
Deep Molecular and In Silico Protein Analysis of p53 Alteration in Myelodysplastic Neoplasia and Acute Myeloid Leukemia

 ,
,  ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Samples
2.2. Immunohistochemistry
2.3. DNA Isolation
2.4. Next-Generation Sequencing
2.5. In Silico Protein Analysis
2.6. Statistical Analysis
3. Results
3.1. Patients Clinicopathological Characteristics
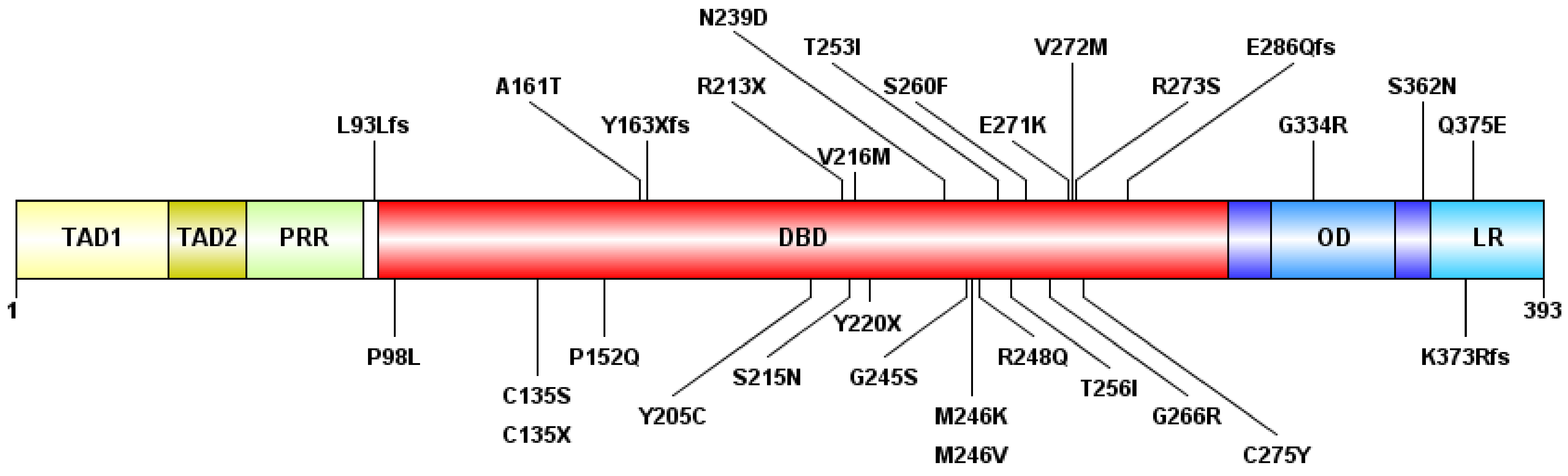
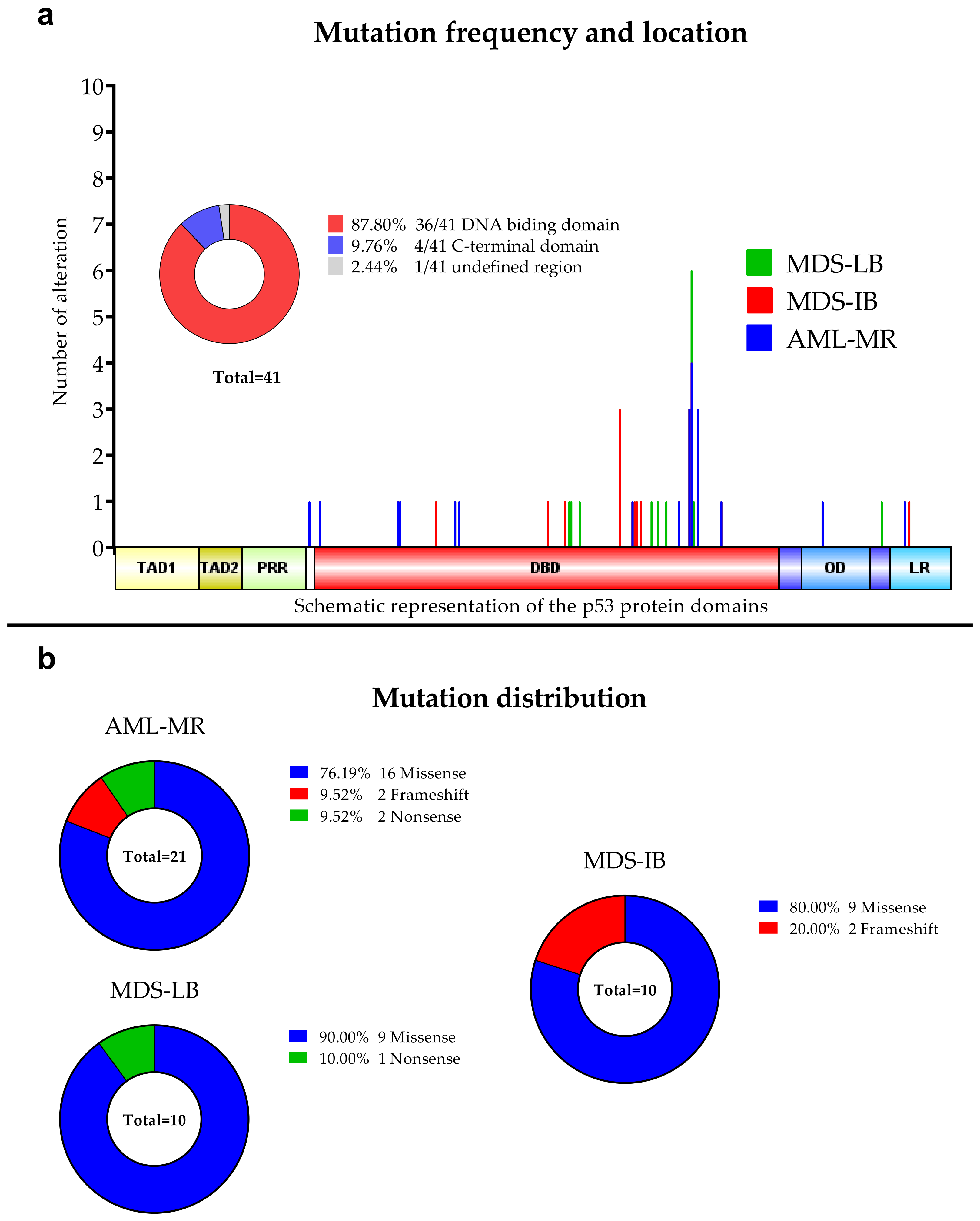
3.2. Next-Generation Sequencing
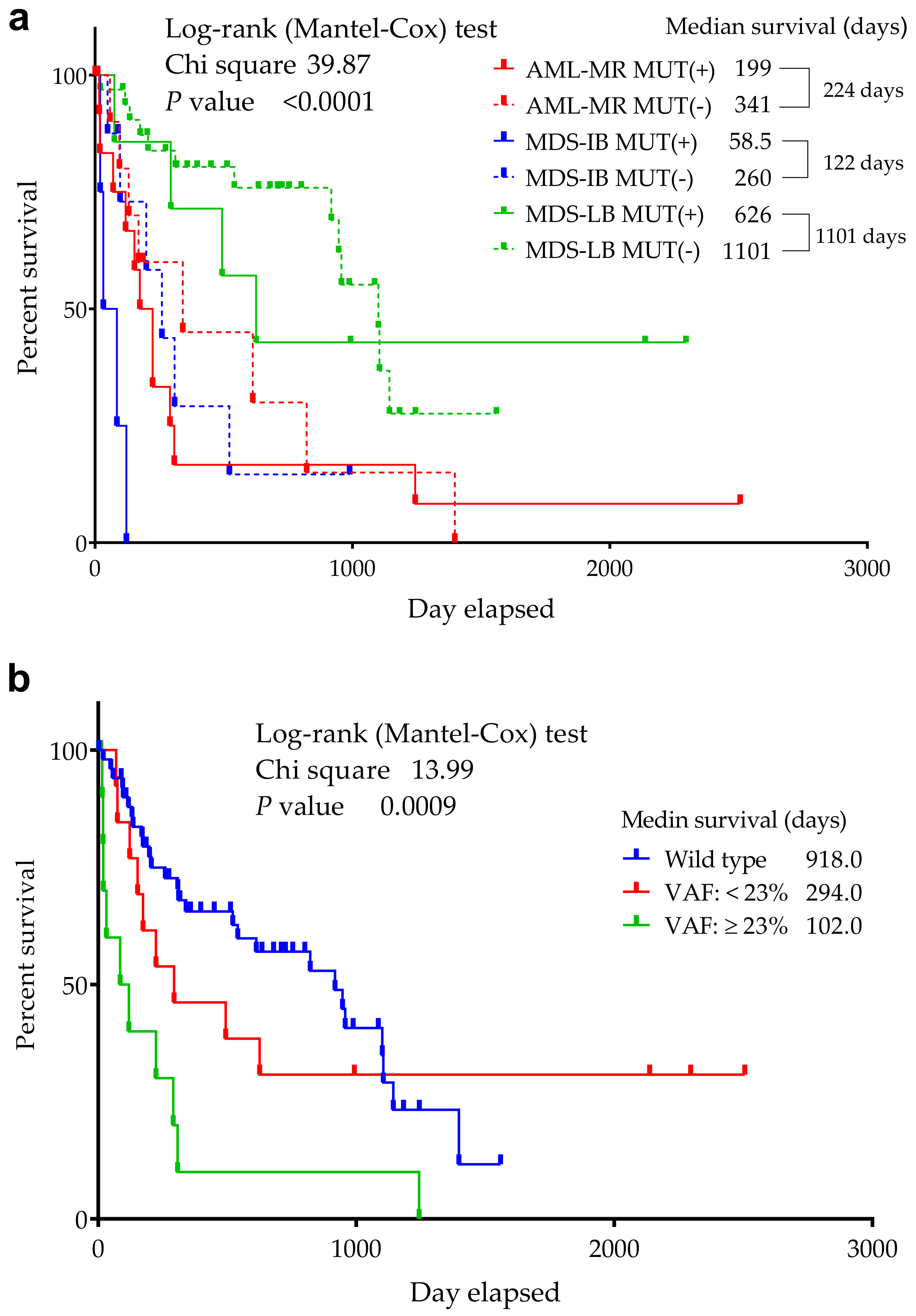
3.3. Comparison of the Cytogenetic, IHC Results, OS, and TP53 Mutation Status
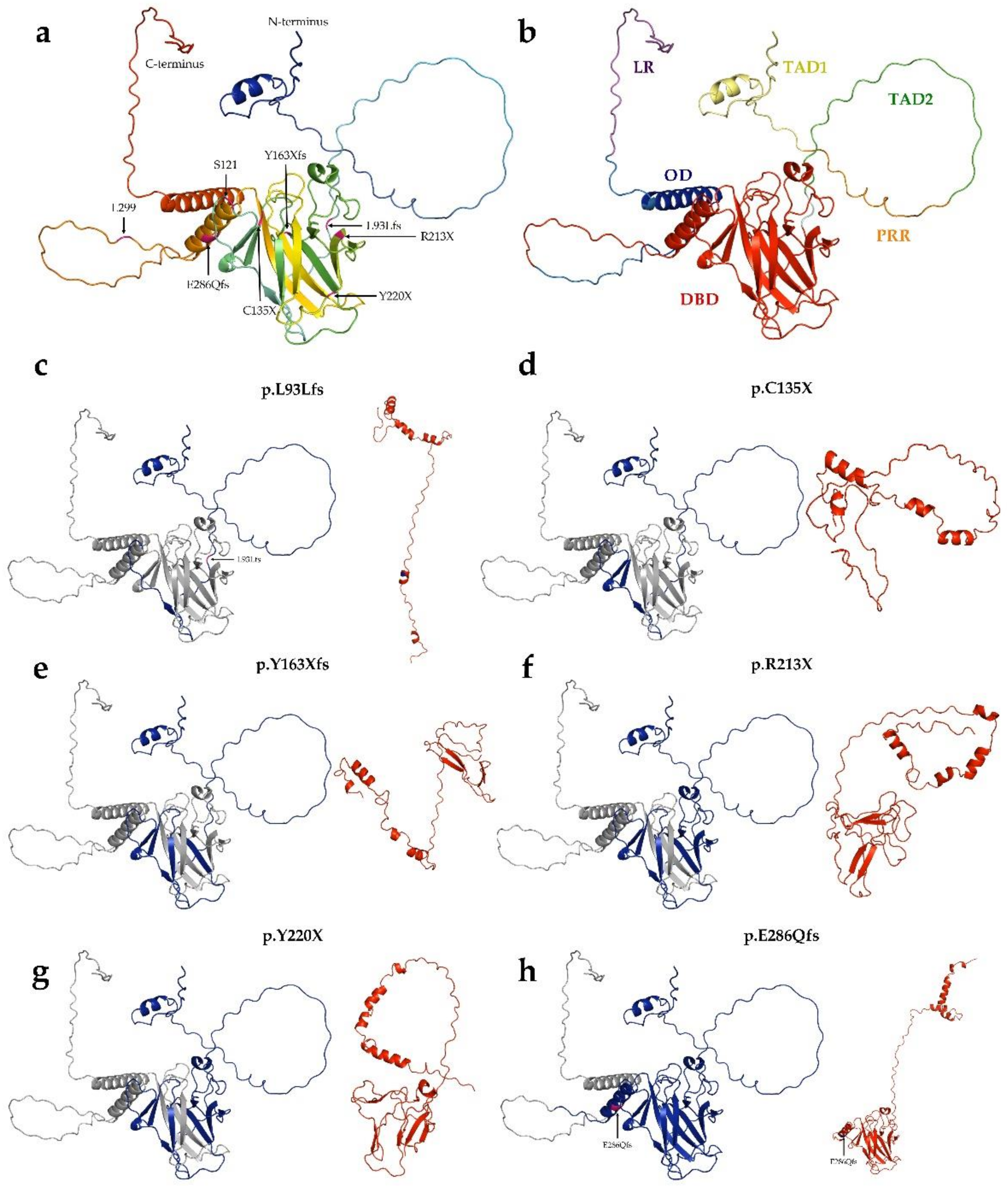
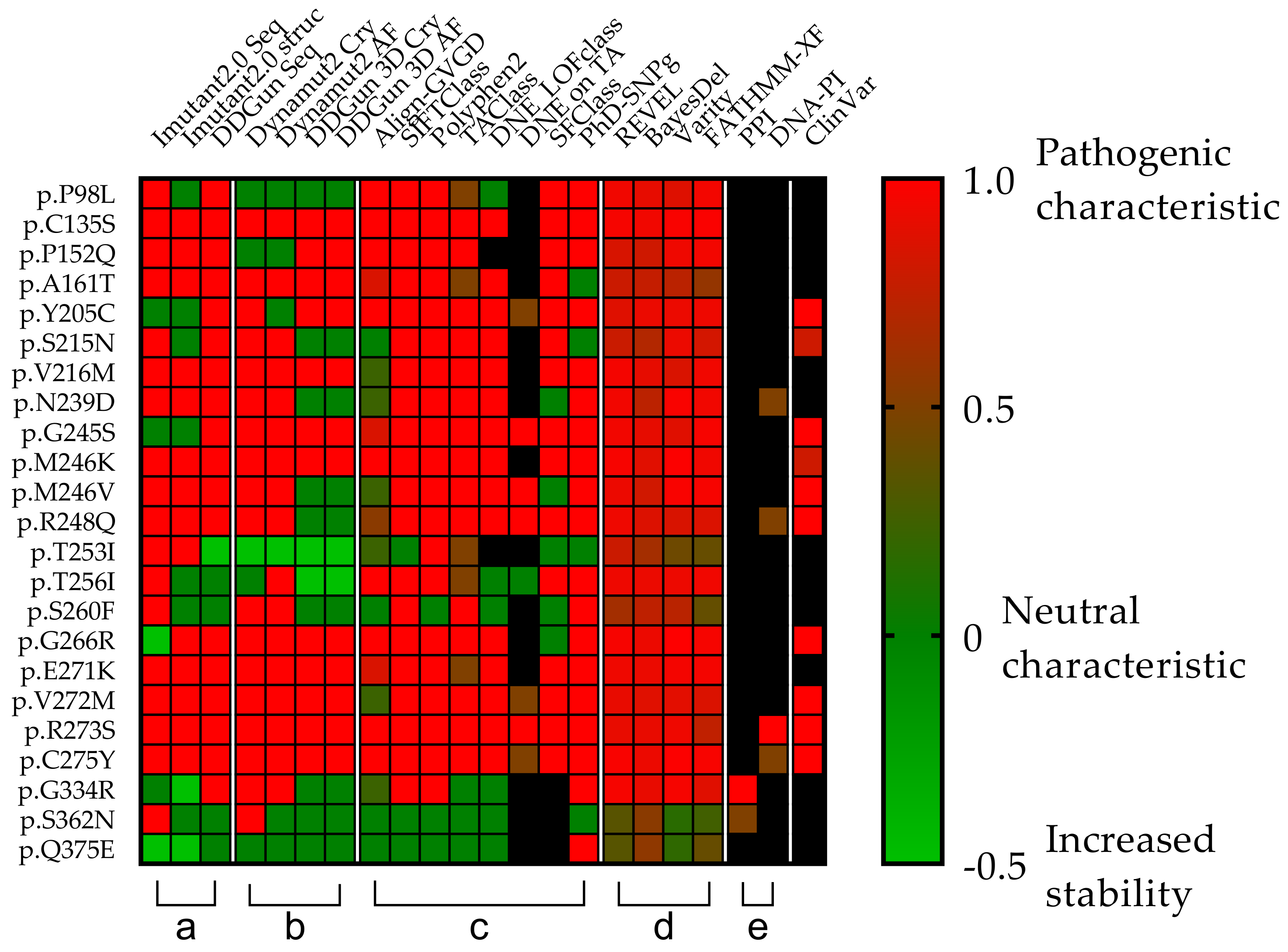
3.4. Mutations’ Pathogenicity
3.5. In Vitro Experiments in the TP53 Mutation Database
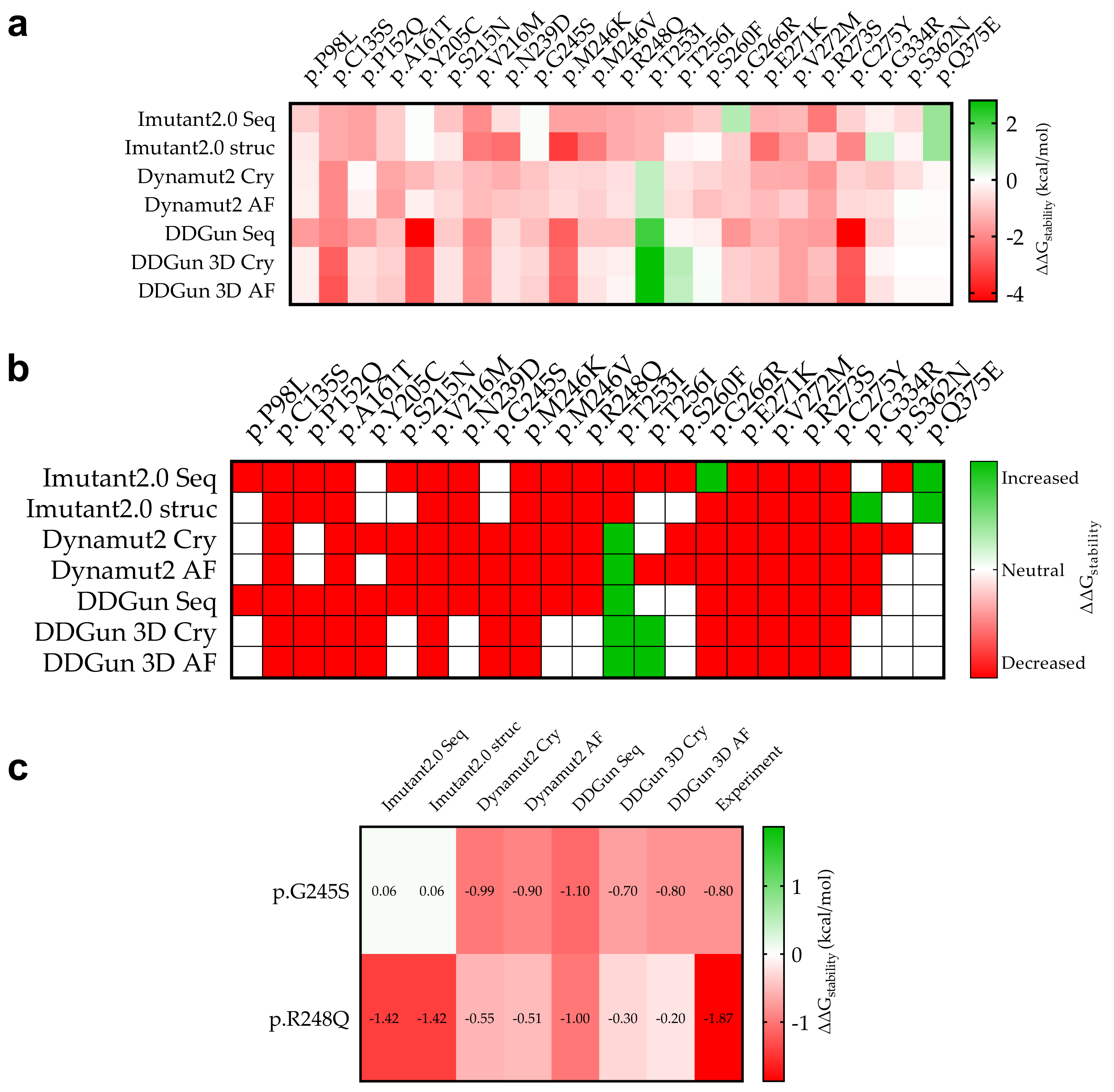
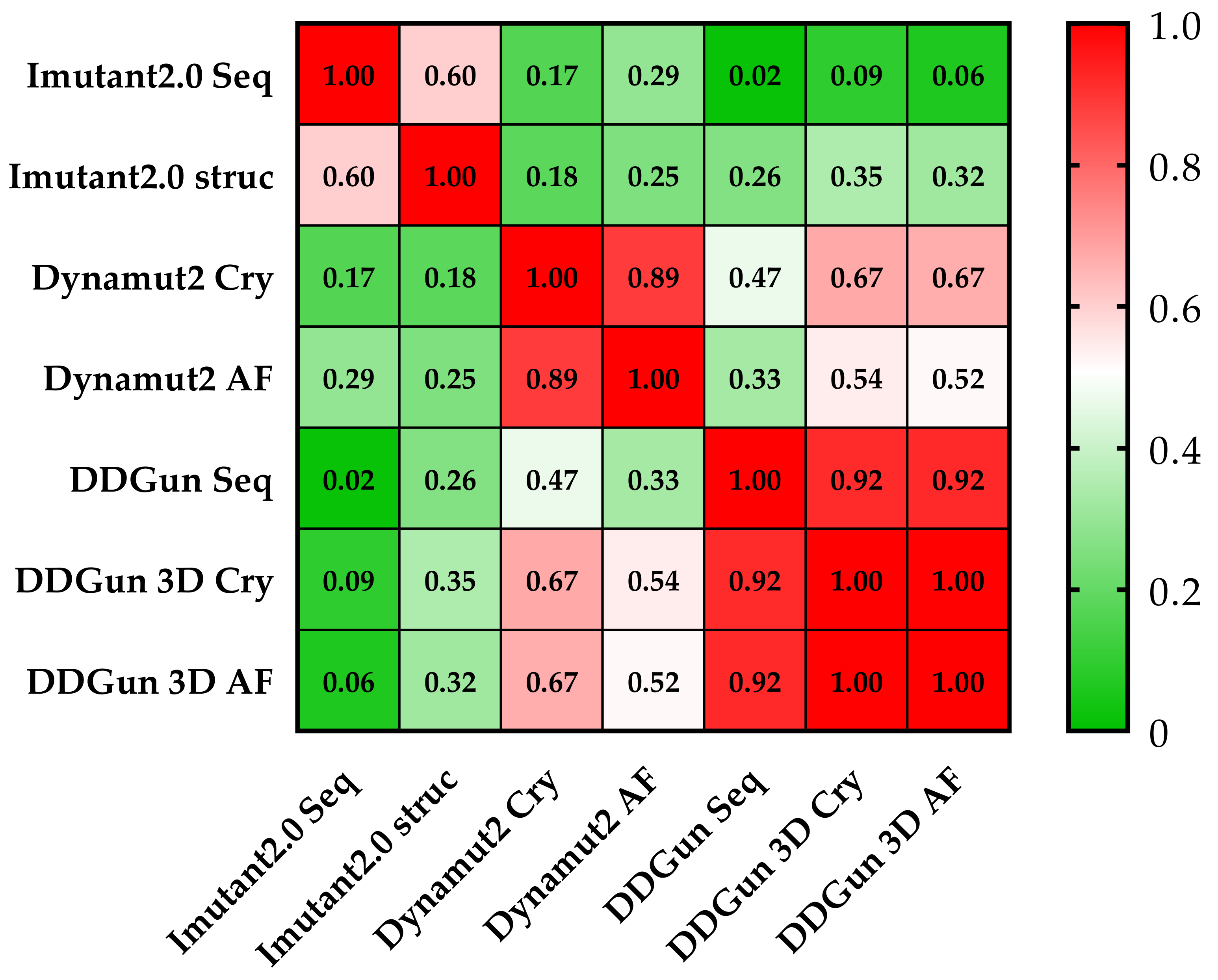
3.6. Mutant p53 Protein Stability Analysis
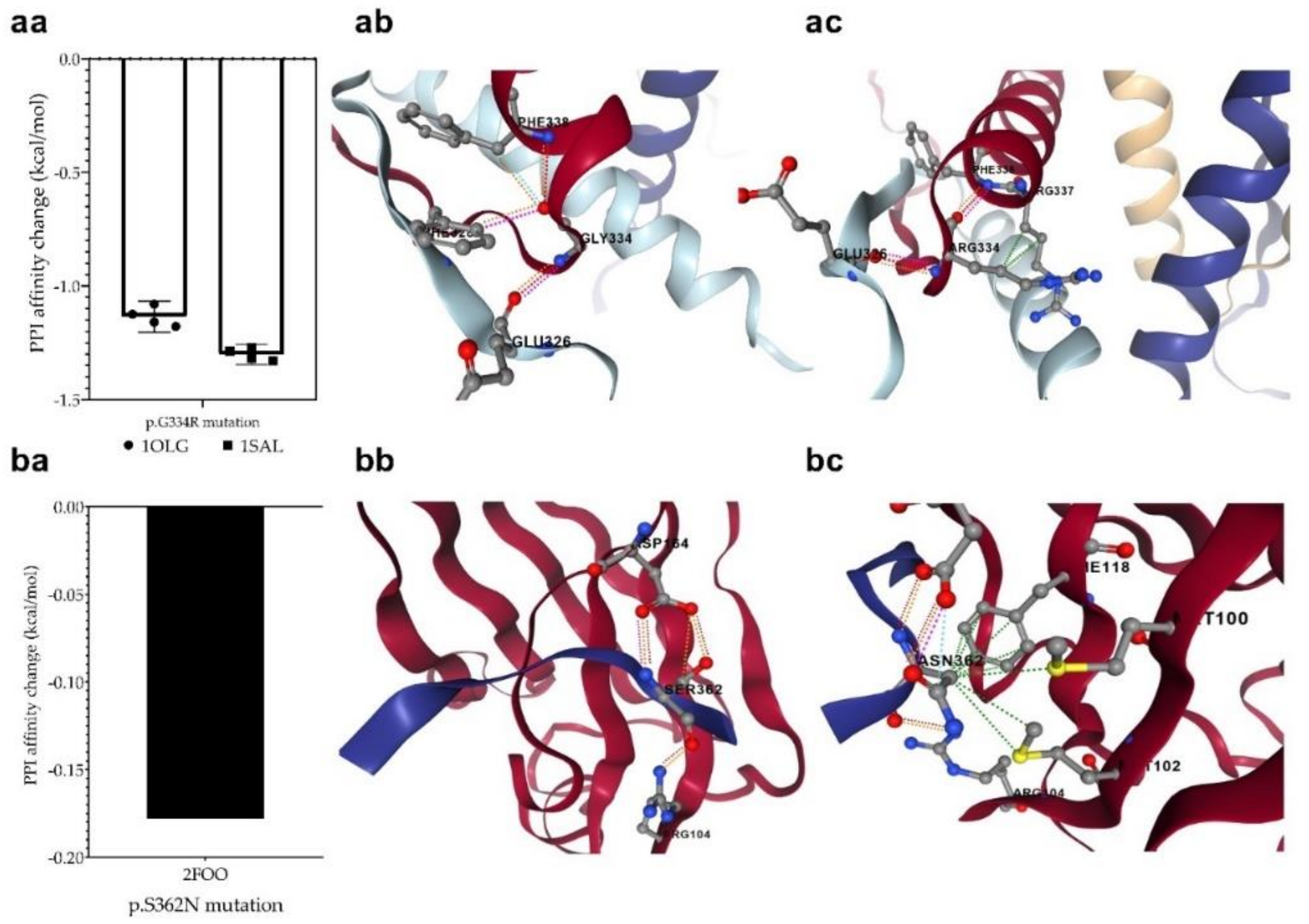
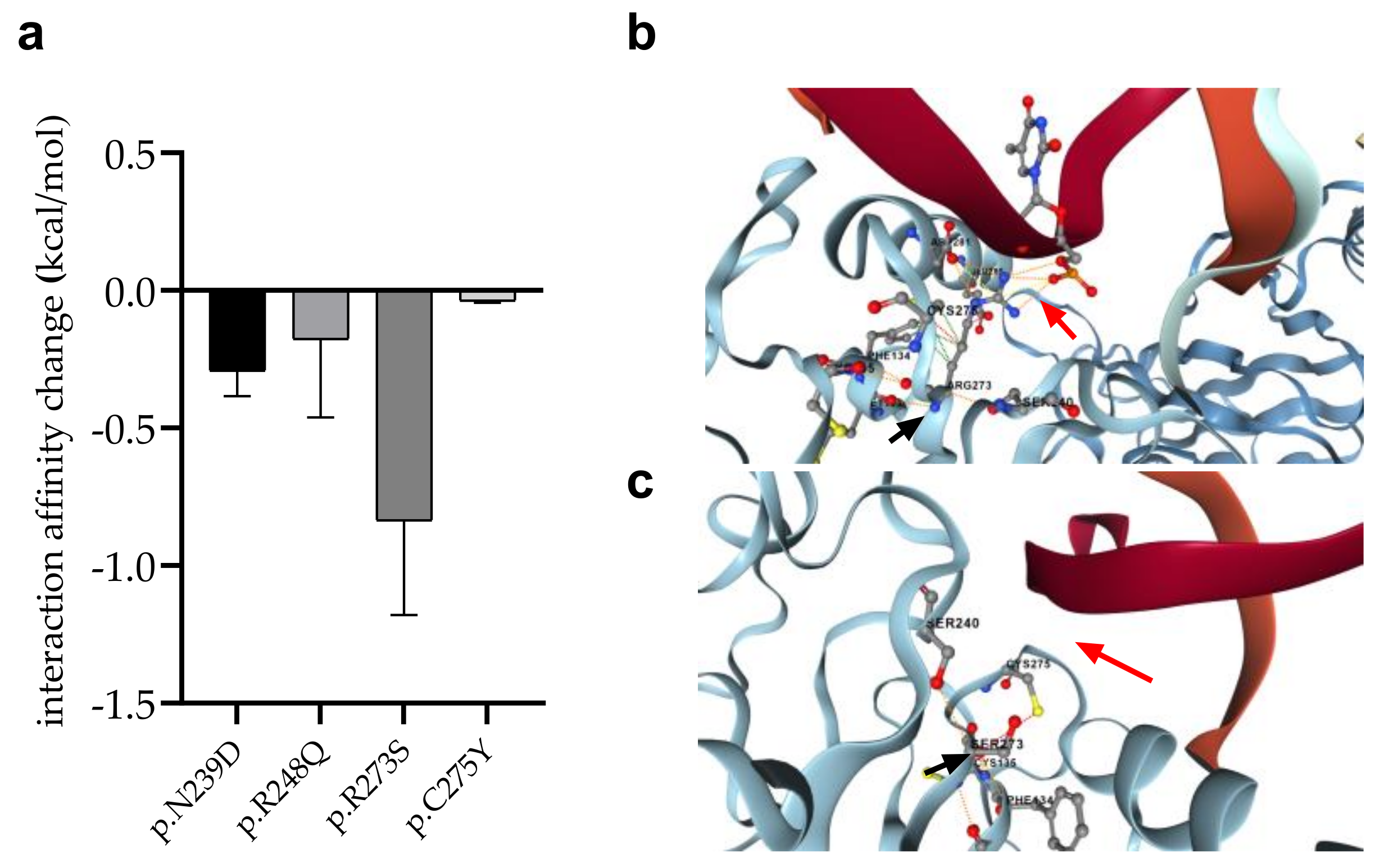
3.7. Changes in Protein-Protein Interactions upon Mutations
3.8. Changes in p53 Protein-DNA Interactions as Affected by Mutations
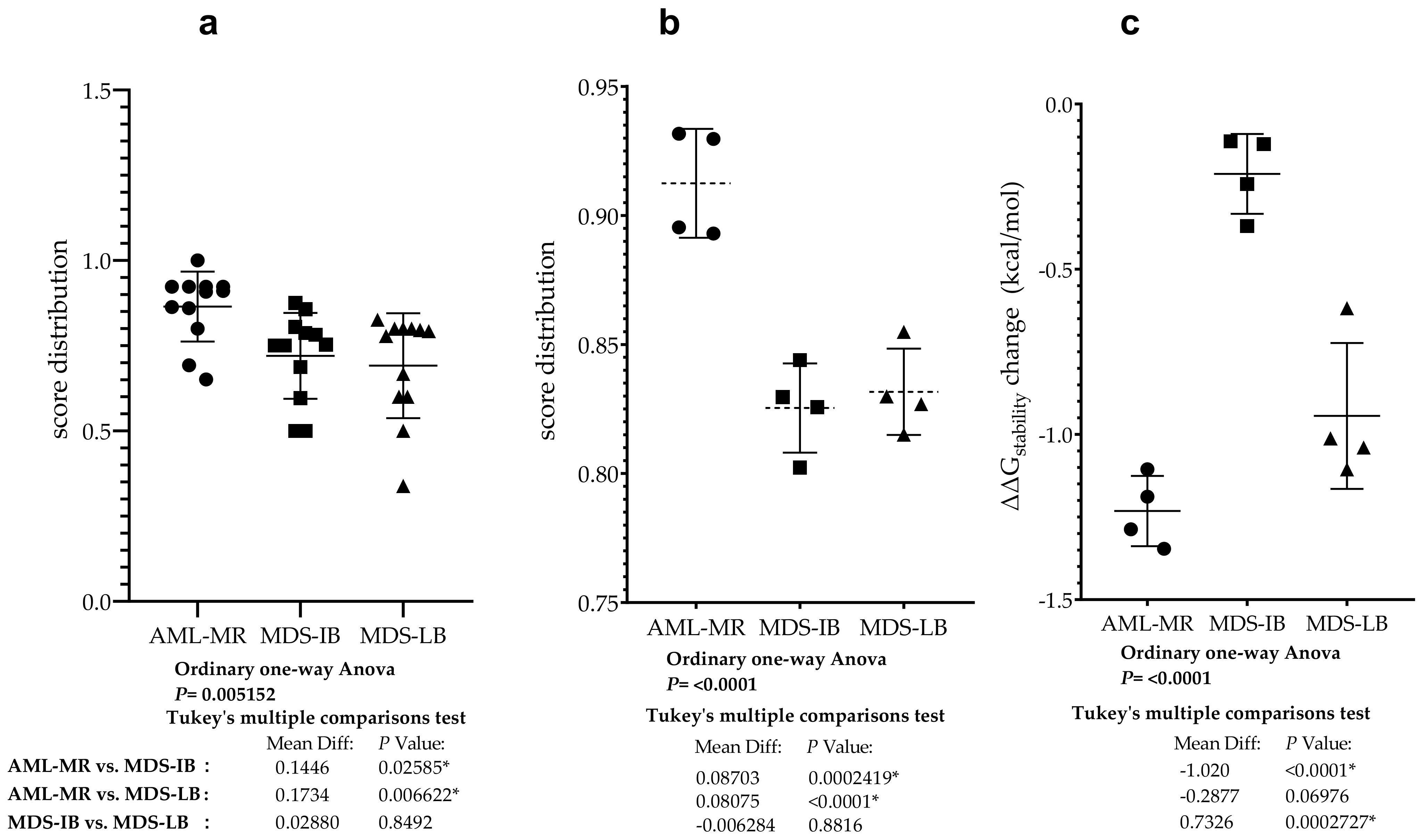
3.9. Statistical Analyses of the Mutant p53 Protein Pathogenicity and Structural Stability in the AML/MDS Patients
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Thol, F.; Platzbecker, U. Do Next-Generation Sequencing Results Drive Diagnostic and Therapeutic Decisions in MDS? Blood Adv. 2019, 3, 3449–3453. [Google Scholar] [CrossRef] [PubMed]
- Brosh, R.; Rotter, V. When Mutants Gain New Powers: News from the Mutant P53 Field. Nat. Rev. Cancer 2009, 9, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Oren, M. The First 30 Years of P53: Growing Ever More Complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Hollstein, M.; Rice, K.; Greenblatt, M.S.; Soussi, T.; Fuchs, R.; Sørlie, T.; Hovig, E.; Smith-Sørensen, B.; Montesano, R.; Harris, C.C. Database of P53 Gene Somatic Mutations in Human Tumors and Cell Lines. Nucleic Acids Res. 1994, 22, 3551–3555. [Google Scholar] [PubMed]
- Levine, A.J. P53, the Cellular Gatekeeper for Growth and Division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef]
- Bouaoun, L.; Sonkin, D.; Ardin, M.; Hollstein, M.; Byrnes, G.; Zavadil, J.; Olivier, M. TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum. Mutat. 2016, 37, 865–876. [Google Scholar] [CrossRef]
- Jenkins, L.M.M.; Durell, S.R.; Mazur, S.J.; Appella, E. P53 N-Terminal Phosphorylation: A Defining Layer of Complex Regulation. Carcinogenesis 2012, 33, 1441–1449. [Google Scholar] [CrossRef]
- Kulasekararaj, A.G.; Smith, A.E.; Mian, S.A.; Mohamedali, A.M.; Krishnamurthy, P.; Lea, N.C.; Gäken, J.; Pennaneach, C.; Ireland, R.; Czepulkowski, B.; et al. TP53 Mutations in Myelodysplastic Syndrome Are Strongly Correlated with Aberrations of Chromosome 5, and Correlate with Adverse Prognosis. Br. J. Haematol. 2013, 160, 660–672. [Google Scholar] [CrossRef]
- Haferlach, C.; Dicker, F.; Herholz, H.; Schnittger, S.; Kern, W.; Haferlach, T. Mutations of the TP53 Gene in Acute Myeloid Leukemia Are Strongly Associated with a Complex Aberrant Karyotype. Leukemia 2008, 22, 1539–1541. [Google Scholar] [CrossRef]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D.; et al. Clinical Effect of Point Mutations in Myelodysplastic Syndromes. N. Engl. J. Med. 2011, 364, 2496–2506. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and Biological Implications of Driver Mutations in Myelodysplastic Syndromes. Blood 2013, 122, 3616–3627. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Anishchenko, I.; Park, H.; Peng, Z.; Ovchinnikov, S.; Baker, D. Improved Protein Structure Prediction Using Predicted Interresidue Orientations. Proc. Natl. Acad. Sci. USA 2020, 117, 1496–1503. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate Prediction of Protein Structures and Interactions Using a Three-Track Neural Network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and Recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef]
- Erdős, G.; Pajkos, M.; Dosztányi, Z. IUPred3: Prediction of Protein Disorder Enhanced with Unambiguous Experimental Annotation and Visualization of Evolutionary Conservation. Nucleic Acids Res. 2021, 49, W297–W303. [Google Scholar] [CrossRef]
- Garnier, J.; Gibrat, J.-F.; Robson, B. [32] GOR Method for Predicting Protein Secondary Structure from Amino Acid Sequence. Methods Enzymol. 1996, 266, 540–553. [Google Scholar]
- Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant2.0: Predicting Stability Changes upon Mutation from the Protein Sequence or Structure. Nucleic Acids Res. 2005, 33, W306–W310. [Google Scholar] [CrossRef]
- Montanucci, L.; Capriotti, E.; Frank, Y.; Ben-Tal, N.; Fariselli, P. DDGun: An Untrained Method for the Prediction of Protein Stability Changes upon Single and Multiple Point Variations. BMC Bioinform. 2019, 20, 335. [Google Scholar] [CrossRef]
- Montanucci, L.; Capriotti, E.; Birolo, G.; Benevenuta, S.; Pancotti, C.; Lal, D.; Fariselli, P. DDGun: An Untrained Predictor of Protein Stability Changes upon Amino Acid Variants. Nucleic Acids Res. 2022, 50, W222–W227. [Google Scholar] [CrossRef]
- Rodrigues, C.H.M.; Pires, D.E.V.; Ascher, D.B. DynaMut2: Assessing Changes in Stability and Flexibility upon Single and Multiple Point Missense Mutations. Protein Sci. 2021, 30, 60–69. [Google Scholar] [CrossRef]
- Golovenko, D.; Bräuning, B.; Vyas, P.; Haran, T.E.; Rozenberg, H.; Shakked, Z. New Insights into the Role of DNA Shape on Its Recognition by P53 Proteins. Structure 2018, 26, 1237–1250.e6. [Google Scholar] [CrossRef]
- Mittl, P.R.; Chène, P.; Grütter, M.G. Crystallization and Structure Solution of P53 (Residues 326–356) by Molecular Replacement Using an NMR Model as Template. Acta Crystallogr. D Biol. Crystallogr. 1998, 54, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, P.D.; Gorina, S.; Pavletich, N.P. Crystal Structure of the Tetramerization Domain of the P53 Tumor Suppressor at 1.7 Angstroms. Science 1995, 267, 1498–1502. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Y.; Saridakis, V.; Sarkari, F.; Duan, S.; Wu, T.; Arrowsmith, C.H.; Frappier, L. Molecular Recognition of P53 and MDM2 by USP7/HAUSP. Nat. Struct. Mol. Biol. 2006, 13, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Avalos, J.L.; Bever, K.M.; Wolberger, C. Mechanism of Sirtuin Inhibition by Nicotinamide: Altering the NAD(+) Cosubstrate Specificity of a Sir2 Enzyme. Mol. Cell 2005, 17, 855–868. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively Expanding the Structural Coverage of Protein-Sequence Space with High-Accuracy Models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Joerger, A.C.; Ang, H.C.; Fersht, A.R. Structural Basis for Understanding Oncogenic P53 Mutations and Designing Rescue Drugs. Proc. Natl. Acad. Sci. USA 2006, 103, 15056–15061. [Google Scholar] [CrossRef]
- Bullock, A.N.; Henckel, J.; Fersht, A.R. Quantitative Analysis of Residual Folding and DNA Binding in Mutant P53 Core Domain: Definition of Mutant States for Rescue in Cancer Therapy. Oncogene 2000, 19, 1245–1256. [Google Scholar] [CrossRef]
- Rodrigues, C.H.M.; Myung, Y.; Pires, D.E.V.; Ascher, D.B. MCSM-PPI2: Predicting the Effects of Mutations on Protein–Protein Interactions. Nucleic Acids Res. 2019, 47, W338–W344. [Google Scholar] [CrossRef]
- Clore, G.M.; Omichinski, J.G.; Sakaguchi, K.; Zambrano, N.; Sakamoto, H.; Appella, E.; Gronenborn, A.M. High-Resolution Structure of the Oligomerization Domain of P53 by Multidimensional NMR. Science 1994, 265, 386–391. [Google Scholar] [CrossRef]
- Clore, G.M.; Ernst, J.; Clubb, R.; Omichinski, J.G.; Kennedy, W.M.; Sakaguchi, K.; Appella, E.; Gronenborn, A.M. Refined Solution Structure of the Oligomerization Domain of the Tumour Suppressor P53. Nat. Struct. Biol. 1995, 2, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Ascher, D.B. MCSM–NA: Predicting the Effects of Mutations on Protein–Nucleic Acids Interactions. Nucleic Acids Res. 2017, 45, W241–W246. [Google Scholar] [CrossRef] [PubMed]
- De Andrade, K.C.; Lee, E.E.; Tookmanian, E.M.; Kesserwan, C.A.; Manfredi, J.J.; Hatton, J.N.; Loukissas, J.K.; Zavadil, J.; Zhou, L.; Olivier, M.; et al. The TP53 Database: Transition from the International Agency for Research on Cancer to the US National Cancer Institute. Cell Death Differ. 2022, 29, 1071–1073. [Google Scholar] [CrossRef] [PubMed]
- Mathe, E.; Olivier, M.; Kato, S.; Ishioka, C.; Hainaut, P.; Tavtigian, S.V. Computational Approaches for Predicting the Biological Effect of P53 Missense Mutations: A Comparison of Three Sequence Analysis Based Methods. Nucleic Acids Res. 2006, 34, 1317–1325. [Google Scholar] [CrossRef]
- Tavtigian, S.V.; Deffenbaugh, A.M.; Yin, L.; Judkins, T.; Scholl, T.; Samollow, P.B.; de Silva, D.; Zharkikh, A.; Thomas, A. Comprehensive Statistical Study of 452 BRCA1 Missense Substitutions with Classification of Eight Recurrent Substitutions as Neutral. J. Med. Genet. 2006, 43, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.-J. PERCH: A Unified Framework for Disease Gene Prioritization. Hum. Mutat. 2017, 38, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Fortuno, C.; James, P.A.; Young, E.L.; Feng, B.; Olivier, M.; Pesaran, T.; Tavtigian, S.V.; Spurdle, A.B. Improved, ACMG-Compliant, in Silico Prediction of Pathogenicity for Missense Substitutions Encoded by TP53 Variants. Hum. Mutat. 2018, 39, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. Predicting Deleterious Amino Acid Substitutions. Genome Res. 2001, 11, 863–874. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A Method and Server for Predicting Damaging Missense Mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Kato, S.; Han, S.-Y.; Liu, W.; Otsuka, K.; Shibata, H.; Kanamaru, R.; Ishioka, C. Understanding the Function-Structure and Function-Mutation Relationships of P53 Tumor Suppressor Protein by High-Resolution Missense Mutation Analysis. Proc. Natl. Acad. Sci. USA 2003, 100, 8424–8429. [Google Scholar] [CrossRef] [PubMed]
- Giacomelli, A.O.; Yang, X.; Lintner, R.E.; McFarland, J.M.; Duby, M.; Kim, J.; Howard, T.P.; Takeda, D.Y.; Ly, S.H.; Kim, E.; et al. Mutational Processes Shape the Landscape of TP53 Mutations in Human Cancer. Nat. Genet. 2018, 50, 1381–1387. [Google Scholar] [CrossRef] [PubMed]
- Dearth, L.R.; Qian, H.; Wang, T.; Baroni, T.E.; Zeng, J.; Chen, S.W.; Yi, S.Y.; Brachmann, R.K. Inactive Full-Length P53 Mutants Lacking Dominant Wild-Type P53 Inhibition Highlight Loss of Heterozygosity as an Important Aspect of P53 Status in Human Cancers. Carcinogenesis 2007, 28, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Monti, P.; Perfumo, C.; Bisio, A.; Ciribilli, Y.; Menichini, P.; Russo, D.; Umbach, D.M.; Resnick, M.A.; Inga, A.; Fronza, G. Dominant-Negative Features of Mutant TP53 in Germline Carriers Have Limited Impact on Cancer Outcomes. Mol. Cancer Res. 2011, 9, 271–279. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public Archive of Relationships among Sequence Variation and Human Phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. ClinVar: Public Archive of Interpretations of Clinically Relevant Variants. Nucleic Acids Res. 2016, 44, D862–D868. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Kattman, B.L. ClinVar at Five Years: Delivering on the Promise. Hum. Mutat. 2018, 39, 1623–1630. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving Access to Variant Interpretations and Supporting Evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Chitipiralla, S.; Brown, G.R.; Chen, C.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; Kaur, K.; Liu, C.; et al. ClinVar: Improvements to Accessing Data. Nucleic Acids Res. 2020, 48, D835–D844. [Google Scholar] [CrossRef]
- Wu, Y.; Liu, H.; Li, R.; Sun, S.; Weile, J.; Roth, F.P. Improved Pathogenicity Prediction for Rare Human Missense Variants. Am. J. Hum. Genet. 2021, 108, 1891–1906. [Google Scholar] [CrossRef]
- Capriotti, E.; Fariselli, P. PhD-SNPg: A Webserver and Lightweight Tool for Scoring Single Nucleotide Variants. Nucleic Acids Res. 2017, 45, W247–W252. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.F.; Shihab, H.A.; Mort, M.; Cooper, D.N.; Gaunt, T.R.; Campbell, C. FATHMM-XF: Accurate Prediction of Pathogenic Point Mutations via Extended Features. Bioinformatics 2018, 34, 511–513. [Google Scholar] [CrossRef]
- Cumbo, C.; Tota, G.; Anelli, L.; Zagaria, A.; Specchia, G.; Albano, F. TP53 in Myelodysplastic Syndromes: Recent Biological and Clinical Findings. Int. J. Mol. Sci. 2020, 21, 3432. [Google Scholar] [CrossRef] [PubMed]
- Bally, C.; Adès, L.; Renneville, A.; Sebert, M.; Eclache, V.; Preudhomme, C.; Mozziconacci, M.-J.; de The, H.; Lehmann-Che, J.; Fenaux, P. Prognostic Value of TP53 Gene Mutations in Myelodysplastic Syndromes and Acute Myeloid Leukemia Treated with Azacitidine. Leuk. Res. 2014, 38, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of Genetic Lesions in 944 Patients with Myelodysplastic Syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Haase, D.; Stevenson, K.E.; Neuberg, D.; Maciejewski, J.P.; Nazha, A.; Sekeres, M.A.; Ebert, B.L.; Garcia-Manero, G.; Haferlach, C.; Haferlach, T.; et al. TP53 Mutation Status Divides Myelodysplastic Syndromes with Complex Karyotypes into Distinct Prognostic Subgroups. Leukemia 2019, 33, 1747–1758. [Google Scholar] [CrossRef]
- Bernard, E.; Nannya, Y.; Hasserjian, R.P.; Devlin, S.M.; Tuechler, H.; Medina-Martinez, J.S.; Yoshizato, T.; Shiozawa, Y.; Saiki, R.; Malcovati, L.; et al. Implications of TP53 Allelic State for Genome Stability, Clinical Presentation and Outcomes in Myelodysplastic Syndromes. Nat. Med. 2020, 26, 1549–1556. [Google Scholar] [CrossRef]
- Sallman, D.A.; Komrokji, R.; Vaupel, C.; Cluzeau, T.; Geyer, S.M.; McGraw, K.L.; Al Ali, N.H.; Lancet, J.; McGinniss, M.J.; Nahas, S.; et al. Impact of TP53 Mutation Variant Allele Frequency on Phenotype and Outcomes in Myelodysplastic Syndromes. Leukemia 2016, 30, 666–673. [Google Scholar] [CrossRef]
- Goel, S.; Hall, J.; Pradhan, K.; Hirsch, C.; Przychodzen, B.; Shastri, A.; Mantzaris, I.; Janakiram, M.; Battini, R.; Kornblum, N.; et al. High Prevalence and Allele Burden-Independent Prognostic Importance of P53 Mutations in an Inner-City MDS/AML Cohort. Leukemia 2016, 30, 1793–1795. [Google Scholar] [CrossRef]
- Sabapathy, K.; Lane, D.P. Therapeutic Targeting of P53: All Mutants Are Equal, but Some Mutants Are More Equal than Others. Nat. Rev. Clin. Oncol. 2018, 15, 13–30. [Google Scholar] [CrossRef]
- Nakano, Y.; Kiyoi, H.; Miyawaki, S.; Asou, N.; Ohno, R.; Saito, H.; Naoe, T. Molecular Evolution of Acute Myeloid Leukaemia in Relapse: Unstable N-ras and FLT3 Genes Compared with P53 Gene. Br. J. Haematol. 1999, 104, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, A.; Inokuchi, K.; Yamaguchi, H.; Mizuki, T.; Tanosaki, S.; Shimada, T.; Dan, K. Near-Triploidy and near-Tetraploidy in Hematological Malignancies and Mutation of the P53 Gene. Clin. Lab. Haematol. 2004, 26, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Padre, R.C.; De Mendoza, T.H.; Bottero, V.; Tergaonkar, V.B.; Verma, I.M. Phosphorylation of P53 by IκB Kinase 2 Promotes Its Degradation by β-TrCP. Proc. Natl. Acad. Sci. USA 2009, 106, 2629–2634. [Google Scholar] [CrossRef]
- Xu, H.-T.; Lai, W.-L.; Liu, H.-F.; Wong, L.L.-Y.; Ng, I.O.-L.; Ching, Y.P. PAK4 Phosphorylates P53 at Serine 215 to Promote Liver Cancer Metastasis. Cancer Res. 2016, 76, 5732–5742. [Google Scholar] [CrossRef]
- Kakudo, Y.; Shibata, H.; Otsuka, K.; Kato, S.; Ishioka, C. Lack of Correlation between P53-Dependent Transcriptional Activity and the Ability to Induce Apoptosis among 179 Mutant P53s. Cancer Res. 2005, 65, 2108–2114. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, T.; Kato, S.; Otsuka, K.; Watanabe, G.; Kumabe, T.; Tominaga, T.; Yoshimoto, T.; Ishioka, C. The Relationship among P53 Oligomer Formation, Structure and Transcriptional Activity Using a Comprehensive Missense Mutation Library. Oncogene 2005, 24, 6976–6981. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cases | Sex | Age (Years) | Diagnosis | OS (Days) | p53 IHC * | TP53 NGS | Karyotype |
|---|---|---|---|---|---|---|---|
| 1 | F | 65 | AML-MR | 0 | + | + | CK |
| 2 | M | 73 | 119 | + | + | n.a. | |
| 3 | M | 35 | 169 | - | - | CK | |
| 4 | F | 47 | 2506 | - | + | normal | |
| 5 | M | 76 | 1244 | - | + | CK | |
| 6 | M | 64 | 308 | + | + | n.a. | |
| 7 | M | 69 | 171 | - | - | CK | |
| 8 | F | 76 | 3 | - | - | n.a. | |
| 9 | M | 33 | 131 | + | - | normal | |
| 10 | M | 78 | 153 | - | + | CK | |
| 11 | F | 53 | 291 | + | + | CK | |
| 12 | M | 64 | 822 | - | - | normal | |
| 13 | F | 73 | 70 | - | + | tetrasomy | |
| 14 | F | 88 | 95 | - | - | normal | |
| 15 | F | 83 | 1 | + | + | CK | |
| 16 | M | 66 | 174 | - | + | CK | |
| 17 | M | 61 | 224 | - | + | normal | |
| 18 | F | 64 | 186 | - | - | 47,XX,+8 | |
| 19 | F | 66 | 341 | - | - | CK | |
| 20 | M | 63 | 19 | + | + | CK | |
| 21 | F | 86 | 224 | + | + | n.a. | |
| 22 | M | 41 | 58 | - | - | normal | |
| 23 | F | 56 | 613 | - | - | CK | |
| 24 | M | 65 | 10 | + | + | CK | |
| 25 | F | 69 | 16 | + | + | CK | |
| 26 | F | 69 | 1398 | - | - | normal | |
| 27 | M | 48 | MDS-IB | 89 | - | - | normal |
| 28 | M | 56 | 32 | - | + | n.a. | |
| 29 | M | 59 | 85 | - | + | CK | |
| 30 | F | 69 | 989 | - | - | normal | |
| 31 | F | 25 | 260 | - | - | 46,XX,t(8;21)/46,XX | |
| 32 | M | 80 | 20 | + | + | CK | |
| 33 | M | 61 | 199 | - | - | normal | |
| 34 | M | 79 | 122 | + | + | n.a. | |
| 35 | M | 59 | 49 | - | - | CK | |
| 36 | F | 90 | 97 | - | - | normal | |
| 37 | M | 65 | 309 | - | - | normal | |
| 38 | F | 66 | 522 | - | - | normal | |
| 39 | F | 52 | MDS-LB | 626 | - | + | normal |
| 40 | F | 80 | 312 | - | - | normal | |
| 41 | F | 71 | 494 | - | + | 46,XX,t(2;12)/46,XX,t(17;17) | |
| 42 | M | 64 | 2296 | - | + | normal | |
| 43 | F | 64 | 754 | - | - | n.a. | |
| 44 | M | 70 | 450 | - | - | normal | |
| 45 | M | 64 | 918 | - | - | normal | |
| 46 | M | 41 | 294 | - | + | n.a. | |
| 47 | F | 74 | 710 | - | - | 46,XX,del(20q) | |
| 48 | F | 61 | 75 | - | + | normal | |
| 49 | F | 58 | 1086 | - | - | normal | |
| 50 | F | 60 | 993 | - | + | normal | |
| 51 | M | 36 | 274 | - | - | normal | |
| 52 | M | 60 | 175 | - | - | normal | |
| 53 | M | 72 | 513 | - | - | normal | |
| 54 | F | 71 | 1105 | - | - | normal | |
| 55 | F | 78 | 1559 | - | - | 46,XX,del(5q)/46,XX | |
| 56 | F | 64 | 20 | - | - | n.a. | |
| 57 | F | 89 | 541 | - | - | n.a. | |
| 58 | M | 68 | 1101 | - | - | CK | |
| 59 | M | 36 | 203 | - | - | n.a. | |
| 60 | F | 64 | 2137 | - | + | normal | |
| 61 | M | 77 | 1144 | - | - | CK | |
| 62 | F | 43 | 726 | - | - | normal | |
| 63 | F | 74 | 947 | - | - | n.a. | |
| 64 | M | 70 | 680 | - | - | normal | |
| 65 | M | 30 | 135 | - | - | normal | |
| 66 | M | 25 | 635 | - | - | normal | |
| 67 | F | 84 | 801 | - | - | normal | |
| 68 | F | 76 | 1245 | - | - | normal | |
| 69 | F | 75 | 1183 | - | - | normal | |
| 70 | F | 69 | 988 | - | - | normal | |
| 71 | F | 73 | 957 | - | - | normal | |
| 72 | M | 58 | 397 | - | - | normal | |
| 73 | F | 85 | 108 | - | - | 46,XX,del(7q) | |
| 74 | F | 46 | 358 | - | - | 46,XX,del(5q)/46,XX | |
| 75 | F | 71 | 206 | + | - | normal | |
| 76 | F | 67 | 317 | - | - | normal | |
| 77 | F | 73 | 117 | - | - | normal |
| Cases | Diagnosis | TP53 Nucleotide Change | p53 AA Change | VAF (%) |
|---|---|---|---|---|
| 1 | AML-MR | c.811G > A | p.E271K | 27.8 |
| c.715A > G | p.N239D | 29.14 | ||
| 2 | c.824G > A | p.C275Y | 88.04 | |
| 4 | c.779C > T | p.S260F | 16.2 | |
| 5 | c.279delG | p.L93Lfs | 36.66 | |
| 6 | c.814G > A | p.V272M | 30.7 | |
| c.660T > G | p.Y220X | 8.38 | ||
| 10 | c.811G > A | p.E271K | 6.08 | |
| c.715A > G | p.N239D | 6.67 | ||
| 11 | c.736A > G | p.M246V | 28.39 | |
| 13 | c.811G > A | p.E271K | 8.28 | |
| c.737T > A | p.M246K | 8.35 | ||
| c.405C > A | p.C135X | 5.65 | ||
| 15 | c.817C > A | p.R273S | 41.02 | |
| 16 | c.856_869del14 | p.E286Qfs | 16.96 | |
| 17 | c.824G > A | p.C275Y | 12 | |
| 20 | c.796G > A | p.G266R | 41.73 | |
| 21 | c.481G > A | p.A161T | 73.42 | |
| 24 | c.403T > A | p.C135S | 45.06 | |
| 25 | c.646G > A | p.V216M | 39.44 | |
| c.733G > A | p.G245S | 38.22 | ||
| 28 | MDS-IB | c.644G > A | p.S215N | 30.65 |
| c.1118delA | p.K373Rfs | 11.59 | ||
| c.767C > T | p.T256I | 6.82 | ||
| c.293C > T | p.P98L | 6.25 | ||
| c.1123C > G | p.Q375E | 5.6 | ||
| c.758C > T | p.T253I | 5.45 | ||
| 29 | c.489delC | p.Y163Xfs | 70.27 | |
| 32 | c.743G > A | p.R248Q | 39.93 | |
| c.455C > A | p.P152Q | 38.21 | ||
| 34 | c.614A > G | p.Y205C | 13.49 | |
| 39 | MDS-LB | c.824G > A | p.C275Y | 12.2 |
| 41 | c.814G > A | p.V272M | 13.06 | |
| 42 | c.1085G > A | p.S362N | 5.19 | |
| c.1000G > A | p.G334R | 5.04 | ||
| c.814G > A | p.V272M | 6.27 | ||
| c.715A > G | p.N239D | 10.81 | ||
| 46 | c.814G > A | p.V272M | 10.42 | |
| 48 | c.814G > A | p.V272M | 7.52 | |
| 50 | c.814G > A | p.V272M | 19.77 | |
| 60 | c.637C > T | p.R213X | 5.94 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Madarász, K.; Mótyán, J.A.; Bedekovics, J.; Miltényi, Z.; Ujfalusi, A.; Méhes, G.; Mokánszki, A. Deep Molecular and In Silico Protein Analysis of p53 Alteration in Myelodysplastic Neoplasia and Acute Myeloid Leukemia. Cells 2022, 11, 3475. https://doi.org/10.3390/cells11213475
Madarász K, Mótyán JA, Bedekovics J, Miltényi Z, Ujfalusi A, Méhes G, Mokánszki A. Deep Molecular and In Silico Protein Analysis of p53 Alteration in Myelodysplastic Neoplasia and Acute Myeloid Leukemia. Cells. 2022; 11(21):3475. https://doi.org/10.3390/cells11213475
Chicago/Turabian StyleMadarász, Kristóf, János András Mótyán, Judit Bedekovics, Zsófia Miltényi, Anikó Ujfalusi, Gábor Méhes, and Attila Mokánszki. 2022. "Deep Molecular and In Silico Protein Analysis of p53 Alteration in Myelodysplastic Neoplasia and Acute Myeloid Leukemia" Cells 11, no. 21: 3475. https://doi.org/10.3390/cells11213475
APA StyleMadarász, K., Mótyán, J. A., Bedekovics, J., Miltényi, Z., Ujfalusi, A., Méhes, G., & Mokánszki, A. (2022). Deep Molecular and In Silico Protein Analysis of p53 Alteration in Myelodysplastic Neoplasia and Acute Myeloid Leukemia. Cells, 11(21), 3475. https://doi.org/10.3390/cells11213475