



Neuroprotection of Rodent and Human Retinal Ganglion Cells In Vitro/Ex Vivo by the Hybrid Small Molecule SA-2

 ,
,  ,
, 
 and
and
Abstract
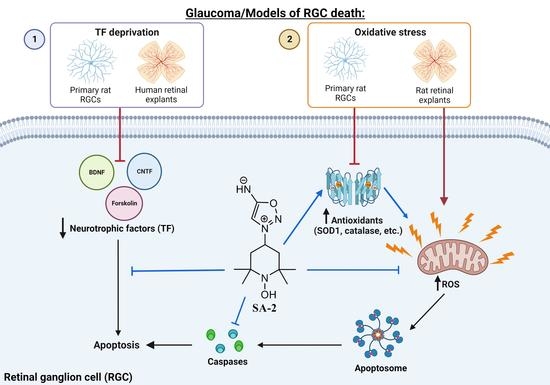
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Compound SA-2
2.2. Animal Care and Protocol Approval
2.3. Trophic Factor Deprivation Model in Primary RGCs
2.4. TBHP-Induced Oxidative Stress Model in Ex Vivo Rat Retinal Explants
2.5. Endothelin-3-Induced Oxidative Stress Model in Primary RGCs
2.6. Trophic Factor Deprivation Model in Ex Vivo Human Retinal Explants
2.7. Statistical Analysis
3. Results
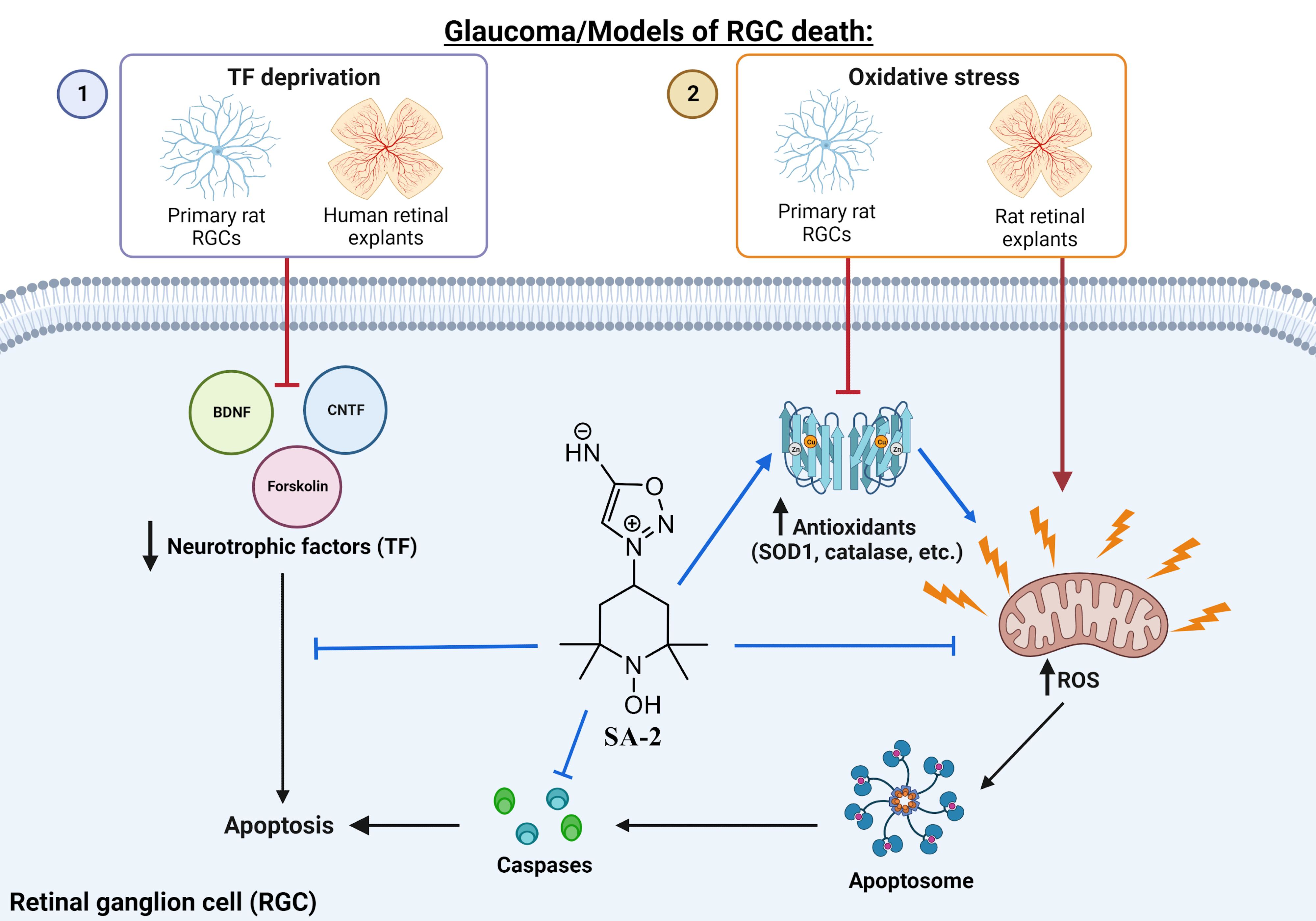
3.1. SA-2 Is Neuroprotective to Primary RGCs in the Trophic Factor Deprivation Model
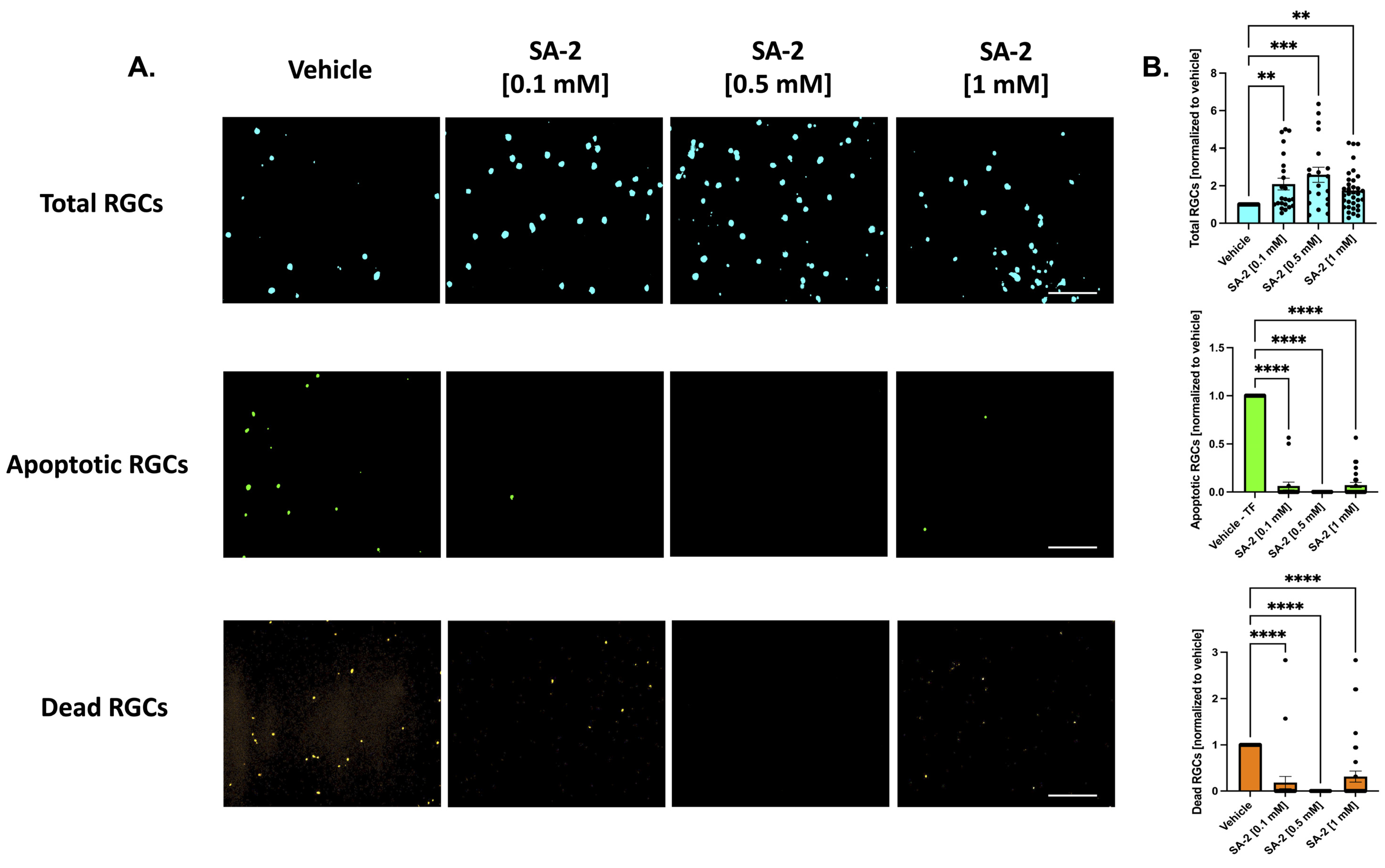
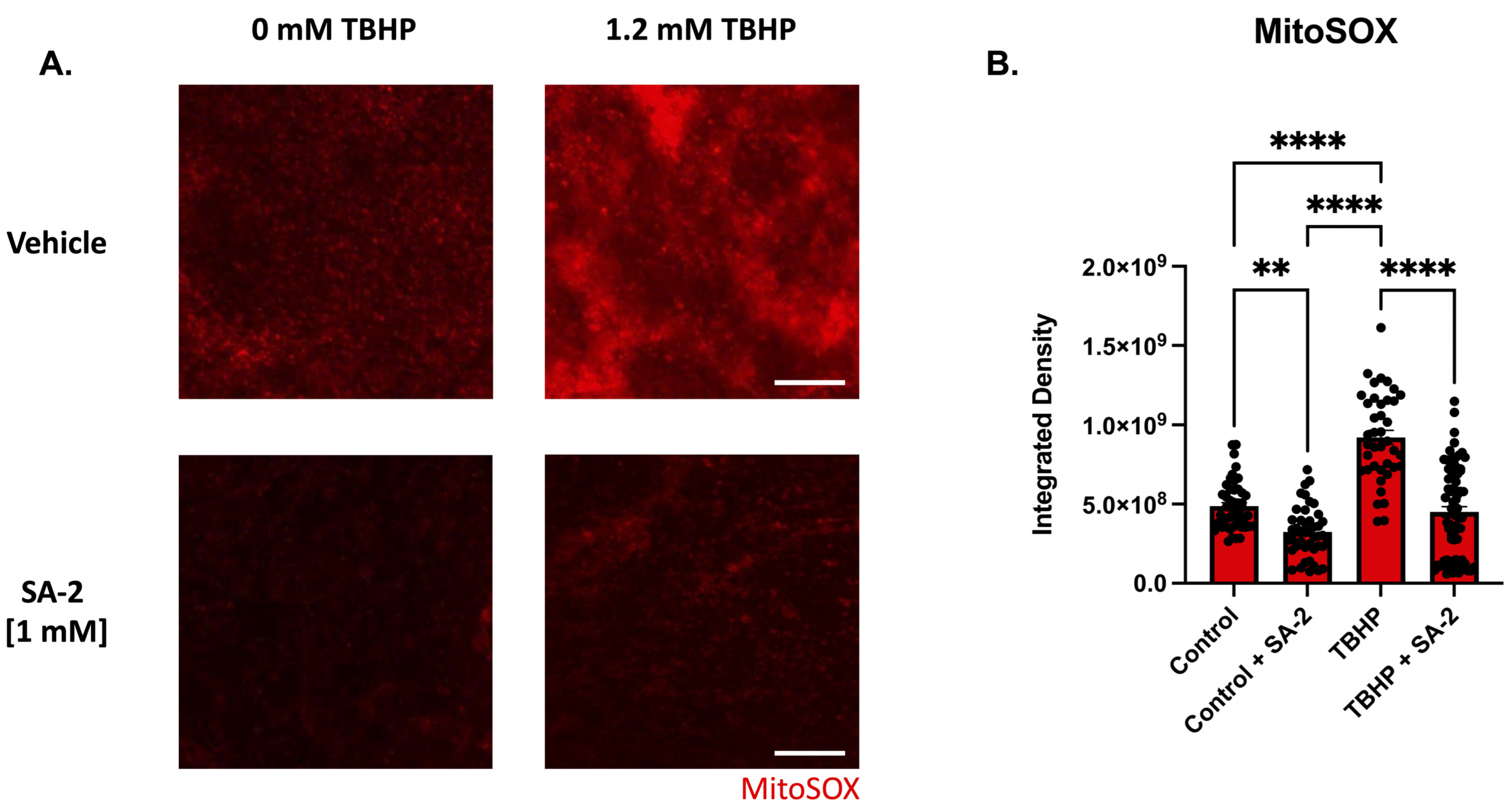
3.2. SA-2 Scavenges Reactive Oxygen Species in TBHP-Induced Oxidative Stress Model in Ex Vivo Rat Retinal Explant
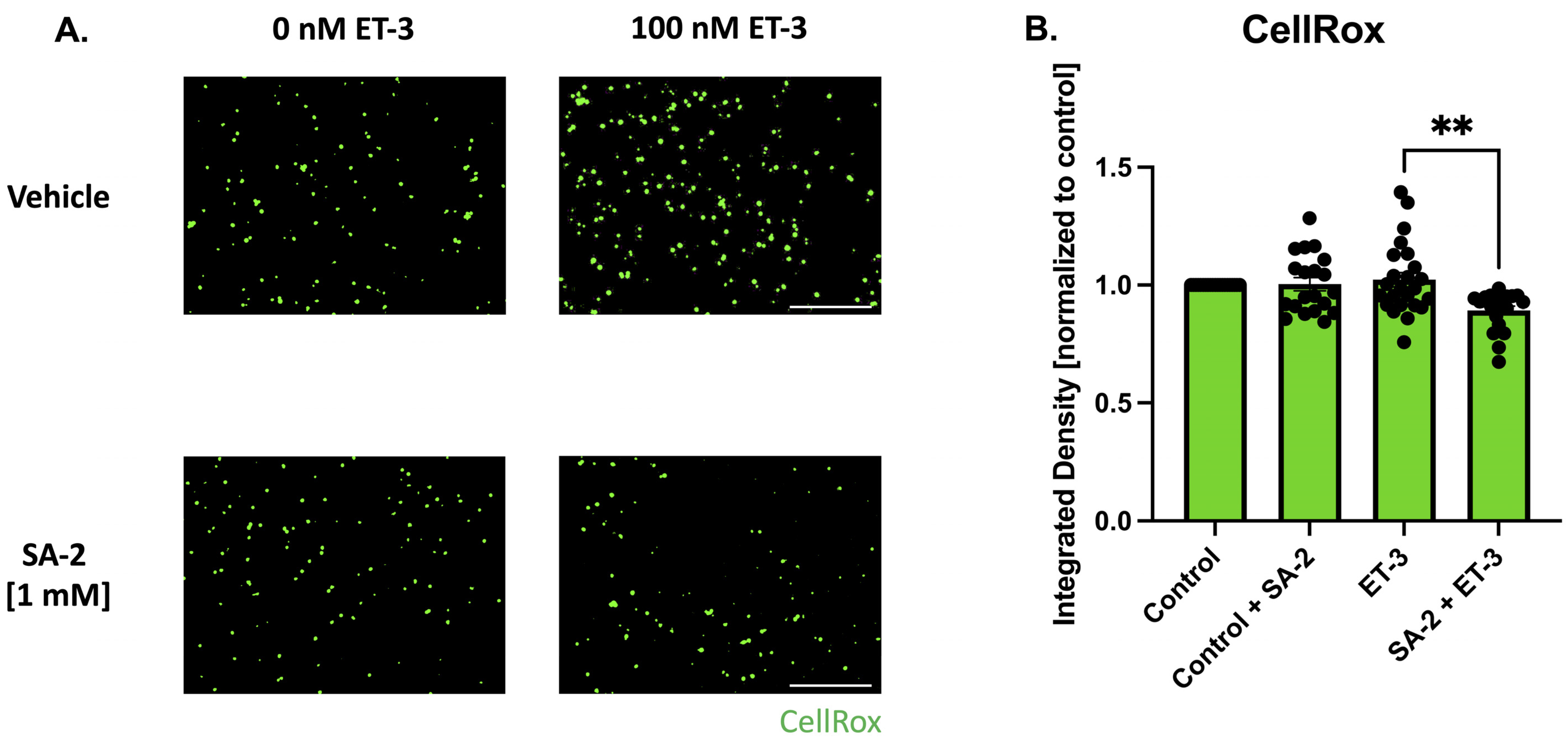
3.3. SA-2 Prevents Reactive Oxygen Species Formation in ET-3-Induced Oxidative Stress in Primary RGCs
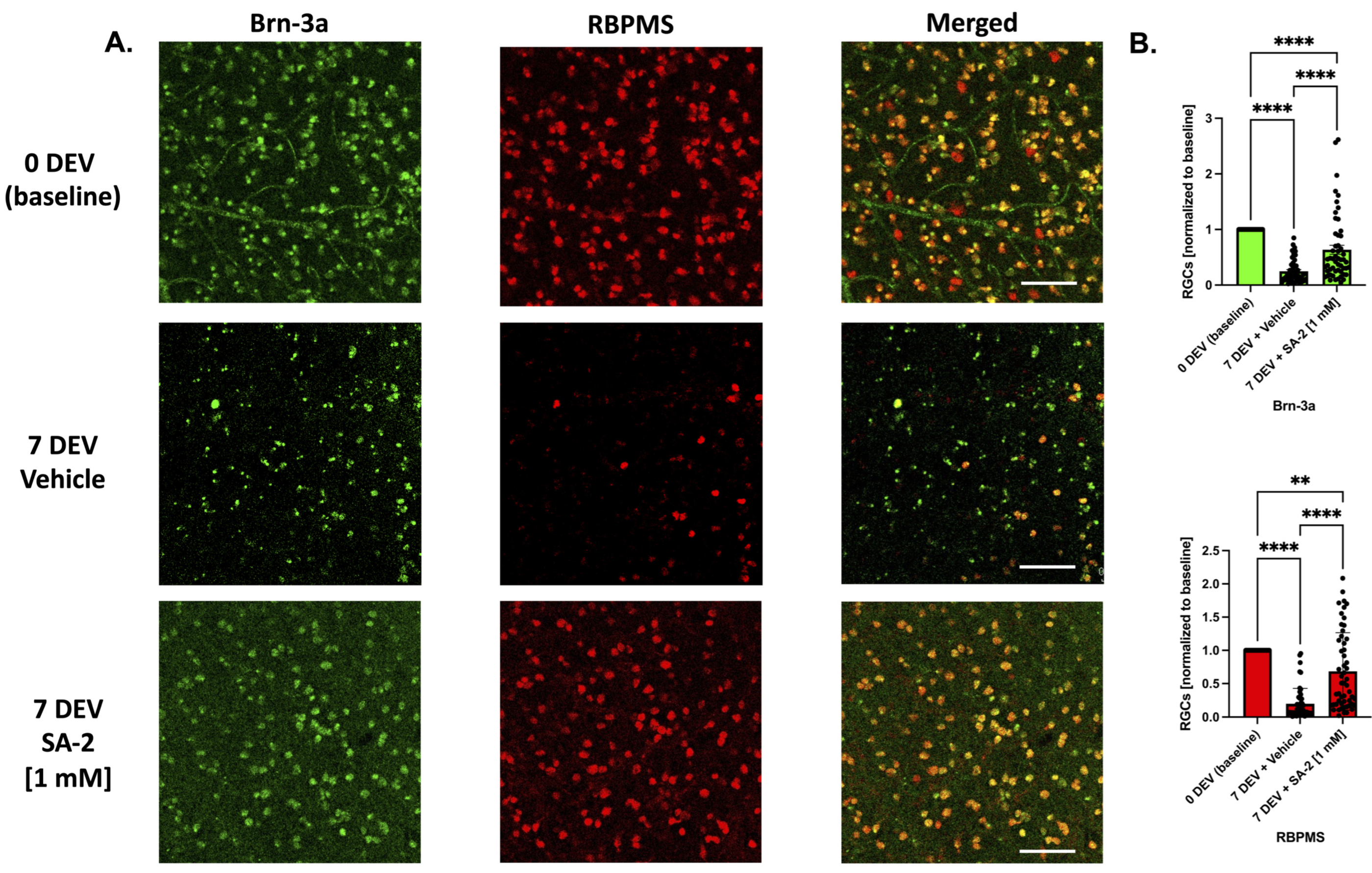
3.4. SA-2 Protects RGCs in Ex Vivo Human Retinal Explant Trophic Factor Deprivation Model
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 4-hydroxy | TEMPOL/TEMPOL 4-hydroxy-2,2,6,6-tetramethyl piperidinyl-1-oxyl |
| BDNF | Brain-derived Neurotrophic Factor |
| CNTF | Ciliary Neurotrophic Factor |
| ET-3 | Endothelin-3 |
| IOP | Intraocular Pressure |
| I/R | Ischemia/Reperfusion |
| NO | Nitric Oxide |
| POAG | Primary Open Angle Glaucoma |
| ROS | Reactive Oxygen Species |
| RGC | Retinal Ganglion Cell |
| SOD | Superoxide Dismutase |
| TBHP | tert-Butyl Hydroperoxide |
| TF | Trophic Factor |
References
- Gupta, V.; Chitranshi, N.; Gupta, V.; You, Y.; Rajput, R.; Paulo, J.A.; Mirzaei, M.; van den Buuse, M.; Graham, S.L. TrkB Receptor Agonist 7,8 Dihydroxyflavone is Protective Against the Inner Retinal Deficits Induced by Experimental Glaucoma. Neuroscience 2022, 490, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Peplow, P.; Martinez, B. MicroRNAs as biomarkers in glaucoma and potential therapeutic targets. Neural Regen. Res. 2022, 17, 2368. [Google Scholar] [CrossRef] [PubMed]
- Allison, K.; Patel, D.; Alabi, O. Epidemiology of Glaucoma: The Past, Present, and Predictions for the Future. Cureus 2020, 12, e11686. [Google Scholar] [CrossRef] [PubMed]
- Topouzis, F.; Coleman, A.L.; Harris, A.; Koskosas, A.; Founti, P.; Gong, G.; Yu, F.; Anastasopoulos, E.; Pappas, T.; Wilson, M.R. Factors Associated with Undiagnosed Open-Angle Glaucoma: The Thessaloniki Eye Study. Am. J. Ophthalmol. 2008, 145, 327–335.e1. [Google Scholar] [CrossRef] [PubMed]
- Tham, Y.C.; Li, X.; Wong, T.Y.; Quigley, H.A.; Aung, T.; Cheng, C.Y. Global prevalence of glaucoma and projections of glaucoma burden through 2040: A systematic review and meta-analysis. Ophthalmology 2014, 121, 2081–2090. [Google Scholar] [CrossRef]
- Sluch, V.M.; Zack, D.J. Stem cells, retinal ganglion cells and glaucoma. Dev. Ophthalmol. 2014, 53, 111–121. [Google Scholar]
- Kang, E.Y.-C.; Liu, P.-K.; Wen, Y.-T.; Quinn, P.M.J.; Levi, S.R.; Wang, N.-K.; Tsai, R.-K. Role of Oxidative Stress in Ocular Diseases Associated with Retinal Ganglion Cells Degeneration. Antioxidants 2021, 10, 1948. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Dong, J.; Cull, G.; Fortune, B.; Cioffi, G.A. Varicosities of intraretinal ganglion cell axons in human and nonhuman primates. Invest. Opthalmol. Vis. Sci. 2003, 44, 2–9. [Google Scholar] [CrossRef]
- Yu, D.-Y.; Cringle, S.J.; Balaratnasingam, C.; Morgan, W.H.; Yu, P.K.; Su, E.-N. Retinal ganglion cells: Energetics, compartmentation, axonal transport, cytoskeletons and vulnerability. Prog. Retin. Eye Res. 2013, 36, 217–246. [Google Scholar] [CrossRef] [PubMed]
- Bouayed, J.; Bohn, T. Exogenous Antioxidants—Double-Edged Swords in Cellular Redox State: Health Beneficial Effects at Physiologic Doses versus Deleterious Effects at High Doses. Oxid. Med. Cell. Longev. 2010, 3, 228–237. [Google Scholar] [CrossRef]
- Hurley, D.J.; Normile, C.; Irnaten, M.; O’Brien, C. The Intertwined Roles of Oxidative Stress and Endoplasmic Reticulum Stress in Glaucoma. Antioxidants 2022, 11, 886. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Huang, Q.; Wang, Q.; Wang, Q.; Cao, X.; Zhao, L.; Xu, N.; Zhuge, Z.; Mao, J.; Fu, X.; et al. Enhanced Renal Afferent Arteriolar Reactive Oxygen Species and Contractility to Endothelin-1 Are Associated with Canonical Wnt Signaling in Diabetic Mice. Kidney Blood Press. Res. 2018, 43, 860–871. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Kanamori, A.; Catrinescu, M.-M.; Mears, K.A.; Beaubien, R.; Levin, L.A. Superoxide is an associated signal for apoptosis in axonal injury. Brain 2010, 133, 2612–2625. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lieven, C.J.; Hoegger, M.J.; Schlieve, C.R.; Levin, L.A. Retinal Ganglion Cell Axotomy Induces an Increase in Intracellular Superoxide Anion. Invest. Opthalmol. Vis. Sci. 2006, 47, 1477–1485. [Google Scholar] [CrossRef] [PubMed]
- Pease, M.E.; McKinnon, S.J.; Quigley, H.A.; Kerrigan-Baumrind, L.A.; Zack, D.J. Obstructed axonal transport of BDNF and its receptor TrkB in experimental glaucoma. Invest. Ophthalmol. Vis. Sci. 2000, 41, 764–774. [Google Scholar]
- Nita, M.; Grzybowski, A. The Role of the Reactive Oxygen Species and Oxidative Stress in the Pathomechanism of the Age-Related Ocular Diseases and Other Pathologies of the Anterior and Posterior Eye Segments in Adults. Oxid. Med. Cell. Longev. 2016, 2016, 3164734. [Google Scholar] [CrossRef]
- Levin, L.A.; Patrick, C.; Choudry, N.B.; Sharif, N.A.; Goldberg, J.L. Neuroprotection in neurodegenerations of the brain and eye: Lessons from the past and directions for the future. Front. Neurol. 2022, 13, 964197. [Google Scholar] [CrossRef]
- Pietrucha-Dutczak, M.; Amadio, M.; Govoni, S.; Lewin-Kowalik, J.; Smedowski, A. The Role of Endogenous Neuroprotective Mechanisms in the Prevention of Retinal Ganglion Cells Degeneration. Front. Neurosci. 2018, 12, 834. [Google Scholar] [CrossRef]
- Yuki, K.; Ozawa, Y.; Yoshida, T.; Kurihara, T.; Hirasawa, M.; Ozeki, N.; Shiba, D.; Noda, K.; Ishida, S.; Tsubota, K. Retinal Ganglion Cell Loss in Superoxide Dismutase 1 Deficiency. Invest. Opthalmol. Vis. Sci. 2011, 52, 4143–4150. [Google Scholar] [CrossRef]
- Nayak, M.S.; Kita, M.; Marmor, M.F. Protection of rabbit retina from ischemic injury by superoxide dismutase and catalase. Investig. Ophthalmol. Vis. Sci. 1993, 34, 2018–2022. [Google Scholar]
- Lindsey, J.D.; Duong-Polk, K.X.; Dai, Y.; Nguyen, D.H.; Leung, C.K.; Weinreb, R.N. Protection by an Oral Disubstituted Hydroxylamine Derivative against Loss of Retinal Ganglion Cell Differentiation following Optic Nerve Crush. PLoS ONE 2013, 8, e6596666. [Google Scholar] [CrossRef]
- Xiao, C.; He, M.; Nan, Y.; Zhang, D.; Chen, B.; Guan, Y.; Pu, M. Physiological Effects of Superoxide Dismutase on Altered Visual Function of Retinal Ganglion Cells in db/db Mice. PLoS ONE 2012, 7, e30343. [Google Scholar] [CrossRef]
- Acharya, S.; Rogers, P.; Krishnamoorthy, R.R.; Stankowska, D.L.; Dias, H.V.R.; Yorio, T. Design and synthesis of novel hybrid sydnonimine and prodrug useful for glaucomatous optic neuropathy. Bioorg. Med. Chem. Lett. 2016, 26, 1490–1494. [Google Scholar] [CrossRef]
- Stankowska, D.L.; Millar, J.C.; Kodati, B.; Behera, S.; Chaphalkar, R.M.; Nguyen, T.; Nguyen, K.T.; Krishnamoorthy, R.R.; Ellis, D.Z.; Acharya, S. Nanoencapsulated hybrid compound SA-2 with long-lasting intraocular pressure–lowering activity in rodent eyes. Mol. Vis. 2021, 27, 37–49. [Google Scholar]
- Stankowska, D.L.; Dibas, A.; Li, L.; Zhang, W.; Krishnamoorthy, V.R.; Chavala, S.H.; Nguyen, T.P.; Yorio, T.; Ellis, D.Z.; Acharya, S. Hybrid Compound SA-2 is Neuroprotective in Animal Models of Retinal Ganglion Cell Death. Invest. Opthalmol. Vis. Sci. 2019, 60, 3064–3073. [Google Scholar] [CrossRef]
- Wareham, L.K.; Buys, E.S.; Sappington, R.M. The nitric oxide-guanylate cyclase pathway and glaucoma. Nitric Oxide 2018, 77, 75–87. [Google Scholar] [CrossRef]
- Doganay, S.; Evereklioglu, C.; Turkoz, Y.; Er, H. Decreased Nitric Oxide Production in Primary Open-Angle Glaucoma. Eur. J. Ophthalmol. 2002, 12, 44–48. [Google Scholar] [CrossRef]
- Nathanson, J.A.; McKee, M. Alterations of ocular nitric oxide synthase in human glaucoma. Invest. Ophthalmol. Vis. Sci. 1995, 36, 1774–1784. [Google Scholar]
- Galassi, F.; Renieri, G.; Sodi, A.; Ucci, F.; Vannozzi, L.; Masini, E. Nitric oxide proxies and ocular perfusion pressure in primary open angle glaucoma. Br. J. Ophthalmol. 2004, 88, 757–760. [Google Scholar] [CrossRef]
- Wang, M.; Zheng, Y. Oxidative stress and antioxidants in the trabecular meshwork. PeerJ 2019, 7, e8121. [Google Scholar] [CrossRef] [PubMed]
- Barres, B.A.; Silverstein, B.E.; Corey, D.P.; Chun, L.L. Immunological, morphological, and electrophysiological variation among retinal ganglion cells purified by panning. Neuron 1988, 1, 791–803. [Google Scholar] [CrossRef] [PubMed]
- Stankowska, D.L.; Ii, B.H.M.; Oku, H.; Ikeda, T.; Dibas, A. Neuroprotective effects of inhibitors of Acid-Sensing ion channels (ASICs) in optic nerve crush model in rodents. Curr. Eye Res. 2017, 43, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Osborne, A.; Sanderson, J.; Martin, K.R. Neuroprotective Effects of Human Mesenchymal Stem Cells and Platelet-Derived Growth Factor on Human Retinal Ganglion Cells. Stem Cells 2017, 36, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Kodati, B.; Stankowska, D.L.; Krishnamoorthy, V.R.; Krishnamoorthy, R.R. Involvement of c-Jun N-terminal kinase 2 (JNK2) in Endothelin-1 (ET-1) Mediated Neurodegeneration of Retinal Ganglion Cells. Invest. Opthalmol. Vis. Sci. 2021, 62, 13. [Google Scholar] [CrossRef]
- Kodati, B.; Merchant, S.A.; Millar, J.C.; Liu, Y. Early-Onset Glaucoma in egl1 Mice Homozygous for Pitx2 Mutation. Biomedicines 2022, 10, 516. [Google Scholar] [CrossRef]
- Chaphalkar, R.M.; Stankowska, D.L.; He, S.; Kodati, B.; Phillips, N.; Prah, J.; Yang, S.; Krishnamoorthy, R.R. Endothelin-1 Mediated Decrease in Mitochondrial Gene Expression and Bioenergetics Contribute to Neurodegeneration of Retinal Ganglion Cells. Sci. Rep. 2020, 10, 3571. [Google Scholar] [CrossRef]
- Yorio, T.; Krishnamoorthy, R.; Prasanna, G. Endothelin: Is it a contributor to glaucoma pathophysiology? J. Glaucoma 2002, 11, 259–270. [Google Scholar] [CrossRef]
- Chauhan, B.C. Endothelin and its potential role in glaucoma. Can. J. Ophthalmol. 2008, 43, 356–360. [Google Scholar] [CrossRef]
- Rosenthal, R.; Fromm, M. Endothelin antagonism as an active principle for glaucoma therapy. J. Cereb. Blood Flow Metab. 2011, 162, 806–816. [Google Scholar] [CrossRef]
- Lo, T.-C.; Chen, Y.-Y.; Hung, M.-C.; Chou, P. Latanoprostene Bunod 0.024% in the Treatment of Open-Angle Glaucoma and Ocular Hypertension: A Meta-Analysis. J. Clin. Med. 2022, 11, 4325. [Google Scholar] [CrossRef]
- Cavet, M.E.; Vittitow, J.L.; Impagnatiello, F.; Ongini, E.; Bastia, E. Nitric Oxide (NO): An Emerging Target for the Treatment of Glaucoma. Invest. Opthalmol. Vis. Sci. 2014, 55, 5005–5015. [Google Scholar] [CrossRef]
- Sharif, N.A. Glaucomatous optic neuropathy treatment options: The promise of novel therapeutics, techniques and tools to help preserve vision. Neural Regen. Res. 2018, 13, 1145–1150. [Google Scholar] [CrossRef]
- Minton, A.Z.; Phatak, N.; Stankowska, D.L.; He, S.; Ma, H.-Y.; Mueller, B.H.; Jiang, M.; Luedtke, R.; Yang, S.; Brownlee, C.; et al. Endothelin B Receptors Contribute to Retinal Ganglion Cell Loss in a Rat Model of Glaucoma. PLoS ONE 2012, 7, e43199. [Google Scholar] [CrossRef]
- Zhang, X.-M.; Liu, D.T.L.; Chiang, S.W.-Y.; Choy, K.-W.; Pang, C.-P.; Lam, D.S.-C.; Yam, G.H.-F. Immunopanning purification and long-term culture of human retinal ganglion cells. Mol. Vis. 2010, 16, 2867–2872. [Google Scholar]
- Ohlemacher, S.K.; Sridhar, A.; Xiao, Y.; Hochstetler, A.E.; Sarfarazi, M.; Cummins, T.R.; Meyer, J.S. Stepwise Differentiation of Retinal Ganglion Cells from Human Pluripotent Stem Cells Enables Analysis of Glaucomatous Neurodegeneration. Stem Cells 2016, 34, 1553–1562. [Google Scholar] [CrossRef]
- Tucker, B.A.; Solivan-Timpe, F.; Roos, B.R.; Anfinson, K.R.; Robin, A.L.; Wiley, L.A.; Mullins, R.F.; Fingert, J.H. Duplication of TBK1 Stimulates Autophagy in iPSC-derived Retinal Cells from a Patient with Normal Tension Glaucoma. J. Stem Cell Res. Ther. 2014, 3, 161. [Google Scholar] [CrossRef]
- Osborne, A.; Hopes, M.; Wright, P.; Broadway, D.C.; Sanderson, J. Human organotypic retinal cultures (HORCs) as a chronic experimental model for investigation of retinal ganglion cell degeneration. Exp. Eye Res. 2016, 143, 28–38. [Google Scholar] [CrossRef]
- Chrysostomou, V.; Rezania, F.; Trounce, I.A.; Crowston, J.G. Oxidative stress and mitochondrial dysfunction in glaucoma. Curr. Opin. Pharmacol. 2013, 13, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Tanaka-Gonome, T.; Xie, Y.; Yamauchi, K.; Maeda-Monai, N.; Tanabu, R.; Kudo, T.; Nakazawa, M. The protective effect of astaxanthin on the ganglion cell complex in glutamate/aspartate transporter deficient mice, a model of normal tension glaucoma, analyzed by spectral domain-optical coherence tomography. Biochem. Biophys. Rep. 2020, 23, 100777. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Pang, Y.; Zhang, Z.; Li, X.; Wang, C.; Lei, Y.; Li, A.; Yu, L.; Ye, J. Mitochondria-targeted antioxidant peptide SS-31 mediates neuroprotection in a rat experimental glaucoma model. Acta Biochim. Biophys. Sin. 2019, 51, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Cao, K.; Ishida, T.; Fang, Y.; Shinohara, K.; Li, X.; Nagaoka, N.; Ohno-Matsui, K.; Yoshida, T. Protection of the Retinal Ganglion Cells: Intravitreal Injection of Resveratrol in Mouse Model of Ocular Hypertension. Invest. Opthalmol. Vis. Sci. 2020, 61, 13. [Google Scholar] [CrossRef]
- Yang, X.; Hondur, G.; Tezel, G. Antioxidant Treatment Limits Neuroinflammation in Experimental Glaucoma. Invest. Opthalmol. Vis. Sci. 2016, 57, 2344–2354. [Google Scholar] [CrossRef]
- Tezel, G.; Yang, X. Caspase-independent component of retinal ganglion cell death, in vitro. Invest. Ophthalmol. Vis. Sci. 2004, 45, 4049–4059. [Google Scholar] [CrossRef]
- Garcia-Medina, J.J.; Rubio-Velazquez, E.; Lopez-Bernal, M.D.; Cobo-Martinez, A.; Zanon-Moreno, V.; Pinazo-Duran, M.D.; Del-Rio-Vellosillo, M. Glaucoma and Antioxidants: Review and Update. Antioxidants 2020, 9, 1031. [Google Scholar] [CrossRef]
- Xu, M.; Ng, Y.-K.; Leong, S.K. Neuroprotective and Neurodestructive Functions of Nitric Oxide after Spinal Cord Hemisection. Exp. Neurol. 2000, 161, 472–480. [Google Scholar] [CrossRef]
- Kashiwagi, K.; Iizuka, Y.; Tanaka, Y.; Mochizuki, S.; Kajiya, F.; Araie, M.; Suzuki, Y.; Iijima, H.; Tsukahara, S. Dual action of nitric oxide on purely isolated retinal ganglion cells. Curr. Eye Res. 2001, 23, 233–239. [Google Scholar] [CrossRef]
- Godínez-Rubí, M.; Rojas-Mayorquín, A.E.; Ortuño-Sahagún, D. Nitric Oxide Donors as Neuroprotective Agents after an Ischemic Stroke-Related Inflammatory Reaction. Oxidative Med. Cell. Longev. 2013, 2013, 297357. [Google Scholar] [CrossRef]
- Figueroa, S.; Cañadas, S.; Arce, C.; Oset-Gasque, M.; González, M. SNAP, a NO donor, induces cortical neuron death by a mechanism in which the caspase pathway is implicated. Brain Res. 2005, 1047, 168–176. [Google Scholar] [CrossRef]
- Li, J.; Billiar, T.R.; Talanian, R.V.; Kim, Y.M. Nitric Oxide Reversibly Inhibits Seven Members of the Caspase Family via S-Nitrosylation. Biochem. Biophys. Res. Commun. 1997, 240, 419–424. [Google Scholar] [CrossRef]
- Koriyama, Y.; Kamiya, M.; Arai, K.; Sugitani, K.; Ogai, K.; Kato, S. Nipradilol Promotes Axon Regeneration Through S-Nitrosylation of PTEN in Retinal Ganglion Cells. Adv. Exp. Med. Biol. 2014, 801, 751–757. [Google Scholar]
- Husain, S.; Abdul, Y.; Singh, S.; Ahmad, A.; Husain, M. Regulation of nitric oxide production by delta-opioid receptors during glaucomatous injury. PLoS ONE 2014, 9, e110397. [Google Scholar] [CrossRef] [PubMed]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef]
- Sarkar, M.; Wang, Y.; Ekpenyong, O.; Liang, D.; Xie, H. Pharmacokinetic behaviors of soft nanoparticulate formulations of chemotherapeutics. WIREs Nanomed. Nanobiotechnol. 2022, e1846. [Google Scholar] [CrossRef]
- Pritchard, N.; Kaitu’U-Lino, T.; Harris, L.; Tong, S.; Hannan, N. Nanoparticles in pregnancy: The next frontier in reproductive therapeutics. Hum. Reprod. Update 2020, 27, 280–304. [Google Scholar] [CrossRef]
- Sharif, N.A. Therapeutic Drugs and Devices for Tackling Ocular Hypertension and Glaucoma, and Need for Neuroprotection and Cytoprotective Therapies. Front. Pharmacol. 2021, 12, 729249. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pham, J.H.; Johnson, G.A.; Rangan, R.S.; Amankwa, C.E.; Acharya, S.; Stankowska, D.L. Neuroprotection of Rodent and Human Retinal Ganglion Cells In Vitro/Ex Vivo by the Hybrid Small Molecule SA-2. Cells 2022, 11, 3741. https://doi.org/10.3390/cells11233741
Pham JH, Johnson GA, Rangan RS, Amankwa CE, Acharya S, Stankowska DL. Neuroprotection of Rodent and Human Retinal Ganglion Cells In Vitro/Ex Vivo by the Hybrid Small Molecule SA-2. Cells. 2022; 11(23):3741. https://doi.org/10.3390/cells11233741
Chicago/Turabian StylePham, Jennifer H., Gretchen A. Johnson, Rajiv S. Rangan, Charles E. Amankwa, Suchismita Acharya, and Dorota L. Stankowska. 2022. "Neuroprotection of Rodent and Human Retinal Ganglion Cells In Vitro/Ex Vivo by the Hybrid Small Molecule SA-2" Cells 11, no. 23: 3741. https://doi.org/10.3390/cells11233741
APA StylePham, J. H., Johnson, G. A., Rangan, R. S., Amankwa, C. E., Acharya, S., & Stankowska, D. L. (2022). Neuroprotection of Rodent and Human Retinal Ganglion Cells In Vitro/Ex Vivo by the Hybrid Small Molecule SA-2. Cells, 11(23), 3741. https://doi.org/10.3390/cells11233741