Repositioning Mifepristone as a Leukaemia Inhibitory Factor Receptor Antagonist for the Treatment of Pancreatic Adenocarcinoma

 ,
,  , , ,
, , ,  ,
,  ,
,  ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. GSE196009 Data Sets
2.2. Pancreatic Cancer Cell Lines
2.3. Real-Time PCR
- Cmyc (for TCGGATTCTCTGCTCTCCTC; rev TTTTCCACAGAAACAACATCG),
- Snail1 (for ACCCACACTGGCGAGAAG; rev TGACATCTGAGTGGGTCTGG),
- Vimentin (for TCAGAGAGAGGAAGCCGAAA; rev ATTCCACTTTGCGTTCAAGG),
- Cxcr4 (for AACGTCAGTGAGGCAGATGA; rev TGGAGTGTGACAGCTTGGAG).
2.4. Immunofluorescence (IF)
2.5. Image Analysis
2.6. Cell Proliferation Assay
2.7. Protein Preparation of hLIFR and hLIF-hLIFR Homology Model
2.8. FDA-Approved Library Preparation
2.9. Docking Procedures
2.10. Molecular Dynamics Simulations (MDs)
2.11. Alpha Screen
2.12. Transactivation
2.13. Flow Cytometry
2.14. Wound Healing Assay
2.15. Western Blot Analysis
2.16. AmpliSeq Transcriptome
2.17. Statistical Analysis
3. Results
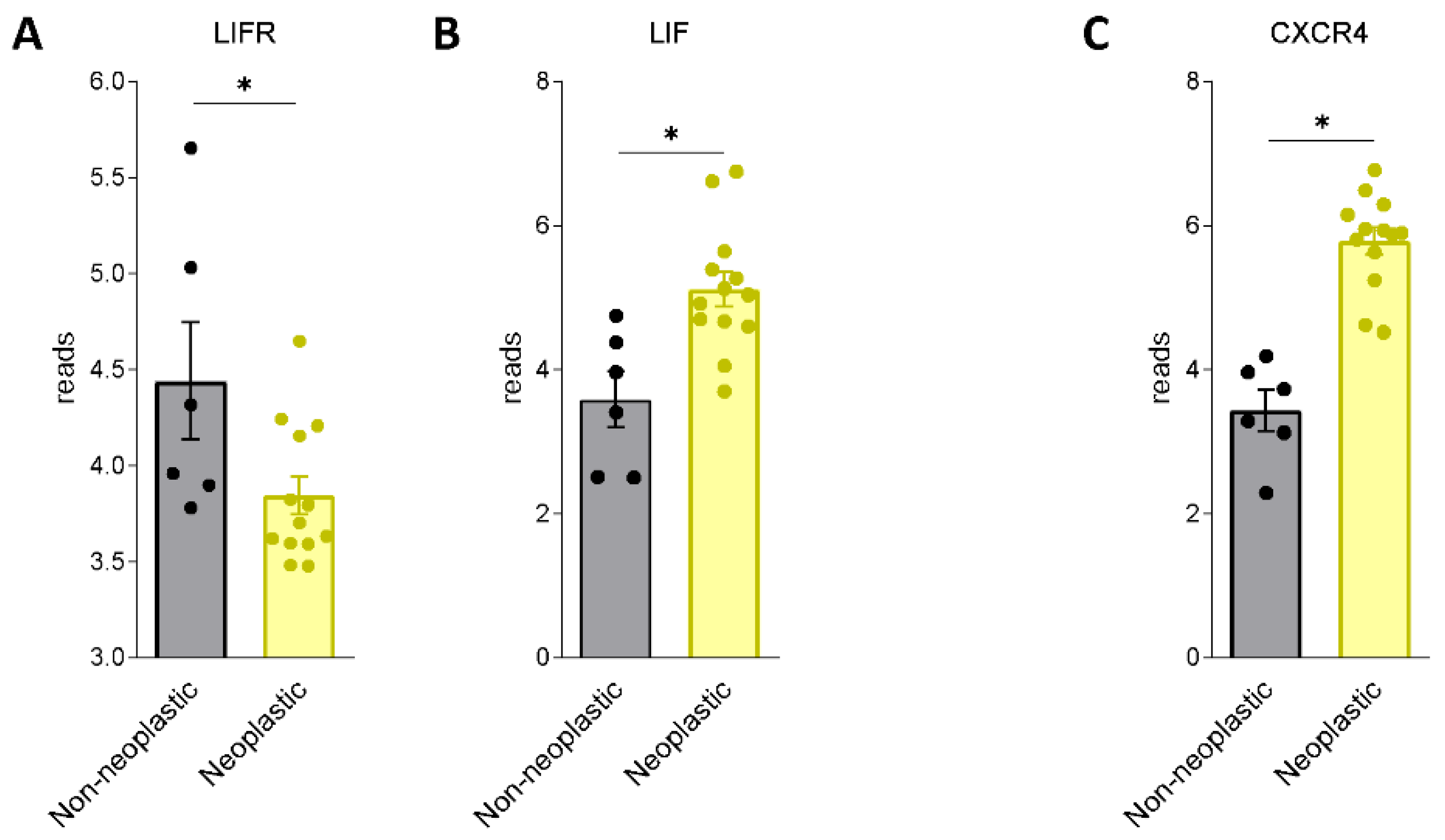
3.1. LIF and LIFR Expression in PDAC
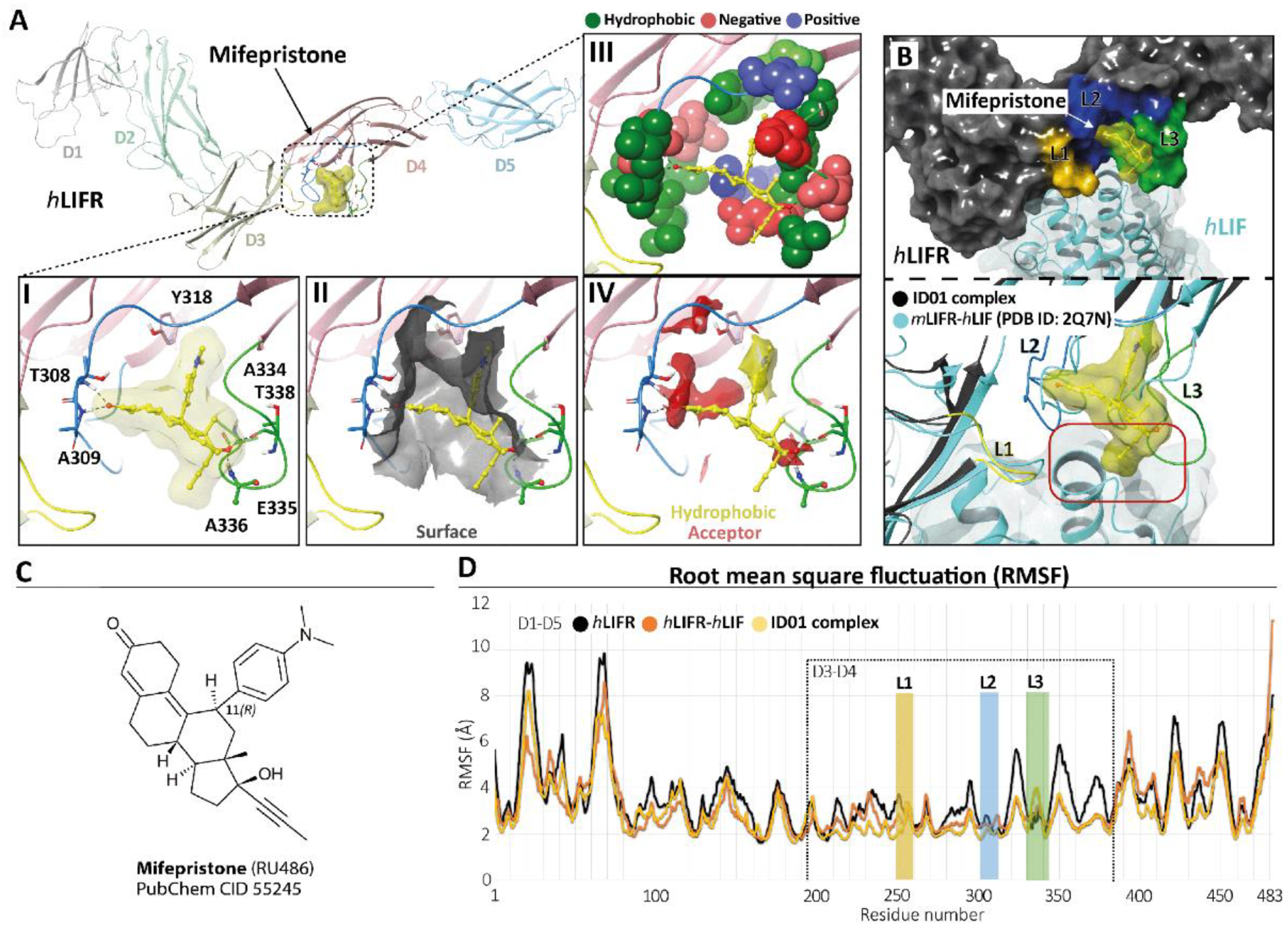
3.2. Mifepristone Putative Binding at the LIFR-LIF Interface
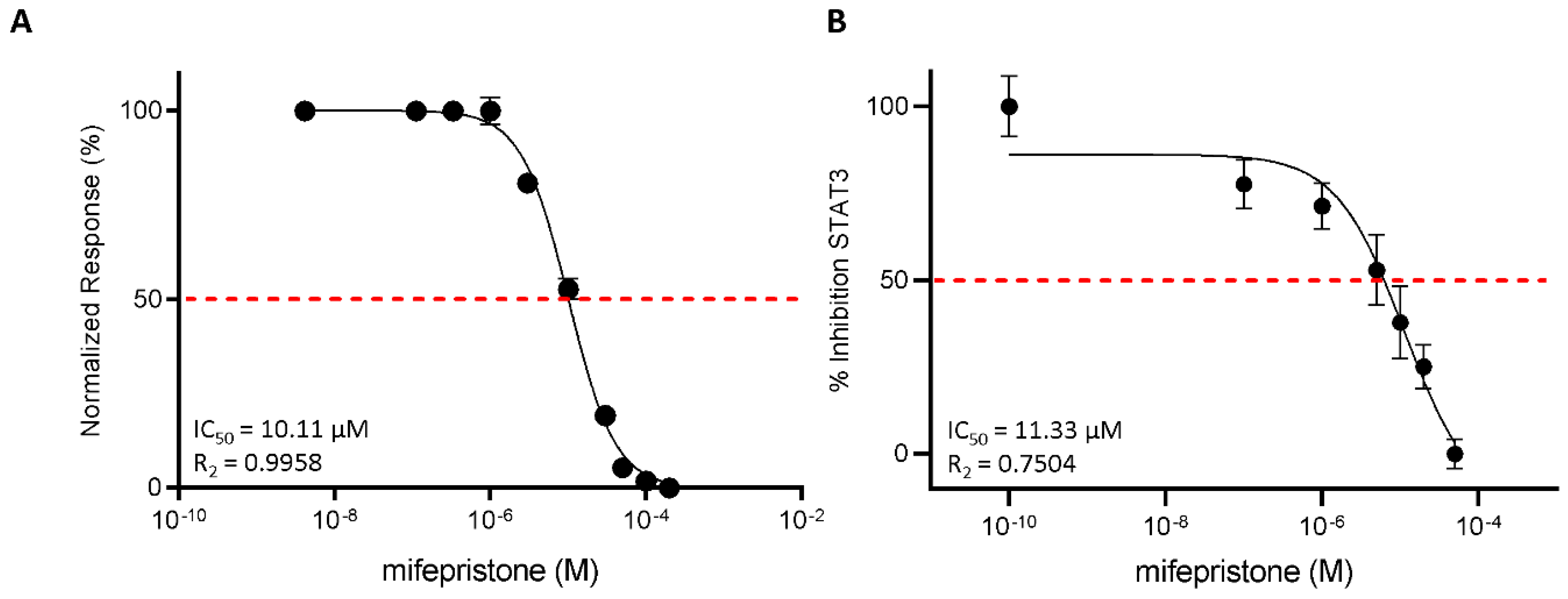
3.3. Functional Characterization of Mifepristone as LIFR Antagonist by Alpha Screen and Transactivation Assay
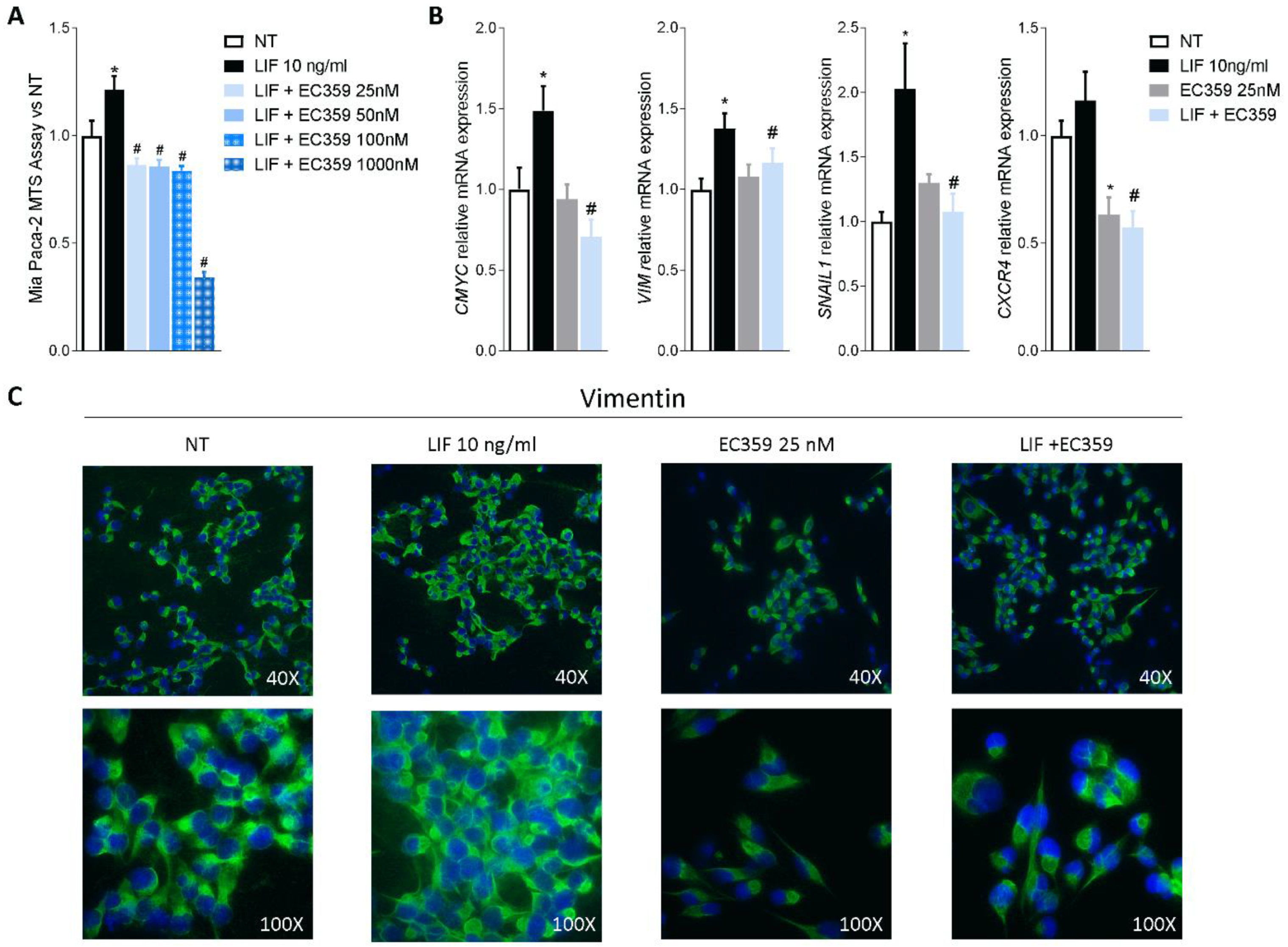
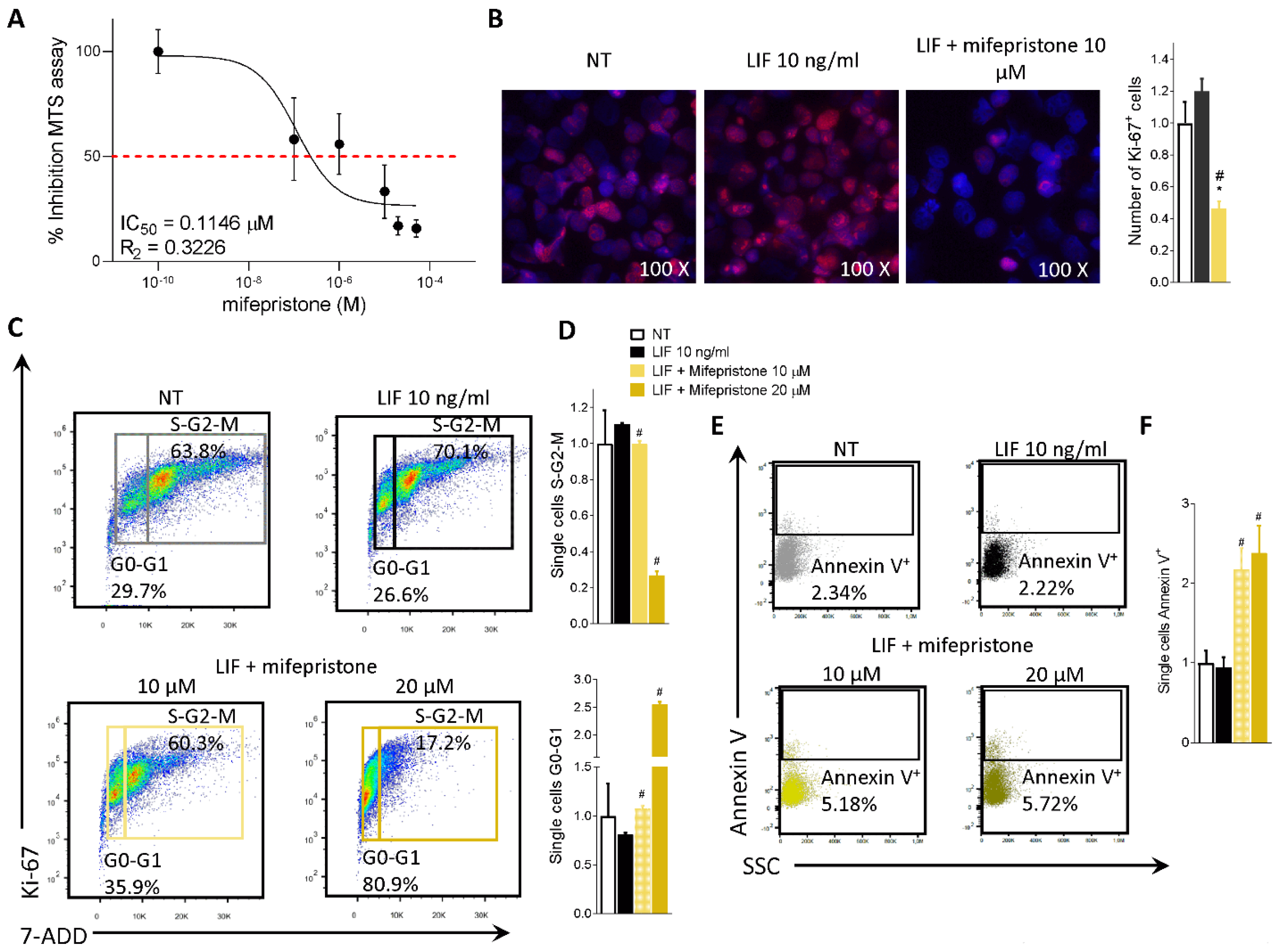
3.4. Effect of Mifepristone on PDAC Cell Proliferation and EMT
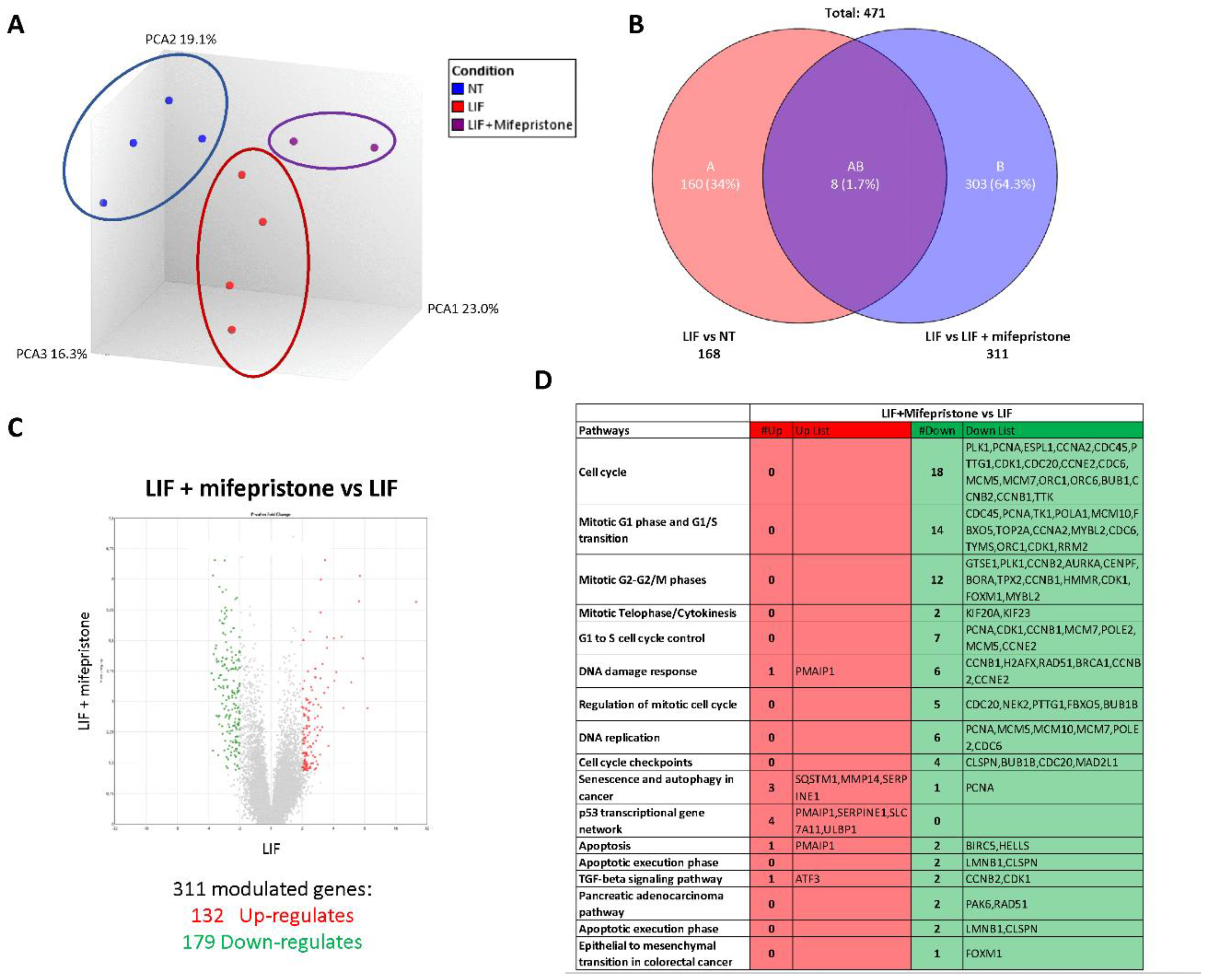
3.5. RNAseq Analysis of the Effects of LIF and Mifepristone on MIA PaCa-2 Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M.; Cascinu, S.; Kleeff, J.; Labianca, R.; Löhr, J.-M.; Neoptolemos, J.; Real, F.X.; Van Laethem, J.-L.; Heinemann, V. Addressing the Challenges of Pancreatic Cancer: Future Directions for Improving Outcomes. Pancreatol. Off. J. Int. Assoc. Pancreatol. 2015, 15, 8–18. [Google Scholar] [CrossRef] [PubMed]
- De La Cruz, M.S.D.; Young, A.P.; Ruffin, M.T. Diagnosis and Management of Pancreatic Cancer. Am. Fam. Physician 2014, 89, 626–632. [Google Scholar] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Páez, D.; Labonte, M.J.; Lenz, H.-J. Pancreatic Cancer: Medical Management (Novel Chemotherapeutics). Gastroenterol. Clin. North Am. 2012, 41, 189–209. [Google Scholar] [CrossRef]
- Bosetti, C.; Lucenteforte, E.; Silverman, D.T.; Petersen, G.; Bracci, P.M.; Ji, B.T.; Negri, E.; Li, D.; Risch, H.A.; Olson, S.H.; et al. Cigarette Smoking and Pancreatic Cancer: An Analysis from the International Pancreatic Cancer Case-Control Consortium (Panc4). Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2012, 23, 1880–1888. [Google Scholar] [CrossRef]
- Wood, L.D.; Canto, M.I.; Jaffee, E.M.; Simeone, D.M. Pancreatic Cancer: Pathogenesis, Screening, Diagnosis, and Treatment. Gastroenterology 2022, 163, 386–402.e1. [Google Scholar] [CrossRef]
- Klein, A.P.; Brune, K.A.; Petersen, G.M.; Goggins, M.; Tersmette, A.C.; Offerhaus, G.J.A.; Griffin, C.; Cameron, J.L.; Yeo, C.J.; Kern, S.; et al. Prospective Risk of Pancreatic Cancer in Familial Pancreatic Cancer Kindreds. Cancer Res. 2004, 64, 2634–2638. [Google Scholar] [CrossRef]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.-L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Birrer, D.L.; Golcher, H.; Casadei, R.; Haile, S.R.; Fritsch, R.; Hussung, S.; Brunner, T.B.; Fietkau, R.; Meyer, T.; Grützmann, R.; et al. Neoadjuvant Therapy for Resectable Pancreatic Cancer: A New Standard of Care. Pooled Data From 3 Randomized Controlled Trials. Ann. Surg. 2021, 274, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Sohal, D.P.S.; Duong, M.; Ahmad, S.A.; Gandhi, N.S.; Beg, M.S.; Wang-Gillam, A.; Wade, J.L., III; Chiorean, E.G.; Guthrie, K.A.; Lowy, A.M.; et al. Efficacy of Perioperative Chemotherapy for Resectable Pancreatic Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2021, 7, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Park, W.; Chawla, A.; O’Reilly, E.M. Pancreatic Cancer: A Review. JAMA 2021, 326, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Nicola, N.A.; Babon, J.J. Leukemia Inhibitory Factor (LIF). Cytokine Growth Factor Rev. 2015, 26, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Kamohara, H.; Ogawa, M.; Ishiko, T.; Sakamoto, K.; Baba, H. Leukemia Inhibitory Factor Functions as a Growth Factor in Pancreas Carcinoma Cells: Involvement of Regulation of LIF and Its Receptor Expression. Int. J. Oncol. 2007, 30, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Di Giorgio, C.; Marchianò, S.; Marino, E.; Biagioli, M.; Roselli, R.; Bordoni, M.; Bellini, R.; Urbani, G.; Zampella, A.; Distrutti, E.; et al. Next-Generation Sequencing Analysis of Gastric Cancer Identifies the Leukemia Inhibitory Factor Receptor as a Driving Factor in Gastric Cancer Progression and as a Predictor of Poor Prognosis. Front. Oncol. 2022, 12, 939969. [Google Scholar] [CrossRef]
- Wang, J.; Xie, C.; Pan, S.; Liang, Y.; Han, J.; Lan, Y.; Sun, J.; Li, K.; Sun, B.; Yang, G.; et al. N-Myc Downstream-Regulated Gene 2 Inhibits Human Cholangiocarcinoma Progression and Is Regulated by Leukemia Inhibitory Factor/MicroRNA-181c Negative Feedback Pathway. Hepatology 2016, 64, 1606–1622. [Google Scholar] [CrossRef]
- Yu, H.; Yue, X.; Zhao, Y.; Li, X.; Wu, L.; Zhang, C.; Liu, Z.; Lin, K.; Xu-Monette, Z.Y.; Young, K.H.; et al. LIF Negatively Regulates Tumour-Suppressor P53 through Stat3/ID1/MDM2 in Colorectal Cancers. Nat. Commun. 2014, 5, 5218. [Google Scholar] [CrossRef]
- Li, X.; Yang, Q.; Yu, H.; Wu, L.; Zhao, Y.; Zhang, C.; Yue, X.; Liu, Z.; Wu, H.; Haffty, B.G.; et al. LIF Promotes Tumorigenesis and Metastasis of Breast Cancer through the AKT-MTOR Pathway. Oncotarget 2014, 5, 788–801. [Google Scholar] [CrossRef]
- Yue, X.; Zhao, Y.; Zhang, C.; Li, J.; Liu, Z.; Liu, J.; Hu, W. Leukemia Inhibitory Factor Promotes EMT through STAT3-Dependent MiR-21 Induction. Oncotarget 2016, 7, 3777–3790. [Google Scholar] [CrossRef]
- Gearing, D.P.; Thut, C.J.; VandeBos, T.; Gimpel, S.D.; Delaney, P.B.; King, J.; Price, V.; Cosman, D.; Beckmann, M.P. Leukemia Inhibitory Factor Receptor Is Structurally Related to the IL-6 Signal Transducer, Gp130. EMBO J. 1991, 10, 2839–2848. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Jing, X.; Shen, B.; Liu, X.; Cheng, X.; Wang, B.; Fu, Z.; Peng, C.; Qiu, W. Leukemia Inhibitory Factor Receptor Negatively Regulates the Metastasis of Pancreatic Cancer Cells in Vitro and in Vivo. Oncol. Rep. 2016, 36, 827–836. [Google Scholar] [CrossRef] [PubMed]
- Viswanadhapalli, S.; Luo, Y.; Sareddy, G.R.; Santhamma, B.; Zhou, M.; Li, M.; Ma, S.; Sonavane, R.; Pratap, U.P.; Altwegg, K.A.; et al. EC359: A First-in-Class Small-Molecule Inhibitor for Targeting Oncogenic LIFR Signaling in Triple-Negative Breast Cancer. Mol. Cancer Ther. 2019, 18, 1341–1354. [Google Scholar] [CrossRef]
- Cadepond, F.; Ulmann, A.; Baulieu, E.E. RU486 (Mifepristone): Mechanisms of Action and Clinical Uses. Annu. Rev. Med. 1997, 48, 129–156. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, J.; Shao, J.; Gao, Y.; Xu, J.; Yu, S.; Liu, Z.; Jia, L. The Unique Pharmacological Characteristics of Mifepristone (RU486): From Terminating Pregnancy to Preventing Cancer Metastasis. Med. Res. Rev. 2014, 34, 979–1000. [Google Scholar] [CrossRef]
- Ashida, R.; Nakata, B.; Shigekawa, M.; Mizuno, N.; Sawaki, A.; Hirakawa, K.; Arakawa, T.; Yamao, K. Gemcitabine Sensitivity-Related MRNA Expression in Endoscopic Ultrasound-Guided Fine-Needle Aspiration Biopsy of Unresectable Pancreatic Cancer. J. Exp. Clin. Cancer Res. 2009, 28, 83. [Google Scholar] [CrossRef]
- Huyton, T.; Zhang, J.-G.; Luo, C.S.; Lou, M.-Z.; Hilton, D.J.; Nicola, N.A.; Garrett, T.P.J. An Unusual Cytokine:Ig-Domain Interaction Revealed in the Crystal Structure of Leukemia Inhibitory Factor (LIF) in Complex with the LIF Receptor. Proc. Natl. Acad. Sci. USA 2007, 104, 12737–12742. [Google Scholar] [CrossRef]
- Case, D.; Babin, V.; Berryman, J.; Betz, R.; Cai, Q.; Cerutti, D.; Cheatham, T.; Darden, T.; Duke, R.; Gohlke, H.; et al. Amber 2014; University of California: San Francisco, CA, USA, 2014. [Google Scholar]
- Lee, T.-S.; Cerutti, D.S.; Mermelstein, D.; Lin, C.; LeGrand, S.; Giese, T.J.; Roitberg, A.; Case, D.A.; Walker, R.C.; York, D.M. GPU-Accelerated Molecular Dynamics and Free Energy Methods in Amber18: Performance Enhancements and New Features. J. Chem. Inf. Model. 2018, 58, 2043–2050. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges: The RESP Model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. G16_C01 2016, Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic Atom Type and Bond Type Perception in Molecular Mechanical Calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Man, V.H.; Yang, W.; Lee, T.-S.; Wang, J. A Fast and High-Quality Charge Model for the next Generation General AMBER Force Field. J. Chem. Phys. 2020, 153, 114502. [Google Scholar] [CrossRef]
- Jorgensen, W.; Chandrasekhar, J.; Madura, J.; Impey, R.; Klein, M. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Li, P.; Roberts, B.P.; Chakravorty, D.K.; Merz, K.M.J. Rational Design of Particle Mesh Ewald Compatible Lennard-Jones Parameters for +2 Metal Cations in Explicit Solvent. J. Chem. Theory Comput. 2013, 9, 2733–2748. [Google Scholar] [CrossRef] [PubMed]
- Roe, D.R.; Cheatham, T.E. 3rd PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA Methods to Estimate Ligand-Binding Affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Chen, Y. Scratch Wound Healing Assay. Bio-Protocol 2012, 2, e100. [Google Scholar] [CrossRef]
- Gradiz, R.; Silva, H.C.; Carvalho, L.; Botelho, M.F.; Mota-Pinto, A. MIA PaCa-2 and PANC-1—Pancreas Ductal Adenocarcinoma Cell Lines with Neuroendocrine Differentiation and Somatostatin Receptors. Sci. Rep. 2016, 6, 21648. [Google Scholar] [CrossRef]
- Seeneevassen, L.; Giraud, J.; Molina-Castro, S.; Sifré, E.; Tiffon, C.; Beauvoit, C.; Staedel, C.; Mégraud, F.; Lehours, P.; Martin, O.C.B.; et al. Leukaemia Inhibitory Factor (LIF) Inhibits Cancer Stem Cells Tumorigenic Properties through Hippo Kinases Activation in Gastric Cancer. Cancers 2020, 12, 2011. [Google Scholar] [CrossRef]
- Urbanova, M.; Buocikova, V.; Trnkova, L.; Strapcova, S.; Kajabova, V.H.; Melian, E.B.; Novisedlakova, M.; Tomas, M.; Dubovan, P.; Earl, J.; et al. DNA Methylation Mediates EMT Gene Expression in Human Pancreatic Ductal Adenocarcinoma Cell Lines. Int. J. Mol. Sci. 2022, 23, 2117. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Li, J.; Lei, L.; He, H.; Chen, E.; Dong, J.; Yang, J. High Vimentin Expression Predicts a Poor Prognosis and Progression in Colorectal Cancer: A Study with Meta-Analysis and TCGA Database. Biomed Res. Int. 2018, 2018, 6387810. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Gao, W.; Lytle, N.K.; Huang, P.; Yuan, X.; Dann, A.M.; Ridinger-Saison, M.; DelGiorno, K.E.; Antal, C.E.; Liang, G.; et al. Targeting LIF-Mediated Paracrine Interaction for Pancreatic Cancer Therapy and Monitoring. Nature 2019, 569, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Zhang, M.; Gui, L.; Zhang, K. Expression Patterns and Prognostic Values of the Cyclin-Dependent Kinase 1 and Cyclin A2 Gene Cluster in Pancreatic Adenocarcinoma. J. Int. Med. Res. 2020, 48, 300060520930113. [Google Scholar] [CrossRef]
- Koppula, P.; Zhuang, L.; Gan, B. Cystine Transporter SLC7A11/XCT in Cancer: Ferroptosis, Nutrient Dependency, and Cancer Therapy. Protein Cell 2021, 12, 599–620. [Google Scholar] [CrossRef]
- Feng, Y.; Shi, C.; Wang, D.; Wang, X.; Chen, Z. Integrated Analysis of DNA Copy Number Changes and Gene Expression Identifies Key Genes in Gastric Cancer. J. Comput. Biol. 2020, 27, 877–887. [Google Scholar] [CrossRef]
- Viswanadhapalli, S.; Dileep, K.V.; Zhang, K.Y.J.; Nair, H.B.; Vadlamudi, R.K. Targeting LIF/LIFR Signaling in Cancer. Genes Dis. 2022, 9, 973–980. [Google Scholar] [CrossRef]
- Sasaki, N.; Hirano, K.; Shichi, Y.; Gomi, F.; Yoshimura, H.; Matsushita, A.; Toyoda, M.; Ishiwata, T. Gp130-Mediated STAT3 Activation Contributes to the Aggressiveness of Pancreatic Cancer through H19 Long Non-Coding RNA Expression. Cancers 2022, 14, 2055. [Google Scholar] [CrossRef]
- Stahl, N.; Boulton, T.G.; Farruggella, T.; Ip, N.Y.; Davis, S.; Witthuhn, B.A.; Quelle, F.W.; Silvennoinen, O.; Barbieri, G.; Pellegrini, S. Association and Activation of Jak-Tyk Kinases by CNTF-LIF-OSM-IL-6 Beta Receptor Components. Science 1994, 263, 92–95. [Google Scholar] [CrossRef]
- Bian, S.-B.; Yang, Y.; Liang, W.-Q.; Zhang, K.-C.; Chen, L.; Zhang, Z.-T. Leukemia Inhibitory Factor Promotes Gastric Cancer Cell Proliferation, Migration, and Invasion via the LIFR-Hippo-YAP Pathway. Ann. N. Y. Acad. Sci. 2021, 1484, 74–89. [Google Scholar] [CrossRef]
- Wrona, E.; Potemski, P.; Sclafani, F.; Borowiec, M. Leukemia Inhibitory Factor: A Potential Biomarker and Therapeutic Target in Pancreatic Cancer. Arch. Immunol. Ther. Exp. 2021, 69, 2. [Google Scholar] [CrossRef] [PubMed]
- Hunter, S.A.; McIntosh, B.J.; Shi, Y.; Sperberg, R.A.P.; Funatogawa, C.; Labanieh, L.; Soon, E.; Wastyk, H.C.; Mehta, N.; Carter, C.; et al. An Engineered Ligand Trap Inhibits Leukemia Inhibitory Factor as Pancreatic Cancer Treatment Strategy. Commun. Biol. 2021, 4, 452. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.-C.; Tsang, N.-M.; Chiang, W.-C.; Chang, K.-P.; Hsueh, C.; Liang, Y.; Juang, J.-L.; Chow, K.-P.N.; Chang, Y.-S. Leukemia Inhibitory Factor Promotes Nasopharyngeal Carcinoma Progression and Radioresistance. J. Clin. Investig. 2013, 123, 5269–5283. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Meng, L.; Wang, X.; Ma, G.; Chen, J. Expression of RRM1 and RRM2 as a Novel Prognostic Marker in Advanced Non-Small Cell Lung Cancer Receiving Chemotherapy. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2014, 35, 1899–1906. [Google Scholar] [CrossRef] [PubMed]
- Duxbury, M.S.; Ito, H.; Zinner, M.J.; Ashley, S.W.; Whang, E.E. RNA Interference Targeting the M2 Subunit of Ribonucleotide Reductase Enhances Pancreatic Adenocarcinoma Chemosensitivity to Gemcitabine. Oncogene 2004, 23, 1539–1548. [Google Scholar] [CrossRef] [PubMed]
- Bhutia, Y.D.; Hung, S.W.; Krentz, M.; Patel, D.; Lovin, D.; Manoharan, R.; Thomson, J.M.; Govindarajan, R. Differential Processing of Let-7a Precursors Influences RRM2 Expression and Chemosensitivity in Pancreatic Cancer: Role of LIN-28 and SET Oncoprotein. PLoS ONE 2013, 8, e53436. [Google Scholar] [CrossRef]
- Shin, S.Y.; Kim, J.H.; Lee, J.H.; Lim, Y.; Lee, Y.H. 2’-Hydroxyflavanone Induces Apoptosis through Egr-1 Involving Expression of Bax, P21, and NAG-1 in Colon Cancer Cells. Mol. Nutr. Food Res. 2012, 56, 761–774. [Google Scholar] [CrossRef]
- Fang, Y.; Yu, H.; Liang, X.; Xu, J.; Cai, X. Chk1-Induced CCNB1 Overexpression Promotes Cell Proliferation and Tumor Growth in Human Colorectal Cancer. Cancer Biol. Ther. 2014, 15, 1268–1279. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, X.; Li, X.; Meng, W.-B.; Bai, Z.-T.; Rui, S.-Z.; Wang, Z.-F.; Zhou, W.-C.; Jin, X.-D. Effect of CCNB1 Silencing on Cell Cycle, Senescence, and Apoptosis through the P53 Signaling Pathway in Pancreatic Cancer. J. Cell. Physiol. 2018, 234, 619–631. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Giorgio, C.; Lupia, A.; Marchianò, S.; Bordoni, M.; Bellini, R.; Massa, C.; Urbani, G.; Roselli, R.; Moraca, F.; Sepe, V.; et al. Repositioning Mifepristone as a Leukaemia Inhibitory Factor Receptor Antagonist for the Treatment of Pancreatic Adenocarcinoma. Cells 2022, 11, 3482. https://doi.org/10.3390/cells11213482
Di Giorgio C, Lupia A, Marchianò S, Bordoni M, Bellini R, Massa C, Urbani G, Roselli R, Moraca F, Sepe V, et al. Repositioning Mifepristone as a Leukaemia Inhibitory Factor Receptor Antagonist for the Treatment of Pancreatic Adenocarcinoma. Cells. 2022; 11(21):3482. https://doi.org/10.3390/cells11213482
Chicago/Turabian StyleDi Giorgio, Cristina, Antonio Lupia, Silvia Marchianò, Martina Bordoni, Rachele Bellini, Carmen Massa, Ginevra Urbani, Rosalinda Roselli, Federica Moraca, Valentina Sepe, and et al. 2022. "Repositioning Mifepristone as a Leukaemia Inhibitory Factor Receptor Antagonist for the Treatment of Pancreatic Adenocarcinoma" Cells 11, no. 21: 3482. https://doi.org/10.3390/cells11213482
APA StyleDi Giorgio, C., Lupia, A., Marchianò, S., Bordoni, M., Bellini, R., Massa, C., Urbani, G., Roselli, R., Moraca, F., Sepe, V., Catalanotti, B., Morretta, E., Monti, M. C., Biagioli, M., Distrutti, E., Zampella, A., & Fiorucci, S. (2022). Repositioning Mifepristone as a Leukaemia Inhibitory Factor Receptor Antagonist for the Treatment of Pancreatic Adenocarcinoma. Cells, 11(21), 3482. https://doi.org/10.3390/cells11213482