


Actin-Binding Proteins in Cardiac Hypertrophy

Abstract
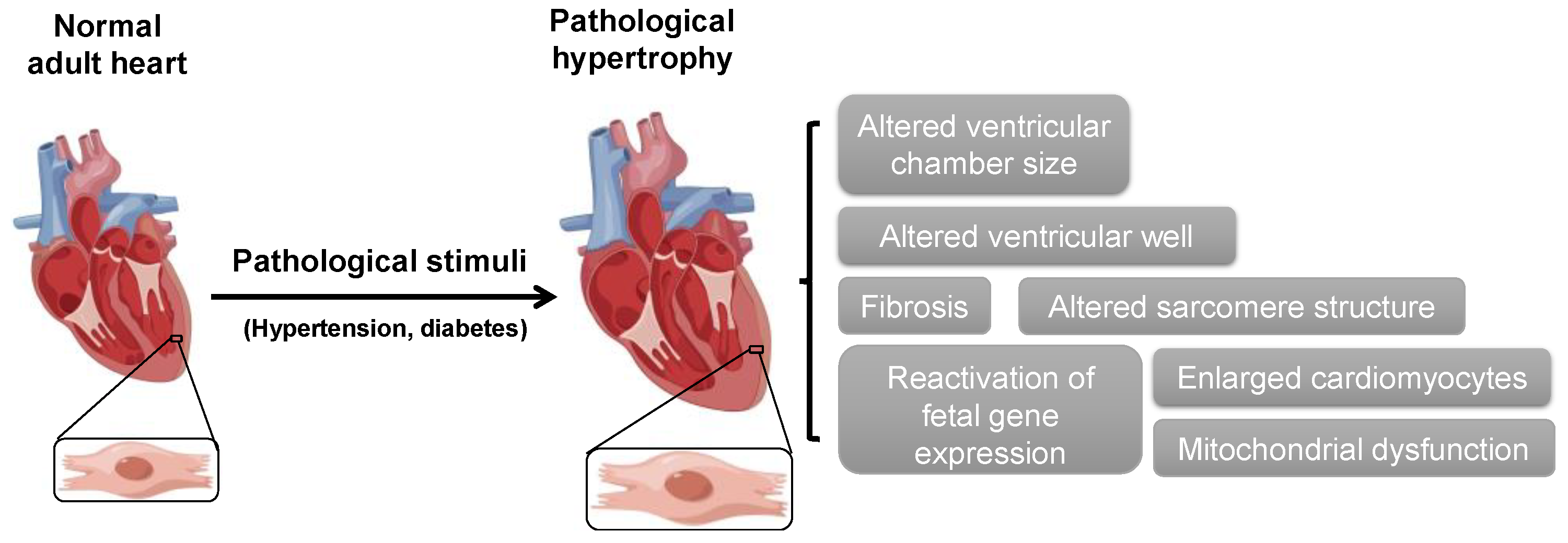
1. Introduction
2. ABPs in Cardiac Hypertrophy
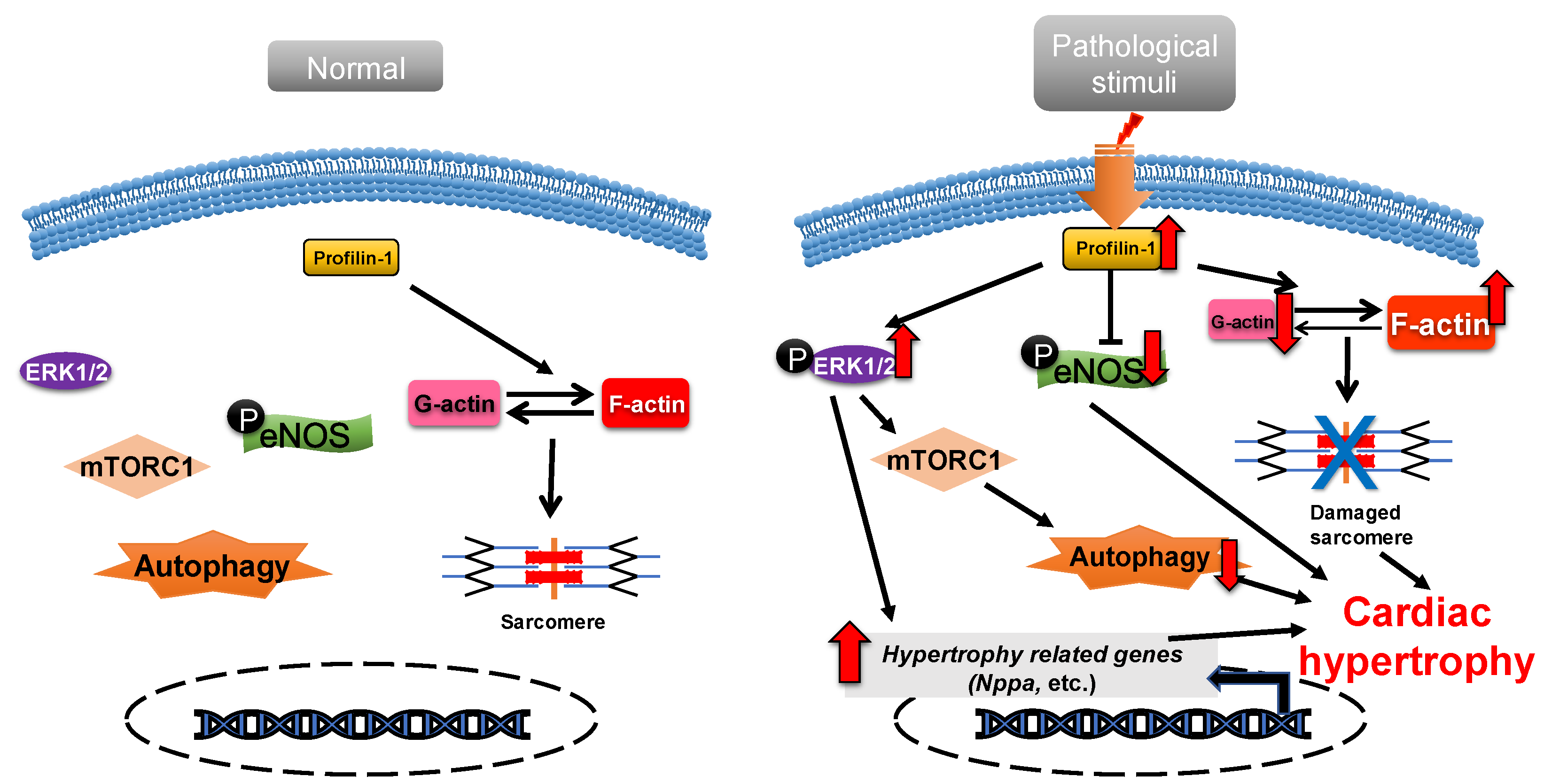
2.1. Profilin-1
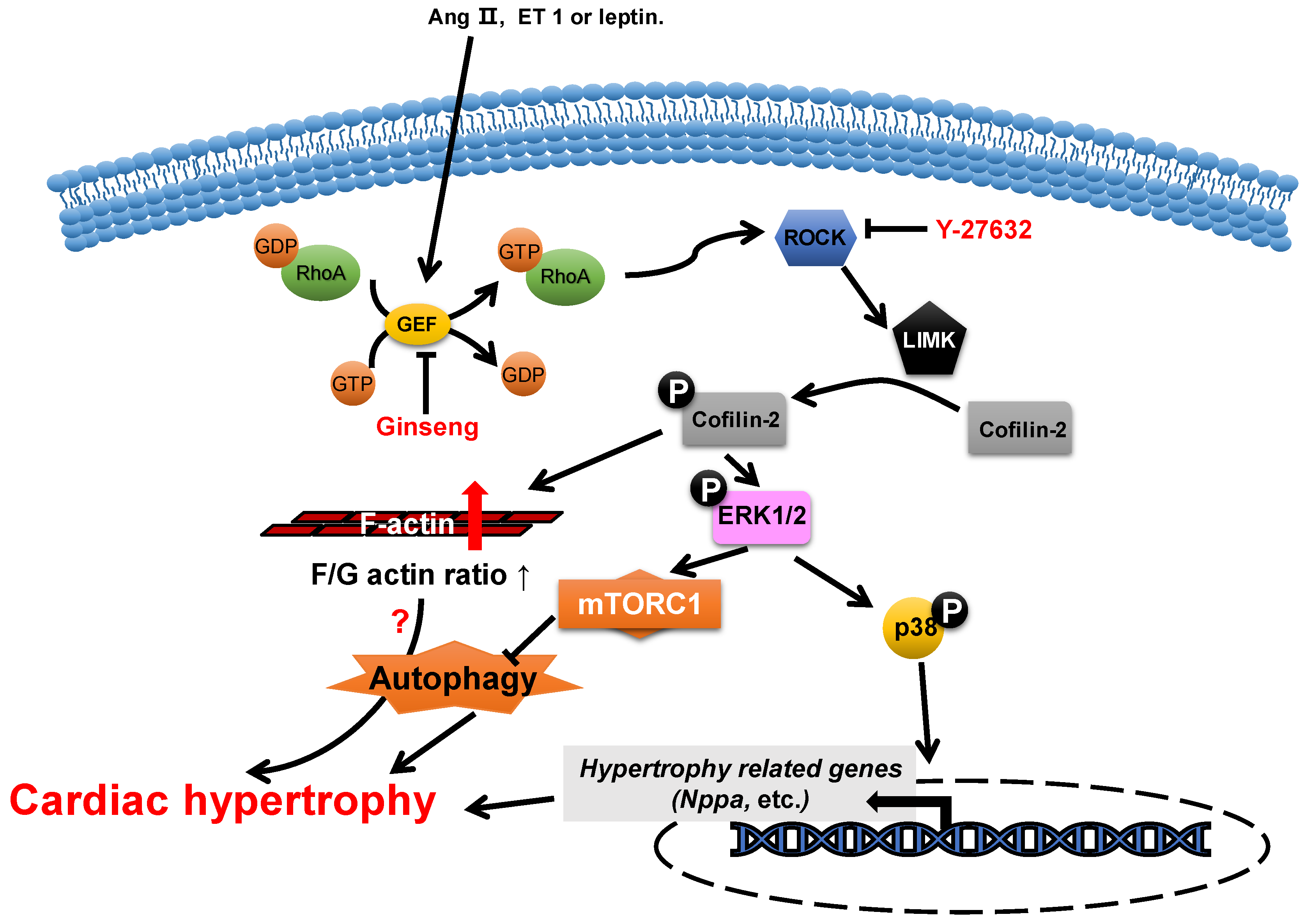
2.2. ADF/Cofilin
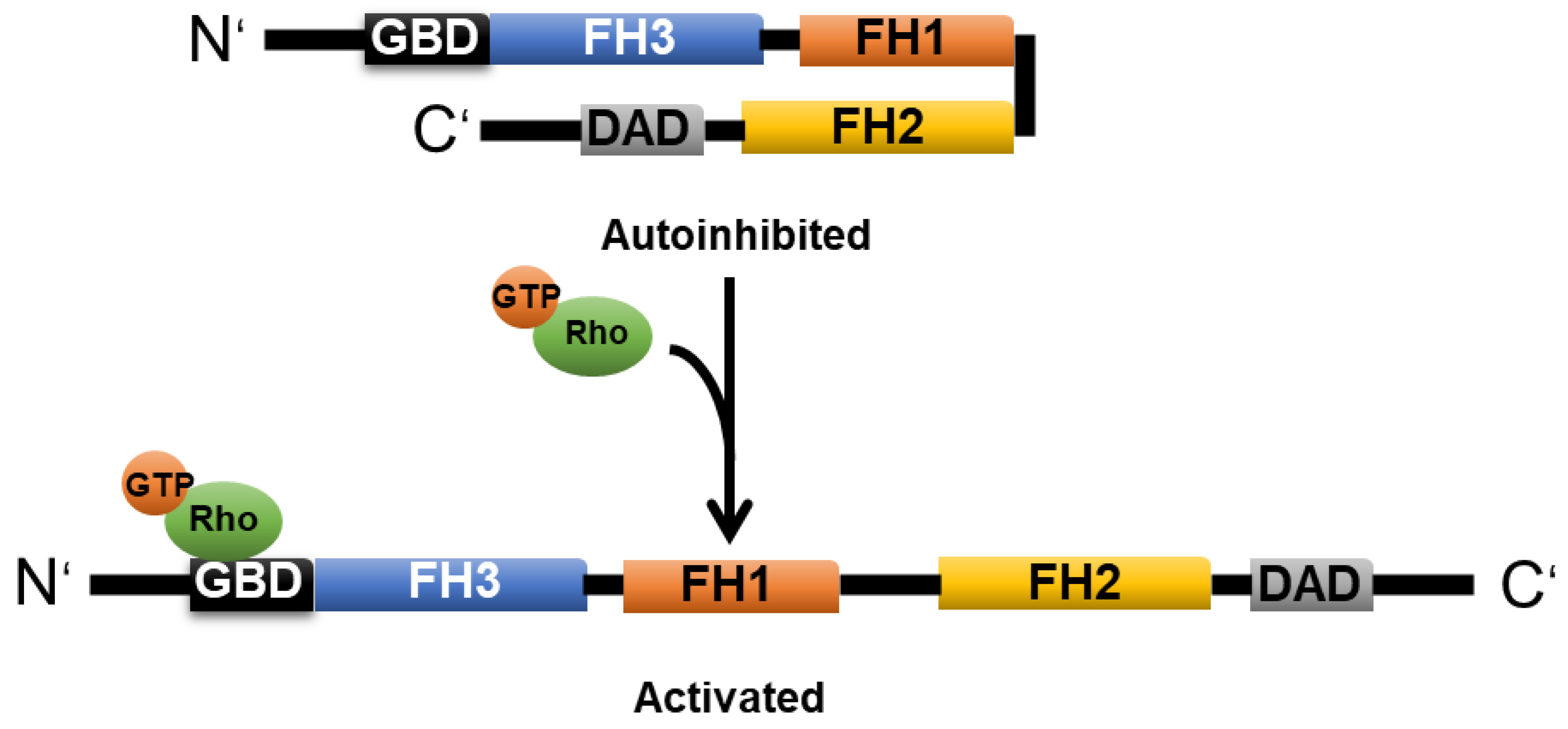
2.3. Formin
2.3.1. mDia1
2.3.2. FHOD3
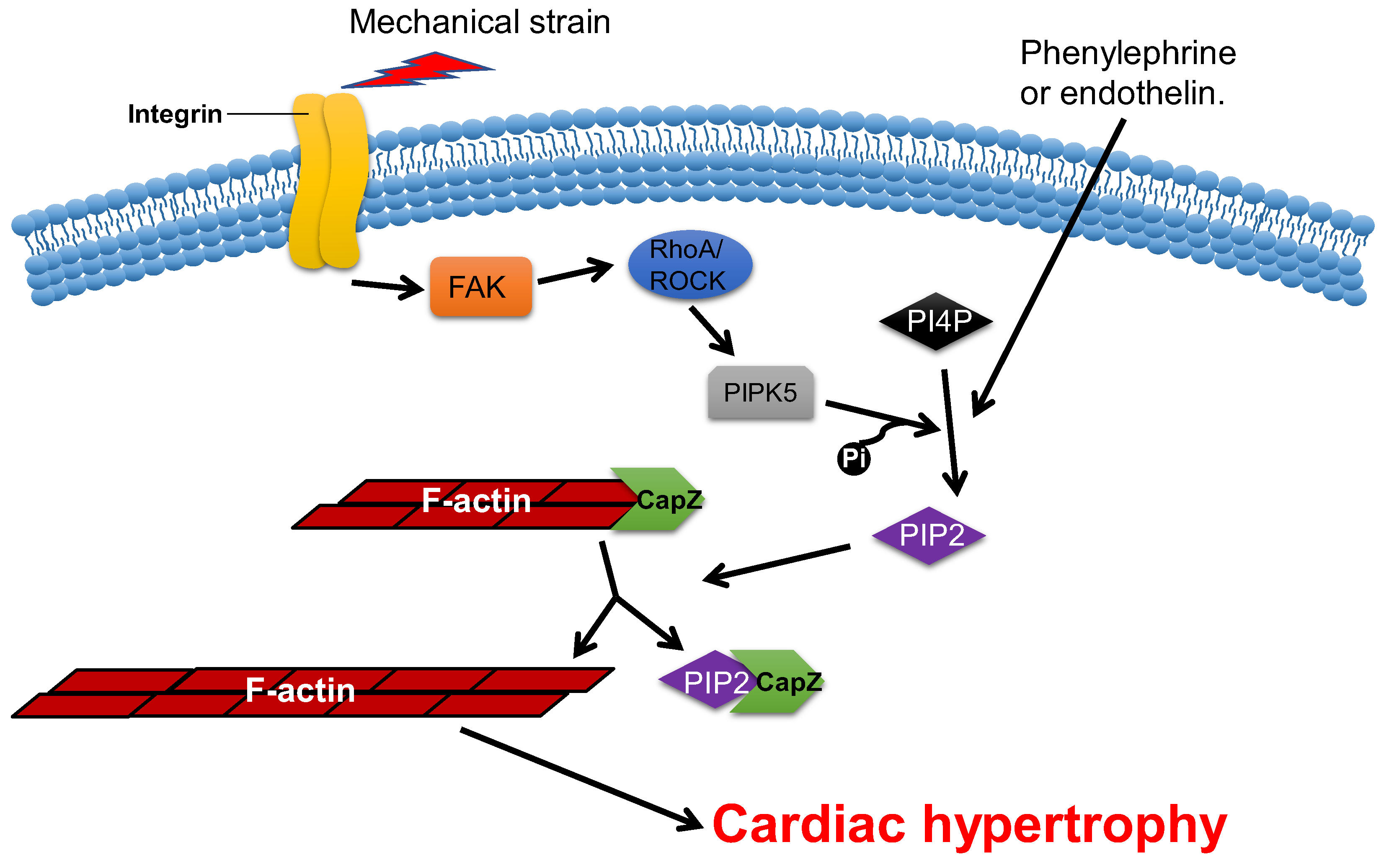
2.4. CapZ
2.5. Gelsolin
2.6. Human Heart LIM Protein
2.7. Myosin
2.8. Dystrophin
2.9. Other ABPs
3. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Heidenreich, P.A.; Bozkurt, B.; Aguilar, D.; Allen, L.A.; Byun, J.J.; Colvin, M.M.; Deswal, A.; Drazner, M.H.; Dunlay, S.M.; Evers, L.R.; et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2022, 145, e895–e1032. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407. [Google Scholar] [CrossRef] [PubMed]
- Kehat, I.; Molkentin, J.D. Molecular pathways underlying cardiac remodeling during pathophysiological stimulation. Circulation 2010, 122, 2727–2735. [Google Scholar] [CrossRef] [PubMed]
- Frey, N.; Olson, E.N. Cardiac hypertrophy: The good, the bad, and the ugly. Annu. Rev. Physiol. 2003, 65, 45–79. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, W.; Deng, K.Q.; Tian, S.; Liu, H.; Shi, H.; Fang, Q.; Liu, Z.; Chen, Z.; Tian, T.; et al. The E3 Ligase TRIM16 Is a Key Suppressor of Pathological Cardiac Hypertrophy. Circ. Res. 2022, 130, 1586–1600. [Google Scholar] [CrossRef]
- Chen, Q.Q.; Ma, G.; Liu, J.F.; Cai, Y.Y.; Zhang, J.Y.; Wei, T.T.; Pan, A.; Jiang, S.; Xiao, Y.; Xiao, P.; et al. Neuraminidase 1 is a driver of experimental cardiac hypertrophy. Eur. Heart J. 2021, 42, 3770–3782. [Google Scholar] [CrossRef]
- Zhang, Y.; Da, Q.; Cao, S.; Yan, K.; Shi, Z.; Miao, Q.; Li, C.; Hu, L.; Sun, S.; Wu, W.; et al. HINT1 (Histidine Triad Nucleotide-Binding Protein 1) Attenuates Cardiac Hypertrophy Via Suppressing HOXA5 (Homeobox A5) Expression. Circulation 2021, 144, 638–654. [Google Scholar] [CrossRef]
- Pollard, T.D.; Cooper, J.A. Actin, a central player in cell shape and movement. Science 2009, 326, 1208–1212. [Google Scholar] [CrossRef]
- Winder, S.J.; Ayscough, K.R. Actin-binding proteins. J. Cell Sci. 2005, 118, 651–654. [Google Scholar] [CrossRef]
- Chalut, K.J.; Paluch, E.K. The Actin Cortex: A Bridge between Cell Shape and Function. Dev. Cell 2016, 38, 571–573. [Google Scholar] [CrossRef]
- Pollard, T.D. Actin and Actin-Binding Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a018226. [Google Scholar] [CrossRef] [PubMed]
- Pluess, M.; Ehler, E. Cardiac Cytoarchitecture in Health and Disease. In Cardiac Cytoarchitecture: How to Maintain a Working Heart; Ehler, E., Ed.; Springer: Cham, Switzerland, 2015; pp. 1–14. [Google Scholar]
- Huang, X.; Li, Z.; Hu, J.; Yang, Z.; Liu, Z.; Zhang, T.; Zhang, C.; Yuan, B. Knockout of Wdr1 results in cardiac hypertrophy and impaired cardiac function in adult mouse heart. Gene 2019, 697, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Kruppa, A.J.; Kendrick-Jones, J.; Buss, F. Myosins, Actin and Autophagy. Traffic 2016, 17, 878–890. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, M.O.; Berón, W.; Colombo, M.I. The actin cytoskeleton participates in the early events of autophagosome formation upon starvation induced autophagy. Autophagy 2012, 8, 1590–1603. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, C.; Ji, Y.; Chen, Z.; Kitazato, K.; Xiang, Y.; Zhong, M.; Wang, Q.; Pei, Y.; Ju, H.; Wang, Y. Proteomics analysis of autophagy-deficient Atg7-/- MEFs reveals a close relationship between F-actin and autophagy. Biochem. Biophys. Res. Commun. 2013, 437, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.H.; Qiu, J.; Wang, Y.; Ji, X.; Liu, X.J.; You, B.A.; Sheng, Y.P.; Li, X.; Gao, H.Q. Profilin-1 promotes the development of hypertension-induced cardiac hypertrophy. J. Hypertens. 2013, 31, 576–586; discussion 586. [Google Scholar] [CrossRef]
- Zeidan, A.; Javadov, S.; Karmazyn, M. Essential role of Rho/ROCK-dependent processes and actin dynamics in mediating leptin-induced hypertrophy in rat neonatal ventricular myocytes. Cardiovasc. Res. 2006, 72, 101–111. [Google Scholar] [CrossRef]
- Hunter, J.C.; Zeidan, A.; Javadov, S.; Kilic, A.; Rajapurohitam, V.; Karmazyn, M. Nitric oxide inhibits endothelin-1-induced neonatal cardiomyocyte hypertrophy via a RhoA-ROCK-dependent pathway. J. Mol. Cell. Cardiol. 2009, 47, 810–818. [Google Scholar] [CrossRef]
- Funk, J.; Merino, F.; Venkova, L.; Heydenreich, L.; Kierfeld, J.; Vargas, P.; Raunser, S.; Piel, M.; Bieling, P. Profilin and formin constitute a pacemaker system for robust actin filament growth. eLife 2019, 8, e50963. [Google Scholar] [CrossRef]
- Safer, D.; Elzinga, M.; Nachmias, V.T. Thymosin beta 4 and Fx, an actin-sequestering peptide, are indistinguishable. J. Biol. Chem. 1991, 266, 4029–4032. [Google Scholar] [CrossRef]
- Rybakova, I.N.; Amann, K.J.; Ervasti, J.M. A new model for the interaction of dystrophin with F-actin. J. Cell Biol. 1996, 135, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Yonis, A.; Vaghela, M.; Barriga, E.H.; Chugh, P.; Smith, M.B.; Maufront, J.; Lavoie, G.; Méant, A.; Ferber, E.; et al. SPIN90 associates with mDia1 and the Arp2/3 complex to regulate cortical actin organization. Nat. Cell Biol. 2020, 22, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, S.; Geyer, M. Formins as effector proteins of Rho GTPases. Small GTPases 2014, 5, e29513. [Google Scholar] [CrossRef] [PubMed]
- Chereau, D.; Boczkowska, M.; Skwarek-Maruszewska, A.; Fujiwara, I.; Hayes, D.B.; Rebowski, G.; Lappalainen, P.; Pollard, T.D.; Dominguez, R. Leiomodin is an actin filament nucleator in muscle cells. Science 2008, 320, 239–243. [Google Scholar] [CrossRef]
- Hosokawa, N.; Kuragano, M.; Yoshino, A.; Shibata, K.; Uyeda, T.Q.P.; Tokuraku, K. Unidirectional cooperative binding of fimbrin actin-binding domain 2 to actin filament. Biochem. Biophys. Res. Commun. 2021, 552, 59–65. [Google Scholar] [CrossRef]
- Sobral, A.F.; Chan, F.Y.; Norman, M.J.; Osorio, D.S.; Dias, A.B.; Ferreira, V.; Barbosa, D.J.; Cheerambathur, D.; Gassmann, R.; Belmonte, J.M.; et al. Plastin and spectrin cooperate to stabilize the actomyosin cortex during cytokinesis. Curr. Biol. 2021, 31, 5415–5428.e10. [Google Scholar] [CrossRef]
- Zheng, B.; Wen, J.K.; Han, M. hhLIM is a novel F-actin binding protein involved in actin cytoskeleton remodeling. FEBS J. 2008, 275, 1568–1578. [Google Scholar] [CrossRef]
- Archer, S.K.; Claudianos, C.; Campbell, H.D. Evolution of the gelsolin family of actin-binding proteins as novel transcriptional coactivators. BioEssays 2005, 27, 388–396. [Google Scholar] [CrossRef]
- Chen, Q.; Courtemanche, N.; Pollard, T.D. Aip1 promotes actin filament severing by cofilin and regulates constriction of the cytokinetic contractile ring. J. Biol. Chem. 2015, 290, 2289–2300. [Google Scholar] [CrossRef]
- Burtnick, L.D.; Urosev, D.; Irobi, E.; Narayan, K.; Robinson, R.C. Structure of the N-terminal half of gelsolin bound to actin: Roles in severing, apoptosis and FAF. EMBO J. 2004, 23, 2713–2722. [Google Scholar] [CrossRef]
- Takács-Kollár, V.; Lőrinczy, D.; Nyitrai, M.; Hild, G. Spectroscopic characterization of the effect of mouse twinfilin-1 on actin filaments at different pH values. J. Photochem. Photobiol. B 2016, 164, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Harris, E.S.; Li, F.; Higgs, H.N. The mouse formin, FRLalpha, slows actin filament barbed end elongation, competes with capping protein, accelerates polymerization from monomers, and severs filaments. J. Biol. Chem. 2004, 279, 20076–20087. [Google Scholar] [CrossRef] [PubMed]
- Gurel, P.S.; Ge, P.; Grintsevich, E.E.; Shu, R.; Blanchoin, L.; Zhou, Z.H.; Reisler, E.; Higgs, H.N. INF2-mediated severing through actin filament encirclement and disruption. Curr. Biol. 2014, 24, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Johnston, A.B.; Hilton, D.M.; McConnell, P.; Johnson, B.; Harris, M.T.; Simone, A.; Amarasinghe, G.K.; Cooper, J.A.; Goode, B.L. A novel mode of capping protein-regulation by twinfilin. eLife 2018, 7, e41313. [Google Scholar] [CrossRef]
- Sun, H.Q.; Yamamoto, M.; Mejillano, M.; Yin, H.L. Gelsolin, a multifunctional actin regulatory protein. J. Biol. Chem. 1999, 274, 33179–33182. [Google Scholar] [CrossRef]
- Bao, Y.; Kake, T.; Hanashima, A.; Nomiya, Y.; Kubokawa, K.; Kimura, S. Actin capping proteins, CapZ (β-actinin) and tropomodulin in amphioxus striated muscle. Gene 2012, 510, 78–86. [Google Scholar] [CrossRef]
- Svitkina, T. The Actin Cytoskeleton and Actin-Based Motility. Cold Spring Harb. Perspect. Biol. 2018, 10, a018267. [Google Scholar] [CrossRef]
- Pinto-Costa, R.; Sousa, M.M. Profilin as a dual regulator of actin and microtubule dynamics. Cytoskeleton 2020, 77, 76–83. [Google Scholar] [CrossRef]
- Jockusch, B.M.; Murk, K.; Rothkegel, M. The profile of profilins. Rev. Physiol. Biochem. Pharmacol. 2007, 159, 131–149. [Google Scholar] [CrossRef] [PubMed]
- Suetsugu, S.; Miki, H.; Takenawa, T. The essential role of profilin in the assembly of actin for microspike formation. EMBO J. 1998, 17, 6516–6526. [Google Scholar] [CrossRef]
- Paul, A.S.; Pollard, T.D. The role of the FH1 domain and profilin in formin-mediated actin-filament elongation and nucleation. Curr. Biol. 2008, 18, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Ferron, F.; Rebowski, G.; Lee, S.H.; Dominguez, R. Structural basis for the recruitment of profilin-actin complexes during filament elongation by Ena/VASP. EMBO J. 2007, 26, 4597–4606. [Google Scholar] [CrossRef] [PubMed]
- Kooij, V.; Viswanathan, M.C.; Lee, D.I.; Rainer, P.P.; Schmidt, W.; Kronert, W.A.; Harding, S.E.; Kass, D.A.; Bernstein, S.I.; van Eyk, J.E.; et al. Profilin modulates sarcomeric organization and mediates cardiomyocyte hypertrophy. Cardiovasc. Res. 2016, 110, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Moustafa-Bayoumi, M.; Alhaj, M.A.; El-Sayed, O.; Wisel, S.; Chotani, M.A.; Abouelnaga, Z.A.; Hassona, M.D.; Rigatto, K.; Morris, M.; Nuovo, G.; et al. Vascular hypertrophy and hypertension caused by transgenic overexpression of profilin 1. J. Biol. Chem. 2007, 282, 37632–37639. [Google Scholar] [CrossRef] [PubMed]
- Elnakish, M.T.; Hassanain, H.H.; Janssen, P.M. Vascular remodeling-associated hypertension leads to left ventricular hypertrophy and contractile dysfunction in profilin-1 transgenic mice. J. Cardiovasc. Pharmacol. 2012, 60, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Joy, M.; Gau, D.; Castellucci, N.; Prywes, R.; Roy, P. The myocardin-related transcription factor MKL co-regulates the cellular levels of two profilin isoforms. J. Biol. Chem. 2017, 292, 11777–11791. [Google Scholar] [CrossRef]
- Oeing, C.U.; Nakamura, T.; Pan, S.; Mishra, S.; Dunkerly-Eyring, B.L.; Kokkonen-Simon, K.M.; Lin, B.L.; Chen, A.; Zhu, G.; Bedja, D.; et al. PKG1alpha Cysteine-42 Redox State Controls mTORC1 Activation in Pathological Cardiac Hypertrophy. Circ. Res. 2020, 127, 522–533. [Google Scholar] [CrossRef]
- Altamirano, F.; Oyarce, C.; Silva, P.; Toyos, M.; Wilson, C.; Lavandero, S.; Uhlen, P.; Estrada, M. Testosterone induces cardiomyocyte hypertrophy through mammalian target of rapamycin complex 1 pathway. J. Endocrinol. 2009, 202, 299–307. [Google Scholar] [CrossRef]
- Ranek, M.J.; Kokkonen-Simon, K.M.; Chen, A.; Dunkerly-Eyring, B.L.; Vera, M.P.; Oeing, C.U.; Patel, C.H.; Nakamura, T.; Zhu, G.; Bedja, D.; et al. PKG1-modified TSC2 regulates mTORC1 activity to counter adverse cardiac stress. Nature 2019, 566, 264–269. [Google Scholar] [CrossRef]
- Yang, D.; Wang, Y.; Jiang, M.; Deng, X.; Pei, Z.; Li, F.; Xia, K.; Zhu, L.; Yang, T.; Chen, M. Downregulation of Profilin-1 Expression Attenuates Cardiomyocytes Hypertrophy and Apoptosis Induced by Advanced Glycation End Products in H9c2 Cells. BioMed Res. Int. 2017, 2017, 9716087. [Google Scholar] [CrossRef]
- Yang, D.; Liu, W.; Ma, L.; Wang, Y.; Ma, J.; Jiang, M.; Deng, X.; Huang, F.; Yang, T.; Chen, M. Profilin-1 contributes to cardiac injury induced by advanced glycation end-products in rats. Mol. Med. Rep. 2017, 16, 6634–6641. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.W.; Bamburg, J.R. ADF/cofilin: A functional node in cell biology. Trends Cell Biol. 2010, 20, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, K.; Sato, N.; Nakagaki, T.; Abe, H.; Ono, S.; Obinata, T. Two mouse cofilin isoforms, muscle-type (MCF) and non-muscle type (NMCF), interact with F-actin with different efficiencies. J. Biochem. 2005, 138, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Mseka, T.; Cramer, L.P. Actin depolymerization-based force retracts the cell rear in polarizing and migrating cells. Curr. Biol. 2011, 21, 2085–2091. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, A.B.; Hall, A. Rho GTPases: Biochemistry and biology. Annu. Rev. Cell Dev. Biol. 2005, 21, 247–269. [Google Scholar] [CrossRef]
- Hild, G.; Kalmár, L.; Kardos, R.; Nyitrai, M.; Bugyi, B. The other side of the coin: Functional and structural versatility of ADF/cofilins. Eur. J. Cell Biol. 2014, 93, 238–251. [Google Scholar] [CrossRef] [PubMed]
- Rangrez, A.Y.; Hoppe, P.; Kuhn, C.; Zille, E.; Frank, J.; Frey, N.; Frank, D. MicroRNA miR-301a is a novel cardiac regulator of Cofilin-2. PLoS ONE 2017, 12, e0183901. [Google Scholar] [CrossRef]
- Aoki, H.; Izumo, S.; Sadoshima, J. Angiotensin II activates RhoA in cardiac myocytes: A critical role of RhoA in angiotensin II-induced premyofibril formation. Circ. Res. 1998, 82, 666–676. [Google Scholar] [CrossRef]
- Aikawa, R.; Komuro, I.; Nagai, R.; Yazaki, Y. Rho plays an important role in angiotensin II-induced hypertrophic responses in cardiac myocytes. Mol. Cell. Biochem. 2000, 212, 177–182. [Google Scholar] [CrossRef]
- Zeidan, A.; Javadov, S.; Chakrabarti, S.; Karmazyn, M. Leptin-induced cardiomyocyte hypertrophy involves selective caveolae and RhoA/ROCK-dependent p38 MAPK translocation to nuclei. Cardiovasc. Res. 2008, 77, 64–72. [Google Scholar] [CrossRef]
- Moey, M.; Rajapurohitam, V.; Zeidan, A.; Karmazyn, M. Ginseng (Panax quinquefolius) attenuates leptin-induced cardiac hypertrophy through inhibition of p115Rho guanine nucleotide exchange factor-RhoA/Rho-associated, coiled-coil containing protein kinase-dependent mitogen-activated protein kinase pathway activation. J. Pharmacol. Exp. Ther. 2011, 339, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, A.; Gan, X.T.; Thomas, A.; Karmazyn, M. Prevention of RhoA activation and cofilin-mediated actin polymerization mediates the antihypertrophic effect of adenosine receptor agonists in angiotensin II- and endothelin-1-treated cardiomyocytes. Mol. Cell. Biochem. 2014, 385, 239–248. [Google Scholar] [CrossRef]
- Lai, D.; Gao, J.; Bi, X.; He, H.; Shi, X.; Weng, S.; Chen, Y.; Yang, Y.; Ye, Y.; Fu, G. The Rho kinase inhibitor, fasudil, ameliorates diabetes-induced cardiac dysfunction by improving calcium clearance and actin remodeling. J. Mol. Med. 2017, 95, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Chesarone, M.A.; DuPage, A.G.; Goode, B.L. Unleashing formins to remodel the actin and microtubule cytoskeletons. Nat. Rev. Mol. Cell Biol. 2010, 11, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Miralles, F.; Posern, G.; Zaromytidou, A.I.; Treisman, R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 2003, 113, 329–342. [Google Scholar] [CrossRef]
- Copeland, J.W.; Copeland, S.J.; Treisman, R. Homo-oligomerization is essential for F-actin assembly by the formin family FH2 domain. J. Biol. Chem. 2004, 279, 50250–50256. [Google Scholar] [CrossRef]
- Abe, I.; Terabayashi, T.; Hanada, K.; Kondo, H.; Teshima, Y.; Ishii, Y.; Miyoshi, M.; Kira, S.; Saito, S.; Tsuchimochi, H.; et al. Disruption of actin dynamics regulated by Rho effector mDia1 attenuates pressure overload-induced cardiac hypertrophic responses and exacerbates dysfunction. Cardiovasc. Res. 2021, 117, 1103–1117. [Google Scholar] [CrossRef]
- Krainer, E.C.; Ouderkirk, J.L.; Miller, E.W.; Miller, M.R.; Mersich, A.T.; Blystone, S.D. The multiplicity of human formins: Expression patterns in cells and tissues. Cytoskeleton 2013, 70, 424–438. [Google Scholar] [CrossRef]
- Taniguchi, K.; Takeya, R.; Suetsugu, S.; Kan-O, M.; Narusawa, M.; Shiose, A.; Tominaga, R.; Sumimoto, H. Mammalian formin fhod3 regulates actin assembly and sarcomere organization in striated muscles. J. Biol. Chem. 2009, 284, 29873–29881. [Google Scholar] [CrossRef]
- Kan-O, M.; Takeya, R.; Abe, T.; Kitajima, N.; Nishida, M.; Tominaga, R.; Kurose, H.; Sumimoto, H. Mammalian formin Fhod3 plays an essential role in cardiogenesis by organizing myofibrillogenesis. Biol. Open 2012, 1, 889–896. [Google Scholar] [CrossRef]
- Iskratsch, T.; Reijntjes, S.; Dwyer, J.; Toselli, P.; Degano, I.R.; Dominguez, I.; Ehler, E. Two distinct phosphorylation events govern the function of muscle FHOD3. Cell. Mol. Life Sci. 2013, 70, 893–908. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Wei, S.S.; Wang, H.; Wang, Q.; Li, W.; Li, G.; Hou, J.W.; Chen, X.M.; Chen, J.; Xu, W.P.; et al. Crucial Role of ROCK2-Mediated Phosphorylation and Upregulation of FHOD3 in the Pathogenesis of Angiotensin II-Induced Cardiac Hypertrophy. Hypertension 2017, 69, 1070–1083. [Google Scholar] [CrossRef] [PubMed]
- Ushijima, T.; Fujimoto, N.; Matsuyama, S.; Kan-O, M.; Kiyonari, H.; Shioi, G.; Kage, Y.; Yamasaki, S.; Takeya, R.; Sumimoto, H. The actin-organizing formin protein Fhod3 is required for postnatal development and functional maintenance of the adult heart in mice. J. Biol. Chem. 2018, 293, 148–162. [Google Scholar] [CrossRef] [PubMed]
- Wooten, E.C.; Hebl, V.B.; Wolf, M.J.; Greytak, S.R.; Orr, N.M.; Draper, I.; Calvino, J.E.; Kapur, N.K.; Maron, M.S.; Kullo, I.J.; et al. Formin homology 2 domain containing 3 variants associated with hypertrophic cardiomyopathy. Circ. Cardiovasc. Genet. 2013, 6, 10–18. [Google Scholar] [CrossRef]
- Schafer, D.A.; Hug, C.; Cooper, J.A. Inhibition of CapZ during myofibrillogenesis alters assembly of actin filaments. J. Cell Biol. 1995, 128, 61–70. [Google Scholar] [CrossRef]
- Hart, M.C.; Cooper, J.A. Vertebrate isoforms of actin capping protein beta have distinct functions In vivo. J. Cell Biol. 1999, 147, 1287–1298. [Google Scholar] [CrossRef]
- Kim, K.; McCully, M.E.; Bhattacharya, N.; Butler, B.; Sept, D.; Cooper, J.A. Structure/function analysis of the interaction of phosphatidylinositol 4,5-bisphosphate with actin-capping protein: Implications for how capping protein binds the actin filament. J. Biol. Chem. 2007, 282, 5871–5879. [Google Scholar] [CrossRef]
- Montgomery, D.E.; Chandra, M.; Huang, Q.; Jin, J.; Solaro, R.J. Transgenic incorporation of skeletal TnT into cardiac myofilaments blunts PKC-mediated depression of force. Am. J. Physiol. Heart. Circ. Physiol. 2001, 280, H1011–H1018. [Google Scholar] [CrossRef]
- Lin, Y.H.; Warren, C.M.; Li, J.; McKinsey, T.A.; Russell, B. Myofibril growth during cardiac hypertrophy is regulated through dual phosphorylation and acetylation of the actin capping protein CapZ. Cell. Signal. 2016, 28, 1015–1024. [Google Scholar] [CrossRef]
- Hartman, T.J.; Martin, J.L.; Solaro, R.J.; Samarel, A.M.; Russell, B. CapZ dynamics are altered by endothelin-1 and phenylephrine via PIP2- and PKC-dependent mechanisms. Am. J. Physiol. Cell Physiol. 2009, 296, C1034–C1039. [Google Scholar] [CrossRef]
- Li, J.; Russell, B. Phosphatidylinositol 4,5-bisphosphate regulates CapZβ1 and actin dynamics in response to mechanical strain. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1614–H1623. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.H.; Li, J.; Swanson, E.R.; Russell, B. CapZ and actin capping dynamics increase in myocytes after a bout of exercise and abates in hours after stimulation ends. J. Appl. Physiol. 2013, 114, 1603–1609. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Mkrtschjan, M.A.; Lin, Y.H.; Russell, B. Variation in stiffness regulates cardiac myocyte hypertrophy via signaling pathways. Can. J. Physiol. Pharmacol. 2016, 94, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Heidings, J.B.; Demosthene, B.; Merlino, T.R.; Castaneda, N.; Kang, E.H. Gelsolin-mediated actin filament severing in crowded environments. Biochem. Biophys. Res. Commun. 2020, 532, 548–554. [Google Scholar] [CrossRef]
- Mosaddeghzadeh, N.; Ahmadian, M.R. The RHO Family GTPases: Mechanisms of Regulation and Signaling. Cells 2021, 10, 1831. [Google Scholar] [CrossRef]
- Hu, W.S.; Ho, T.J.; Pai, P.; Chung, L.C.; Kuo, C.H.; Chang, S.H.; Tsai, F.J.; Tsai, C.H.; Jie, Y.C.; Liou, Y.M.; et al. Gelsolin (GSN) induces cardiomyocyte hypertrophy and BNP expression via p38 signaling and GATA-4 transcriptional factor activation. Mol. Cell. Biochem. 2014, 390, 263–270. [Google Scholar] [CrossRef]
- Li, G.H.; Shi, Y.; Chen, Y.; Sun, M.; Sader, S.; Maekawa, Y.; Arab, S.; Dawood, F.; Chen, M.; de Couto, G.; et al. Gelsolin regulates cardiac remodeling after myocardial infarction through DNase I-mediated apoptosis. Circ. Res. 2009, 104, 896–904. [Google Scholar] [CrossRef]
- Dai, B.; Li, H.; Fan, J.; Zhao, Y.; Yin, Z.; Nie, X.; Wang, D.W.; Chen, C. MiR-21 protected against diabetic cardiomyopathy induced diastolic dysfunction by targeting gelsolin. Cardiovasc. Diabetol. 2018, 17, 123. [Google Scholar] [CrossRef]
- Putinski, C.; Abdul-Ghani, M.; Brunette, S.; Burgon, P.G.; Megeney, L.A. Caspase Cleavage of Gelsolin Is an Inductive Cue for Pathologic Cardiac Hypertrophy. J. Am. Heart Assoc. 2018, 7, e010404. [Google Scholar] [CrossRef]
- Zheng, B.; Han, M.; Wen, J.K.; Zhang, R. Human heart LIM protein activates atrial-natriuretic-factor gene expression by interacting with the cardiac-restricted transcription factor Nkx2.5. Biochem. J. 2008, 409, 683–690. [Google Scholar] [CrossRef]
- Zheng, B.; Wen, J.K.; Han, M.; Zhou, A.R. hhLIM protein is involved in cardiac hypertrophy. Biochim. Biophys. Acta 2004, 1690, 1–10. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Riaz, M.; Park, J.; Sewanan, L.R.; Ren, Y.; Schwan, J.; Das, S.K.; Pomianowski, P.T.; Huang, Y.; Ellis, M.W.; Luo, J.; et al. Muscle LIM Protein Force-Sensing Mediates Sarcomeric Biomechanical Signaling in Human Familial Hypertrophic Cardiomyopathy. Circulation 2022, 145, 1238–1253. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.F.; Lyu, J.L.; Fang, J.; Chen, J.; Chen, W.W.; Huang, J.Q.; Xia, S.D.; Jin, J.M.; Dong, F.H.; Cheng, H.Q.; et al. Impact of LDB3 gene polymorphisms on clinical presentation and implantable cardioverter defibrillator (ICD) implantation in Chinese patients with idiopathic dilated cardiomyopathy. J. Zhejiang Univ. Sci. B 2019, 20, 766–775. [Google Scholar] [CrossRef] [PubMed]
- Soares, D.D.S.; Pinto, G.H.; Lopes, A.; Caetano, D.S.L.; Nascimento, T.G.; Andrades, M.E.; Clausell, N.; Rohde, L.E.P.; Leitao, S.A.T.; Biolo, A. Cardiac hypertrophy in mice submitted to a swimming protocol: Influence of training volume and intensity on myocardial renin-angiotensin system. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2019, 316, R776–R782. [Google Scholar] [CrossRef]
- Marston, S. The Molecular Mechanisms of Mutations in Actin and Myosin that Cause Inherited Myopathy. Int. J. Mol. Sci. 2018, 19, 2020. [Google Scholar] [CrossRef]
- Lehman, S.J.; Crocini, C.; Leinwand, L.A. Targeting the sarcomere in inherited cardiomyopathies. Nat. Rev. Cardiol. 2022, 19, 353–363. [Google Scholar] [CrossRef]
- Sedaghat-Hamedani, F.; Kayvanpour, E.; Tugrul, O.F.; Lai, A.; Amr, A.; Haas, J.; Proctor, T.; Ehlermann, P.; Jensen, K.; Katus, H.A.; et al. Clinical outcomes associated with sarcomere mutations in hypertrophic cardiomyopathy: A meta-analysis on 7675 individuals. Clin. Res. Cardiol. 2018, 107, 30–41. [Google Scholar] [CrossRef]
- Mori, A.A.; Castro, L.R.; Bortolin, R.H.; Bastos, G.M.; Oliveira, V.F.; Ferreira, G.M.; Hirata, T.D.C.; Fajardo, C.M.; Sampaio, M.F.; Moreira, D.A.R.; et al. Association of variants in MYH7, MYBPC3 and TNNT2 with sudden cardiac death-related risk factors in Brazilian patients with hypertrophic cardiomyopathy. Forensic Sci. Int. Genet. 2021, 52, 102478. [Google Scholar] [CrossRef]
- Morelli, C.; Ingrasciotta, G.; Jacoby, D.; Masri, A.; Olivotto, I. Sarcomere protein modulation: The new frontier in cardiovascular medicine and beyond. Eur. J. Intern. Med. 2022, 102, 1–7. [Google Scholar] [CrossRef]
- Zampieri, M.; Argiro, A.; Marchi, A.; Berteotti, M.; Targetti, M.; Fornaro, A.; Tomberli, A.; Stefano, P.; Marchionni, N.; Olivotto, I. Mavacamten, a Novel Therapeutic Strategy for Obstructive Hypertrophic Cardiomyopathy. Curr. Cardiol. Rep. 2021, 23, 79. [Google Scholar] [CrossRef]
- Henderson, D.M.; Lin, A.Y.; Thomas, D.D.; Ervasti, J.M. The carboxy-terminal third of dystrophin enhances actin binding activity. J. Mol. Biol. 2012, 416, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Garbincius, J.F.; Merz, L.E.; Cuttitta, A.J.; Bayne, K.V.; Schrade, S.; Armstead, E.A.; Converso-Baran, K.L.; Whitesall, S.E.; D’Alecy, L.G.; Michele, D.E. Enhanced dimethylarginine degradation improves coronary flow reserve and exercise tolerance in Duchenne muscular dystrophy carrier mice. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H582–H603. [Google Scholar] [CrossRef] [PubMed]
- Crilley, J.G.; Boehm, E.A.; Rajagopalan, B.; Blamire, A.M.; Styles, P.; Muntoni, F.; Hilton-Jones, D.; Clarke, K. Magnetic resonance spectroscopy evidence of abnormal cardiac energetics in Xp21 muscular dystrophy. J. Am. Coll. Cardiol. 2000, 36, 1953–1958. [Google Scholar] [CrossRef]
- Prado, F.P.; Dos Santos, D.O.; Blefari, V.; Silva, C.A.; Machado, J.; Kettelhut, I.D.C.; Ramos, S.G.; Baruffi, M.D.; Salgado, H.C.; Prado, C.M. Early dystrophin loss is coincident with the transition of compensated cardiac hypertrophy to heart failure. PLoS ONE 2017, 12, e0189469. [Google Scholar] [CrossRef] [PubMed]
- Jearawiriyapaisarn, N.; Moulton, H.M.; Sazani, P.; Kole, R.; Willis, M.S. Long-term improvement in mdx cardiomyopathy after therapy with peptide-conjugated morpholino oligomers. Cardiovasc. Res. 2010, 85, 444–453. [Google Scholar] [CrossRef]
- Parente, J.M.; de Mello, M.M.B.; Silva, P.; Omoto, A.C.M.; Pernomian, L.; Oliveira, I.S.; Mahmud, Z.; Fazan, R., Jr.; Arantes, E.C.; Schulz, R.; et al. MMP inhibition attenuates hypertensive eccentric cardiac hypertrophy and dysfunction by preserving troponin I and dystrophin. Biochem. Pharmacol. 2021, 193, 114744. [Google Scholar] [CrossRef]
- Dwyer, J.; Pluess, M.; Iskratsch, T.; dos Remedios, C.G.; Ehler, E. The formin FHOD1 in cardiomyocytes. Anat. Rec. 2014, 297, 1560–1570. [Google Scholar] [CrossRef]
- Pappas, C.T.; Mayfield, R.M.; Henderson, C.; Jamilpour, N.; Cover, C.; Hernandez, Z.; Hutchinson, K.R.; Chu, M.; Nam, K.H.; Valdez, J.M.; et al. Knockout of Lmod2 results in shorter thin filaments followed by dilated cardiomyopathy and juvenile lethality. Proc. Natl. Acad. Sci. USA 2015, 112, 13573–13578. [Google Scholar] [CrossRef]
- Peche, V.S.; Holak, T.A.; Burgute, B.D.; Kosmas, K.; Kale, S.P.; Wunderlich, F.T.; Elhamine, F.; Stehle, R.; Pfitzer, G.; Nohroudi, K.; et al. Ablation of cyclase-associated protein 2 (CAP2) leads to cardiomyopathy. Cell. Mol. Life Sci. 2013, 70, 527–543. [Google Scholar] [CrossRef]
- Welch, S.; Plank, D.; Witt, S.; Glascock, B.; Schaefer, E.; Chimenti, S.; Andreoli, A.M.; Limana, F.; Leri, A.; Kajstura, J.; et al. Cardiac-specific IGF-1 expression attenuates dilated cardiomyopathy in tropomodulin-overexpressing transgenic mice. Circ. Res. 2002, 90, 641–648. [Google Scholar] [CrossRef]
- Schultheiss, H.-P.; Fairweather, D.; Caforio, A.L.P.; Escher, F.; Hershberger, R.E.; Lipshultz, S.E.; Liu, P.P.; Matsumori, A.; Mazzanti, A.; McMurray, J.; et al. Dilated cardiomyopathy. Nat. Rev. Dis. Prim. 2019, 5, 32. [Google Scholar] [CrossRef] [PubMed]
- Narumiya, S.; Tanji, M.; Ishizaki, T. Rho signaling, ROCK and mDia1, in transformation, metastasis and invasion. Cancer Metastasis Rev. 2009, 28, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Burtnick, L.D.; Koepf, E.K.; Grimes, J.; Jones, E.Y.; Stuart, D.I.; McLaughlin, P.J.; Robinson, R.C. The crystal structure of plasma gelsolin: Implications for actin severing, capping, and nucleation. Cell 1997, 90, 661–670. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Types | ABPs | Basic Function | Refs. |
|---|---|---|---|
| G-actin-binding | Profilin, thymosin β4, cofilin | Bound to G-actin | [11,20,21] |
| F-actin-binding | Dystrophin, tropomyosin | Bound to F-actin | [9,11,22] |
| Actin-nucleating | Formin, Arp2/3 complex, proteins with tandem WH2 domains, leiomodin | Nucleation to initiate actin polymerization | [11,23,24,25] |
| Actin-elongating | Formin, tetramers of Ena/VASP | Regulation of actin assembly | [11,24] |
| Actin-bundling | Fimbrin/Plastin, hhLIM, gelsolin | Causes parallel F-actin filaments to closely pack together | [26,27,28,29] |
| Severing | ADF/cofilin, gelsolin, twinfilin, FRL-α, INF-2 | Severs F-actin | [30,31,32,33,34] |
| Capping | Twinfilin, gelsolin, tropomodulin, CapZ, Arp2/3 complex | Caps F-actin to inhibit actin polymerization | [11,35,36,37] |
| Motor | Myosin | Cargo transfer | [38] |
| ABPs | Function | Protein Synthesis/Phosphorylation | Refs. | |
|---|---|---|---|---|
| Over Expression | Knock Down/Out | |||
| Profilin-1 | Polymerization | ANP, BNP and α-SMA↑; p-eNOS↓. | ANP, BNP and p-ERK1/2↓; p-eNOS↑. | [17,44,51,52] |
| Cofilin-2 | Severing | / | / | [11] |
| mDia1 | Nucleation | / | SRF, MRTF, pERK1/2 and pFAK↓ | [68,113] |
| FHOD3 | Nucleation | / | ANP, BNP and MYH7↑. | [69,74] |
| CapZ | Capping | / | / | [77] |
| Gelsolin | Severing, Capping | / | ANP and BNP↓. | [85,89,114] |
| hhLIM | Bundling | / | BNP and α-SMA↓. | [28,92] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, C.; Wang, S.; Liu, C.; Ren, Z. Actin-Binding Proteins in Cardiac Hypertrophy. Cells 2022, 11, 3566. https://doi.org/10.3390/cells11223566
Pan C, Wang S, Liu C, Ren Z. Actin-Binding Proteins in Cardiac Hypertrophy. Cells. 2022; 11(22):3566. https://doi.org/10.3390/cells11223566
Chicago/Turabian StylePan, Congbin, Siqi Wang, Chao Liu, and Zhanhong Ren. 2022. "Actin-Binding Proteins in Cardiac Hypertrophy" Cells 11, no. 22: 3566. https://doi.org/10.3390/cells11223566
APA StylePan, C., Wang, S., Liu, C., & Ren, Z. (2022). Actin-Binding Proteins in Cardiac Hypertrophy. Cells, 11(22), 3566. https://doi.org/10.3390/cells11223566