | Phase 1 Phase 2 |
| NCT04262154 | Study of Abiraterone Acetate, Atezolizumab, GnRH Analog and Radiation Therapy in Men With Newly Diagnosed Hormone- sensitive Prostate Cancer | Metastatic prostate cancer | Atezolizumab Abiraterone acetate Prednisone Lupron® (leuprolide) SBRT Enzalutamide | Phase 2 |
| NCT04190056 | Pembrolizumab and Tamoxifen With or Without Vorinostat for the Treatment of Estrogen Receptor Positive Breast Cancer | Breast cancer | Pembrolizumab Tamoxifen Vorinostat | Phase 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fabian, K.P.; Kowalczyk, J.T.; Reynolds, S.T.; Hodge, J.W. Dying of Stress: Chemotherapy, Radiotherapy, and Small-Molecule Inhibitors in Immunogenic Cell Death and Immunogenic Modulation. Cells 2022, 11, 3826. https://doi.org/10.3390/cells11233826
Fabian KP, Kowalczyk JT, Reynolds ST, Hodge JW. Dying of Stress: Chemotherapy, Radiotherapy, and Small-Molecule Inhibitors in Immunogenic Cell Death and Immunogenic Modulation. Cells. 2022; 11(23):3826. https://doi.org/10.3390/cells11233826
Chicago/Turabian StyleFabian, Kellsye P., Joshua T. Kowalczyk, Sandy T. Reynolds, and James W. Hodge. 2022. "Dying of Stress: Chemotherapy, Radiotherapy, and Small-Molecule Inhibitors in Immunogenic Cell Death and Immunogenic Modulation" Cells 11, no. 23: 3826. https://doi.org/10.3390/cells11233826
APA StyleFabian, K. P., Kowalczyk, J. T., Reynolds, S. T., & Hodge, J. W. (2022). Dying of Stress: Chemotherapy, Radiotherapy, and Small-Molecule Inhibitors in Immunogenic Cell Death and Immunogenic Modulation. Cells, 11(23), 3826. https://doi.org/10.3390/cells11233826