



Destabilizing COXIV in Müller Glia Increases Retinal Glycolysis and Alters Scotopic Electroretinogram


Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. PCR on Genomic DNA
2.3. Cell Culture
2.4. Intraocular Pressure (IOP) Measurements
2.5. Cryosectioning
2.6. Immunofluorescence (IF)
2.7. Histochemistry
2.8. Protein Analysis by Capillary-Based Electrophoresis
2.9. Electroretinography (ERG)
2.9.1. Scotopic
2.9.2. Photopic Negative Response
2.10. Biochemical Assays
2.11. Ocular Hypertension (OHT) Model
2.12. Statistics
3. Results
3.1. Characterization of COX10 Knockout
3.2. Stress Effect of COX10 Knockout and ERG
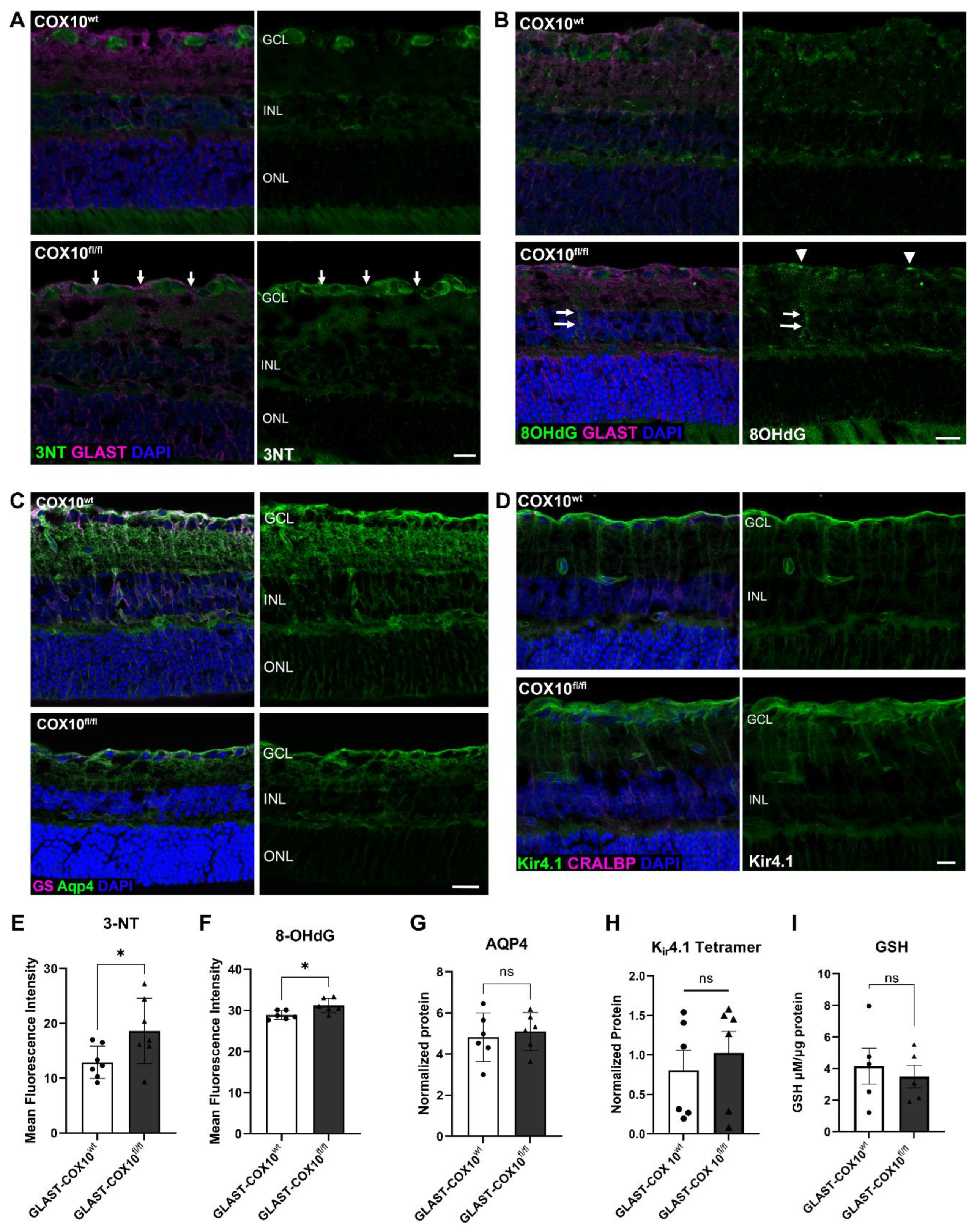
3.3. Oxidative Stress and MG Homeostasis
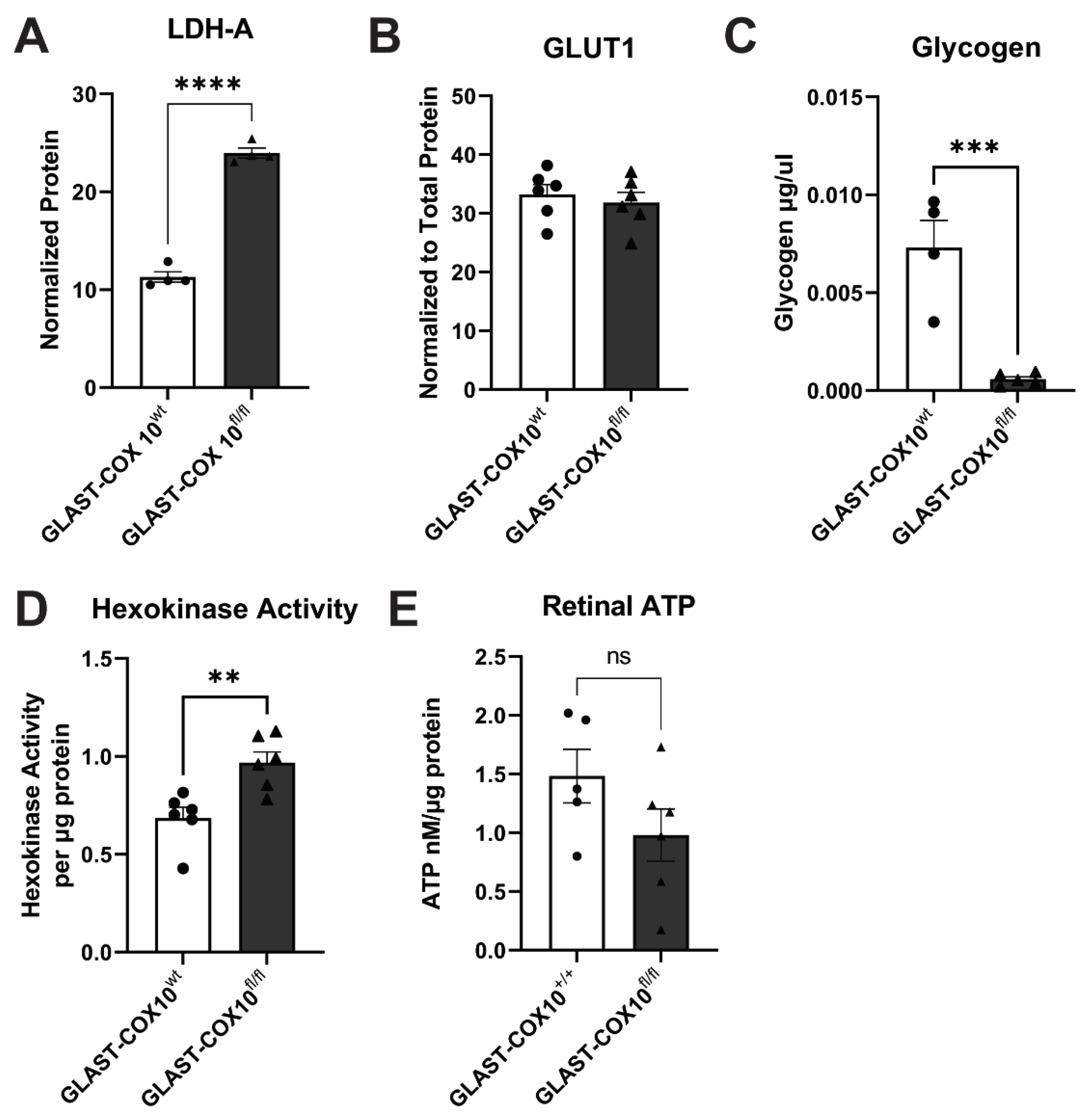
3.4. Changes in Retinal Metabolism
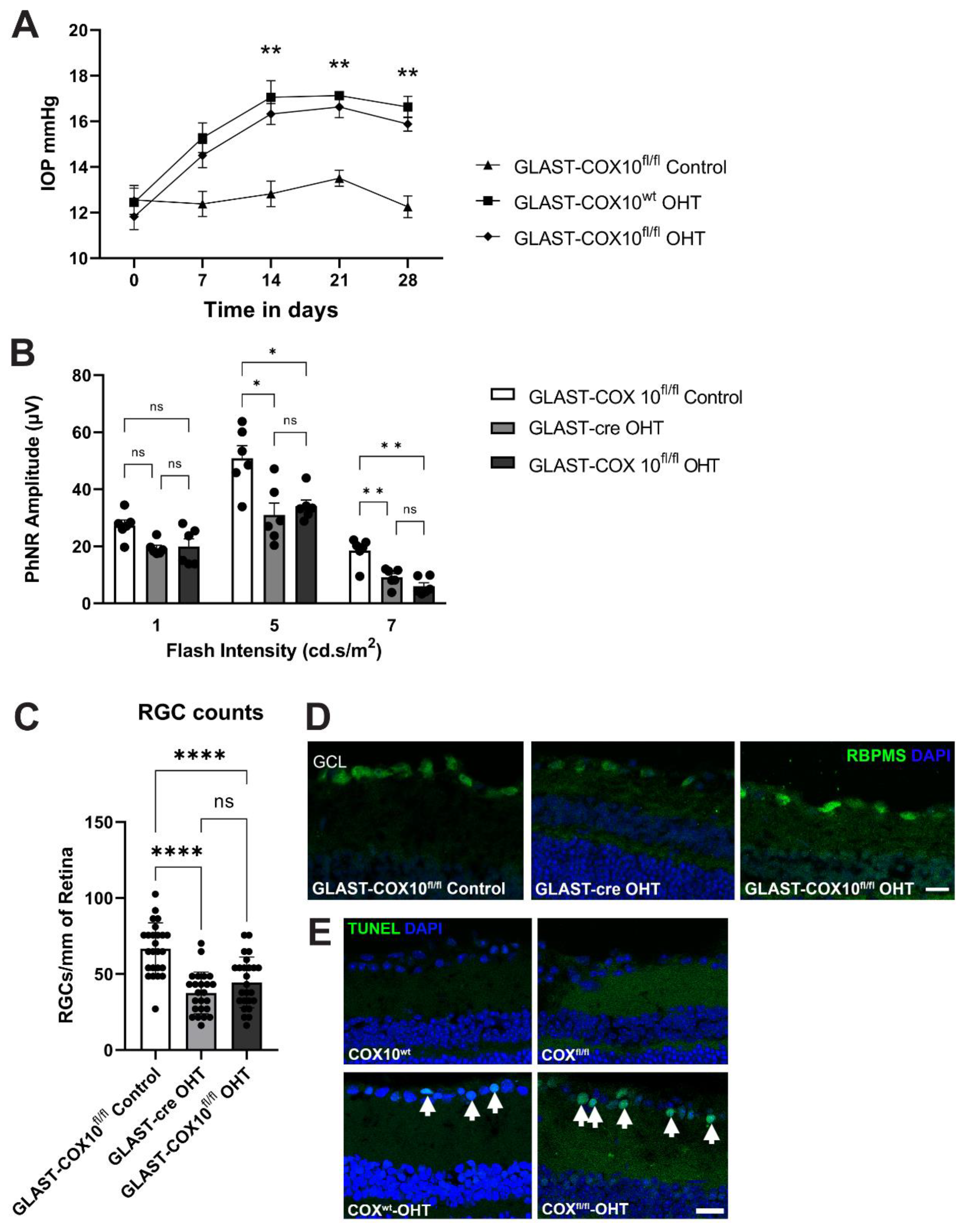
3.5. COX10 Knockout and Ocular Hypertension
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Casson, R.J.; Chidlow, G.; Crowston, J.G.; Williams, P.A.; Wood, J.P.M. Retinal energy metabolism in health and glaucoma. Prog. Retin. Eye Res. 2021, 81, 100881. [Google Scholar] [CrossRef] [PubMed]
- Kamel, K.; O’Brien, C.J.; Zhdanov, A.V.; Papkovsky, D.B.; Clark, A.F.; Stamer, W.D.; Irnaten, M. Reduced Oxidative Phosphorylation and Increased Glycolysis in Human Glaucoma Lamina Cribrosa Cells. Investig. Ophthalmol. Vis. Sci. 2020, 61, 4. [Google Scholar] [CrossRef] [PubMed]
- Kisilevsky, E.; Freund, P.; Margolin, E. Mitochondrial disorders and the eye. Surv. Ophthalmol. 2020, 65, 294–311. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.A.; Harder, J.M.; Foxworth, N.E.; Cochran, K.E.; Philip, V.M.; Porciatti, V.; Smithies, O.; John, S.W.M. Vitamin B3 modulates mitochondrial vulnerability and prevents glaucoma in aged mice. Science 2017, 355, 756–760. [Google Scholar] [CrossRef]
- Baltan, S.; Inman, D.M.; Danilov, C.A.; Morrison, R.S.; Calkins, D.J.; Horner, P.J. Metabolic Vulnerability Disposes Retinal Ganglion Cell Axons to Dysfunction in a Model of Glaucomatous Degeneration. J. Neurosci. 2010, 30, 5644–5652. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.J.; Cascio, M.A.; Rosca, M.G. Diabetic Retinopathy: The Role of Mitochondria in the Neural Retina and Microvascular Disease. Antioxidants 2020, 9, 905. [Google Scholar] [CrossRef]
- Tsacopoulos, M.; Poitry-Yamate, C.L.; MacLeish, P.R.; Poitry, S. Trafficking of molecules and metabolic signals in the retina. Prog. Retin. Eye Res. 1998, 17, 429–442. [Google Scholar] [CrossRef]
- Viegas, F.O.; Neuhauss, S.C.F. A Metabolic Landscape for Maintaining Retina Integrity and Function. Front. Mol. Neurosci. 2021, 14. Available online: https://www.frontiersin.org/articles/10.3389/fnmol.2021.656000 (accessed on 30 September 2022). [CrossRef]
- Magistretti, P.J.; Allaman, I. A Cellular Perspective on Brain Energy Metabolism and Functional Imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef]
- Fünfschilling, U.; Supplie, L.M.; Mahad, D.; Boretius, S.; Saab, A.S.; Edgar, J.; Brinkmann, B.G.; Kassmann, C.M.; Tzvetanova, I.D.; Möbius, W.; et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 2012, 485, 517–521. [Google Scholar] [CrossRef]
- Supplie, L.M.; Düking, T.; Campbell, G.; Diaz, F.; Moraes, C.T.; Götz, M.; Hamprecht, B.; Boretius, S.; Mahad, D.; Nave, K.-A. Respiration-Deficient Astrocytes Survive As Glycolytic Cells In Vivo. J. Neurosci. 2017, 37, 4231–4242. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Lee, S.-R.; Mathai, A.E.; Zhang, R.; Du, J.; Yam, M.X.; Pye, V.; Barnett, N.L.; Rayner, C.L.; Zhu, L.; et al. Effect of selectively knocking down key metabolic genes in Müller glia on photoreceptor health. Glia 2021, 69, 1966–1986. [Google Scholar] [CrossRef] [PubMed]
- Kanow, M.A.; Giarmarco, M.M.; Jankowski, C.S.; Tsantilas, K.; Engel, A.L.; Du, J.; Linton, J.D.; Farnsworth, C.C.; Sloat, S.R.; Rountree, A.; et al. Biochemical adaptations of the retina and retinal pigment epithelium support a metabolic ecosystem in the vertebrate eye. eLife 2017, 6, e28899. [Google Scholar] [CrossRef] [PubMed]
- Poitry-Yamate, C.L.; Poitry, S.; Tsacopoulos, M. Lactate Released by Miiller Glial Cells Is Metabolized by Photoreceptors from Mammalian Retina. J. Neurosci. 1995, 15 Pt 2, 5179–5191. [Google Scholar] [CrossRef] [PubMed]
- Winkler, B.S.; Arnold, M.J.; Brassell, M.A.; Puro, D.G. Energy Metabolism in Human Retinal Müller Cells. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3183–3190. [Google Scholar]
- Xu, Y.; Ola, M.S.; Berkich, D.A.; Gardner, T.W.; Barber, A.J.; Palmieri, F.; Hutson, S.M.; LaNoue, K.F. Energy sources for glutamate neurotransmission in the retina: Absence of the aspartate/glutamate carrier produces reliance on glycolysis in glia. J. Neurochem. 2007, 101, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Toft-Kehler, A.K.; Skytt, D.M.; Svare, A.; Lefevere, E.; Van Hove, I.; Moons, L.; Waagepetersen, H.S.; Kolko, M. Mitochondrial function in Müller cells—Does it matter? Mitochondrion 2017, 36, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Bringmann, A.; Pannicke, T.; Grosche, J.; Francke, M.; Wiedemann, P.; Skatchkov, S.N.; Osborne, N.N.; Reichenbach, A. Müller cells in the healthy and diseased retina. Prog. Retin. Eye Res. 2006, 25, 397–424. [Google Scholar] [CrossRef] [PubMed]
- Reichenbach, A.; Bringmann, A. New functions of Müller cells. Glia 2013, 61, 651–678. [Google Scholar] [CrossRef] [PubMed]
- Pow, D.V.; Crook, D.K. Immunocytochemical evidence for the presence of high levels of reduced glutathione in radial glial cells and horizontal cells in the rabbit retina. Neurosci. Lett. 1995, 193, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Da, T.; Verkman, A.S. Aquaporin-4 Gene Disruption in Mice Protects against Impaired Retinal Function and Cell Death after Ischemia. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4477–4483. [Google Scholar] [CrossRef] [PubMed]
- Sarthy, V.P.; Pignataro, L.; Pannicke, T.; Weick, M.; Reichenbach, A.; Harada, T.; Tanaka, K.; Marc, R. Glutamate transport by retinal Müller cells in glutamate/aspartate transporter-knockout mice. Glia 2005, 49, 184–196. [Google Scholar] [CrossRef] [PubMed]
- Bringmann, A.; Pannicke, T.; Biedermann, B.; Francke, M.; Iandiev, I.; Grosche, J.; Wiedemann, P.; Albrecht, J.; Reichenbach, A. Role of retinal glial cells in neurotransmitter uptake and metabolism. Neurochem. Int. 2009, 54, 143–160. [Google Scholar] [CrossRef]
- Bringmann, A.; Iandiev, I.; Pannicke, T.; Wurm, A.; Hollborn, M.; Wiedemann, P.; Osborne, N.N.; Reichenbach, A. Cellular signaling and factors involved in Müller cell gliosis: Neuroprotective and detrimental effects. Prog. Retin. Eye Res. 2009, 28, 423–451. [Google Scholar] [CrossRef] [PubMed]
- Unterlauft, J.D.; Eichler, W.; Kuhne, K.; Yang, X.M.; Yafai, Y.; Wiedemann, P.; Reichenbach, A.; Claudepierre, T. Pigment Epithelium-Derived Factor Released by Müller Glial Cells Exerts Neuroprotective Effects on Retinal Ganglion Cells. Neurochem. Res. 2012, 37, 1524–1533. [Google Scholar] [CrossRef] [PubMed]
- Schütte, M.; Werner, P. Redistribution of glutathione in the ischemic rat retina. Neurosci. Lett. 1998, 246, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Bringmann, A.; Wiedemann, P. Müller Glial Cells in Retinal Disease. Ophthalmologica 2012, 227, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Sarantis, M.; Attwell, D. Glutamate uptake in mammalian retinal glia is voltage- and potassium-dependent. Brain Res. 1990, 516, 322–325. [Google Scholar] [CrossRef] [PubMed]
- Pannicke, T.; Stabel, J.; Heinemann, U.; Reichelt, W. Alpha-aminoadipic acid blocks the Na+-dependent glutamate transport into acutely isolated Müller glial cells from guinea pig retina. Pflüg. Arch. 1994, 429, 140–142. [Google Scholar] [CrossRef] [PubMed]
- Pannicke, T.; Iandiev, I.; Uckermann, O.; Biedermann, B.; Kutzera, F.; Wiedemann, P.; Wolburg, H.; Reichenbach, A.; Bringmann, A. A potassium channel-linked mechanism of glial cell swelling in the postischemic retina. Mol. Cell Neurosci. 2004, 26, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Jassim, A.H.; Inman, D.M. Evidence of Hypoxic Glial Cells in a Model of Ocular Hypertension. Investig. Ophthalmol. Vis. Sci. 2019, 60, 1–15. [Google Scholar] [CrossRef]
- Jassim, A.H.; Nsiah, N.Y.; Inman, D.M. Ocular Hypertension Results in Hypoxia within Glia and Neurons throughout the Visual Projection. Antioxidants 2022, 11, 888. [Google Scholar] [CrossRef] [PubMed]
- Zeevalk, G.D.; Nicklas, W.J. Contribution of Glial Metabolism to Neuronal Damage Caused by Partial Inhibition of Energy Metabolism in Retina. Exp. Eye Res. 1997, 65, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Vohra, R.; Gurubaran, I.S.; Henriksen, U.; Bergersen, L.H.; Rasmussen, L.J.; Desler, C.; Skytt, D.M.; Kolko, M. Disturbed mitochondrial function restricts glutamate uptake in the human Müller glia cell line, MIO-M1. Mitochondrion 2017, 36, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, K.J.; Du, J.; Sloat, S.R.; Contreras, L.; Linton, J.D.; Turner, S.J.; Sadilek, M.; Satrústegui, J.; Hurley, J.B. Pyruvate kinase and aspartate-glutamate carrier distributions reveal key metabolic links between neurons and glia in retina. Proc. Natl. Acad. Sci. USA 2014, 111, 15579–15584. [Google Scholar] [CrossRef] [PubMed]
- Rueda, E.M.; Johnson, J.E.; Giddabasappa, A.; Swaroop, A.; Brooks, M.J.; Sigel, I.; Chaney, S.Y.; Fox, D.A. The cellular and compartmental profile of mouse retinal glycolysis, tricarboxylic acid cycle, oxidative phosphorylation, and ~P transferring kinases. Mol. Vis. 2016, 22, 847–885. [Google Scholar] [PubMed]
- Diaz, F.; Thomas, C.K.; Garcia, S.; Hernandez, D.; Moraes, C.T. Mice lacking COX10 in skeletal muscle recapitulate the phenotype of progressive mitochondrial myopathies associated with cytochrome c oxidase deficiency. Hum. Mol. Genet. 2005, 14, 2737–2748. [Google Scholar] [CrossRef]
- Pitceathly, R.D.S.; Taanman, J.-W.; Rahman, S.; Meunier, B.; Sadowski, M.; Cirak, S.; Hargreaves, I.; Land, J.M.; Nanji, T.; Polke, J.M.; et al. COX10 Mutations Resulting in Complex Multisystem Mitochondrial Disease That Remains Stable Into Adulthood. JAMA Neurol. 2013, 70, 1556–1561. [Google Scholar] [CrossRef]
- Ross, J.M. Visualization of Mitochondrial Respiratory Function using Cytochrome C Oxidase/Succinate Dehydrogenase (COX/SDH) Double-labeling Histochemistry. J. Vis. Exp. 2011, 23, e3266. [Google Scholar] [CrossRef]
- Abu-Amero, K.K.; Morales, J.; Bosley, T.M. Mitochondrial Abnormalities in Patients with Primary Open-Angle Glaucoma. Investig. Ophthalmol. Vis. Sci. 2006, 47, 2533–2541. [Google Scholar] [CrossRef]
- McElnea, E.M.; Quill, B.; Docherty, N.G.; Irnaten, M.; Siah, W.F.; Clark, A.F.; O’Brien, C.J.; Wallace, D.M. Oxidative stress, mitochondrial dysfunction and calcium overload in human lamina cribrosa cells from glaucoma donors. Mol. Vis. 2011, 17, 1182–1191. [Google Scholar] [PubMed]
- Lee, S.; Sheck, L.; Crowston, J.G.; Van Bergen, N.J.; O’Neill, E.C.; O’Hare, F.; Kong, Y.X.G.; Chrysostomou, V.; Vincent, A.L.; Trounce, I.A. Impaired Complex-I-Linked Respiration and ATP Synthesis in Primary Open-Angle Glaucoma Patient Lymphoblasts. Investig. Ophthalmol. Vis. Sci. 2012, 53, 2431–2437. [Google Scholar] [CrossRef] [PubMed]
- Dahl, D. The radial glia of Müller in the rat retina and their response to injury. An immunofluorescence study with antibodies to the glial fibrillary acidic (GFA) protein. Exp. Eye Res. 1979, 28, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Erickson, P.A.; Fisher, S.K.; Guérin, C.J.; Anderson, D.H.; Kaska, D.D. Glial fibrillary acidic protein increases in Müller cells after retinal detachment. Exp. Eye Res. 1987, 44, 37–48. [Google Scholar] [CrossRef]
- Inman, D.M.; Horner, P.J. Reactive nonproliferative gliosis predominates in a chronic mouse model of glaucoma. Glia 2007, 55, 942–953. [Google Scholar] [CrossRef]
- Behrens, C.; Schubert, T.; Haverkamp, S.; Euler, T.; Berens, P. Connectivity map of bipolar cells and photoreceptors in the mouse retina. eLife 2016, 5, e20041. [Google Scholar] [CrossRef]
- Kaylor, J.J.; Cook, J.D.; Makshanoff, J.; Bischoff, N.; Yong, J.; Travis, G.H. Identification of the 11-cis-specific retinyl-ester synthase in retinal Müller cells as multifunctional O-acyltransferase (MFAT). Proc. Natl. Acad. Sci. USA 2014, 111, 7302–7307. [Google Scholar] [CrossRef]
- Connors, N.C.; Kofuji, P. Potassium channel Kir4.1 macromolecular complex in retinal glial cells. Glia 2006, 53, 124–131. [Google Scholar] [CrossRef]
- Kofuji, P.; Ceelen, P.; Zahs, K.R.; Surbeck, L.W.; Lester, H.A.; Newman, E.A. Genetic Inactivation of an Inwardly Rectifying Potassium Channel (Kir4.1 Subunit) in Mice: Phenotypic Impact in Retina. J. Neurosci. 2000, 20, 5733–5740. [Google Scholar] [CrossRef]
- Choi, S.-H.; Kim, K.-Y.; Perkins, G.A.; Phan, S.; Edwards, G.; Xia, Y.; Kim, J.; Skowronska-Krawczyk, D.; Weinreb, R.N.; Ellisman, M.H.; et al. AIBP protects retinal ganglion cells against neuroinflammation and mitochondrial dysfunction in glaucomatous neurodegeneration. Redox Biol. 2020, 37, 101703. [Google Scholar] [CrossRef]
- Ozawa, Y.; Toda, E.; Kawashima, H.; Homma, K.; Osada, H.; Nagai, N.; Abe, Y.; Yasui, M.; Tsubota, K. Aquaporin 4 Suppresses Neural Hyperactivity and Synaptic Fatigue and Fine-Tunes Neurotransmission to Regulate Visual Function in the Mouse Retina. Mol. Neurobiol. 2019, 56, 8124–8135. [Google Scholar] [CrossRef] [PubMed]
- Chinchore, Y.; Begaj, T.; Wu, D.; Drokhlyansky, E.; Cepko, C.L. Glycolytic reliance promotes anabolism in photoreceptors. eLife 2017, 6, e25946. [Google Scholar] [CrossRef] [PubMed]
- KUWABARA, T.; COGAN, D.G. Retinal Glycogen. Arch. Ophthalmol. 1961, 66, 680–688. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer-Guglielmi, B.; Francke, M.; Reichenbach, A.; Fleckenstein, B.; Jung, G.; Hamprecht, B. Glycogen phosphorylase isozyme pattern in mammalian retinal Müller (glial) cells and in astrocytes of retina and optic nerve. Glia 2005, 49, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Tseng, Y.-C.; Huang, C.-J.; Chang, J.C.-H.; Teng, W.-Y.; Baba, O.; Fann, M.-J.; Hwang, P.-P. Glycogen phosphorylase in glycogen-rich cells is involved in the energy supply for ion regulation in fish gill epithelia. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2007, 293, R482–R491. [Google Scholar] [CrossRef] [PubMed]
- Goldman, D. Müller glial cell reprogramming and retina regeneration. Nat. Rev. Neurosci. 2014, 15, 431–442. [Google Scholar] [CrossRef]
- Hurley, J.B.; Lindsay, K.J.; Du, J. Glucose, Lactate, and Shuttling of Metabolites in Vertebrate Retinas. J. Neurosci. Res. 2015, 93, 1079–1092. [Google Scholar] [CrossRef]
- Nagelhus, E.A.; Veruki, M.L.; Torp, R.; Haug, F.-M.; Laake, J.H.; Nielsen, S.; Agre, P.; Ottersen, O.P. Aquaporin-4 Water Channel Protein in the Rat Retina and Optic Nerve: Polarized Expression in Müller Cells and Fibrous Astrocytes. J. Neurosci. 1998, 18, 2506–2519. [Google Scholar] [CrossRef]
- Brasko, C.; Hawkins, V.; De La Rocha, I.C.; Butt, A.M. Expression of Kir4.1 and Kir5.1 inwardly rectifying potassium channels in oligodendrocytes, the myelinating cells of the CNS. Brain Struct. Funct. 2017, 222, 41–59. [Google Scholar] [CrossRef]
- Neely, J.D.; Christensen, B.M.; Nielsen, S.; Agre, P. Heterotetrameric Composition of Aquaporin-4 Water Channels. Biochemistry 1999, 38, 11156–11163. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen | Species | Manufacturer | Catalog Number | Dilution |
|---|---|---|---|---|
| AQP4 | Rabbit | Abcam | ab61392 | 1:250 IF; 1:50 CE |
| COX 10 | Rabbit | Novus Biologicals | NBP1-59554 | 1:100 IF |
| COX IV | Rabbit | Cell Signaling | 4850 | 1:100 IF; 1:25 CE |
| CRALBP | Mouse | Novus Biologicals | NB 100-74392 | 1:200; 1:50 CE |
| GFAP | Goat | Abcam | ab53554 | 1:500 |
| GLAST | Rabbit | Abcam | ab416 | 1:200 IF; 1:50 CE |
| Glutamine Synthetase | Mouse | Santa Cruz | SC-74430 | 1:150 |
| MTCO1 | Mouse | Invitrogen | 459600 | 1:100 |
| RBPMS | Rabbit | Genetex | 118619 | 1:300 |
| Vimentin | Chicken | Novus Biologicals | NB300-233 | 1:250 |
| GLUT1 | Rabbit | Novus Biologicals | NB110-39113 | 1:50 CE |
| LDH-A | Rabbit | Novus Biologicals | NBP1-48336 | 1:50 CE |
| Kir4.1 | Rabbit | ProteinTech | 12503-1-AP | 1:50 CE |
| 8-Hydroxy-deoxy-guanosine (8OHdG) | Rabbit | QED Bioscience | 12501 | 1:200 |
| 3-Nitrotyrosine | Mouse | Abcam | ab61392 | 1:200 |
| Parameter | Manufacturer | Catalog Number |
|---|---|---|
| Glycogen | Millipore-Sigma | MAK016-1KT |
| Glutathione | Cayman Chemical | 703002 |
| Hexokinase | Millipore-Sigma | MAK091-1KT |
| ATP | Invitrogen | A22066 |
| TUNEL | Promega | G7130 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nsiah, N.Y.; Inman, D.M. Destabilizing COXIV in Müller Glia Increases Retinal Glycolysis and Alters Scotopic Electroretinogram. Cells 2022, 11, 3756. https://doi.org/10.3390/cells11233756
Nsiah NY, Inman DM. Destabilizing COXIV in Müller Glia Increases Retinal Glycolysis and Alters Scotopic Electroretinogram. Cells. 2022; 11(23):3756. https://doi.org/10.3390/cells11233756
Chicago/Turabian StyleNsiah, Nana Yaa, and Denise M. Inman. 2022. "Destabilizing COXIV in Müller Glia Increases Retinal Glycolysis and Alters Scotopic Electroretinogram" Cells 11, no. 23: 3756. https://doi.org/10.3390/cells11233756
APA StyleNsiah, N. Y., & Inman, D. M. (2022). Destabilizing COXIV in Müller Glia Increases Retinal Glycolysis and Alters Scotopic Electroretinogram. Cells, 11(23), 3756. https://doi.org/10.3390/cells11233756