
Mitochondria Transfer in Brain Injury and Disease




Abstract
1. Introduction
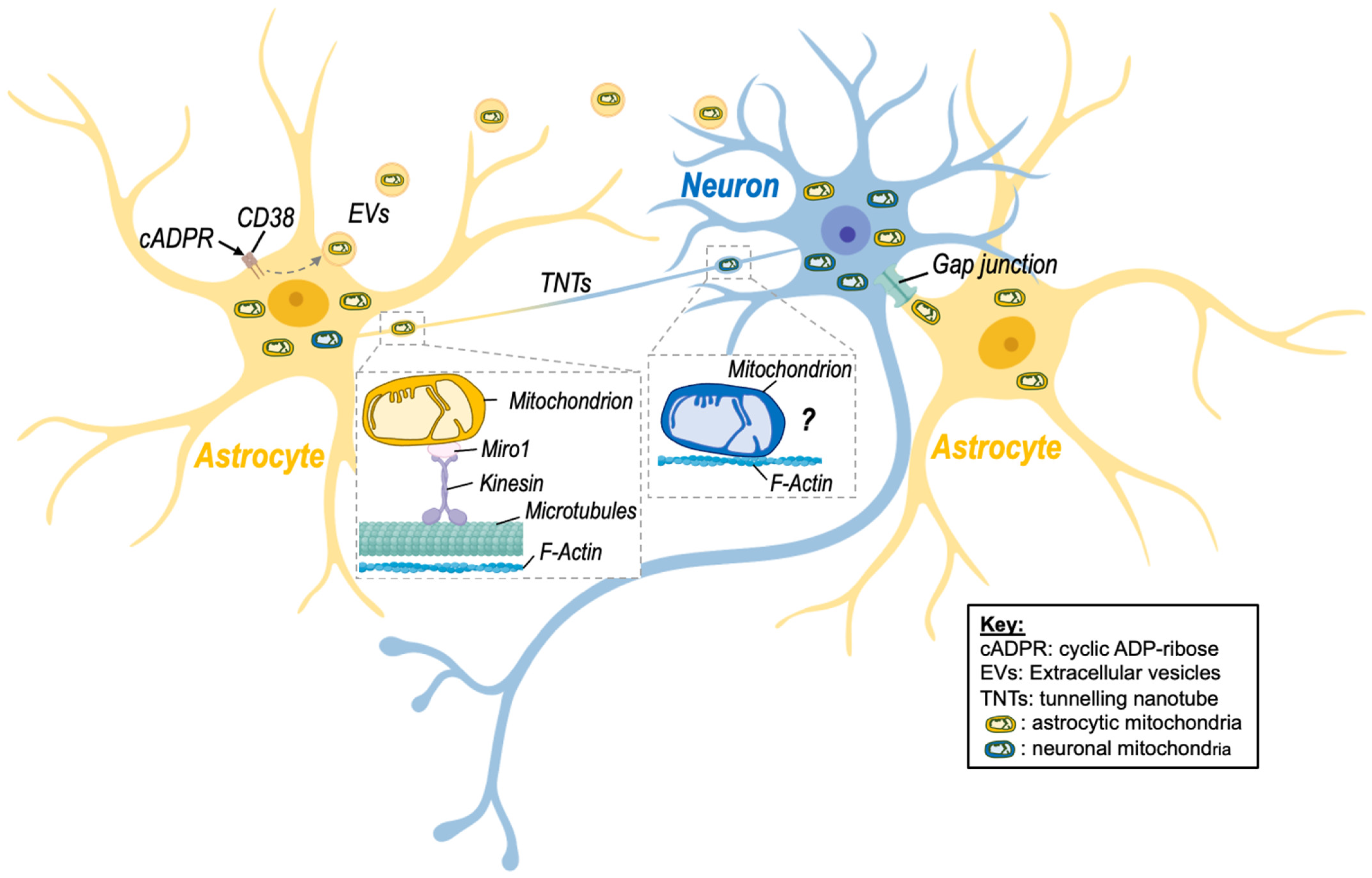
2. Evidence of Intercellular Mitochondrial Transfer in the Brain
3. Structural Mechanisms of Mitochondrial Transfer
3.1. Extracellular Vesicles
3.2. Tunneling Nanotubes
3.3. Other Mechanisms

{kind=link}
{kind=link}
{kind=link}
| Method of Transfer | Cell Type | Disease Model/Stressor | Effects of Mito Transfer | Ref. | ||
|---|---|---|---|---|---|---|
| Donor | Recipient | Donor | Recipient | |||
| EVs | Neural stem cells | BMDM | LPS | N/A | -Increased mitochondrial fusion | [56] |
| -Increased cellular respiration | ||||||
| -Reduced inflammatory gene profiles | ||||||
| EVs | Primary human | N/A | ATP released from neighbouring cells | N/A | N/A | [39] |
| Astrocytes | ||||||
| EVs | Primary rat astrocytes | Primary rat neurons | Oxygen-glucose deprivation | N/A | -Increase ATP levels | [20] |
| -Increased cell viability | ||||||
| TNTs | PC12 cells | PC12 cells | UV light | N/A | -Decreased apoptosis | [48] |
| TNTs | MMSC | Primary astrocytes | Oxygen-glucose deprivation | -Increased transfer | -Restored bioenergetics | [49] |
| and | -Increased proliferation | |||||
| PC12 cells | ||||||
| TNTs | MSC | Neural stem cells | Cisplatin | N/A | -Decreased apoptosis | [50] |
| -Increased MMP | ||||||
| TNTs | Primary mouse astrocytes | Primary mouse neurons | Compressed nitrogen–oxygen mixed gas | N/A | -Increased dendrite length | [19] |
| -Increased transcription of mitochondrial synthesis-related genes | ||||||
| TNTs | Primary mouse microglia | Primary mouse microglia | α-syn | N/A | -Decreased ROS levels | [44] |
| -Decreased apoptotic signalling | ||||||
| TNTs | Primary rat astrocytes | Primary rat astrocytes and neurons | H2O2 or serum depletion | N/A | [45] | |
| TNTs | primary mouse neurons | primary mouse astrocytes | 5xFAD | N/A | -Increased transmitophagy | [33] |
| TNTs | Glioblastoma stem-like cells | Glioblastoma stem-like cells | Irradiation | C1: no effect | N/A | [36] |
| C2: increased transfer | ||||||
4. Effects of Mitochondrial Transfer in the Brain
- To enhance cell viability via transferring healthy mitochondria to stressed/injured cells;
- To enhance degradation of dysfunctional mitochondria via transferring unhealthy mitochondria to healthy cells;
- To modulate glia-mediated neuroinflammation.
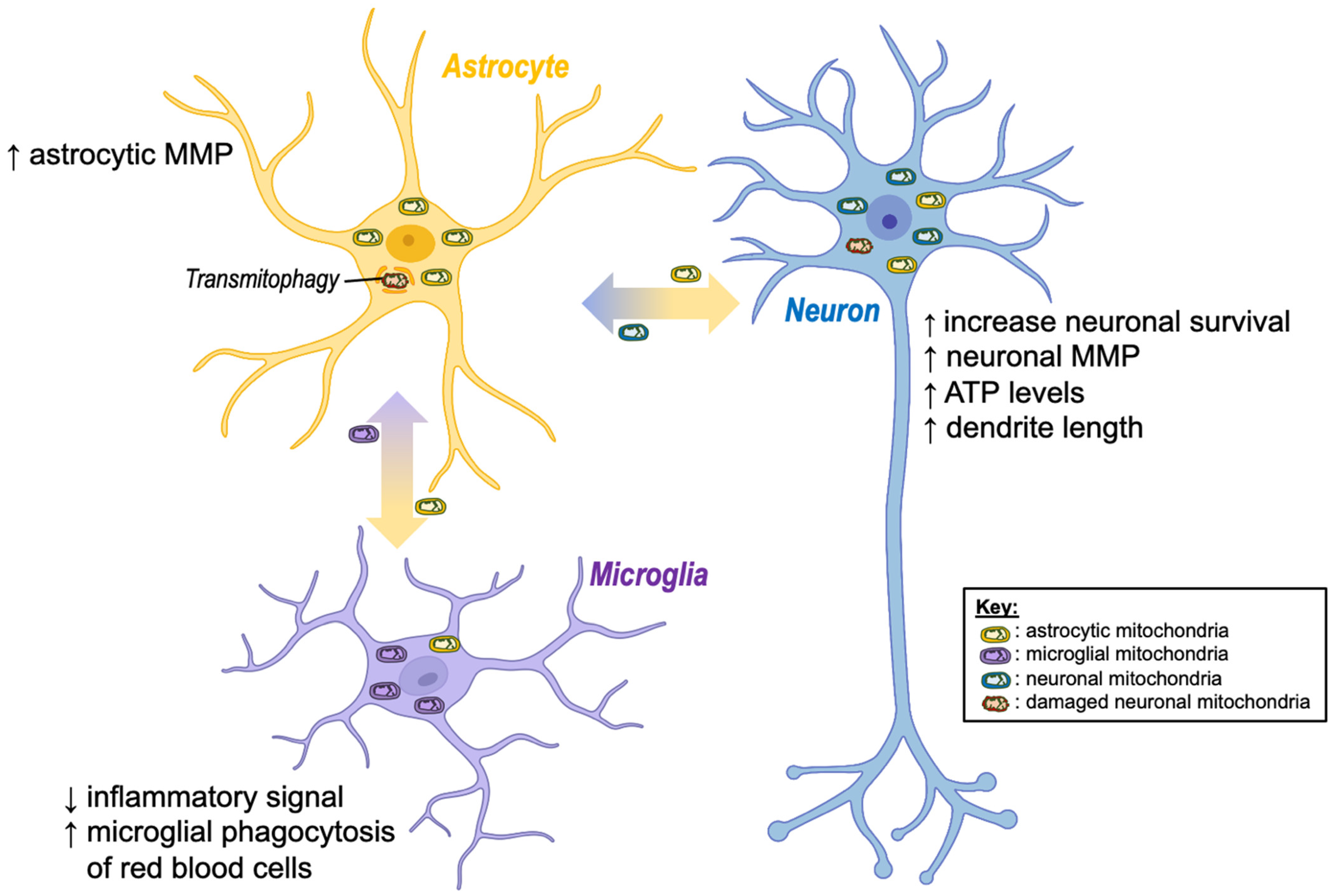
4.1. Enhancement of Cell Viability
4.2. Enhancement of Mitochondrial Degradation
4.3. Modulation of Glia-Mediated Neuroinflammation
4.4. Deleterious Effects
5. Mitochondrial Transfer in Brain Injury and Disease
5.1. Brain Injury
5.2. Neurodegenerative Diseases
5.3. Neurodevelopmental Disease
5.4. Chemotherapy
6. Therapeutic Strategies Targeting Mitochondrial Transfer
6.1. Pharmacologic Approaches
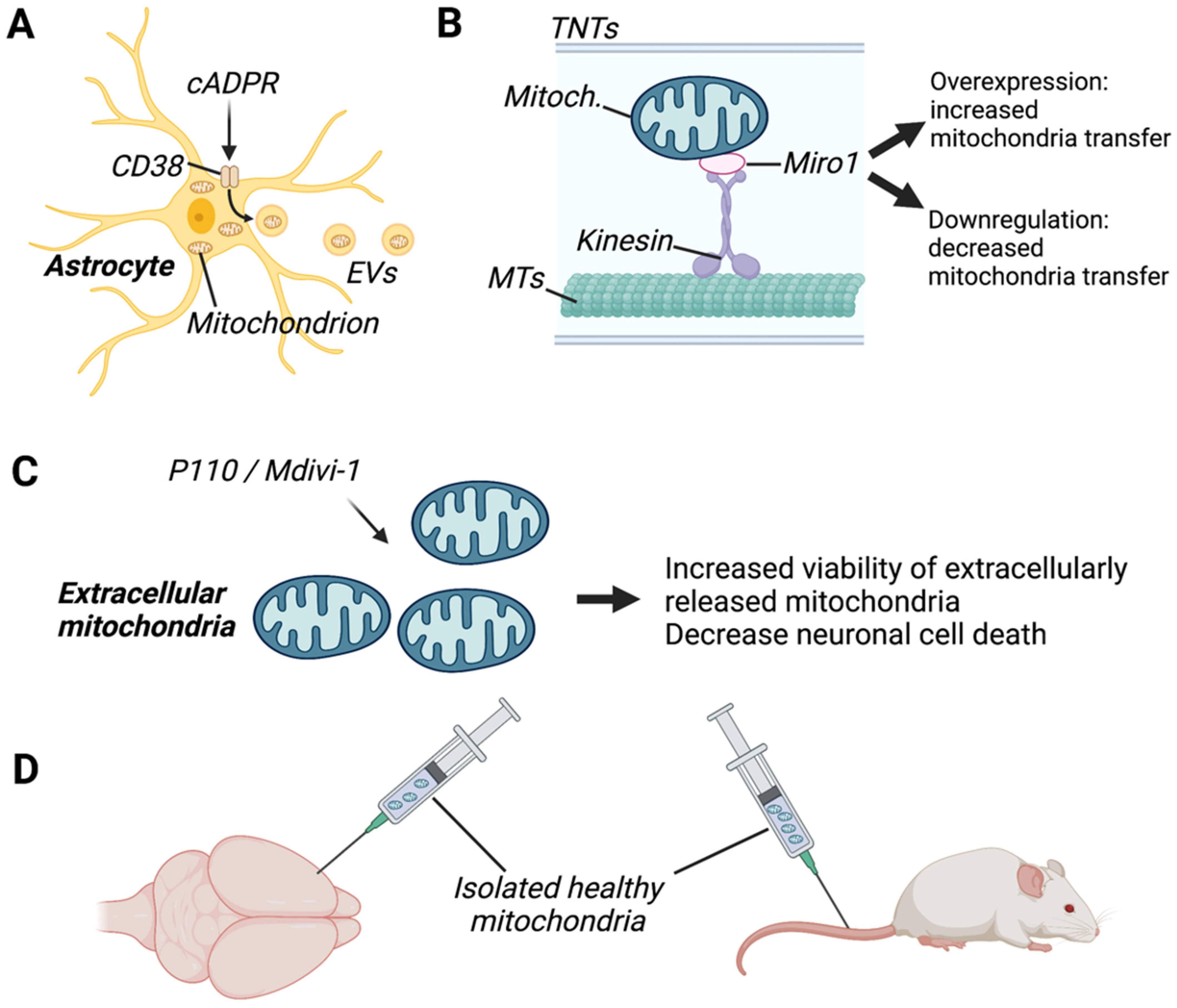
6.1.1. CD38
6.1.2. MIRO-1
6.1.3. CX43
6.1.4. Mitochondrial Fission/Fusion
6.2. Mitochondrial Transplantation
| Disease Model | Source of Mitochondria | Method of Delivery for AMT | Effects | References |
|---|---|---|---|---|
| Stroke | MMSC with overexpressed Miro-1 | I.V. injection | -Increased neurological function | [49] |
| -MCAO model of focal ischemia | ||||
| Stroke | Primary mouse astrocytes | Local injection into peri-infarct cortex | -Upregulation of cell survival signals | [20] |
| -Focal cerebral ischaemia | ||||
| Stroke | Baby hamster kidney fibroblast (BHK-21) | ICV or systemic intra-arterial injection | -Increased motor performance | [78] |
| -MCAO model of focal ischemia | -Decreased brain infarct area | |||
| -Decreased neuronal death | ||||
| Stroke | Mouse placenta | I.V. injection | -Decreased brain infarct area | [79] |
| -Focal cerebral ischaemia | ||||
| Stroke | Primary mouse astrocytes | I.V. injection | -Increased neuronal viability | [80] |
| -Intracerebral haemorrhage | -Reduced neurologic deficits | |||
| -Restored Mn-SOD levels | ||||
| Stroke | Human umbilical-cord-derived mesenchymal stem cells | ICV | -Decreased apoptosis | [81] |
| -MCAO model of focal ischemia | -Decreased gliosis | |||
| -Improved motor function | ||||
| -Decreased brain infarct area | ||||
| Chemotherapy-induced neurotoxicity | MSC | Intranasal | -Reduced apoptosis | [50] |
| -Cisplatin treatment | ||||
| Alzheimer’s disease | HeLa cells | I.V. injection | -Improved cognitive function | [85] |
| -Amyloid-β intracerebroventricularly injected | -Decreased neuronal loss | |||
| -Decreased gliosis | ||||
| -Increased citrate-synthase and cytochrome c oxidase activities | ||||
| Parkinson’s disease | PC12 cells | Local injection into MFB | -Improved locomotive activity | [86] |
| -6-OHDA-lesioned rat model | or | -Increased neuronal survival | ||
| Human osteosarcoma cybrids | -Restored mitochondrial dynamics | |||
| Parkinson’s disease | Rat liver | Intranasal | -Improved locomotive activity | [87] |
| -6-OHDA-lesioned rat model | -Increased neuronal survival | |||
| -Decreased oxidative damage | ||||
| Parkinson’s disease | HepG2 cells | I.V. injection | -Improved locomotive activity | [88] |
| -MPTP-induced mouse model | -Increased ATP levels | |||
| -Decreased ROS levels | ||||
| Multiple sclerosis | Neural stem cells | ICV | -Ameliorated EAE severity | [56] |
| -MOG35-55-induced EAE | ||||
| Schizophrenia | Human lymphoblasts | Intra-prefrontal cortex injection | -Rescued attentional deficits | [96] |
| -Prenatal poly-I:C exposure | Or | -Increased MMP | ||
| Rat brain | ||||
| Traumatic brain injury | Mouse liver | Local injection into cerebral cortex | -Increased ATP levels | [84] |
| -Controlled cortical impact | Mouse muscle | -Upregulated astrocytic BDNF | ||
| Improved spatial memory and cognitive function | ||||
| Traumatic brain injury | Mouse brain | Local injection into cerebral cortex | -Decreased apoptosis | [83] |
| Controlled cortical impact | -Increased angiogenesis | |||
| -Decreased brain oedema | ||||
| -Decreased blood brain barrier leakage |
7. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Grimm, A.; Eckert, A. Brain aging and neurodegeneration: From a mitochondrial point of view. J. Neurochem. 2017, 143, 418–431. [Google Scholar] [CrossRef] [PubMed]
- Eshraghi, M.; Adlimoghaddam, A.; Mahmoodzadeh, A.; Sharifzad, F.; Yasavoli-Sharahi, H.; Lorzadeh, S.; Albensi, B.C.; Ghavami, S. Alzheimer’s Disease Pathogenesis: Role of Autophagy and Mitophagy Focusing in Microglia. Int. J. Mol. Sci. 2021, 22, 3330. [Google Scholar] [CrossRef] [PubMed]
- Grimm, A. Impairments in Brain Bioenergetics in Aging and Tau Pathology: A Chicken and Egg Situation? Cells 2021, 10, 2531. [Google Scholar] [CrossRef] [PubMed]
- Watts, M.E.; Pocock, R.; Claudianos, C. Brain Energy and Oxygen Metabolism: Emerging Role in Normal Function and Disease. Front. Mol. Neurosci. 2018, 11, 216. [Google Scholar] [CrossRef] [PubMed]
- Dagda, R.K. Role of Mitochondrial Dysfunction in Degenerative Brain Diseases, an Overview. Brain Sci. 2018, 8, 178. [Google Scholar] [CrossRef] [PubMed]
- Norat, P.; Soldozy, S.; Sokolowski, J.D.; Gorick, C.M.; Kumar, J.S.; Chae, Y.; Yagmurlu, K.; Prada, F.; Walker, M.; Levitt, M.R.; et al. Mitochondrial dysfunction in neurological disorders: Exploring mitochondrial transplantation. NPJ Regen. Med. 2020, 5, 22. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef]
- Schmitt, K.; Grimm, A.; Kazmierczak, A.; Strosznajder, J.B.; Götz, J.; Eckert, A. Insights into Mitochondrial Dysfunction: Aging, Amyloid-β, and Tau–A Deleterious Trio. Antioxid. Redox Signal. 2012, 16, 1456–1466. [Google Scholar] [CrossRef]
- Gaignard, P.; Liere, P.; Therond, P.; Schumacher, M.; Slama, A.; Guennoun, R. Role of Sex Hormones on Brain Mitochondrial Function, with Special Reference to Aging and Neurodegenerative Diseases. Front. Aging Neurosci. 2017, 9, 406. [Google Scholar] [CrossRef]
- Skulachev, V.P. Mitochondria-targeted antioxidants as promising drugs for treatment of age-related brain diseases. J. Alzheimers Dis. 2012, 28, 283–289. [Google Scholar] [CrossRef]
- Fairley, L.H.; Lai, K.O.; Wong, J.H.; Salvatore, A.V.; D’Agostino, G.; Wu, X.; Jayaraman, A.; Langley, S.; Ruedl, C.; Barron, A. Mitochondrial control of microglial phagocytosis in Alzheimer’s disease. bioRxiv 2021. [Google Scholar] [CrossRef]
- Fairley, L.H.; Sahara, N.; Aoki, I.; Ji, B.; Suhara, T.; Higuchi, M.; Barron, A.M. Neuroprotective effect of mitochondrial translocator protein ligand in a mouse model of tauopathy. J. Neuroinflamm. 2021, 18, 76. [Google Scholar] [CrossRef] [PubMed]
- Grimm, A.; Biliouris, E.E.; Lang, U.E.; Götz, J.; Mensah-Nyagan, A.G.; Eckert, A. Sex hormone-related neurosteroids differentially rescue bioenergetic deficits induced by amyloid-β or hyperphosphorylated tau protein. Cell. Mol. Life Sci. 2015, 73, 201–215. [Google Scholar] [CrossRef] [PubMed]
- Grimm, A.; Schmitt, K.; Lang, U.E.; Mensah-Nyagan, A.G.; Eckert, A. Improvement of neuronal bioenergetics by neurosteroids: Implications for age-related neurodegenerative disorders. Biochim. Biophys. Acta 2014, 1842, 2427–2438. [Google Scholar] [CrossRef] [PubMed]
- Varghese, N.; Werner, S.; Grimm, A.; Eckert, A. Dietary Mitophagy Enhancer: A Strategy for Healthy Brain Aging? Antioxidants 2020, 9, 932. [Google Scholar] [CrossRef] [PubMed]
- Cunnane, S.C.; Trushina, E.; Morland, C.; Prigione, A.; Casadesus, G.; Andrews, Z.B.; Beal, M.F.; Bergersen, L.H.; Brinton, R.D.; de la Monte, S.; et al. Brain energy rescue: An emerging therapeutic concept for neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2020, 19, 609–633. [Google Scholar] [CrossRef]
- Lightowlers, R.N.; Chrzanowska-Lightowlers, Z.M.A.; Russell, O.M. Mitochondrial transplantation—A possible therapeutic for mitochondrial dysfunction? EMBO Rep. 2020, 21, e50964. [Google Scholar] [CrossRef]
- English, K.; Shepherd, A.; Uzor, N.E.; Trinh, R.; Kavelaars, A.; Heijnen, C.J. Astrocytes rescue neuronal health after cisplatin treatment through mitochondrial transfer. Acta Neuropathol. Commun. 2020, 8, 36. [Google Scholar] [CrossRef]
- Ren, D.; Zheng, P.; Zou, S.; Gong, Y.; Wang, Y.; Duan, J.; Deng, J.; Chen, H.; Feng, J.; Zhong, C.; et al. GJA1-20K Enhances Mitochondria Transfer from Astrocytes to Neurons via Cx43-TnTs After Traumatic Brain Injury. Cell. Mol. Neurobiol. 2022, 42, 1887–1895. [Google Scholar] [CrossRef]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef]
- Allen, N.J.; Lyons, D.A. Glia as architects of central nervous system formation and function. Science 2018, 362, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Jakel, S.; Dimou, L. Glial Cells and Their Function in the Adult Brain: A Journey through the History of Their Ablation. Front. Cell Neurosci. 2017, 11, 24. [Google Scholar] [CrossRef] [PubMed]
- Fairley, L.H.; Wong, J.H.; Barron, A.M. Mitochondrial Regulation of Microglial Immunometabolism in Alzheimer’s Disease. Front. Immunol. 2021, 12, 624538. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.N.; Das, S.R.; Emin, M.T.; Wei, M.; Sun, L.; Westphalen, K.; Rowlands, D.J.; Quadri, S.K.; Bhattacharya, S.; Bhattacharya, J. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 2012, 18, 759–765. [Google Scholar] [CrossRef]
- Jackson, M.V.; Morrison, T.J.; Doherty, D.F.; McAuley, D.F.; Matthay, M.A.; Kissenpfennig, A.; O’Kane, C.M.; Krasnodembskaya, A.D. Mitochondrial Transfer via Tunneling Nanotubes is an Important Mechanism by Which Mesenchymal Stem Cells Enhance Macrophage Phagocytosis in the In Vitro and In Vivo Models of ARDS. Stem Cells 2016, 34, 2210–2223. [Google Scholar] [CrossRef]
- Morrison, T.J.; Jackson, M.V.; Cunningham, E.K.; Kissenpfennig, A.; McAuley, D.F.; O’Kane, C.M.; Krasnodembskaya, A.D. Mesenchymal Stromal Cells Modulate Macrophages in Clinically Relevant Lung Injury Models by Extracellular Vesicle Mitochondrial Transfer. Am. J. Respir. Crit. Care Med. 2017, 196, 1275–1286. [Google Scholar] [CrossRef]
- Goldberg, M.P.; Choi, D.W. Combined oxygen and glucose deprivation in cortical cell culture: Calcium-dependent and calcium-independent mechanisms of neuronal injury. J. Neurosci. 1993, 13, 3510–3524. [Google Scholar] [CrossRef]
- Xu, L.; Sapolsky, R.M.; Giffard, R.G. Differential sensitivity of murine astrocytes and neurons from different brain regions to injury. Exp. Neurol. 2001, 169, 416–424. [Google Scholar] [CrossRef]
- Lee, D.R.; Helps, S.C.; Gibbins, I.L.; Nilsson, M.; Sims, N.R. Losses of NG2 and NeuN immunoreactivity but not astrocytic markers during early reperfusion following severe focal cerebral ischemia. Brain Res. 2003, 989, 221–230. [Google Scholar] [CrossRef]
- Hudson, G.; Chinnery, P.F. Mitochondrial DNA polymerase-gamma and human disease. Hum. Mol. Genet. 2006, 15, R244–R252. [Google Scholar] [CrossRef]
- Gao, L.; Zhang, Z.; Lu, J.; Pei, G. Mitochondria Are Dynamically Transferring Between Human Neural Cells and Alexander Disease-Associated GFAP Mutations Impair the Astrocytic Transfer. Front. Cell. Neurosci. 2019, 13, 316. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.-h.O.; Kim, K.-Y.; Bushong, E.A.; Mills, E.A.; Boassa, D.; Shih, T.; Kinebuchi, M.; Phan, S.; Zhou, Y.; Bihlmeyer, N.A.; et al. Transcellular degradation of axonal mitochondria. Proc. Natl. Acad. Sci. USA 2014, 111, 9633–9638. [Google Scholar] [CrossRef] [PubMed]
- Lampinen, R.; Belaya, I.; Saveleva, L.; Liddell, J.R.; Rait, D.; Huuskonen, M.T.; Giniatullina, R.; Sorvari, A.; Soppela, L.; Mikhailov, N.; et al. Neuron-astrocyte transmitophagy is altered in Alzheimer’s disease. Neurobiol. Dis. 2022, 170, 105753. [Google Scholar] [CrossRef]
- Jung, J.E.; Sun, G.; Bautista Garrido, J.; Obertas, L.; Mobley, A.S.; Ting, S.M.; Zhao, X.; Aronowski, J. The Mitochondria-Derived Peptide Humanin Improves Recovery from Intracerebral Hemorrhage: Implication of Mitochondria Transfer and Microglia Phenotype Change. J. Neurosci. 2020, 40, 2154–2165. [Google Scholar] [CrossRef]
- Joshi, A.U.; Minhas, P.S.; Liddelow, S.A.; Haileselassie, B.; Andreasson, K.I.; Dorn, G.W., II; Mochly-Rosen, D. Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat. Neurosci. 2019, 22, 1635–1648. [Google Scholar] [CrossRef] [PubMed]
- Pinto, G.; Saenz-de-Santa-Maria, I.; Chastagner, P.; Perthame, E.; Delmas, C.; Toulas, C.; Moyal-Jonathan-Cohen, E.; Brou, C.; Zurzolo, C. Patient-derived glioblastoma stem cells transfer mitochondria through tunneling nanotubes in tumor organoids. Biochem. J. 2021, 478, 21–39. [Google Scholar] [CrossRef]
- Valdebenito, S.; Malik, S.; Luu, R.; Loudig, O.; Mitchell, M.; Okafo, G.; Bhat, K.; Prideaux, B.; Eugenin, E.A. Tunneling nanotubes, TNT, communicate glioblastoma with surrounding non-tumor astrocytes to adapt them to hypoxic and metabolic tumor conditions. Sci. Rep. 2021, 11, 14556. [Google Scholar] [CrossRef]
- Doyle, L.M.; Wang, M.Z. Overview of Extracellular Vesicles, Their Origin, Composition, Purpose, and Methods for Exosome Isolation and Analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef]
- Falchi, A.M.; Sogos, V.; Saba, F.; Piras, M.; Congiu, T.; Piludu, M. Astrocytes shed large membrane vesicles that contain mitochondria, lipid droplets and ATP. Histochem. Cell Biol. 2012, 139, 221–231. [Google Scholar] [CrossRef]
- D’Acunzo, P.; Perez-Gonzalez, R.; Kim, Y.; Hargash, T.; Miller, C.; Alldred, M.J.; Erdjument-Bromage, H.; Penikalapati, S.C.; Pawlik, M.; Saito, M.; et al. Mitovesicles are a novel population of extracellular vesicles of mitochondrial origin altered in Down syndrome. Sci. Adv. 2021, 7, eabe5085. [Google Scholar] [CrossRef]
- Sugiura, A.; McLelland, G.L.; Fon, E.A.; McBride, H.M. A new pathway for mitochondrial quality control: Mitochondrial-derived vesicles. EMBO J. 2014, 33, 2142–2156. [Google Scholar] [CrossRef] [PubMed]
- Rustom, A.; Saffrich, R.; Markovic, I.; Walther, P.; Gerdes, H.H. Nanotubular highways for intercellular organelle transport. Science 2004, 303, 1007–1010. [Google Scholar] [CrossRef] [PubMed]
- Dupont, M.; Souriant, S.; Lugo-Villarino, G.; Maridonneau-Parini, I.; Verollet, C. Tunneling Nanotubes: Intimate Communication between Myeloid Cells. Front. Immunol. 2018, 9, 43. [Google Scholar] [CrossRef] [PubMed]
- Scheiblich, H.; Dansokho, C.; Mercan, D.; Schmidt, S.V.; Bousset, L.; Wischhof, L.; Eikens, F.; Odainic, A.; Spitzer, J.; Griep, A.; et al. Microglia jointly degrade fibrillar alpha-synuclein cargo by distribution through tunneling nanotubes. Cell 2021, 184, 5089–5106.e5021. [Google Scholar] [CrossRef]
- Wang, Y.; Cui, J.; Sun, X.; Zhang, Y. Tunneling-nanotube development in astrocytes depends on p53 activation. Cell Death Differ. 2010, 18, 732–742. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Y. Tunneling nanotubes between rat primary astrocytes and C6 glioma cells alter proliferation potential of glioma cells. Neurosci. Bull. 2015, 31, 371–378. [Google Scholar] [CrossRef]
- Zhu, D.; Tan, K.S.; Zhang, X.; Sun, A.Y.; Sun, G.Y.; Lee, J.C.M. Hydrogen peroxide alters membrane and cytoskeleton properties and increases intercellular connections in astrocytes. J. Cell Sci. 2005, 118, 3695–3703. [Google Scholar] [CrossRef]
- Wang, X.; Gerdes, H.H. Transfer of mitochondria via tunneling nanotubes rescues apoptotic PC12 cells. Cell Death Differ. 2015, 22, 1181–1191. [Google Scholar] [CrossRef]
- Babenko, V.A.; Silachev, D.N.; Popkov, V.A.; Zorova, L.D.; Pevzner, I.B.; Plotnikov, E.Y.; Sukhikh, G.T.; Zorov, D.B. Miro1 Enhances Mitochondria Transfer from Multipotent Mesenchymal Stem Cells (MMSC) to Neural Cells and Improves the Efficacy of Cell Recovery. Molecules 2018, 23, 687. [Google Scholar] [CrossRef]
- Boukelmoune, N.; Chiu, G.S.; Kavelaars, A.; Heijnen, C.J. Mitochondrial transfer from mesenchymal stem cells to neural stem cells protects against the neurotoxic effects of cisplatin. Acta Neuropathol. Commun. 2018, 6, 139. [Google Scholar] [CrossRef]
- Sartori-Rupp, A.; Cordero Cervantes, D.; Pepe, A.; Gousset, K.; Delage, E.; Corroyer-Dulmont, S.; Schmitt, C.; Krijnse-Locker, J.; Zurzolo, C. Correlative cryo-electron microscopy reveals the structure of TNTs in neuronal cells. Nat. Commun. 2019, 10, 342. [Google Scholar] [CrossRef] [PubMed]
- Torralba, D.; Baixauli, F.; Sanchez-Madrid, F. Mitochondria Know No Boundaries: Mechanisms and Functions of Intercellular Mitochondrial Transfer. Front. Cell Dev. Biol. 2016, 4, 107. [Google Scholar] [CrossRef] [PubMed]
- Goodenough, D.A.; Paul, D.L. Gap junctions. Cold Spring Harb. Perspect. Biol. 2009, 1, a002576. [Google Scholar] [CrossRef] [PubMed]
- Norris, R.P. Transfer of mitochondria and endosomes between cells by gap junction internalization. Traffic 2021, 22, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, C.; He, T.; Zhao, T.; Chen, Y.Y.; Shen, Y.L.; Zhang, X.; Wang, L.L. Mitochondrial Transfer from Bone Marrow Mesenchymal Stem Cells to Motor Neurons in Spinal Cord Injury Rats via Gap Junction. Theranostics 2019, 9, 2017–2035. [Google Scholar] [CrossRef] [PubMed]
- Peruzzotti-Jametti, L.; Bernstock, J.D.; Willis, C.M.; Manferrari, G.; Rogall, R.; Fernandez-Vizarra, E.; Williamson, J.C.; Braga, A.; van den Bosch, A.; Leonardi, T.; et al. Neural stem cells traffic functional mitochondria via extracellular vesicles. PLoS Biol. 2021, 19, e3001166. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, P.A.; Aizenman, E. Hundred-fold increase in neuronal vulnerability to glutamate toxicity in astrocyte-poor cultures of rat cerebral cortex. Neurosci. Lett. 1989, 103, 162–168. [Google Scholar] [CrossRef]
- Voloboueva, L.A.; Suh, S.W.; Swanson, R.A.; Giffard, R.G. Inhibition of mitochondrial function in astrocytes: Implications for neuroprotection. J. Neurochem. 2007, 102, 1383–1394. [Google Scholar] [CrossRef]
- Gao, L.; Liu, F.; Hou, P.P.; Manaenko, A.; Xiao, Z.P.; Wang, F.; Xu, T.L.; Hu, Q. Neurons Release Injured Mitochondria as “Help-Me” Signaling After Ischemic Stroke. Front. Aging Neurosci. 2022, 14, 785761. [Google Scholar] [CrossRef]
- Morales, I.; Sanchez, A.; Puertas-Avendaño, R.; Rodriguez-Sabate, C.; Perez-Barreto, A.; Rodriguez, M. Neuroglial transmitophagy and Parkinson’s disease. Glia 2020, 68, 2277–2299. [Google Scholar] [CrossRef]
- Sharma, M.; Ramirez Jarquin, U.N.; Rivera, O.; Kazantzis, M.; Eshraghi, M.; Shahani, N.; Sharma, V.; Tapia, R.; Subramaniam, S. Rhes, a striatal-enriched protein, promotes mitophagy via Nix. Proc. Natl. Acad. Sci. USA 2019, 116, 23760–23771. [Google Scholar] [CrossRef] [PubMed]
- Mistry, J.J.; Marlein, C.R.; Moore, J.A.; Hellmich, C.; Wojtowicz, E.E.; Smith, J.G.W.; Macaulay, I.; Sun, Y.; Morfakis, A.; Patterson, A.; et al. ROS-mediated PI3K activation drives mitochondrial transfer from stromal cells to hematopoietic stem cells in response to infection. Proc. Natl. Acad. Sci. USA 2019, 116, 24610–24619. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Boza-Serrano, A.; Dunning, C.J.R.; Clausen, B.H.; Lambertsen, K.L.; Deierborg, T. Inflammation leads to distinct populations of extracellular vesicles from microglia. J. Neuroinflamm. 2018, 15, 168. [Google Scholar] [CrossRef] [PubMed]
- Valdinocci, D.; Simoes, R.F.; Kovarova, J.; Cunha-Oliveira, T.; Neuzil, J.; Pountney, D.L. Intracellular and Intercellular Mitochondrial Dynamics in Parkinson’s Disease. Front. Neurosci. 2019, 13, 930. [Google Scholar] [CrossRef]
- Valdinocci, D.; Kovarova, J.; Neuzil, J.; Pountney, D.L. Alpha-Synuclein Aggregates Associated with Mitochondria in Tunnelling Nanotubes. Neurotox. Res. 2021, 39, 429–443. [Google Scholar] [CrossRef]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science 2004, 304, 448–452. [Google Scholar] [CrossRef]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef]
- Devi, L.; Prabhu, B.M.; Galati, D.F.; Avadhani, N.G.; Anandatheerthavarada, H.K. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J. Neurosci. 2006, 26, 9057–9068. [Google Scholar] [CrossRef]
- Tardivel, M.; Begard, S.; Bousset, L.; Dujardin, S.; Coens, A.; Melki, R.; Buee, L.; Colin, M. Tunneling nanotube (TNT)-mediated neuron-to neuron transfer of pathological Tau protein assemblies. Acta Neuropathol. Commun. 2016, 4, 117. [Google Scholar] [CrossRef]
- Zhang, K.; Sun, Z.; Chen, X.; Zhang, Y.; Guo, A.; Zhang, Y. Intercellular transport of Tau protein and beta-amyloid mediated by tunneling nanotubes. Am. J. Transl. Res. 2021, 13, 12509–12522. [Google Scholar]
- Sun, J.; Liu, Q.; Zhang, X.; Dun, S.; Liu, L. Mitochondrial hijacking: A potential mechanism for SARS-CoV-2 to impair female fertility. Med. Hypotheses 2022, 160, 110778. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.E.; Paek, S.H. Mitochondrial Dysfunction in Parkinson’s Disease. Exp. Neurobiol. 2015, 24, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Barcelos, I.P.; Troxell, R.M.; Graves, J.S. Mitochondrial Dysfunction and Multiple Sclerosis. Biology 2019, 8, 37. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Lu, J.; Manaenko, A.; Tang, J.; Hu, Q. Mitochondria in Ischemic Stroke: New Insight and Implications. Aging Dis. 2018, 9, 924–937. [Google Scholar] [CrossRef] [PubMed]
- Hiebert, J.B.; Shen, Q.; Thimmesch, A.R.; Pierce, J.D. Traumatic brain injury and mitochondrial dysfunction. Am. J. Med. Sci. 2015, 350, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Valenti, D.; Vacca, R.A.; Moro, L.; Atlante, A. Mitochondria Can Cross Cell Boundaries: An Overview of the Biological Relevance, Pathophysiological Implications and Therapeutic Perspectives of Intercellular Mitochondrial Transfer. Int. J. Mol. Sci. 2021, 22, 8312. [Google Scholar] [CrossRef]
- Rajasekaran, A.; Venkatasubramanian, G.; Berk, M.; Debnath, M. Mitochondrial dysfunction in schizophrenia: Pathways, mechanisms and implications. Neurosci. Biobehav. Rev. 2015, 48, 10–21. [Google Scholar] [CrossRef]
- Huang, P.J.; Kuo, C.C.; Lee, H.C.; Shen, C.I.; Cheng, F.C.; Wu, S.F.; Chang, J.C.; Pan, H.C.; Lin, S.Z.; Liu, C.S.; et al. Transferring Xenogenic Mitochondria Provides Neural Protection Against Ischemic Stress in Ischemic Rat Brains. Cell Transpl. 2016, 25, 913–927. [Google Scholar] [CrossRef]
- Nakamura, Y.; Lo, E.H.; Hayakawa, K. Placental Mitochondria Therapy for Cerebral Ischemia-Reperfusion Injury in Mice. Stroke 2020, 51, 3142–3146. [Google Scholar] [CrossRef]
- Tashiro, R.; Bautista-Garrido, J.; Ozaki, D.; Sun, G.; Obertas, L.; Mobley, A.S.; Kim, G.S.; Aronowski, J.; Jung, J.E. Transplantation of astrocytic mitochondria modulates neuronal antioxidant defense and neuroplasticity and promotes functional recovery after intracerebral hemorrhage. J. Neurosci. 2022, 42, 7001–7014. [Google Scholar] [CrossRef]
- Pourmohammadi-Bejarpasi, Z.; Roushandeh, A.M.; Saberi, A.; Rostami, M.K.; Toosi, S.M.R.; Jahanian-Najafabadi, A.; Tomita, K.; Kuwahara, Y.; Sato, T.; Roudkenar, M.H. Mesenchymal stem cells-derived mitochondria transplantation mitigates I/R-induced injury, abolishes I/R-induced apoptosis, and restores motor function in acute ischemia stroke rat model. Brain Res. Bull. 2020, 165, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Lippert, T.; Borlongan, C.V. Prophylactic treatment of hyperbaric oxygen treatment mitigates inflammatory response via mitochondria transfer. CNS Neurosci. 2019, 25, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Gao, Y.; Li, Q.; Sun, D.; Dong, X.; Li, X.; Xin, W.; Zhang, J. Effects of Brain-Derived Mitochondria on the Function of Neuron and Vascular Endothelial Cell After Traumatic Brain Injury. World Neurosurg. 2020, 138, e1–e9. [Google Scholar] [CrossRef]
- Zhao, J.; Qu, D.; Xi, Z.; Huan, Y.; Zhang, K.; Yu, C.; Yang, D.; Kang, J.; Lin, W.; Wu, S.; et al. Mitochondria transplantation protects traumatic brain injury via promoting neuronal survival and astrocytic BDNF. Transl. Res. 2021, 235, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Nitzan, K.; Benhamron, S.; Valitsky, M.; Kesner, E.E.; Lichtenstein, M.; Ben-Zvi, A.; Ella, E.; Segalstein, Y.; Saada, A.; Lorberboum-Galski, H.; et al. Mitochondrial Transfer Ameliorates Cognitive Deficits, Neuronal Loss, and Gliosis in Alzheimer’s Disease Mice. J. Alzheimers Dis. 2019, 72, 587–604. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.C.; Wu, S.L.; Liu, K.H.; Chen, Y.H.; Chuang, C.S.; Cheng, F.C.; Su, H.L.; Wei, Y.H.; Kuo, S.J.; Liu, C.S. Allogeneic/xenogeneic transplantation of peptide-labeled mitochondria in Parkinson’s disease: Restoration of mitochondria functions and attenuation of 6-hydroxydopamine-induced neurotoxicity. Transl. Res. 2016, 170, 40–56.e43. [Google Scholar] [CrossRef]
- Chang, J.C.; Chao, Y.C.; Chang, H.S.; Wu, Y.L.; Chang, H.J.; Lin, Y.S.; Cheng, W.L.; Lin, T.T.; Liu, C.S. Intranasal delivery of mitochondria for treatment of Parkinson’s Disease model rats lesioned with 6-hydroxydopamine. Sci. Rep. 2021, 11, 10597. [Google Scholar] [CrossRef]
- Shi, X.; Zhao, M.; Fu, C.; Fu, A. Intravenous administration of mitochondria for treating experimental Parkinson’s disease. Mitochondrion 2017, 34, 91–100. [Google Scholar] [CrossRef]
- Cheng, X.Y.; Biswas, S.; Li, J.; Mao, C.J.; Chechneva, O.; Chen, J.; Li, K.; Li, J.; Zhang, J.R.; Liu, C.F.; et al. Human iPSCs derived astrocytes rescue rotenone-induced mitochondrial dysfunction and dopaminergic neurodegeneration in vitro by donating functional mitochondria. Transl. Neurodegener. 2020, 9, 13. [Google Scholar] [CrossRef]
- Rosenfeld, M.; Brenner-Lavie, H.; Ari, S.G.; Kavushansky, A.; Ben-Shachar, D. Perturbation in mitochondrial network dynamics and in complex I dependent cellular respiration in schizophrenia. Biol. Psychiatry 2011, 69, 980–988. [Google Scholar] [CrossRef]
- Prabakaran, S.; Swatton, J.E.; Ryan, M.M.; Huffaker, S.J.; Huang, J.T.; Griffin, J.L.; Wayland, M.; Freeman, T.; Dudbridge, F.; Lilley, K.S.; et al. Mitochondrial dysfunction in schizophrenia: Evidence for compromised brain metabolism and oxidative stress. Mol. Psychiatry 2004, 9, 684–697. [Google Scholar] [CrossRef] [PubMed]
- Bergman, O.; Ben-Shachar, D. Mitochondrial Oxidative Phosphorylation System (OXPHOS) Deficits in Schizophrenia: Possible Interactions with Cellular Processes. Can. J. Psychiatry 2016, 61, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Dror, N.; Klein, E.; Karry, R.; Sheinkman, A.; Kirsh, Z.; Mazor, M.; Tzukerman, M.; Ben-Shachar, D. State-dependent alterations in mitochondrial complex I activity in platelets: A potential peripheral marker for schizophrenia. Mol. Psychiatry 2002, 7, 995–1001. [Google Scholar] [CrossRef]
- Faizi, M.; Salimi, A.; Rasoulzadeh, M.; Naserzadeh, P.; Pourahmad, J. Schizophrenia induces oxidative stress and cytochrome C release in isolated rat brain mitochondria: A possible pathway for induction of apoptosis and neurodegeneration. Iran. J. Pharm. Res. 2014, 13, 93–100. [Google Scholar]
- D’Antoni, S.; de Bari, L.; Valenti, D.; Borro, M.; Bonaccorso, C.M.; Simmaco, M.; Vacca, R.A.; Catania, M.V. Aberrant mitochondrial bioenergetics in the cerebral cortex of the Fmr1 knockout mouse model of fragile X syndrome. Biol. Chem. 2020, 401, 497–503. [Google Scholar] [CrossRef]
- Robicsek, O.; Ene, H.M.; Karry, R.; Ytzhaki, O.; Asor, E.; McPhie, D.; Cohen, B.M.; Ben-Yehuda, R.; Weiner, I.; Ben-Shachar, D. Isolated Mitochondria Transfer Improves Neuronal Differentiation of Schizophrenia-Derived Induced Pluripotent Stem Cells and Rescues Deficits in a Rat Model of the Disorder. Schizophr. Bull. 2018, 44, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Ha, B.G.; Heo, J.Y.; Jang, Y.J.; Park, T.S.; Choi, J.Y.; Jang, W.Y.; Jeong, S.J. Depletion of Mitochondrial Components from Extracellular Vesicles Secreted from Astrocytes in a Mouse Model of Fragile X Syndrome. Int. J. Mol. Sci. 2021, 22, 410. [Google Scholar] [CrossRef]
- Andres, A.L.; Gong, X.; Di, K.; Bota, D.A. Low-doses of cisplatin injure hippocampal synapses: A mechanism for ‘chemo’ brain? Exp. Neurol. 2014, 255, 137–144. [Google Scholar] [CrossRef]
- Guerreiro, S.; Privat, A.L.; Bressac, L.; Toulorge, D. CD38 in Neurodegeneration and Neuroinflammation. Cells 2020, 9, 471. [Google Scholar] [CrossRef]
- Marlein, C.R.; Piddock, R.E.; Mistry, J.J.; Zaitseva, L.; Hellmich, C.; Horton, R.H.; Zhou, Z.; Auger, M.J.; Bowles, K.M.; Rushworth, S.A. CD38-Driven Mitochondrial Trafficking Promotes Bioenergetic Plasticity in Multiple Myeloma. Cancer Res. 2019, 79, 2285–2297. [Google Scholar] [CrossRef]
- Bruzzone, S.; Verderio, C.; Schenk, U.; Fedele, E.; Zocchi, E.; Matteoli, M.; De Flora, A. Glutamate-mediated overexpression of CD38 in astrocytes cultured with neurones. J. Neurochem. 2004, 89, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Nakamura, Y.; Li, W.; Hamanaka, G.; Arai, K.; Lo, E.H.; Hayakawa, K. Effects of O-GlcNAcylation on functional mitochondrial transfer from astrocytes. J. Cereb. Blood Flow Metab. 2021, 41, 1523–1535. [Google Scholar] [CrossRef] [PubMed]
- Mueller, T.; Ouyang, X.; Johnson, M.S.; Qian, W.J.; Chatham, J.C.; Darley-Usmar, V.; Zhang, J. New Insights Into the Biology of Protein O-GlcNAcylation: Approaches and Observations. Front. Aging 2020, 1, 620382. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Qian, K. Protein O-GlcNAcylation: Emerging mechanisms and functions. Nat. Rev. Mol. Cell. Biol. 2017, 18, 452–465. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Feng, Z.; Yang, X.; Liu, J. The regulatory roles of O-GlcNAcylation in mitochondrial homeostasis and metabolic syndrome. Free Radic. Res. 2016, 50, 1080–1088. [Google Scholar] [CrossRef]
- Ahmad, T.; Mukherjee, S.; Pattnaik, B.; Kumar, M.; Singh, S.; Kumar, M.; Rehman, R.; Tiwari, B.K.; Jha, K.A.; Barhanpurkar, A.P.; et al. Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J. 2014, 33, 994–1010. [Google Scholar] [CrossRef]
- Mahrouf-Yorgov, M.; Augeul, L.; Da Silva, C.C.; Jourdan, M.; Rigolet, M.; Manin, S.; Ferrera, R.; Ovize, M.; Henry, A.; Guguin, A.; et al. Mesenchymal stem cells sense mitochondria released from damaged cells as danger signals to activate their rescue properties. Cell Death Differ. 2017, 24, 1224–1238. [Google Scholar] [CrossRef]
- Clark, M.A.; Shay, J.W. Mitochondrial transformation of mammalian cells. Nature 1982, 295, 605–607. [Google Scholar] [CrossRef]
- Masuzawa, A.; Black, K.M.; Pacak, C.A.; Ericsson, M.; Barnett, R.J.; Drumm, C.; Seth, P.; Bloch, D.B.; Levitsky, S.; Cowan, D.B.; et al. Transplantation of autologously derived mitochondria protects the heart from ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H966–H982. [Google Scholar] [CrossRef]
- Lin, H.C.; Lai, I.R. Isolated mitochondria infusion mitigates ischemia-reperfusion injury of the liver in rats: Reply. Shock 2013, 39, 543. [Google Scholar] [CrossRef]
- Moschoi, R.; Imbert, V.; Nebout, M.; Chiche, J.; Mary, D.; Prebet, T.; Saland, E.; Castellano, R.; Pouyet, L.; Collette, Y.; et al. Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood 2016, 128, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Paliwal, S.; Chaudhuri, R.; Agrawal, A.; Mohanty, S. Human tissue-specific MSCs demonstrate differential mitochondria transfer abilities that may determine their regenerative abilities. Stem Cell Res. 2018, 9, 298. [Google Scholar] [CrossRef] [PubMed]
- Gollihue, J.L.; Patel, S.P.; Eldahan, K.C.; Cox, D.H.; Donahue, R.R.; Taylor, B.K.; Sullivan, P.G.; Rabchevsky, A.G. Effects of Mitochondrial Transplantation on Bioenergetics, Cellular Incorporation, and Functional Recovery after Spinal Cord Injury. J. Neurotrauma 2018, 35, 1800–1818. [Google Scholar] [CrossRef] [PubMed]
- Heydari, K.; Shamshirian, A.; Lotfi-Foroushani, P.; Aref, A.; Hedayatizadeh-Omran, A.; Ahmadi, M.; Janbabei, G.; Keyhanian, S.; Zaboli, E.; Ghasemzadeh, S.M.; et al. The risk of malignancies in patients receiving hematopoietic stem cell transplantation: A systematic review and meta-analysis. Clin. Transl. Oncol. 2020, 22, 1825–1837. [Google Scholar] [CrossRef]
- Morris, M.C.; Depollier, J.; Mery, J.; Heitz, F.; Divita, G. A peptide carrier for the delivery of biologically active proteins into mammalian cells. Nat. Biotechnol. 2001, 19, 1173–1176. [Google Scholar] [CrossRef]
- Chang, J.C.; Liu, K.H.; Chuang, C.S.; Su, H.L.; Wei, Y.H.; Kuo, S.J.; Liu, C.S. Treatment of human cells derived from MERRF syndrome by peptide-mediated mitochondrial delivery. Cytotherapy 2013, 15, 1580–1596. [Google Scholar] [CrossRef]
- Emani, S.M.; Piekarski, B.L.; Harrild, D.; Del Nido, P.J.; McCully, J.D. Autologous mitochondrial transplantation for dysfunction after ischemia-reperfusion injury. J. Thorac. Cardiovasc. Surg. 2017, 154, 286–289. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fairley, L.H.; Grimm, A.; Eckert, A. Mitochondria Transfer in Brain Injury and Disease. Cells 2022, 11, 3603. https://doi.org/10.3390/cells11223603
Fairley LH, Grimm A, Eckert A. Mitochondria Transfer in Brain Injury and Disease. Cells. 2022; 11(22):3603. https://doi.org/10.3390/cells11223603
Chicago/Turabian StyleFairley, Lauren H., Amandine Grimm, and Anne Eckert. 2022. "Mitochondria Transfer in Brain Injury and Disease" Cells 11, no. 22: 3603. https://doi.org/10.3390/cells11223603
APA StyleFairley, L. H., Grimm, A., & Eckert, A. (2022). Mitochondria Transfer in Brain Injury and Disease. Cells, 11(22), 3603. https://doi.org/10.3390/cells11223603