
Overlapping Machinery in Lysosome-Related Organelle Trafficking: A Lesson from Rare Multisystem Disorders


Abstract
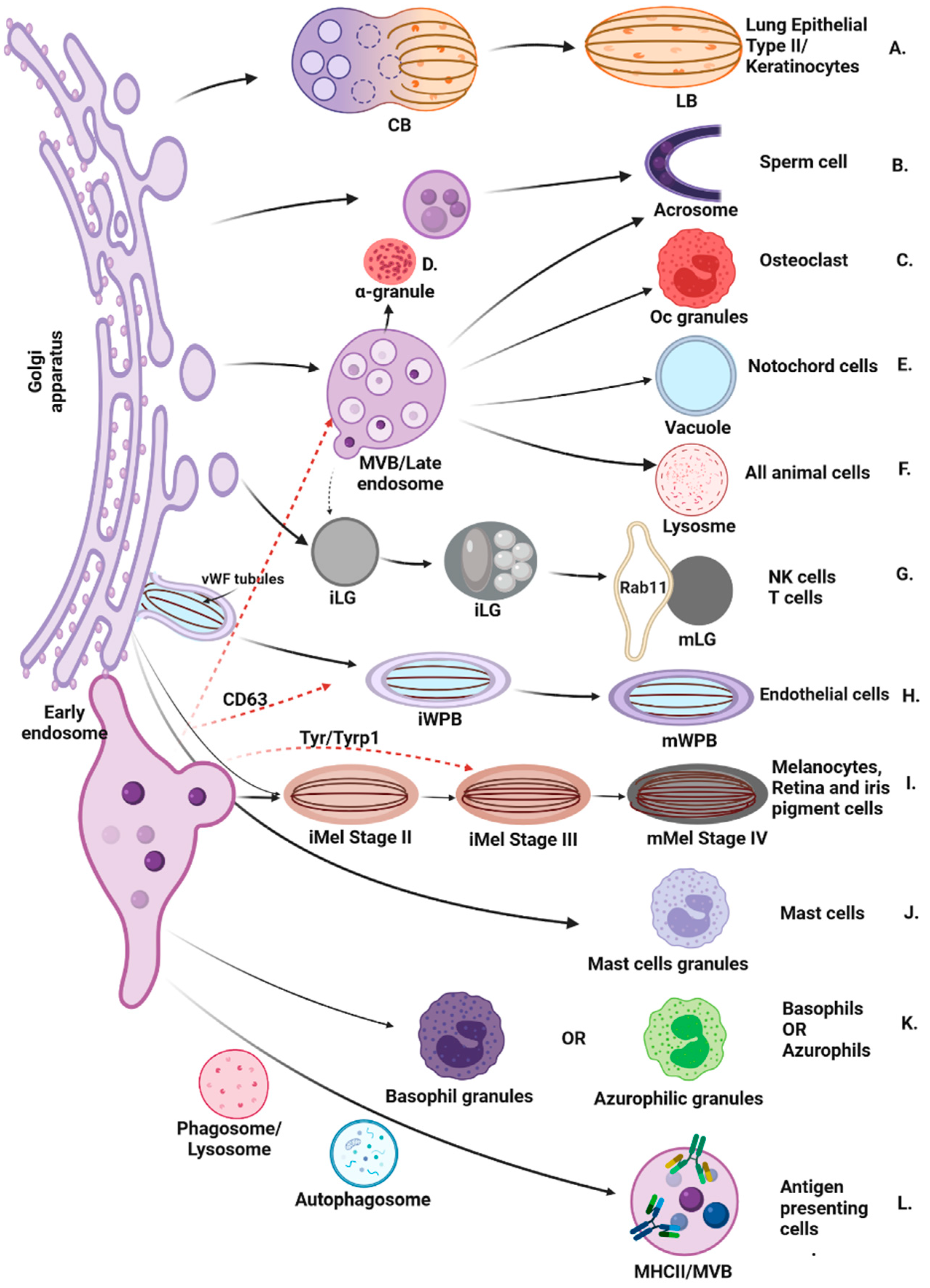
1. Introduction
2. Dysfunction of LROs Trafficking Proteins Is Associated with Rare Multisystem Disorders
3. Griscelli Syndrome Reveals Common Regulators Required for LRO Motility
4. GS1 and MYH9RD Are Caused by Defective Myosin-Mediated LRO Trafficking
5. Rab27a and Its Effectors in LRO Trafficking
6. CHS Is Caused by Impaired LYST-Mediated LRO Fission
7. Defects in LRO Biogenesis: The Hermansky–Pudlak Syndrome
8. AP3 Sorts Different Cargoes in Different LROs and Its Deficient Function Associates with HPS2 and HPS10
9. BLOC1, -2 and -3 Sort Different Cargoes in Different LROs and Its Deficient Function Associates with HPS1 and HPS3-9
10. The CHEVI Complex in Arthrogryposis, Renal Dysfunction and Cholestasis Syndrome
11. Transcriptional Regulation of Proteins within the Same Complex Could Constitute a Cell-Specific Mechanism for Protein Function Versatility
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ARC | Arthrogryposis Renal dysfunction and Cholestasis |
| BLOC | Biogenesis of Lysosome-related Organelles Complex |
| BSEP | Bile Salt Export Pump |
| CEA | CarcinoEmbryonic Antigen |
| CHS | Chediak–Higashi Syndrome |
| CIVC | Collagen IV Carriers |
| CORVET | class c core vacuole/endosome tethering complex |
| FHL3 | Four and a half LIM domains protein 3 |
| FHL3 | Familial Hemophagocytic Lymphohistiocytosis |
| GABA | Gamma-AminoButyric Acid |
| GS | Griscelli Syndrome |
| HOPS | HOmotypic fusion and vacuole Protein Sorting complex |
| HPS | Hermansky–Pudlak Syndrome |
| HS | Hemophagocytic Syndrome |
| KLK5 | Kallikrein-Related Peptidase 5 |
| LB | Lamellar Bodies |
| LH3 | Lysyl Hydroxylase 3 |
| LROs | Lysosome-related organelles |
| MTOC | MicroTubule Organising Centre |
| MYH9 | Myosin9 |
| MYH9RD | MYH9-related thrombocytopenia |
| MYH9RD | MYH-9–Related Disease |
| NK | Natural Killer |
| PRDX6 | Peroxiredoxin 6 |
| TYR | Tyrosinase |
| vWF | von Willebrand Factor |
| WPB | Weibel-Palade Bodies |
References
- Sachse, M.; Ramm, G.; Strous, G.; Klumperman, J. Endosomes: Multipurpose designs for integrating housekeeping and specialized tasks. Histochem. Cell Biol. 2002, 117, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Götz, T.W.; Puchkov, D.; Lysiuk, V.; Lützkendorf, J.; Nikonenko, A.G.; Quentin, C.; Lehmann, M.; Sigrist, S.J.; Petzoldt, A.G. Rab2 regulates presynaptic precursor vesicle biogenesis at the trans-Golgi. J. Cell Biol. 2021, 220. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Marks, M.S. The Dark Side of Lysosome-Related Organelles: Specialization of the Endocytic Pathway for Melanosome Biogenesis. Traffic 2002, 3, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Marks, M.S.; Cutler, D.F. Lysosome-related organelles: Driving post-Golgi compartments into specialisation. Curr. Opin. Cell Biol. 2007, 19, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Marks, M.S.; Heijnen, H.F.; Raposo, G. Lysosome-related organelles: Unusual compartments become mainstream. Curr. Opin. Cell Biol. 2013, 25, 495–505. [Google Scholar] [CrossRef]
- Bonifacino, J.S. Insights into the Biogenesis of Lysosome-Related Organelles from the Study of the Hermansky-Pudlak Syndrome. Ann. N. Y. Acad. Sci. 2004, 1038, 103–114. [Google Scholar] [CrossRef]
- Dell’Angelica, E.C.; Mullins, C.; Caplan, S.; Bonifacino, J.S. Lysosome-related organelles. Faseb j 2000, 14, 1265–1278. [Google Scholar]
- Bowman, S.L.; Bi-Karchin, J.; Le, L.; Marks, M.S. The road to lysosome-related organelles: Insights from Hermansky-Pudlak syndrome and other rare diseases. Traffic 2019, 20, 404–435. [Google Scholar] [CrossRef]
- Wei, A.-H.; Li, W. Hermansky-Pudlak syndrome: Pigmentary and non-pigmentary defects and their pathogenesis. Pigment. Cell Melanoma Res. 2013, 26, 176–192. [Google Scholar] [CrossRef]
- Cullinane, A.R.; Straatman-Iwanowska, A.; Zaucker, A.; Wakabayashi, Y.; Bruce, C.K.; Luo, G.; Rahman, F.; Gürakan, F.; Utine, E.; Özkan, T.B.; et al. Mutations in VIPAR cause an arthrogryposis, renal dysfunction and cholestasis syndrome phenotype with defects in epithelial polarization. Nat. Genet. 2010, 42, 303–312. [Google Scholar] [CrossRef]
- Shiflett, S.L.; Kaplan, J.; Ward, D.M. Chediak-Higashi Syndrome: A Rare Disorder of Lysosomes and Lysosome Related Organelles. Pigment Cell Res. 2002, 15, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Griscelli, C.; Prunieras, M. Pigment dilution and immunodeficiency: A new syndrome. Int. J. Dermatol. 1978, 17, 788–791. [Google Scholar] [CrossRef] [PubMed]
- Bohn, G.; Allroth, A.; Brandes, G.; Thiel, J.; Glocker, E.; A Schäffer, A.; Rathinam, C.; Taub, N.; Teis, D.; Zeidler, C.; et al. A novel human primary immunodeficiency syndrome caused by deficiency of the endosomal adaptor protein p14. Nat. Med. 2007, 13, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Luzio, J.P.; Hackmann, Y.; Dieckmann, N.M.G.; Griffiths, G.M. The Biogenesis of Lysosomes and Lysosome-Related Organelles. Cold Spring Harb. Perspect. Biol. 2014, 6, a016840. [Google Scholar] [CrossRef]
- Jani, R.A.; Mahanty, S.; Setty, S.R.G. SNAREs in the maturation and function of LROs. BioArchitecture 2016, 6, 1–11. [Google Scholar] [CrossRef]
- Blott, E.J.; Griffiths, G.M. Secretory lysosomes. Nat. Rev. Mol. Cell Biol. 2002, 3, 122–131. [Google Scholar] [CrossRef]
- Sitaram, A.; Marks, M.S. Mechanisms of Protein Delivery to Melanosomes in Pigment Cells. Physiology 2012, 27, 85–99. [Google Scholar] [CrossRef]
- Raposo, G.; Marks, M.S. Melanosomes—Dark organelles enlighten endosomal membrane transport. Nat. Rev. Mol. Cell Biol. 2007, 8, 786–797. [Google Scholar] [CrossRef]
- Tamura, K.; Ohbayashi, N.; Maruta, Y.; Kanno, E.; Itoh, T.; Fukuda, M. Varp Is a Novel Rab32/38-binding Protein That Regulates Tyrp1 Trafficking in Melanocytes. Mol. Biol. Cell 2009, 20, 2900–2908. [Google Scholar] [CrossRef]
- Bultema, J.J.; Ambrosio, A.L.; Burek, C.L.; Di Pietro, S.M. BLOC-2, AP-3, and AP-1 Proteins Function in Concert with Rab38 and Rab32 Proteins to Mediate Protein Trafficking to Lysosome-related Organelles. J. Biol. Chem. 2012, 287, 19550–19563. [Google Scholar] [CrossRef]
- Kloer, D.P.; Rojas, R.; Ivan, V.; Moriyama, K.; van Vlijmen, T.; Murthy, N.; Ghirlando, R.; van der Sluijs, P.; Hurley, J.H.; Bonifacino, J.S. Assembly of the Biogenesis of Lysosome-related Organelles Complex-3 (BLOC-3) and Its Interaction with Rab9. J. Biol. Chem. 2010, 285, 7794–7804. [Google Scholar] [CrossRef] [PubMed]
- Jani, R.A.; Purushothaman, L.K.; Rani, S.; Bergam, P.; Setty, S.R.G. STX13 regulates cargo delivery from recycling endosomes during melanosome biogenesis. J. Cell Sci. 2015, 128, 3263–3276. [Google Scholar] [CrossRef] [PubMed]
- de Saint Basile, G.; Menasche, G.; Fischer, A. Molecular mechanisms of biogenesis and exocytosis of cytotoxic granules. Nat. Rev. Immunol. 2010, 10, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Dustin, M.; Chakraborty, A.K.; Shaw, A.S. Understanding the Structure and Function of the Immunological Synapse. Cold Spring Harb. Perspect. Biol. 2010, 2, a002311. [Google Scholar] [CrossRef]
- Stinchcombe, J.C.; Majorovits, E.; Bossi, G.; Fuller, S.; Griffiths, G.M. Centrosome polarization delivers secretory granules to the immunological synapse. Nature 2006, 443, 462–465. [Google Scholar] [CrossRef]
- Van Der Sluijs, P.; Zibouche, M.; Van Kerkhof, P. Late Steps in Secretory Lysosome Exocytosis in Cytotoxic Lymphocytes. Front. Immunol. 2013, 4, 359. [Google Scholar] [CrossRef]
- Offenhäuser, C.; Lei, N.; Roy, S.; Collins, B.M.; Stow, J.L.; Murray, R.Z. Syntaxin 11 Binds Vti1b and Regulates Late Endosome to Lysosome Fusion in Macrophages. Traffic 2011, 12, 762–773. [Google Scholar] [CrossRef]
- Griscelli, C.; Durandy, A.; Guy-Grand, D.; Daguillard, F.; Herzog, C.; Prunieras, M. A syndrome associating partial albinism and immunodeficiency. Am. J. Med. 1978, 65, 691–702. [Google Scholar] [CrossRef]
- Langford, G.M.; Molyneaux, B.J. Myosin V in the brain: Mutations lead to neurological defects. Brain Res. Rev. 1998, 28, 1–8. [Google Scholar] [CrossRef]
- Pastural, E.; Barrat, F.J.; Dufourcq-Lagelouse, R.; Certain, S.; Sanal, O.; Jabado, N.; Seger, R.; Griscelli, C.; Fischer, A.; Basile, G.D.S. Griscelli disease maps to chromosome 15q21 and is associated with mutations in the Myosin-Va gene. Nat. Genet. 1997, 16, 289–292. [Google Scholar] [CrossRef]
- Menasche, G.; Pastural, E.; Feldmann, J.; Certain, S.; Ersoy, F.; Dupuis, S.; Wulffraat, N.; Bianchi, D.; Fischer, A.; Le Deist, F.; et al. Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat. Genet. 2000, 25, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Bahadoran, P.; Aberdam, E.; Mantoux, F.; Buscà, R.; Bille, K.; Yalman, N.; de Saint-Basile, G.; Casaroli-Marano, R.; Ortonne, J.P.; Ballotti, R. Rab27a: A key to melanosome transport in human melanocytes. J. Cell Biol. 2001, 152, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Bahadoran, P.; Busca, R.; Chiaverini, C.; Westbroek, W.; Lambert, J.; Bille, K.; Valony, G.; Fukuda, M.; Naeyaert, J.-M.; Ortonne, J.-P.; et al. Characterization of the Molecular Defects in Rab27a, Caused by RAB27A Missense Mutations Found in Patients with Griscelli Syndrome. J. Biol. Chem. 2003, 278, 11386–11392. [Google Scholar] [CrossRef] [PubMed]
- Shah, B.J.; Jagati, A.K.; Katrodiya, N.K.; Patel, S.M. Griscelli syndrome type-3. Indian Dermatol. Online J. 2016, 7, 506–508. [Google Scholar] [CrossRef] [PubMed]
- Seabra, M.C.; Coudrier, E. Rab GTPases and Myosin Motors in Organelle Motility. Traffic 2004, 5, 393–399. [Google Scholar] [CrossRef]
- Van Den Bossche, K.; Naeyaert, J.-M.; Lambert, J. The Quest for the Mechanism of Melanin Transfer. Traffic 2006, 7, 769–778. [Google Scholar] [CrossRef]
- Blanche, S.; Caniglia, M.; Girault, D.; Landman, J.; Griscelli, C.; Fischer, A. Treatment of hemophagocytic lymphohistiocytosis with chemotherapy and bone marrow transplantation: A single-center study of 22 cases. Blood 1991, 78, 51–54. [Google Scholar] [CrossRef]
- Menasche, G.; Ho, C.H.; Sanal, O.; Feldmann, J.; Tezcan, I.; Ersoy, F.; Houdusse, A.; Fischer, A.; de Saint Basile, G. Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1). J. Clin. Investig. 2003, 112, 450–456. [Google Scholar] [CrossRef]
- Jenkins, N.A.; Copeland, N.G.; Taylor, B.A.; Lee, B.K. Dilute (d) coat colour mutation of DBA/2J mice is associated with the site of integration of an ecotropic MuLV genome. Nature 1981, 293, 370–374. [Google Scholar] [CrossRef]
- Wilson, S.M.; Yip, R.; Swing, D.A.; O’Sullivan, T.N.; Zhang, Y.; Novak, E.K.; Swank, R.T.; Russell, L.B.; Copeland, N.G.; Jenkins, N.A. A mutation in Rab27a causes the vesicle transport defects observed in ashen mice. Proc. Natl. Acad. Sci. USA 2000, 97, 7933–7938. [Google Scholar] [CrossRef]
- Hume, A.N.; Collinson, L.M.; Hopkins, C.R.; Strom, M.; Barral, D.C.; Bossi, G.; Griffiths, G.M.; Seabra, M.C. The leaden gene product is required with Rab27a to recruit myosin Va to melanosomes in melanocytes. Traffic 2002, 3, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Conti, M.A.; Malide, D.; Dong, F.; Wang, A.; Shmist, Y.A.; Liu, C.; Zerfas, P.; Daniels, M.P.; Chan, C.-C.; et al. Mouse models of MYH9-related disease: Mutations in nonmuscle myosin II-A. Blood 2012, 119, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Ren, Q.; Wimmer, C.; Chicka, M.C.; Ye, S.; Ren, Y.; Hughson, F.M.; Whiteheart, S.W. Munc13-4 is a limiting factor in the pathway required for platelet granule release and hemostasis. Blood 2010, 116, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Prieur, D.J.; Collier, L.L. Animal model of human disease: Chédiak-Higashi syndrome. Am. J. Pathol. 1978, 90, 533–536. [Google Scholar] [PubMed]
- Gardner, J.M.; Wildenberg, S.C.; Keiper, N.M.; Novak, E.K.; Rusiniak, M.E.; Swank, R.T.; Puri, N.; Finger, J.N.; Hagiwara, N.; Lehman, A.L.; et al. The mouse pale ear ( ep ) mutation is the homologue of human Hermansky–Pudlak syndrome. Proc. Natl. Acad. Sci. USA 1997, 94, 9238–9243. [Google Scholar] [CrossRef] [PubMed]
- Lane, P.W. Linkage Groups III and XVII in the Mouse and the Position of the Light-Ear Locus. J. Hered. 1967, 58, 21–24. [Google Scholar] [CrossRef]
- Sarvella, P.A. Pearl, a new spontaneous coat and eye color mutation in the house mouse. J. Hered. 1954, 45, 19–20. [Google Scholar] [CrossRef]
- Novak, E.K.; Sweet, H.O.; Prochazka, M.; Parentis, M.; Soble, R.; Reddington, M.; Cairo, A.; Swank, R.T. Cocoa: A new mouse model for platelet storage pool deficiency. Br. J. Haematol. 1988, 69, 371–378. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhao, B.; Li, W.; Oiso, N.; Novak, E.K.; Rusiniak, M.E.; Gautam, R.; Chintala, S.; O’Brien, E.P.; Zhang, Y.; et al. Ru2 and Ru encode mouse orthologs of the genes mutated in human Hermansky-Pudlak syndrome types 5 and 6. Nat. Genet. 2003, 33, 145–153. [Google Scholar] [CrossRef]
- Swank, R.T.; Sweet, H.O.; Davisson, M.T.; Reddington, M.; Novak, E.K. Sandy: A new mouse model for platelet storage pool deficiency. Genet. Res. 1991, 58, 51–62. [Google Scholar] [CrossRef]
- Gibb, S.; Håkansson, E.M.; Lundin, L.-G.; Shire, J.G.M. Reduced pigmentation (rp), a new coat colour gene with effects on kidney lysosomal glycosidases in the mouse. Genet. Res. 1981, 37, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E. A new mutation in the house mouse (Mus musculus). Science 1931, 74, 569. [Google Scholar] [CrossRef] [PubMed]
- Lane, P.W.; Deol, M.S. Mocha, a New Coat Color and Behavior Mutation on Chromosome 10 of the Mouse. J. Hered. 1974, 65, 362–364. [Google Scholar] [CrossRef] [PubMed]
- Hanley, J.; Dhar, D.K.; Mazzacuva, F.; Fiadeiro, R.; Burden, J.J.; Lyne, A.-M.; Smith, H.; Straatman-Iwanowska, A.; Banushi, B.; Virasami, A.; et al. Vps33b is crucial for structural and functional hepatocyte polarity. J. Hepatol. 2017, 66, 1001–1011. [Google Scholar] [CrossRef]
- Bem, D.; Smith, H.; Banushi, B.; Burden, J.J.; White, I.J.; Hanley, J.; Jeremiah, N.; Rieux-Laucat, F.; Bettels, R.; Ariceta, G.; et al. VPS33B regulates protein sorting into and maturation of α-granule progenitor organelles in mouse megakaryocytes. Blood 2015, 126, 133–143. [Google Scholar] [CrossRef]
- Banushi, B.; Forneris, F.; Straatman-Iwanowska, A.; Strange, A.; Lyne, A.-M.; Rogerson, C.; Burden, J.J.; Heywood, W.E.; Hanley, J.; Doykov, I.; et al. Regulation of post-Golgi LH3 trafficking is essential for collagen homeostasis. Nat. Commun. 2016, 7, 12111. [Google Scholar] [CrossRef]
- Eichler, T.; Kögel, T.; Bukoreshtliev, N.; Gerdes, H.-H. The role of myosin Va in secretory granule trafficking and exocytosis. Biochem. Soc. Trans. 2006, 34, 671–674. [Google Scholar] [CrossRef]
- Huang, J.-D.; Brady, S.T.; Richards, B.W.; Stenoien, D.; Resau, J.H.; Copeland, N.G.; Jenkins, N.A. Direct interaction of microtubule- and actin-based transport motors. Nature 1999, 397, 267–270. [Google Scholar] [CrossRef]
- Espreafico, E.M.; Cheney, R.; Matteoli, M.; Nascimento, A.A.C.; De Camilli, P.V.; Larson, R.E.; Mooseker, M.S. Primary structure and cellular localization of chicken brain myosin-V (p190), an unconventional myosin with calmodulin light chains. J. Cell Biol. 1992, 119, 1541–1557. [Google Scholar] [CrossRef]
- Pulido, I.R.; Nightingale, T.; Darchen, F.; Seabra, M.; Cutler, D.; Gerke, V. Myosin Va Acts in Concert with Rab27a and MyRIP to Regulate Acute Von-Willebrand Factor Release from Endothelial Cells. Traffic 2011, 12, 1371–1382. [Google Scholar] [CrossRef]
- Novak, E.K.; Gautam, R.; Reddington, M.; Collinson, L.M.; Copeland, N.G.; Jenkins, N.A.; McGarry, M.P.; Swank, R.T. The regulation of platelet-dense granules by Rab27a in the ashen mouse, a model of Hermansky-Pudlak and Griscelli syndromes, is granule-specific and dependent on genetic background. Blood 2002, 100, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Kögel, T.; Bittins, C.M.; Rudolf, R.; Gerdes, H.-H. Versatile roles for myosin Va in dense core vesicle biogenesis and function. Biochem. Soc. Trans. 2010, 38, 199–204. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sanborn, K.B.; Rak, G.D.; Maru, S.Y.; Demers, K.; Difeo, A.; Martignetti, J.A.; Betts, M.R.; Favier, R.; Banerjee, P.P.; Orange, J.S. Myosin IIA Associates with NK Cell Lytic Granules to Enable Their Interaction with F-Actin and Function at the Immunological Synapse. J. Immunol. 2009, 182, 6969–6984. [Google Scholar] [CrossRef]
- Vijayan, K.V. Myosin IIa signal von Willebrand factor release. Blood 2018, 131, 592–593. [Google Scholar] [CrossRef] [PubMed]
- Balduini, C.L.; Pecci, A.; Savoia, A. Recent advances in the understanding and management of MYH9-related inherited thrombocytopenias. Br. J. Haematol. 2011, 154, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, R.; Higashi, T.; Kondo, H.; Yoshioka, A.; Kita, T.; Horiuchi, H. Purification and Functional Analysis of a Rab27 Effector Munc13-4 Using a Semiintact Platelet Dense-Granule Secretion Assay. Methods Enzymol. 2005, 403, 778–788. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, J.; Callebaut, I.; Raposo, G.; Certain, S.; Bacq, D.; Dumont, C.; Lambert, N.; Ouachée-Chardin, M.; Chedeville, G.; Tamary, H.; et al. Munc13-4 Is Essential for Cytolytic Granules Fusion and Is Mutated in a Form of Familial Hemophagocytic Lymphohistiocytosis (FHL3). Cell 2003, 115, 461–473. [Google Scholar] [CrossRef]
- Henter, J.-I.; Horne, A.; Aricó, M.; Egeler, R.M.; Filipovich, A.H.; Imashuku, S.; Ladisch, S.; McClain, K.; Webb, D.; Winiarski, J.; et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr. Blood Cancer 2007, 48, 124–131. [Google Scholar] [CrossRef]
- Izumi, T.; Gomi, H.; Kasai, K.; Mizutani, S.; Torii, S. The Roles of Rab27 and Its Effectors in the Regulated Secretory Pathways. Cell Struct. Funct. 2003, 28, 465–474. [Google Scholar] [CrossRef]
- Wang, H.; Ishizaki, R.; Xu, J.; Kasai, K.; Kobayashi, E.; Gomi, H.; Izumi, T. The Rab27a effector exophilin7 promotes fusion of secretory granules that have not been docked to the plasma membrane. Mol. Biol. Cell 2013, 24, 319–330. [Google Scholar] [CrossRef]
- Stinchcombe, J.C.; Barral, D.; Mules, E.H.; Booth, S.; Hume, A.; Machesky, L.; Seabra, M.; Griffiths, G.M. Rab27a Is Required for Regulated Secretion in Cytotoxic T Lymphocytes. J. Cell Biol. 2001, 152, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Brzezinska, A.A.; Johnson, J.L.; Munafo, D.B.; Crozat, K.; Beutler, B.; Kiosses, W.B.; Ellis, B.A.; Catz, S.D. The Rab27a Effectors JFC1/Slp1 and Munc13-4 Regulate Exocytosis of Neutrophil Granules. Traffic 2008, 9, 2151–2164. [Google Scholar] [CrossRef] [PubMed]
- Yi, Z.; Yokota, H.; Torii, S.; Aoki, T.; Hosaka, M.; Zhao, S.; Takata, K.; Takeuchi, T.; Izumi, T. The Rab27a/Granuphilin Complex Regulates the Exocytosis of Insulin-Containing Dense-Core Granules. Mol. Cell. Biol. 2002, 22, 1858–1867. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, R.; Higashi, T.; Tabuchi, A.; Yoshioka, A.; Nishioka, H.; Fukuda, M.; Kita, T.; Horiuchi, H. Munc13-4 Is a GTP-Rab27-binding Protein Regulating Dense Core Granule Secretion in Platelets. J. Biol. Chem. 2004, 279, 10730–10737. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, T.; Fukuda, M. The Slp4-a Linker Domain Controls Exocytosis through Interaction with Munc18-1·Syntaxin-1a Complex. Mol. Biol. Cell 2006, 17, 2101–2112. [Google Scholar] [CrossRef]
- Kurowska, M.; Goudin, N.; Nehme, N.T.; Court, M.; Garin, J.; Fischer, A.; de Saint Basile, G.; Ménasché, G. Terminal transport of lytic granules to the immune synapse is mediated by the kinesin-1/Slp3/Rab27a complex. Blood 2012, 119, 3879–3889. [Google Scholar] [CrossRef]
- Munafó, D.B.; Johnson, J.L.; Ellis, B.A.; Rutschmann, S.; Beutler, B.; Catz, S.D. Rab27a is a key component of the secretory machinery of azurophilic granules in granulocytes. Biochem. J. 2007, 402, 229–239. [Google Scholar] [CrossRef]
- Kim, J.D.; Willetts, L.; Ochkur, S.; Srivastava, N.; Hamburg, R.; Shayeganpour, A.; Seabra, M.C.; Lee, J.J.; Moqbel, R.; Lacy, P. An essential role for Rab27a GTPase in eosinophil exocytosis. J. Leukoc. Biol. 2013, 94, 1265–1274. [Google Scholar] [CrossRef]
- Singh, R.K.; Mizuno, K.; Wasmeier, C.; Wavre-Shapton, S.T.; Recchi, C.; Catz, S.D.; Futter, C.; Tolmachova, T.; Hume, A.N.; Seabra, M.C. Distinct and opposing roles for Rab27a/Mlph/MyoVa and Rab27b/Munc13-4 in mast cell secretion. FEBS J. 2013, 280, 892–903. [Google Scholar] [CrossRef]
- Munoz, I.; Danelli, L.; Claver, J.; Goudin, N.; Kurowska, M.; Madera-Salcedo, I.K.; Huang, J.-D.; Fischer, A.; González-Espinosa, C.; Basile, G.D.S.; et al. Kinesin-1 controls mast cell degranulation and anaphylaxis through PI3K-dependent recruitment to the granular Slp3/Rab27b complex. J. Cell Biol. 2016, 215, 203–216. [Google Scholar] [CrossRef]
- Itzstein, C.; Coxon, F.P.; Rogers, M.J. The regulation of osteoclast function and bone resorption by small GTPases. Small GTPases 2011, 2, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Shimada-Sugawara, M.; Sakai, E.; Okamoto, K.; Fukuda, M.; Izumi, T.; Yoshida, N.; Tsukuba, T. Rab27A Regulates Transport of Cell Surface Receptors Modulating Multinucleation and Lysosome-Related Organelles in Osteoclasts. Sci. Rep. 2015, 5, 9620. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; Ellis, B.A.; Noack, D.; Seabra, M.C.; Catz, S.D. The Rab27a-binding protein, JFC1, regulates androgen-dependent secretion of prostate-specific antigen and prostatic-specific acid phosphatase. Biochem. J. 2005, 391, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Arimura, N.; Kimura, T.; Nakamuta, S.; Taya, S.; Funahashi, Y.; Hattori, A.; Shimada, A.; Ménager, C.; Kawabata, S.; Fujii, K.; et al. Anterograde Transport of TrkB in Axons Is Mediated by Direct Interaction with Slp1 and Rab27. Dev. Cell 2009, 16, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Saegusa, C.; Kanno, E.; Itohara, S.; Fukuda, M. Expression of Rab27B-binding protein Slp1 in pancreatic acinar cells and its involvement in amylase secretion. Arch. Biochem. Biophys. 2008, 475, 87–92. [Google Scholar] [CrossRef]
- Saegusa, C.; Tanaka, T.; Tani, S.; Itohara, S.; Mikoshiba, K.; Fukuda, M. Decreased basal mucus secretion by Slp2-a-deficient gastric surface mucous cells. Genes Cells 2006, 11, 623–631. [Google Scholar] [CrossRef]
- Yu, M.; Kasai, K.; Nagashima, K.; Torii, S.; Yokota-Hashimoto, H.; Okamoto, K.; Takeuchi, T.; Gomi, H.; Izumi, T. Exophilin4/Slp2-a Targets Glucagon Granules to the Plasma Membrane through Unique Ca2+-inhibitory Phospholipid-binding Activity of the C2A Domain. Mol. Biol. Cell 2007, 18, 688–696. [Google Scholar] [CrossRef]
- Fukuda, M. Rab27 Effectors, Pleiotropic Regulators in Secretory Pathways. Traffic 2013, 14, 949–963. [Google Scholar] [CrossRef]
- Ajitkumar, A.; Yarrarapu, S.N.S.; Ramphul, K. Chediak Higashi Syndrome. In StatPearls; StatPearls Publishing Copyright © 2022, StatPearls Publishing LLC.: Treasure Island, FL, USA, 2022. [Google Scholar]
- Durchfort, N.; Verhoef, S.; Vaughn, M.B.; Shrestha, R.; Adam, D.; Kaplan, J.; Ward, D.M. The Enlarged Lysosomes in beigej Cells Result From Decreased Lysosome Fission and Not Increased Lysosome Fusion. Traffic 2012, 13, 108–119. [Google Scholar] [CrossRef]
- Duarte, P.V.; Hardenberg, R.; Mari, M.; Walter, S.; Reggiori, F.; Fröhlich, F.; Montoro, A.G.; Ungermann, C. The yeast LYST homolog Bph1 is a Rab5 effector and prevents Atg8 lipidation at endosomes. J. Cell Sci. 2022, 135. [Google Scholar] [CrossRef]
- Kypri, E.; Falkenstein, K.; De Lozanne, A. Antagonistic Control of Lysosomal Fusion by Rab14 and the Lyst-Related Protein LvsB. Traffic 2013, 14, 599–609. [Google Scholar] [CrossRef]
- Westphal, A.; Cheng, W.; Yu, J.; Grassl, G.; Krautkrämer, M.; Holst, O.; Föger, N.; Lee, K.-H. Lysosomal trafficking regulator Lyst links membrane trafficking to toll-like receptor–mediated inflammatory responses. J. Exp. Med. 2017, 214, 227–244. [Google Scholar] [CrossRef] [PubMed]
- Hermansky, F.; Pudlak, P. Albinism Associated with Hemorrhagic Diathesis and Unusual Pigmented Reticular Cells in the Bone Marrow: Report of Two Cases with Histochemical Studies. Blood 1959, 14, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.L. Hermansky-Pudlak syndrome: A disease of protein trafficking and organelle function. Pigment Cell Res. 2006, 19, 19–42. [Google Scholar] [CrossRef] [PubMed]
- Young, L.R.; Gulleman, P.M.; Bridges, J.P.; Weaver, T.E.; Deutsch, G.H.; Blackwell, T.S.; McCormack, F.X. The Alveolar Epithelium Determines Susceptibility to Lung Fibrosis in Hermansky-Pudlak Syndrome. Am. J. Respir. Crit. Care Med. 2012, 186, 1014–1024. [Google Scholar] [CrossRef]
- Hengst, M.; Naehrlich, L.; Mahavadi, P.; Grosse-Onnebrink, J.; Terheggen-Lagro, S.; Skanke, L.H.; Schuch, L.A.; Brasch, F.; Guenther, A.; Reu, S.; et al. Hermansky-Pudlak syndrome type 2 manifests with fibrosing lung disease early in childhood. Orphanet J. Rare Dis. 2018, 13, 42. [Google Scholar] [CrossRef]
- Kirshenbaum, A.S.; Cruse, G.; Desai, A.; Bandara, G.; Leerkes, M.; Lee, C.-C.; Fischer, E.R.; O’Brien, K.J.; Gochuico, B.R.; Stone, K.; et al. Immunophenotypic and Ultrastructural Analysis of Mast Cells in Hermansky-Pudlak Syndrome Type-1: A Possible Connection to Pulmonary Fibrosis. PLoS ONE 2016, 11, e0159177. [Google Scholar] [CrossRef]
- White, J.G.; Witkop, C.J.; Gerritsen, S.M. The Hermansky-Pudlak Syndrome: Inclusions in Circulating Leucocytes. Br. J. Haematol. 1973, 24, 761–765. [Google Scholar] [CrossRef]
- Sakuma, T.; Monma, N.; Satodate, R.; Satoh, T.; Takeda, R.; Kuriya, S.-I. Ceroid pigment deposition in circulating blood monocytes and T lymphocytes in Hermansky-Pudlak syndrome: An ultrastructural study. Pathol. Int. 1995, 45, 866–870. [Google Scholar] [CrossRef]
- Clark, R.H.; Stinchcombe, J.C.; Day, A.; Blott, E.; Booth, S.; Bossi, G.; Hamblin, T.; Davies, E.G.; Griffiths, G. Adaptor protein 3–dependent microtubule-mediated movement of lytic granules to the immunological synapse. Nat. Immunol. 2003, 4, 1111–1120. [Google Scholar] [CrossRef]
- Fontana, S.; Parolini, S.; Vermi, W.; Booth, S.; Gallo, F.; Donini, M.; Benassi, M.; Gentili, F.; Ferrari, D.; Notarangelo, L.D.; et al. Innate immunity defects in Hermansky-Pudlak type 2 syndrome. Blood 2006, 107, 4857–4864. [Google Scholar] [CrossRef] [PubMed]
- Simpson, F.; Peden, A.; Christopoulou, L.; Robinson, M.S. Characterization of the Adaptor-related Protein Complex, AP-3. J. Cell Biol. 1997, 137, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Oiso, N.; Gautam, R.; Novak, E.K.; Panthier, J.-J.; Suprabha, P.G.; Vida, T.; Swank, R.T.; Spritz, R.A. The mouse organellar biogenesis mutant buff results from a mutation in Vps33a, a homologue of yeast vps33 and Drosophila carnation. Proc. Natl. Acad. Sci. USA 2003, 100, 1146–1150. [Google Scholar] [CrossRef]
- Wasmeier, C.; Romao, M.; Plowright, L.; Bennett, D.; Raposo, G.; Seabra, M.C. Rab38 and Rab32 control post-Golgi trafficking of melanogenic enzymes. J. Cell Biol. 2006, 175, 271–281. [Google Scholar] [CrossRef]
- Odorizzi, G.; Cowles, C.R.; Emr, S.D. The AP-3 complex: A coat of many colours. Trends Cell Biol. 1998, 8, 282–288. [Google Scholar] [CrossRef]
- Field, M.C.; Dacks, J.B. First and last ancestors: Reconstructing evolution of the endomembrane system with ESCRTs, vesicle coat proteins, and nuclear pore complexes. Curr. Opin. Cell Biol. 2009, 21, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Llinares, E.; Barry, A.O.; André, B. The AP-3 adaptor complex mediates sorting of yeast and mammalian PQ-loop-family basic amino acid transporters to the vacuolar/lysosomal membrane. Sci. Rep. 2015, 5, 16665. [Google Scholar] [CrossRef]
- Cowles, C.R.; Odorizzi, G.; Payne, G.S.; Emr, S.D. The AP-3 Adaptor Complex Is Essential for Cargo-Selective Transport to the Yeast Vacuole. Cell 1997, 91, 109–118. [Google Scholar] [CrossRef]
- Mardones, G.A.; Burgos, P.V.; Lin, Y.; Kloer, D.P.; Magadán, J.G.; Hurley, J.H.; Bonifacino, J.S. Structural Basis for the Recognition of Tyrosine-based Sorting Signals by the μ3A Subunit of the AP-3 Adaptor Complex. J. Biol. Chem. 2013, 288, 9563–9571. [Google Scholar] [CrossRef]
- Robinson, M.S.; Bonifacino, J.S. Adaptor-related proteins. Curr. Opin. Cell Biol. 2001, 13, 444–453. [Google Scholar] [CrossRef]
- Peden, A.; Oorschot, V.; Hesser, B.A.; Austin, C.D.; Scheller, R.H.; Klumperman, J. Localization of the AP-3 adaptor complex defines a novel endosomal exit site for lysosomal membrane proteins. J. Cell Biol. 2004, 164, 1065–1076. [Google Scholar] [CrossRef] [PubMed]
- Ammann, S.; Schulz, A.; Krägeloh-Mann, I.; Dieckmann, N.M.G.; Niethammer, K.; Fuchs, S.; Eckl, K.M.; Plank, R.; Werner, R.; Altmüller, J.; et al. Mutations in AP3D1 associated with immunodeficiency and seizures define a new type of Hermansky-Pudlak syndrome. Blood 2016, 127, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Newell-Litwa, K.; Salazar, G.; Smith, Y.; Faundez, V. Roles of BLOC-1 and Adaptor Protein-3 Complexes in Cargo Sorting to Synaptic Vesicles. Mol. Biol. Cell 2009, 20, 1441–1453. [Google Scholar] [CrossRef]
- Salazar, G.; Love, R.; Werner, E.; Doucette, M.M.; Cheng, S.; Levey, A.; Faundez, V. The Zinc Transporter ZnT3 Interacts with AP-3 and It Is Preferentially Targeted to a Distinct Synaptic Vesicle Subpopulation. Mol. Biol. Cell 2004, 15, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Nakatsu, F.; Okada, M.; Mori, F.; Kumazawa, N.; Iwasa, H.; Zhu, G.; Kasagi, Y.; Kamiya, H.; Harada, A.; Nishimura, K.; et al. Defective function of GABA-containing synaptic vesicles in mice lacking the AP-3B clathrin adaptor. J. Cell Biol. 2004, 167, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Dell’Angelica, E.C.; Shotelersuk, V.; Aguilar, R.C.; Gahl, W.A.; Bonifacino, J.S. Altered trafficking of lysosomal proteins in Hermansky-Pudlak syndrome due to mutations in the beta 3A subunit of the AP-3 adaptor. Mol. Cell 1999, 3, 11–21. [Google Scholar] [CrossRef]
- Theos, A.C.; Tenza, D.; Martina, J.; Hurbain, I.; Peden, A.; Sviderskaya, E.V.; Stewart, A.; Robinson, M.S.; Bennett, D.; Cutler, D.; et al. Functions of Adaptor Protein (AP)-3 and AP-1 in Tyrosinase Sorting from Endosomes to Melanosomes. Mol. Biol. Cell 2005, 16, 5356–5372. [Google Scholar] [CrossRef]
- Huizing, M.; Pederson, B.; A Hess, R.; Griffin, A.; Helip-Wooley, A.; Westbroek, W.; Dorward, H.; O’Brien, K.J.; Golas, G.; Tsilou, E.; et al. Clinical and cellular characterisation of Hermansky-Pudlak syndrome type 6. J. Med. Genet. 2009, 46, 803–810. [Google Scholar] [CrossRef]
- Kook, S.; Wang, P.; Young, L.R.; Schwake, M.; Saftig, P.; Weng, X.; Meng, Y.; Neculai, D.; Marks, M.S.; Gonzales, L.; et al. Impaired Lysosomal Integral Membrane Protein 2-dependent Peroxiredoxin 6 Delivery to Lamellar Bodies Accounts for Altered Alveolar Phospholipid Content in Adaptor Protein-3-deficient pearl Mice. J. Biol. Chem. 2016, 291, 8414–8427. [Google Scholar] [CrossRef]
- Jung, J.; Bohn, G.; Allroth, A.; Boztug, K.; Brandes, G.; Sandrock, I.; Schaffer, A.A.; Rathinam, C.; Kollner, I.; Beger, C.; et al. Identification of a homozygous deletion in the AP3B1 gene causing Hermansky-Pudlak syndrome, type 2. Blood 2006, 108, 362–369. [Google Scholar] [CrossRef]
- Benson, K.F.; Li, F.-Q.; E Person, R.; Albani, D.; Duan, Z.; Wechsler, J.; Meade-White, K.; Williams, K.; Acland, G.M.; Niemeyer, G.; et al. Mutations associated with neutropenia in dogs and humans disrupt intracellular transport of neutrophil elastase. Nat. Genet. 2003, 35, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, M.; Benson, K.F.; Duan, Z.; Li, F.-Q.; Person, R.E. Hereditary neutropenia: Dogs explain human neutrophil elastase mutations. Trends Mol. Med. 2004, 10, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Mantegazza, A.; Guttentag, S.; El-Benna, J.; Sasai, M.; Iwasaki, A.; Shen, H.; Laufer, T.M.; Marks, M.S. Adaptor Protein-3 in Dendritic Cells Facilitates Phagosomal Toll-like Receptor Signaling and Antigen Presentation to CD4+ T Cells. Immunity 2012, 36, 782–794. [Google Scholar] [CrossRef] [PubMed]
- Sugita, M.; Cao, X.; Watts, G.F.; Rogers, R.A.; Bonifacino, J.S.; Brenner, M.B. Failure of Trafficking and Antigen Presentation by CD1 in AP-3-Deficient Cells. Immunity 2002, 16, 697–706. [Google Scholar] [CrossRef]
- Elewaut, D.; Lawton, A.P.; Nagarajan, N.A.; Maverakis, E.; Khurana, A.; Höning, S.; Benedict, C.A.; Sercarz, E.; Bakke, O.; Kronenberg, M.; et al. The Adaptor Protein AP-3 Is Required for CD1d-Mediated Antigen Presentation of Glycosphingolipids and Development of Vα14i NKT Cells. J. Exp. Med. 2003, 198, 1133–1146. [Google Scholar] [CrossRef]
- Briken, V.; Jackman, R.; Dasgupta, S.; Hoening, S.; Porcelli, S. Intracellular trafficking pathway of newly synthesized CD1b molecules. EMBO J. 2002, 21, 825–834. [Google Scholar] [CrossRef]
- da Silva, E.Z.M.; Freitas-Filho, E.G.; de Souza-Júnior, D.A.; Dasilva, L.L.P.; Jamur, M.C.; Oliver, C. Adaptor protein-3: A key player in RBL-2H3 mast cell mediator release. PLoS ONE 2017, 12, e0173462. [Google Scholar] [CrossRef]
- Di Pietro, S.M.; Falcon-Perez, J.M.; Tenza, D.; Setty, S.R.G.; Marks, M.; Raposo, G.; Angelica, E.C.D. BLOC-1 Interacts with BLOC-2 and the AP-3 Complex to Facilitate Protein Trafficking on Endosomes. Mol. Biol. Cell 2006, 17, 4027–4038. [Google Scholar] [CrossRef]
- Hoyle, D.J.; A Rodriguez-Fernandez, I.; Dell’Angelica, E.C. Functional interactions between OCA2 and the protein complexes BLOC-1, BLOC-2, and AP-3 inferred from epistatic analyses of mouse coat pigmentation. Pigment Cell Melanoma Res. 2011, 24, 275–281. [Google Scholar] [CrossRef]
- Sitaram, A.; Dennis, M.K.; Chaudhuri, R.; De Jesus-Rojas, W.; Tenza, D.; Setty, S.R.G.; Wood, C.S.; Sviderskaya, E.V.; Bennett, D.C.; Raposo, G.; et al. Differential recognition of a dileucine-based sorting signal by AP-1 and AP-3 reveals a requirement for both BLOC-1 and AP-3 in delivery of OCA2 to melanosomes. Mol. Biol. Cell 2012, 23, 3178–3192. [Google Scholar] [CrossRef]
- Setty, S.R.G.; Tenza, D.; Sviderskaya, E.V.; Bennett, D.C.; Raposo, G.; Marks, M.S. Cell-specific ATP7A transport sustains copper-dependent tyrosinase activity in melanosomes. Nature 2008, 454, 1142–1146. [Google Scholar] [CrossRef] [PubMed]
- Setty, S.R.G.; Tenza, D.; Truschel, S.T.; Chou, E.; Sviderskaya, E.V.; Theos, A.C.; Lamoreux, M.L.; Di Pietro, S.M.; Starcevic, M.; Bennett, D.C.; et al. BLOC-1 Is Required for Cargo-specific Sorting from Vacuolar Early Endosomes toward Lysosome-related Organelles. Mol. Biol. Cell 2007, 18, 768–780. [Google Scholar] [CrossRef] [PubMed]
- Huizing, M.; Sarangarajan, R.; Strovel, E.; Zhao, Y.; Gahl, W.A.; Boissy, R.E. AP-3 Mediates Tyrosinase but Not TRP-1 Trafficking in Human Melanocytes. Mol. Biol. Cell 2001, 12, 2075–2085. [Google Scholar] [CrossRef]
- Gokhale, A.; Vrailas-Mortimer, A.; Larimore, J.; Comstra, H.S.; Zlatic, S.A.; Werner, E.; Manvich, D.F.; Iuvone, P.M.; Weinshenker, D.; Faundez, V. Neuronal copper homeostasis susceptibility by genetic defects in dysbindin, a schizophrenia susceptibility factor. Hum. Mol. Genet. 2015, 24, 5512–5523. [Google Scholar] [CrossRef]
- Newell-Litwa, K.; Chintala, S.; Jenkins, S.; Pare, J.-F.; McGaha, L.; Smith, Y.; Faundez, V. Hermansky-Pudlak Protein Complexes, AP-3 and BLOC-1, Differentially Regulate Presynaptic Composition in the Striatum and Hippocampus. J. Neurosci. 2010, 30, 820–831. [Google Scholar] [CrossRef]
- Chen, X.-W.; Feng, Y.-Q.; Hao, C.-J.; Guo, X.-L.; He, X.; Zhou, Z.-Y.; Guo, N.; Huang, H.-P.; Xiong, W.; Zheng, H.; et al. DTNBP1, a schizophrenia susceptibility gene, affects kinetics of transmitter release. J. Cell Biol. 2008, 181, 791–801. [Google Scholar] [CrossRef]
- Salazar, G.; Craige, B.; Styers, M.L.; Newell-Litwa, K.; Doucette, M.M.; Wainer, B.H.; Falcon-Perez, J.M.; Dell’Angelica, E.C.; Peden, A.; Werner, E.; et al. BLOC-1 Complex Deficiency Alters the Targeting of Adaptor Protein Complex-3 Cargoes. Mol. Biol. Cell 2006, 17, 4014–4026. [Google Scholar] [CrossRef]
- Salazar, G.; Zlatic, S.; Craige, B.; Peden, A.A.; Pohl, J.; Faundez, V. Hermansky-Pudlak Syndrome Protein Complexes Associate with Phosphatidylinositol 4-Kinase Type II α in Neuronal and Non-neuronal Cells. J. Biol. Chem. 2009, 284, 1790–1802. [Google Scholar] [CrossRef]
- Ryder, P.V.; Vistein, R.; Gokhale, A.; Seaman, M.N.; Puthenveedu, M.A.; Faundez, V. The WASH complex, an endosomal Arp2/3 activator, interacts with the Hermansky–Pudlak syndrome complex BLOC-1 and its cargo phosphatidylinositol-4-kinase type IIα. Mol. Biol. Cell 2013, 24, 2269–2284. [Google Scholar] [CrossRef] [PubMed]
- Dennis, M.K.; Delevoye, C.; Acosta-Ruiz, A.; Hurbain, I.; Romao, M.; Hesketh, G.G.; Goff, P.S.; Sviderskaya, E.V.; Bennett, D.C.; Luzio, J.P.; et al. BLOC-1 and BLOC-3 regulate VAMP7 cycling to and from melanosomes via distinct tubular transport carriers. J. Cell Biol. 2016, 214, 293–308. [Google Scholar] [CrossRef]
- Delevoye, C.; Heiligenstein, X.; Ripoll, L.; Gilles-Marsens, F.; Dennis, M.; Linares, R.; Derman, L.; Gokhale, A.; Morel, E.; Faundez, V.; et al. BLOC-1 Brings Together the Actin and Microtubule Cytoskeletons to Generate Recycling Endosomes. Curr. Biol. 2016, 26, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Delevoye, C.; Miserey-Lenkei, S.; Montagnac, G.; Gilles-Marsens, F.; Paul-Gilloteaux, P.; Giordano, F.; Waharte, F.; Marks, M.S.; Goud, B.; Raposo, G. Recycling Endosome Tubule Morphogenesis from Sorting Endosomes Requires the Kinesin Motor KIF13A. Cell Rep. 2014, 6, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Falcón-Pérez, J.M.; Starcevic, M.; Gautam, R.; Dell’Angelica, E.C. BLOC-1, a Novel Complex Containing the Pallidin and Muted Proteins Involved in the Biogenesis of Melanosomes and Platelet-dense Granules. J. Biol. Chem. 2002, 277, 28191–28199. [Google Scholar] [CrossRef] [PubMed]
- Ghiani, C.A.; Starcevic, M.; Rodriguez-Fernandez, I.A.; Nazarian, R.; Cheli, V.T.; Chan, L.N.; Malvar, J.S.; de Vellis, J.; Sabatti, C.; Dell’Angelica, E.C. The dysbindin-containing complex (BLOC-1) in brain: Developmental regulation, interaction with SNARE proteins and role in neurite outgrowth. Mol. Psychiatry 2010, 15, 204–215. [Google Scholar] [CrossRef]
- Zhang, A.; He, X.; Zhang, L.; Yang, L.; Woodman, P.; Li, W. Biogenesis of Lysosome-related Organelles Complex-1 Subunit 1 (BLOS1) Interacts with Sorting Nexin 2 and the Endosomal Sorting Complex Required for Transport-I (ESCRT-I) Component TSG101 to Mediate the Sorting of Epidermal Growth Factor Receptor into Endosomal Compartments. J. Biol. Chem. 2014, 289, 29180–29194. [Google Scholar] [CrossRef] [PubMed]
- Bowman, S.L.; Le, L.; Zhu, Y.; Harper, D.C.; Sitaram, A.; Theos, A.C.; Sviderskaya, E.V.; Bennett, D.C.; Raposo-Benedetti, G.; Owen, D.J.; et al. A BLOC-1–AP-3 super-complex sorts a cis-SNARE complex into endosome-derived tubular transport carriers. J. Cell Biol. 2021, 220, e202005173. [Google Scholar] [CrossRef] [PubMed]
- Huizing, M.; Anikster, Y.; Fitzpatrick, D.L.; Jeong, A.B.; D’Souza, M.; Rausche, M.; Toro, J.R.; Kaiser-Kupfer, M.I.; White, J.G.; Gahl, W.A. Hermansky-Pudlak Syndrome Type 3 in Ashkenazi Jews and Other Non–Puerto Rican Patients with Hypopigmentation and Platelet Storage-Pool Deficiency. Am. J. Hum. Genet. 2001, 69, 1022–1032. [Google Scholar] [CrossRef]
- Michaud, V.; Lasseaux, E.; Plaisant, C.; Verloes, A.; Perdomo-Trujillo, Y.; Hamel, C.; Elcioglu, N.H.; Leroy, B.; Kaplan, J.; Jouk, P.-S.; et al. Clinico-molecular analysis of eleven patients with Hermansky-Pudlak type 5 syndrome, a mild form of HPS. Pigment Cell Melanoma Res. 2017, 30, 563–570. [Google Scholar] [CrossRef]
- Ma, J.; Zhang, Z.; Yang, L.; Kriston-Vizi, J.; Cutler, D.F.; Li, W. BLOC-2 subunit HPS6 deficiency affects the tubulation and secretion of von Willebrand factor from mouse endothelial cells. J. Genet. Genom. 2016, 43, 686–693. [Google Scholar] [CrossRef]
- Dennis, M.; Mantegazza, A.; Snir, O.L.; Tenza, D.; Acosta-Ruiz, A.; Delevoye, C.; Zorger, R.; Sitaram, A.; De Jesus-Rojas, W.; Ravichandran, K.; et al. BLOC-2 targets recycling endosomal tubules to melanosomes for cargo delivery. J. Cell Biol. 2015, 209, 563–577. [Google Scholar] [CrossRef]
- Gerondopoulos, A.; Langemeyer, L.; Liang, J.-R.; Linford, A.; Barr, F.A. BLOC-3 Mutated in Hermansky-Pudlak Syndrome Is a Rab32/38 Guanine Nucleotide Exchange Factor. Curr. Biol. 2012, 22, 2135–2139. [Google Scholar] [CrossRef] [PubMed]
- Ohishi, Y.; Kinoshita, R.; Marubashi, S.; Ishida, M.; Fukuda, M. The BLOC-3 subunit HPS4 is required for activation of Rab32/38 GTPases in melanogenesis, but its Rab9 activity is dispensable for melanogenesis. J. Biol. Chem. 2019, 294, 6912–6922. [Google Scholar] [CrossRef] [PubMed]
- Ambrosio, A.L.; Boyle, J.A.; Di Pietro, S.M. Mechanism of platelet dense granule biogenesis: Study of cargo transport and function of Rab32 and Rab38 in a model system. Blood 2012, 120, 4072–4081. [Google Scholar] [CrossRef] [PubMed]
- Ninkovic, I.; White, J.G.; Rangel-Filho, A.; Datta, Y.H. The role of Rab38 in platelet dense granule defects. J. Thromb. Haemost. 2008, 6, 2143–2151. [Google Scholar] [CrossRef]
- Zhang, L.; Yu, K.; Robert, K.W.; DeBolt, K.M.; Hong, N.; Tao, J.-Q.; Fukuda, M.; Fisher, A.B.; Huang, S. Rab38 targets to lamellar bodies and normalizes their sizes in lung alveolar type II epithelial cells. Am. J. Physiol. Cell. Mol. Physiol. 2011, 301, L461–L477. [Google Scholar] [CrossRef]
- Osanai, K.; Higuchi, J.; Oikawa, R.; Kobayashi, M.; Tsuchihara, K.; Iguchi, M.; Huang, J.; Voelker, D.R.; Toga, H. Altered lung surfactant system in a Rab38-deficient rat model of Hermansky-Pudlak syndrome. Am. J. Physiol. Cell. Mol. Physiol. 2010, 298, L243–L251. [Google Scholar] [CrossRef]
- Ellis, K.; Bagwell, J.; Bagnat, M. Notochord vacuoles are lysosome-related organelles that function in axis and spine morphogenesis. J. Cell Biol. 2013, 200, 667–679. [Google Scholar] [CrossRef]
- Gissen, P.; Tee, L.; Johnson, C.A.; Genin, E.; Caliebe, A.; Chitayat, D.; Clericuzio, C.; Denecke, J.; Di Rocco, M.; Fischler, B.; et al. Clinical and molecular genetic features of ARC syndrome. Qual. Life Res. 2006, 120, 396–409. [Google Scholar] [CrossRef]
- Jang, J.Y.; Kim, K.M.; Kim, G.-H.; Yu, E.; Lee, J.-J.; Park, Y.S.; Yoo, H.-W. Clinical Characteristics and VPS33B Mutations in Patients With ARC Syndrome. J. Pediatr. Gastroenterol. Nutr. 2009, 48, 348–354. [Google Scholar] [CrossRef]
- Smith, H.; Galmes, R.; Gogolina, E.; Straatman-Iwanowska, A.; Reay, K.; Banushi, B.; Bruce, C.K.; Cullinane, A.R.; Romero, R.; Chang, R.; et al. Associations among genotype, clinical phenotype, and intracellular localization of trafficking proteins in ARC syndrome. Hum. Mutat. 2012, 33, 1656–1664. [Google Scholar] [CrossRef]
- Cullinane, A.R.; Straatman-Iwanowska, A.; Seo, J.K.; Ko, J.S.; Song, K.S.; Giżewska, M.; Gruszfeld, D.; Gliwicz, D.; Tuysuz, B.; Erdemir, G.; et al. Molecular investigations to improve diagnostic accuracy in patients with ARC syndrome. Hum. Mutat. 2009, 30, E330–E337. [Google Scholar] [CrossRef] [PubMed]
- Gissen, P.; Johnson, C.A.; Morgan, N.V.; Stapelbroek, J.M.; Forshew, T.; Cooper, W.N.; McKiernan, P.J.; Klomp, L.W.; Morris, A.A.; E Wraith, J.E.; et al. Mutations in VPS33B, encoding a regulator of SNARE-dependent membrane fusion, cause arthrogryposis–renal dysfunction–cholestasis (ARC) syndrome. Nat. Genet. 2004, 36, 400–404. [Google Scholar] [CrossRef]
- Carim, L.; Sumoy, L.; Andreu, N.; Estivill, X.; Escarceller, M. Cloning, mapping and expression analysis of VPS33B, the human orthologue of rat Vps33b. Cytogenet. Genome Res. 2000, 89, 92–95. [Google Scholar] [CrossRef]
- Akbar, M.A.; Ray, S.; Krämer, H. The SM Protein Car/Vps33A Regulates SNARE-mediated Trafficking to Lysosomes and Lysosome-related Organelles. Mol. Biol. Cell 2009, 20, 1705–1714. [Google Scholar] [CrossRef] [PubMed]
- Hunter, M.R.; Hesketh, G.G.; Benedyk, T.H.; Gingras, A.-C.; Graham, S.C. Proteomic and Biochemical Comparison of the Cellular Interaction Partners of Human VPS33A and VPS33B. J. Mol. Biol. 2018, 430, 2153–2163. [Google Scholar] [CrossRef]
- Balderhaar, H.J.K.; Ungermann, C. CORVET and HOPS tethering complexes–coordinators of endosome and lysosome fusion. J. Cell Sci. 2013, 126, 1307–1316. [Google Scholar] [CrossRef]
- Sato, T.K.; Rehling, P.; Peterson, M.R.; Emr, S.D. Class C Vps Protein Complex Regulates Vacuolar SNARE Pairing and Is Required for Vesicle Docking/Fusion. Mol. Cell 2000, 6, 661–671. [Google Scholar] [CrossRef]
- Graham, S.C.; Wartosch, L.; Gray, S.R.; Scourfield, E.J.; Deane, J.E.; Luzio, J.P.; Owen, D.J. Structural basis of Vps33A recruitment to the human HOPS complex by Vps16. Proc. Natl. Acad. Sci. USA 2013, 110, 13345–13350. [Google Scholar] [CrossRef] [PubMed]
- van der Kant, R.; Jonker, C.T.; Wijdeven, R.H.; Bakker, J.; Janssen, L.; Klumperman, J.; Neefjes, J. Characterization of the Mammalian CORVET and HOPS Complexes and Their Modular Restructuring for Endosome Specificity. J. Biol. Chem. 2015, 290, 30280–30290. [Google Scholar] [CrossRef] [PubMed]
- Rogerson, C.; Gissen, P. The CHEVI tethering complex: Facilitating special deliveries. J. Pathol. 2016, 240, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Spang, A. Membrane Tethering Complexes in the Endosomal System. Front. Cell Dev. Biol. 2016, 4, 35. [Google Scholar] [CrossRef] [PubMed]
- van der Beek, J.; Jonker, C.; van der Welle, R.; Liv, N.; Klumperman, J. CORVET, CHEVI and HOPS—Multisubunit tethers of the endo-lysosomal system in health and disease. J. Cell Sci. 2019, 132, jcs189134. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Wang, R.; Lam, S.M.; Zhang, C.; Shui, G.; Li, W. Plasma lipidomic profiling in murine mutants of Hermansky–Pudlak syndrome reveals differential changes in pro- and anti-atherosclerotic lipids. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef]
- Lo, B.; Li, L.; Gissen, P.; Christensen, H.; McKiernan, P.J.; Ye, C.; Abdelhaleem, M.; Hayes, J.A.; Williams, M.D.; Chitayat, D.; et al. Requirement of VPS33B, a member of the Sec1/Munc18 protein family, in megakaryocyte and platelet -granule biogenesis. Blood 2005, 106, 4159–4166. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Lu, Y.; Wang, C.; Chen, X.; Fan, X.; Gu, H.; Wu, X.; Wang, K.; Gartner, T.K.; Zheng, J.; et al. Vps33b regulates Vwf-positive vesicular trafficking in megakaryocytes. J. Pathol. 2016, 240, 108–119. [Google Scholar] [CrossRef]
- Xiang, B.; Zhang, G.; Ye, S.; Zhang, R.; Huang, C.; Liu, J.; Tao, M.; Ruan, C.; Smyth, S.S.; Whiteheart, S.; et al. Characterization of a Novel Integrin Binding Protein, VPS33B, Which Is Important for Platelet Activation and In Vivo Thrombosis and Hemostasis. Circulation 2015, 132, 2334–2344. [Google Scholar] [CrossRef]
- Rogerson, C.; Gissen, P. VPS33B and VIPAR are essential for epidermal lamellar body biogenesis and function. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2018, 1864, 1609–1621. [Google Scholar] [CrossRef]
- Hershkovitz, D.; Mandel, H.; Ishida-Yamamoto, A.; Chefetz, I.; Hino, B.; Luder, A.; Indelman, M.; Bergman, R.; Sprecher, E. Defective Lamellar Granule Secretion in Arthrogryposis, Renal Dysfunction, and Cholestasis Syndrome Caused by a Mutation in VPS33B. Arch. Dermatol. 2008, 144, 334–340. [Google Scholar] [CrossRef]
- Reynier, M.; Allart, S.; Gaspard, E.; Moga, A.; Goudounèche, D.; Serre, G.; Simon, M.; Leprince, C. Rab11a Is Essential for Lamellar Body Biogenesis in the Human Epidermis. J. Investig. Dermatol. 2016, 136, 1199–1209. [Google Scholar] [CrossRef]
- Gruber, R.; Rogerson, C.; Windpassinger, C.; Banushi, B.; Straatman-Iwanowska, A.; Hanley, J.; Forneris, F.; Strohal, R.; Ulz, P.; Crumrine, D.; et al. Autosomal Recessive Keratoderma-Ichthyosis-Deafness (ARKID) Syndrome Is Caused by VPS33B Mutations Affecting Rab Protein Interaction and Collagen Modification. J. Investig. Dermatol. 2017, 137, 845–854. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Z.; Wang, H.; Tang, Z.; Liu, Y.; Liang, Z.; Deng, X.; Zhao, M.; Fu, Q.; Li, L.; et al. VPS33B negatively modulated by nicotine functions as a tumor suppressor in colorectal cancer. Int. J. Cancer 2019, 146, 496–509. [Google Scholar] [CrossRef]
- Liang, Z.; Liu, Z.; Cheng, C.; Wang, H.; Deng, X.; Liu, J.; Liu, C.; Li, Y.; Fang, W. VPS33B interacts with NESG1 to modulate EGFR/PI3K/AKT/c-Myc/P53/miR-133a-3p signaling and induce 5-fluorouracil sensitivity in nasopharyngeal carcinoma. Cell Death Dis. 2019, 10, 305. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Cheng, Y.; Zhang, X.; Li, N.; Zhang, L.; Wang, S.; Tong, X.; Xu, Y.; Chen, G.-Q.; Cheng, S.; et al. Vacuolar Protein Sorting 33B Is a Tumor Suppressor in Hepatocarcinogenesis. Hepatology 2018, 68, 2239–2253. [Google Scholar] [CrossRef] [PubMed]
- Manke, T.; Bringas, R.; Vingron, M. Correlating Protein–DNA and Protein–Protein Interaction Networks. J. Mol. Biol. 2003, 333, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.; Shlomi, T.; Feizi, H.; Ideker, T.; Sharan, R. Transcriptional regulation of protein complexes within and across species. Proc. Natl. Acad. Sci. USA 2007, 104, 1283–1288. [Google Scholar] [CrossRef]
- Simonis, N.; Gonze, D.; Orsi, C.; van Helden, J.; Wodak, S.J. Modularity of the Transcriptional Response of Protein Complexes in Yeast. J. Mol. Biol. 2006, 363, 589–610. [Google Scholar] [CrossRef]
- Holme, A.; Hurcombe, J.; Straatman-Iwanowska, A.; Inward, C.I.; Gissen, P.; Coward, R.J. Glomerular involvement in the arthrogryposis, renal dysfunction and cholestasis syndrome. Clin. Kidney J. 2013, 6, 183–188. [Google Scholar] [CrossRef][Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Syndrome | Mutations | Clinical Phenotype | Cellular Phenotype | Mouse Model |
|---|---|---|---|---|
| Griscelli Type 1 | MYO5A | Hypopigmentation of skin and hair, Neurological disorder | Abnormal accumulation of melanosomes in melanocytes, impaired movement of synaptic vesicles, potential inability of ER to move into the dendritic spines | dilute [39] |
| Griscelli Type 2 | RAB27A | Hypopigmentation of skin and hair Immunological disorder * Increased bleeding (Ashen mice) | Abnormal accumulation of melanosomes in melanocytes, impaired natural-killer cell function, uncontrolled lymphocyte, and macrophage activation * Reduction in the number of platelet dense granules (Ashen mice). | ashen [40] |
| Griscelli Type 3 | MLPH | Hypomelanosis with no immunological and neurological manifestation. | Perinuclear aggregation of melanosomes in melanocytes | leaden [41] |
| MYH9RD | MYH9 | Macrothrombocytopenia with mild bleeding tendency, may develop kidney dysfunction, deafness, and cataracts | Enlarged platelets, Döhle-like bodies in the cytoplasm of neutrophils | R702C, D1424N, E1841K [42] |
| FHL3 | UNC13D | Typical FHL with early onset of uncontrolled activation of T lymphocytes and macrophages, * bleeding | Lytic granules of NK cells and CTLs fail to fuse with the plasma membrane, * abnormalities in platelet secretion | Unc13d(Jinx) [43] |
| Chediak–Higashi Syndrome | LYST | Immunodeficiency, oculocutaneous albinism, bleeding, recurrent infections, neurologic defects, lymphohistiocytosis | Giant intracellular granules in different cell types including neurons, immune cells, melanocytes, platelets | beige [44] |
| HPS1 HPS4 | HPS1 HPS4 | Restrictive lung disease, pulmonary fibrosis, granulomatous colitis, prolonged bleeding (variable), hypopigmentation (variable), inflammatory bowel disease, acanthosis nigricans and hypertrichosis. | Cells filled with phospholipids droplets; enlarged lamellar bodies; absence of platelet dense granules | pale ear [45] light ear [46] |
| HPS2 | AP3B1 | Immunodeficiency, neutropenia, recurrent infections, hypopigmentation, acanthosis nigricans and hypertrichosis, dysplastic ace tabulae, poor balance, conductive hearing loss, haemorrhage | Diminished amounts of β3A protein, melanocytes with abundant multivesicular structures, CTL with increased size of the endosomal network fail to induce polarization of lytic granules to the IS and decreased ability to kill targets, enlarged LB. | pearl [47] |
| HPS3 | HPS3 | Elevated creatinine clearance, ocular albinism, bruising and epistaxis | Mislocalisation of LAMP-1 and LAMP-3 as well as melanosome targeted proteins, decreased levels of melanin in melanocytes | cocoa [48] |
| HPS5 | HPS5 | Elevated creatinine clearance, nystagmus, bruising, hypercholesterolemia | Absent platelet dense bodies, LAMP-3 distributed in granules in a perinuclear region and not in the dendritic processes like normal. | ruby-eye2 [49] |
| HPS6 | HPS6 | T creatinine clearance, respiratory and urinary tract infections, epistaxis, bleeding, oculocutaneous albinism, urinary and rectal incontinence global developmental delay hearing loss | Mislocalisation of melanosome targeted proteins Deficient platelet dense granules | ruby-eye [49] |
| HPS7 | DTNBP1 | Oculocutaneous albinism, bleeding tendency, mild shortness of breath with exertion, reduced lung compliance | Abnormal melanosomes * | sandy [50] |
| HPS8 | BLOC1S3 | Incomplete oculocutaneous albinism, mild platelet dysfunction, easy bruising, hematomas after venesection, frequent epistaxis and prolonged bleeding after surgery or childbearing | Increased kidney lysosomal glycosidase activities, decreased platelet dense bodies, immature melanosomes, decreased melanin levels, abnormal intracellular tyrosinase distribution * | reduced pigmentation [51] |
| HPS9 | BLOC1S6 | Oculocutaneous albinism, nystagmus, recurrent cutaneous infections, thrombocytopenia, and leukopenia | NK cells have an increased surface levels of LAMP1 and CD63, decreased granulation, and decreased cytolytic activity | pallid [52] |
| HPS10 | AP3D1 | Immunodeficiency, oculocutaneous albinism, impaired hearing, severe neurologic impairment, delayed development and seizures, recurrent infections | Reduced levels of renal lysosomal enzymes, deficiency in the dense granules of platelets. Lack of AP3 in mocha tissues and reduced levels of the zinc transporter Znt3 in brain | mocha [53] |
| ARC | VPS33B VIPAS39 | Arthrogryposis, renal tubule dysfunction neonatal cholestasis, ichthyosis, sensorineural deafness, central nervous system malformation, bleeding, recurrent infections, and severe failure to thrive. | Abnormal platelet α-granule and LB biogenesis, mislocalisation of apical proteins in hepatocytes, defects in collagen homeostasis. | Vps33bfl/fl [54,55] Vipas39fl/fl [56] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banushi, B.; Simpson, F. Overlapping Machinery in Lysosome-Related Organelle Trafficking: A Lesson from Rare Multisystem Disorders. Cells 2022, 11, 3702. https://doi.org/10.3390/cells11223702
Banushi B, Simpson F. Overlapping Machinery in Lysosome-Related Organelle Trafficking: A Lesson from Rare Multisystem Disorders. Cells. 2022; 11(22):3702. https://doi.org/10.3390/cells11223702
Chicago/Turabian StyleBanushi, Blerida, and Fiona Simpson. 2022. "Overlapping Machinery in Lysosome-Related Organelle Trafficking: A Lesson from Rare Multisystem Disorders" Cells 11, no. 22: 3702. https://doi.org/10.3390/cells11223702
APA StyleBanushi, B., & Simpson, F. (2022). Overlapping Machinery in Lysosome-Related Organelle Trafficking: A Lesson from Rare Multisystem Disorders. Cells, 11(22), 3702. https://doi.org/10.3390/cells11223702