
Development of Human Pituitary Neuroendocrine Tumor Organoids to Facilitate Effective Targeted Treatments of Cushing’s Disease

 ,
,  , ,
, , 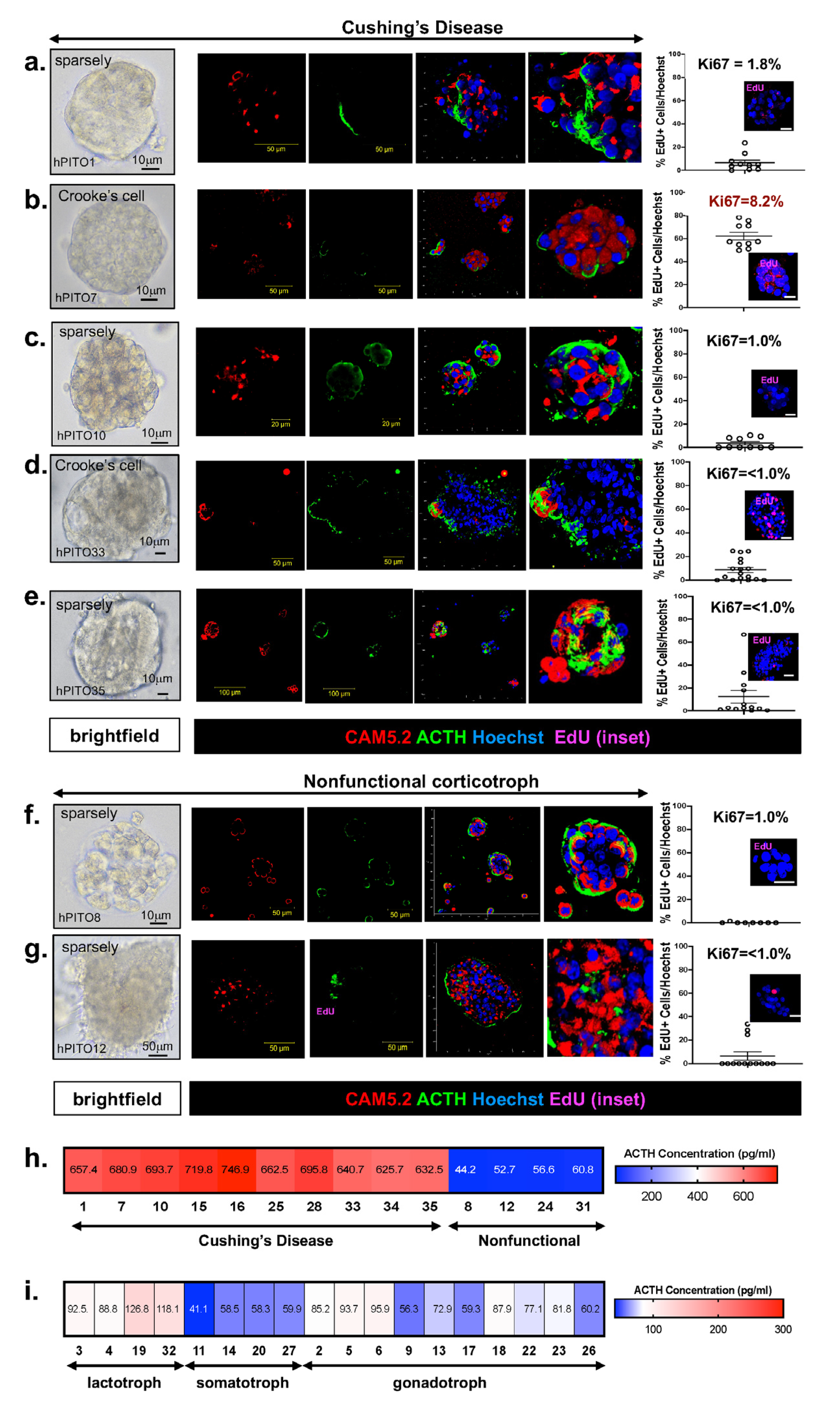{kind=link}
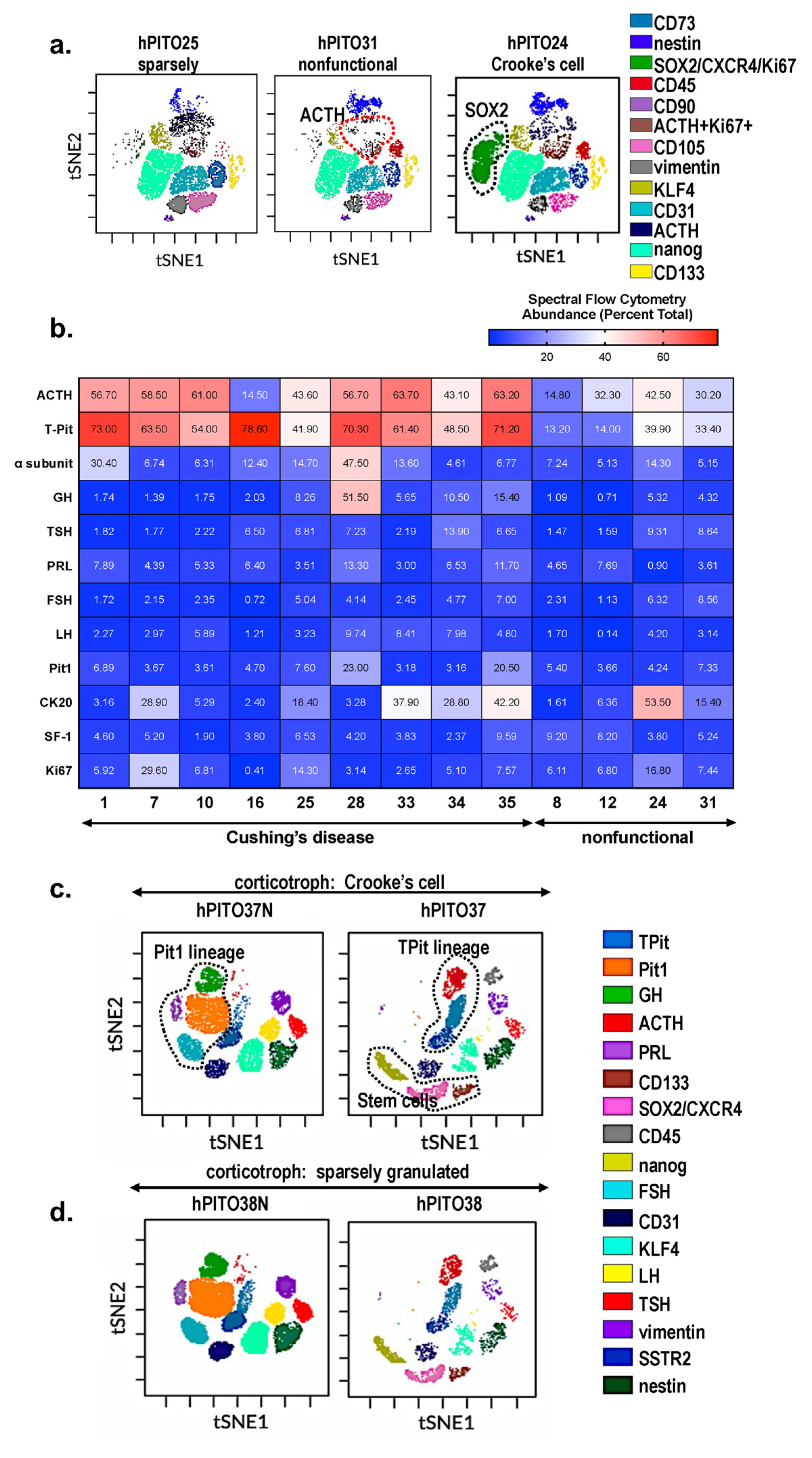{kind=link}
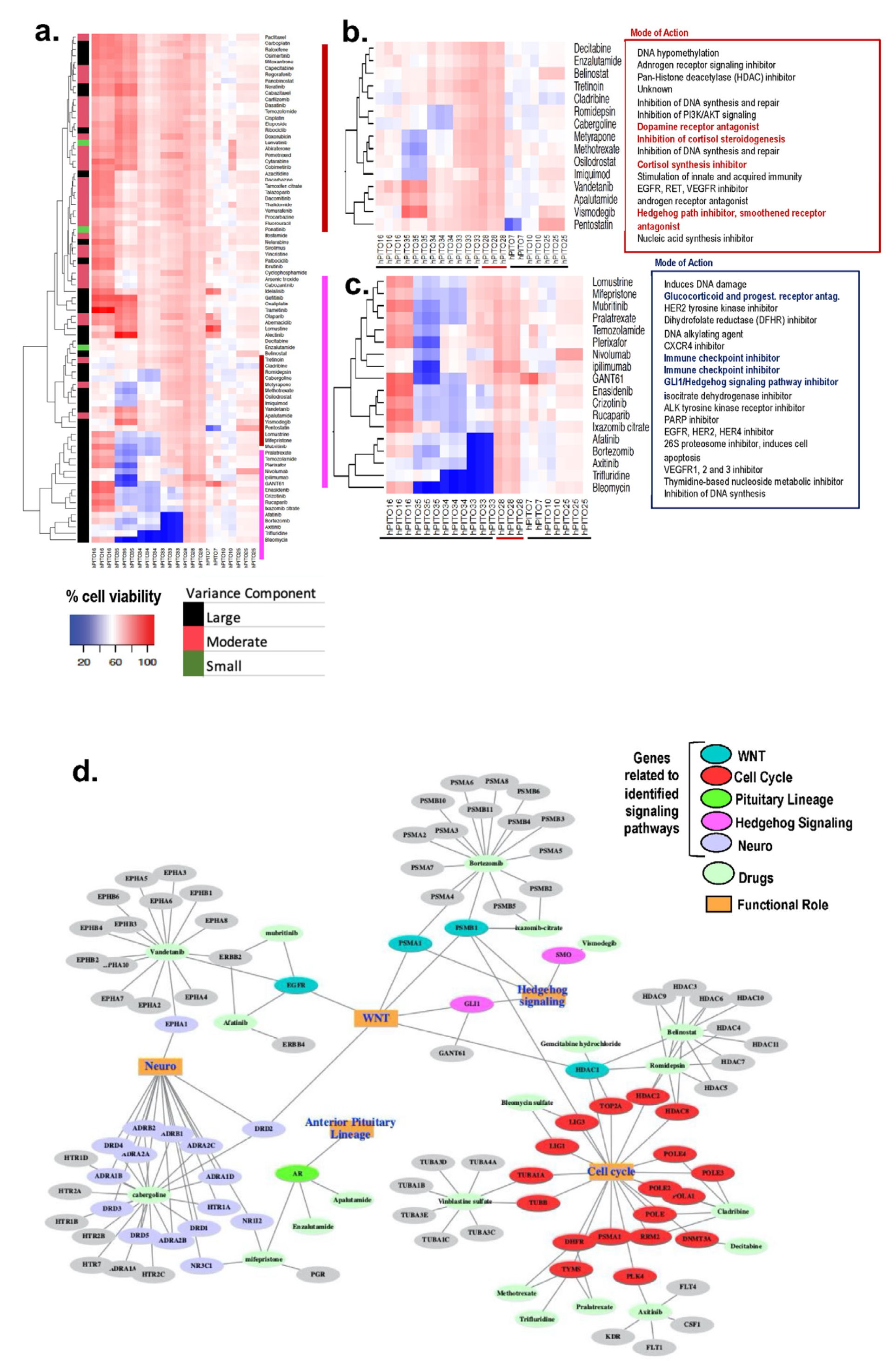{kind=link}
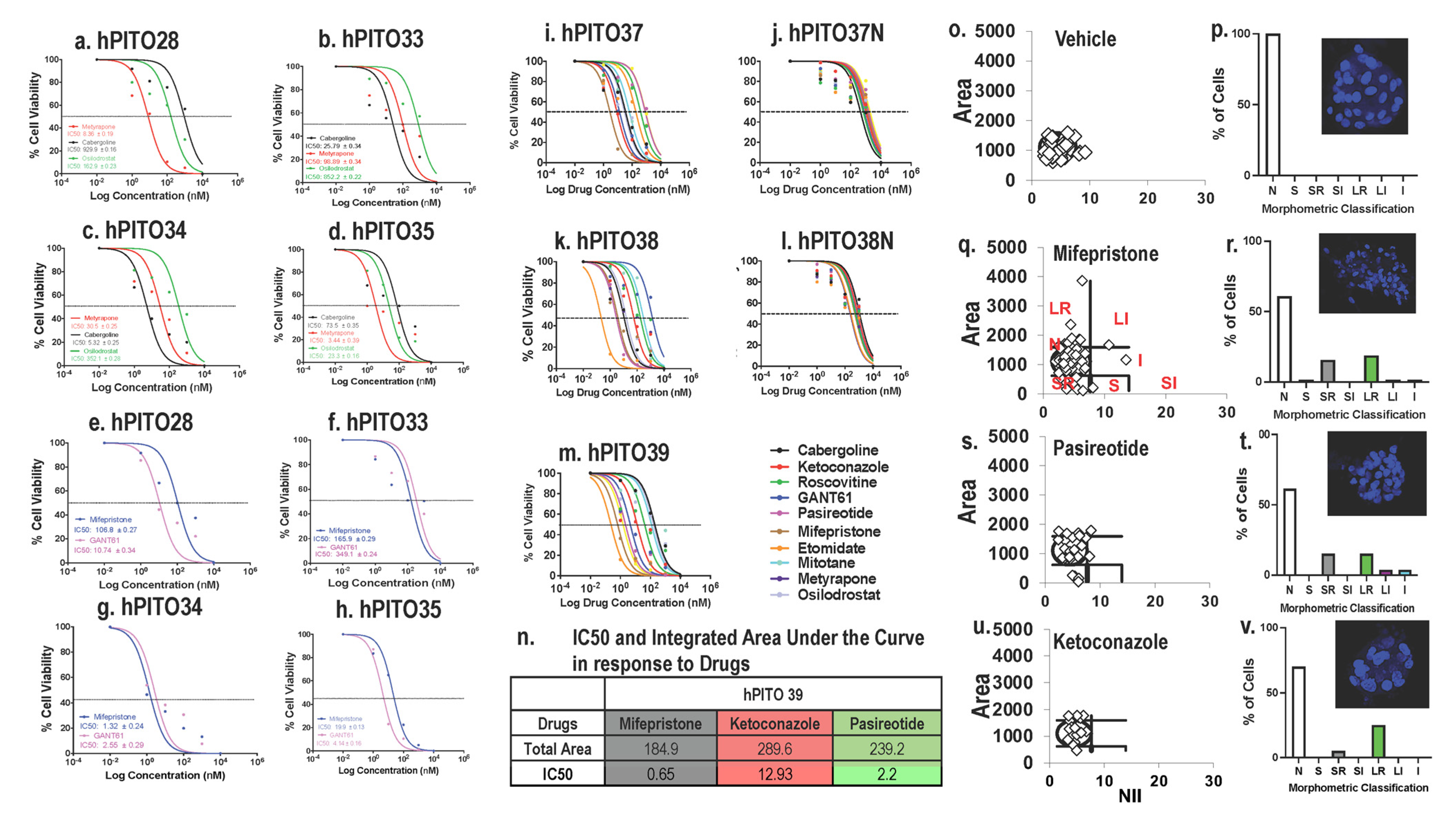{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Generation and Culture of Human Pituitary Neuroendocrine Tumor (PitNET) Organoids
2.2. Generation of Induced Pluripotent Stem Cells (iPSCs)
2.3. Pituitary Organoids Generated from iPSCs
2.4. Spectral Flow Cytometry (Cytek™ Aurora)
2.5. Whole Mount Immunofluorescence
2.6. Nuclear Morphometric Analysis (NMA)
2.7. ELISA
2.8. Drug Assay
2.9. Drug Dose Responses
2.10. Calculation of Area under the Curve (AUC)
2.11. Quantitative RT PCR (qRT-PCR)
2.12. Whole Exome Sequencing
2.13. Single Cell RNA Sequencing (scRNA-Seq)
2.14. Statistical Analyses
3. Results
3.1. Generation and Validation of Human PitNET Tissue Derived Organoids
3.2. Characterization of Cell Lineages in Pituitary Adenoma-Derived Organoids by Spectral Cytek™ Aurora Analysis
3.3. Inherent Patient Differences to Drug Response Is Reflected in the Organoid Culture
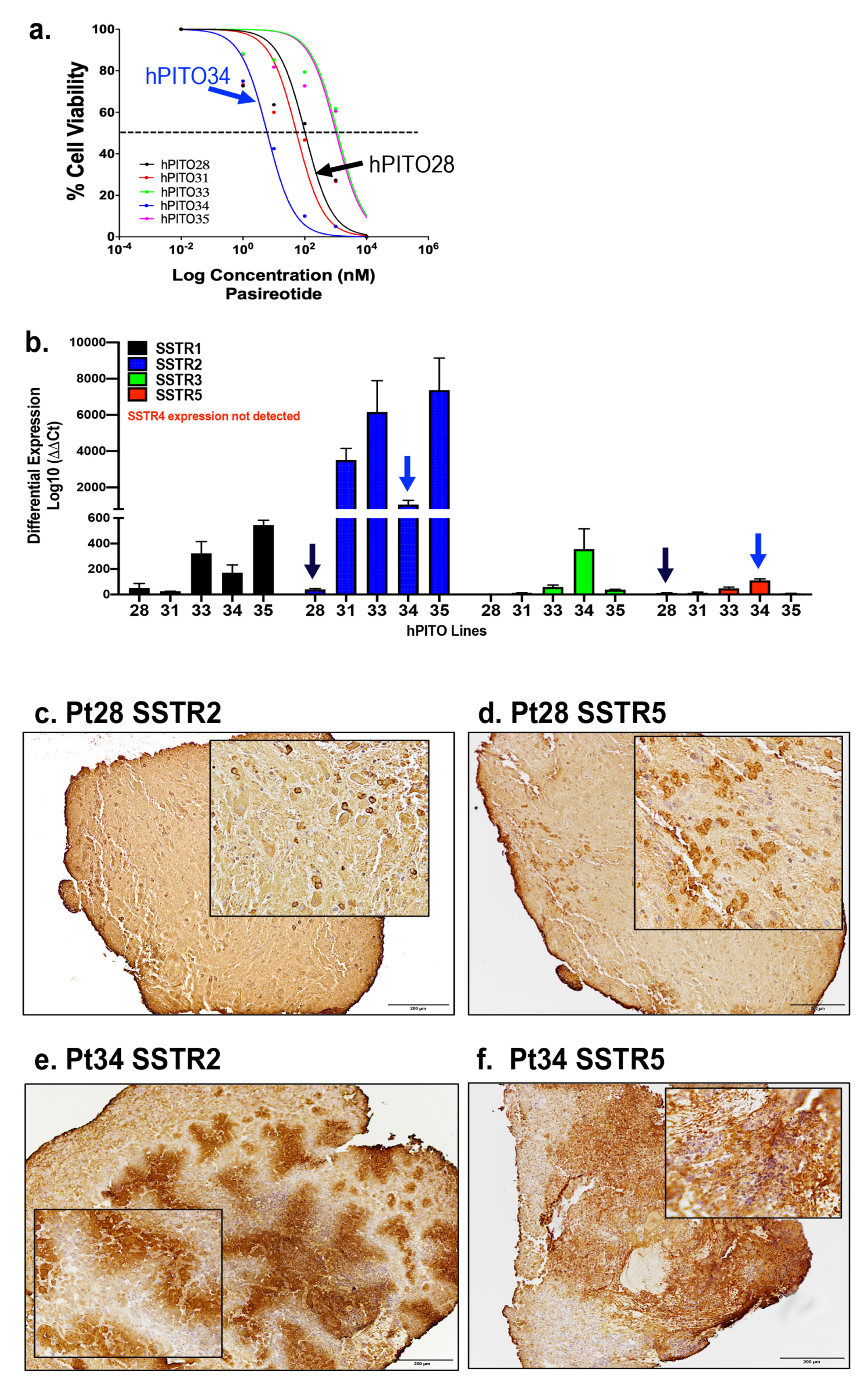
3.4. Organoid Responsiveness to Pasireotide Correlates with SSTR2 and SSTR5 Expression
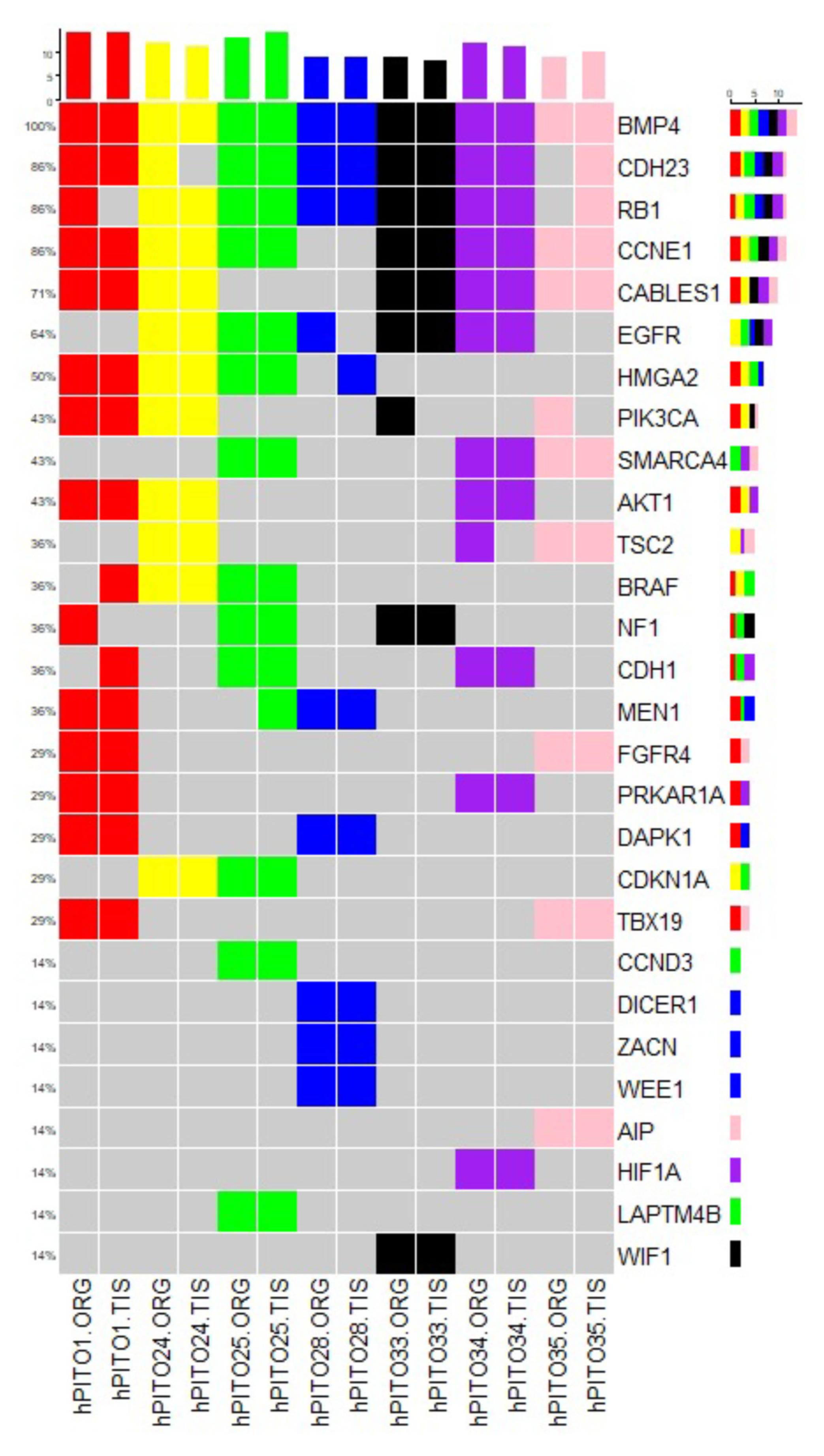
3.5. Organoids Derived from Pituitary Corticotroph Adenomas Retain the Genetic Alterations of the Patient’s Primary Tumor
3.6. IPSC Pituitary Organoids Generated from a CD Patients Expressing Familial Mutations Reveal Corticotroph Adenoma Pathology In Vitro
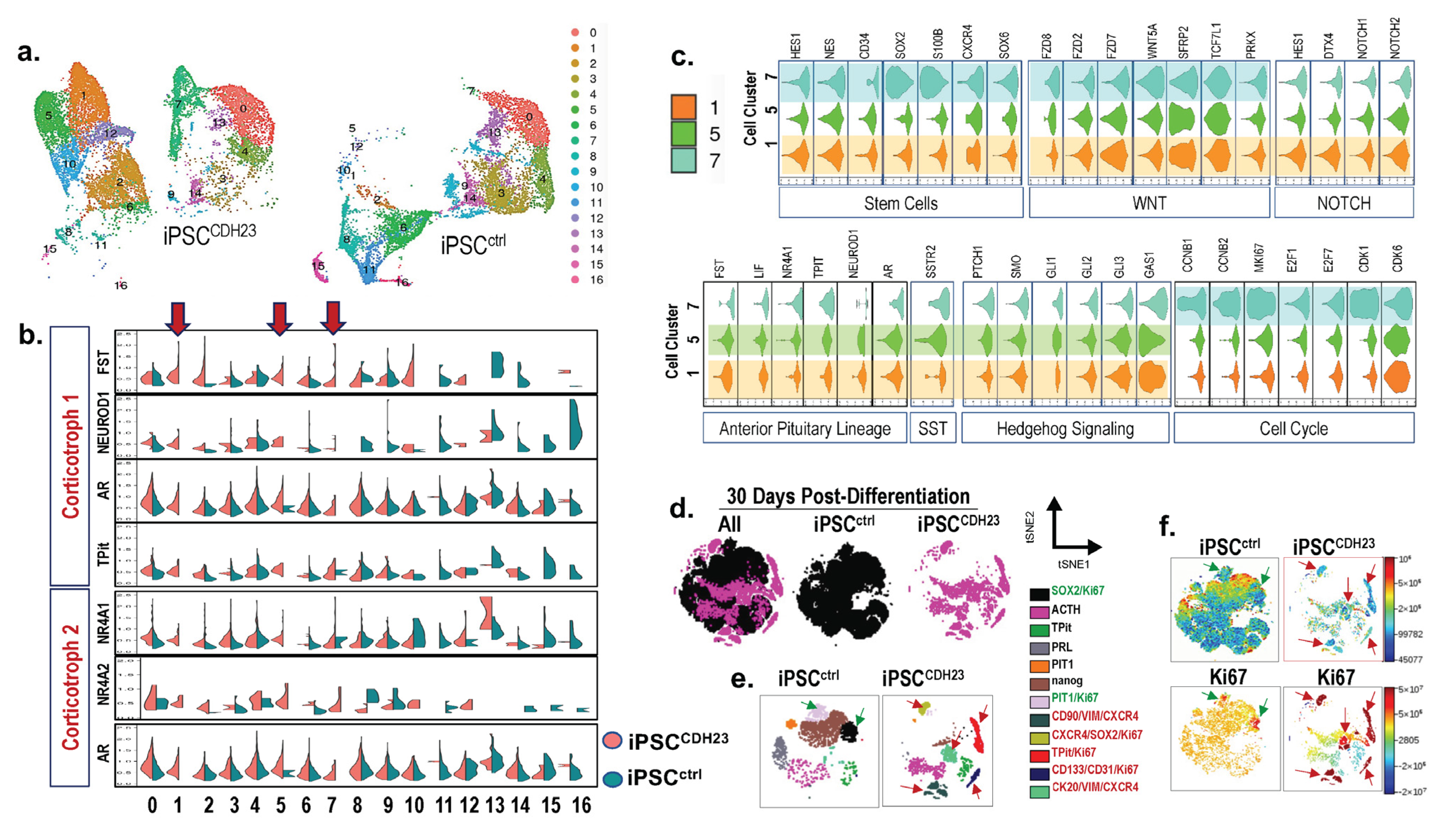
3.7. ScRNA-seq Reveals the Existence of Unique Proliferative Cell Populations in iPSCCDH23 Cultures When Compared to iPSCsctrl
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cushing, H. Posterior Pituitary Activity from an Anatomical Standpoint. Am. J. Pathol. 1933, 9, 539–548.19. [Google Scholar] [PubMed]
- Cushing, H. The basophil adenomas of the pituitary body and their clinical manifestations (pituitary basophilism) 1932. Obes. Res. 1994, 2, 486–508. [Google Scholar] [CrossRef] [PubMed]
- Ironside, N.; Chen, C.J.; Lee, C.C.; Trifiletti, D.M.; Vance, M.L.; Sheehan, J.P. Outcomes of Pituitary Radiation for Cushing’s Disease. Endocrinol. Metab. Clin. N. Am. 2018, 47, 349–365. [Google Scholar] [CrossRef]
- Loriaux, D.L. Diagnosis and Differential Diagnosis of Cushing’s Syndrome. N. Engl. J. Med. 2017, 377, e3. [Google Scholar] [CrossRef]
- Asa, S.L.; Mete, O.; Perry, A.; Osamura, R.Y. Overview of the 2022 WHO Classification of Pituitary Tumors. Endocr. Pathol. 2022, 33, 6–26. [Google Scholar] [CrossRef]
- Nishioka, H.; Yamada, S. Cushing’s Disease. J. Clin. Med. 2019, 8, 1951. [Google Scholar] [CrossRef] [PubMed]
- Feelders, R.A.; Hofland, L.J. Medical treatment of Cushing’s disease. J. Clin. Endocrinol. Metab. 2013, 98, 425–438. [Google Scholar] [CrossRef]
- Limumpornpetch, P.; Morgan, A.W.; Tiganescu, A.; Baxter, P.D.; Nyawira Nyaga, V.; Pujades-Rodriguez, M.; Stewart, P.M. The Effect of Endogenous Cushing Syndrome on All-cause and Cause-specific Mortality. J. Clin. Endocrinol. Metab. 2022, 107, 2377–2388. [Google Scholar] [CrossRef]
- Ciric, I.; Zhao, J.C.; Du, H.; Findling, J.W.; Molitch, M.E.; Weiss, R.E.; Refetoff, S.; Kerr, W.D.; Meyer, J. Transsphenoidal surgery for Cushing disease: Experience with 136 patients. Neurosurgery 2012, 70, 70–80; discussion 71–80. [Google Scholar] [CrossRef]
- Alexandraki, K.I.; Kaltsas, G.A.; Isidori, A.M.; Storr, H.L.; Afshar, F.; Sabin, I.; Akker, S.A.; Chew, S.L.; Drake, W.M.; Monson, J.P.; et al. Long-term remission and recurrence rates in Cushing’s disease: Predictive factors in a single-centre study. Eur. J. Endocrinol. 2013, 168, 639–648. [Google Scholar] [CrossRef]
- Sonino, N.; Zielezny, M.; Fava, G.A.; Fallo, F.; Boscaro, M. Risk factors and long-term outcome in pituitary-dependent Cushing’s disease. J. Clin. Endocrinol. Metab. 1996, 81, 2647–2652. [Google Scholar] [CrossRef] [PubMed]
- Van der Pas, R.; Feelders, R.A.; Gatto, F.; De Bruin, C.; Pereira, A.M.; Van Koetsveld, P.M.; Sprij-Mooij, D.M.; Waaijers, A.M.; Dogan, F.; Schulz, S.; et al. Preoperative normalization of cortisol levels in Cushing’s disease after medical treatment: Consequences for somatostatin and dopamine receptor subtype expression and in vitro response to somatostatin analogs and dopamine agonists. J. Clin. Endocrinol. Metab. 2013, 98, E1880–E1890. [Google Scholar] [CrossRef]
- Kondziolka, D. Cushing’s disease and stereotactic radiosurgery. J. Neurosurg. 2013, 119, 1484–1485; discussion 1485. [Google Scholar] [CrossRef] [PubMed]
- Mehta, G.U.; Sheehan, J.P.; Vance, M.L. Effect of stereotactic radiosurgery before bilateral adrenalectomy for Cushing’s disease on the incidence of Nelson’s syndrome. J. Neurosurg. 2013, 119, 1493–1497. [Google Scholar] [CrossRef] [PubMed]
- Tritos, N.A. Adrenally Directed Medical Therapies for Cushing Syndrome. J. Clin. Endocrinol. Metab. 2021, 106, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Gheorghiu, M.L.; Negreanu, F.; Fleseriu, M. Updates in the Medical Treatment of Pituitary Adenomas. Horm. Metab. Res. 2020, 52, 8–24. [Google Scholar] [CrossRef]
- Kaiser, U.B. Cushing’s disease: Towards precision medicine. Cell. Res. 2015, 25, 649–650. [Google Scholar] [CrossRef][Green Version]
- Bissell, M.S.a.M.J. Organoids: A historical perspective of thinking in three dimensions. J. Cell Biol. 2017, 216, 31–40. [Google Scholar] [CrossRef]
- Danila, D.C.; Zhang, X.; Zhou, Y.; Dickersin, G.R.; Fletcher, J.A.; Hedley-Whyte, E.T.; Selig, M.K.; Johnson, S.R.; Klibanski, A. A human pituitary tumor-derived folliculostellate cell line. J. Clin. Endocrinol. Metab. 2000, 85, 1180–1187. [Google Scholar] [CrossRef]
- Bjoro, T.; Torjesen, P.A.; Ostberg, B.C.; Sand, O.; Iversen, J.G.; Gautvik, K.M.; Haug, E. Bombesin stimulates prolactin secretion from cultured rat pituitary tumour cells (GH4C1) via activation of phospholipase C. Regul. Pept. 1987, 19, 169–182. [Google Scholar] [CrossRef]
- Bjoro, T.; Sand, O.; Ostberg, B.C.; Gordeladze, J.O.; Torjesen, P.; Gautvik, K.M.; Haug, E. The mechanisms by which vasoactive intestinal peptide (VIP) and thyrotropin releasing hormone (TRH) stimulate prolactin release from pituitary cells. Biosci. Rep. 1990, 10, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Cox, B.; Laporte, E.; Vennekens, A.; Kobayashi, H.; Nys, C.; Van Zundert, I.; Uji, I.H.; Vercauteren Drubbel, A.; Beck, B.; Roose, H.; et al. Organoids from pituitary as a novel research model toward pituitary stem cell exploration. J. Endocrinol. 2019, 240, 287–308. [Google Scholar] [CrossRef] [PubMed]
- Vennekens, A.; Laporte, E.; Hermans, F.; Cox, B.; Modave, E.; Janiszewski, A.; Nys, C.; Kobayashi, H.; Malengier-Devlies, B.; Chappell, J.; et al. Interleukin-6 is an activator of pituitary stem cells upon local damage, a competence quenched in the aging gland. Proc. Natl. Acad. Sci. USA 2021, 118, e2100052118. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Hugo, W.; Redublo, P.; Miao, H.; Bergsneider, M.; Wang, M.B.; Kim, W.; Yong, W.H.; Heaney, A.P. A human ACTH-secreting corticotroph tumoroid model: Novel Human ACTH-Secreting Tumor Cell in vitro Model. EBioMedicine 2021, 66, 103294. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, T.; Kouki, T.; Fujiwara, K.; Ramadhani, D.; Horiguchi, K.; Kikuchi, M.; Yashiro, T. Reassembly of anterior pituitary organization by hanging drop three-dimensional cell culture. Acta. Histochem. Cytochem. 2013, 46, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Narsinh, K.H.; Jia, F.; Robbins, R.C.; Kay, M.A.; Longaker, M.T.; Wu, J.C. Generation of adult human induced pluripotent stem cells using nonviral minicircle DNA vectors. Nat. Protoc. 2011, 6, 78–88. [Google Scholar] [CrossRef]
- Steele, N.G.; Chakrabarti, J.; Wang, J.; Biesiada, J.; Holokai, L.; Chang, J.; Nowacki, L.M.; Hawkins, J.; Mahe, M.; Sundaram, N.; et al. An Organoid-Based Preclinical Model of Human Gastric Cancer. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 161–184. [Google Scholar] [CrossRef]
- Bertaux-Skeirik, N.; Feng, R.; Schumacher, M.A.; Li, J.; Mahe, M.M.; Engevik, A.C.; Javier, J.E.; Peek, R.M.J.; Ottemann, K.; Orian-Rousseau, V.; et al. CD44 plays a functional role in Helicobacter pylori-induced epithelial cell proliferation. PLoS Pathog. 2015, 11, e1004663. [Google Scholar] [CrossRef]
- Feng, R.; Aihara, E.; Kenny, S.; Yang, L.; Li, J.; Varro, A.; Montrose, M.H.; Shroyer, N.F.; Wang, T.C.; Shivdasani, R.A.; et al. Indian Hedgehog mediates gastrin-induced proliferation in stomach of adult mice. Gastroenterology 2014, 147, 655–666.e9. [Google Scholar] [CrossRef]
- Filippi-Chiela, E.C.; Oliveira, M.M.; Jurkovski, B.; Callegari-Jacques, S.M.; da Silva, V.D.; Lenz, G. Nuclear morphometric analysis (NMA): Screening of senescence, apoptosis and nuclear irregularities. PLoS ONE 2012, 7, e42522. [Google Scholar] [CrossRef]
- Gagnon, R.C.; Peterson, J.J. Estimation of confidence intervals for area under the curve from destructively obtained pharmacokinetic data. J. Pharm. Biopharm. 1998, 26, 87–102. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.; Schmittgen, T. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Hinojosa-Amaya, J.M.; Varlamov, E.V.; McCartney, S.; Fleseriu, M. Hypercortisolemia Recurrence in Cushing’s Disease; a Diagnostic Challenge. Front. Endocrinol. 2019, 10, 740. [Google Scholar] [CrossRef] [PubMed]
- Patil, C.G.; Prevedello, D.M.; Lad, S.P.; Vance, M.L.; Thorner, M.O.; Katznelson, L.; Laws, E.R., Jr. Late recurrences of Cushing’s disease after initial successful transsphenoidal surgery. J. Clin. Endocrinol. Metab. 2008, 93, 358–362. [Google Scholar] [CrossRef]
- Katznelson, L. Bilateral adrenalectomy for Cushing’s disease. Pituitary 2015, 18, 269–273. [Google Scholar] [CrossRef]
- Reincke, M.; Sbiera, S.; Hayakawa, A.; Theodoropoulou, M.; Osswald, A.; Beuschlein, F.; Meitinger, T.; Mizuno-Yamasaki, E.; Kawaguchi, K.; Saeki, Y.; et al. Mutations in the deubiquitinase gene USP8 cause Cushing’s disease. Nat. Genet. 2015, 47, 31–38. [Google Scholar] [CrossRef]
- Chen, J.; Jian, X.; Deng, S.; Ma, Z.; Shou, X.; Shen, Y.; Zhang, Q.; Song, Z.; Li, Z.; Peng, H.; et al. Identification of recurrent USP48 and BRAF mutations in Cushing’s disease. Nat. Commun. 2018, 9, 3171. [Google Scholar] [CrossRef]
- Zhang, Q.; Peng, C.; Song, J.; Zhang, Y.; Chen, J.; Song, Z.; Shou, X.; Ma, Z.; Peng, H.; Jian, X.; et al. Germline Mutations in CDH23, Encoding Cadherin-Related 23, Are Associated with Both Familial and Sporadic Pituitary Adenomas. Am. J. Hum. Genet. 2017, 100, 817–823. [Google Scholar] [CrossRef]
- Ikeda, H.; Mitsuhashi, T.; Kubota, K.; Kuzuya, N.; Uchimura, H. Epidermal growth factor stimulates growth hormone secretion from superfused rat adenohypophyseal fragments. Endocrinology 1984, 115, 556–558. [Google Scholar] [CrossRef]
- Baek, N.; Seo, O.W.; Kim, M.; Hulme, J.; An, S.S. Monitoring the effects of doxorubicin on 3D-spheroid tumor cells in real-time. Onco. Targets 2016, 9, 7207–7218. [Google Scholar] [CrossRef]
- Laporte, E.; Nys, C.; Vankelecom, H. Development of Organoids from Mouse Pituitary as In Vitro Model to Explore Pituitary Stem Cell Biology. J. Vis. Exp. 2022. [Google Scholar] [CrossRef] [PubMed]
- Nys, C.; Lee, Y.L.; Roose, H.; Mertens, F.; De Pauw, E.; Kobayashi, H.; Sciot, R.; Bex, M.; Versyck, G.; De Vleeschouwer, S.; et al. Exploring stem cell biology in pituitary tumors and derived organoids. Endocr. Relat. Cancer 2022, 29, 427–450. [Google Scholar] [CrossRef]
- Available online: https://www.cancer.gov/publications/dictionaries/cancer-terms/def/organoid (accessed on 20 September 2022).
- Mahe, M.M.; Aihara, E.; Schumacher, M.A.; Zavros, Y.; Montrose, M.H.; Helmrath, M.A.; Sato, T.; Shroyer, N.F. Establishment of Gastrointestinal Epithelial Organoids. Curr. Protoc. Mouse Biol. 2013, 3, 217–240. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, M.A.; Aihara, E.; Feng, R.; Engevik, A.; Shroyer, N.F.; Ottemann, K.M.; Worrell, R.T.; Montrose, M.H.; Shivdasani, R.A.; Zavros, Y. The use of murine-derived fundic organoids in studies of gastric physiology. J. Physiol. 2015, 593, 1809–1827. [Google Scholar] [CrossRef] [PubMed]
- Holokai, L.; Chakrabarti, J.; Lundy, J.; Croagh, D.; Adhikary, P.; Richards, S.S.; Woodson, C.; Steele, N.; Kuester, R.; Scott, A.; et al. Murine- and Human-Derived Autologous Organoid/Immune Cell Co-Cultures as Pre-Clinical Models of Pancreatic Ductal Adenocarcinoma. Cancers 2020, 12, 3816. [Google Scholar] [CrossRef] [PubMed]
- Boj, S.F.; Hwang, C.I.; Baker, L.A.; Chio, I.I.C.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef]
- Tiriac, H.; Belleau, P.; Engle, D.D.; Plenker, D.; Deschenes, A.; Somerville, T.D.D.; Froeling, F.E.M.; Burkhart, R.A.; Denroche, R.E.; Jang, G.H.; et al. Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer. Cancer Discov. 2018, 8, 1112–1129. [Google Scholar] [CrossRef]
- Driehuis, E.; van Hoeck, A.; Moore, K.; Kolders, S.; Francies, H.E.; Gulersonmez, M.C.; Stigter, E.C.A.; Burgering, B.; Geurts, V.; Gracanin, A.; et al. Pancreatic cancer organoids recapitulate disease and allow personalized drug screening. Proc. Natl. Acad. Sci. USA 2019 116, 26580–26590. [CrossRef]
- Jung, Y.H.; Choi, D.H.; Park, K.; Lee, S.B.; Kim, J.; Kim, H.; Jeong, H.W.; Yang, J.H.; Kim, J.A.; Chung, S.; et al. Drug screening by uniform patient derived colorectal cancer hydro-organoids. Biomaterials 2021, 276, 121004. [Google Scholar] [CrossRef]
- Chen, D.; Tan, Y.; Li, Z.; Li, W.; Yu, L.; Chen, W.; Liu, Y.; Liu, L.; Guo, L.; Huang, W.; et al. Organoid Cultures Derived From Patients With Papillary Thyroid Cancer. J. Clin. Endocrinol. Metab. 2021, 106, 1410–1426. [Google Scholar] [CrossRef]
- Reincke, M.; Theodoropoulou, M. Genomics in Cushing’s Disease: The Dawn of a New Era. J. Clin. Endocrinol. Metab. 2021, 106, e2455–e2456. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.Y.; Song, Z.J.; Chen, J.H.; Wang, Y.F.; Li, S.Q.; Zhou, L.F.; Mao, Y.; Li, Y.M.; Hu, R.G.; Zhang, Z.Y.; et al. Recurrent gain-of-function USP8 mutations in Cushing’s disease. Cell. Res. 2015, 25, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Melmed, S. Pathogenesis of pituitary tumors. Nat. Rev. Endocrinol. 2011, 7, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Stratakis, C.A.; Tichomirowa, M.A.; Boikos, S.; Azevedo, M.F.; Lodish, M.; Martari, M.; Verma, S.; Daly, A.F.; Raygada, M.; Keil, M.F.; et al. The role of germline AIP, MEN1, PRKAR1A, CDKN1B and CDKN2C mutations in causing pituitary adenomas in a large cohort of children, adolescents, and patients with genetic syndromes. Clin. Genet. 2010, 78, 457–463. [Google Scholar] [CrossRef]
- Mouchtouris, N.; Smit, R.D.; Piper, K.; Prashant, G.; Evans, J.J.; Karsy, M. A review of multiomics platforms in pituitary adenoma pathogenesis. Front. Biosci. 2022, 27, 77. [Google Scholar] [CrossRef] [PubMed]
- Bolz, H.; von Brederlow, B.; Ramirez, A.; Bryda, E.C.; Kutsche, K.; Nothwang, H.G.; Seeliger, M.; del, C.S.C.M.; Vila, M.C.; Molina, O.P.; et al. Mutation of CDH23, encoding a new member of the cadherin gene family, causes Usher syndrome type 1D. Nat. Genet. 2001, 27, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Cui, Y.; Ma, X.; Yong, J.; Yan, L.; Yang, M.; Ren, J.; Tang, F.; Wen, L.; Qiao, J. Single-cell transcriptomics identifies divergent developmental lineage trajectories during human pituitary development. Nat. Commun. 2020, 11, 5275. [Google Scholar] [CrossRef]
- Cheung, L.Y.; Davis, S.W.; Brinkmeier, M.L.; Camper, S.A.; Perez-Millan, M.I. Regulation of pituitary stem cells by epithelial to mesenchymal transition events and signaling pathways. Mol. Cell. Endocrinol. 2017, 445, 14–26. [Google Scholar] [CrossRef]
- Shintani, A.; Higuchi, M. Isolation of PRRX1-positive adult pituitary stem/progenitor cells from the marginal cell layer of the mouse anterior lobe. Stem Cell. Res. 2021, 52, 102223. [Google Scholar] [CrossRef]
- Yoshida, S.; Nishimura, N.; Ueharu, H.; Kanno, N.; Higuchi, M.; Horiguchi, K.; Kato, T.; Kato, Y. Isolation of adult pituitary stem/progenitor cell clusters located in the parenchyma of the rat anterior lobe. Stem Cell. Res. 2016, 17, 318–329. [Google Scholar] [CrossRef]
- Laporte, E.; Vennekens, A.; Vankelecom, H. Pituitary Remodeling Throughout Life: Are Resident Stem Cells Involved? Front. Endocrinol. 2020, 11, 604519. [Google Scholar] [CrossRef] [PubMed]
- Vankelecom, H.; Roose, H. The Stem Cell Connection of Pituitary Tumors. Front. Endocrinol. 2017, 8, 339. [Google Scholar] [CrossRef] [PubMed]
- Mertens, F.; Gremeaux, L.; Chen, J.; Fu, Q.; Willems, C.; Roose, H.; Govaere, O.; Roskams, T.; Cristina, C.; Becu-Villalobos, D.; et al. Pituitary tumors contain a side population with tumor stem cell-associated characteristics. Endocr. Relat. Cancer 2015, 22, 481–504. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Z.; Tsai, S.Y.; Leone, G. Emerging roles of E2Fs in cancer: An exit from cell cycle control. Nat. Rev. Cancer 2009, 9, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Yuan, X.; Tunici, P.; Liu, G.; Fan, X.; Xu, M.; Hu, J.; Hwang, J.Y.; Farkas, D.L.; Black, K.L.; et al. Isolation of tumour stem-like cells from benign tumours. Br. J. Cancer 2009, 101, 303–311. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chakrabarti, J.; Pandey, R.; Churko, J.M.; Eschbacher, J.; Mallick, S.; Chen, Y.; Hermes, B.; Mallick, P.; Stansfield, B.N.; Pond, K.W.; et al. Development of Human Pituitary Neuroendocrine Tumor Organoids to Facilitate Effective Targeted Treatments of Cushing’s Disease. Cells 2022, 11, 3344. https://doi.org/10.3390/cells11213344
Chakrabarti J, Pandey R, Churko JM, Eschbacher J, Mallick S, Chen Y, Hermes B, Mallick P, Stansfield BN, Pond KW, et al. Development of Human Pituitary Neuroendocrine Tumor Organoids to Facilitate Effective Targeted Treatments of Cushing’s Disease. Cells. 2022; 11(21):3344. https://doi.org/10.3390/cells11213344
Chicago/Turabian StyleChakrabarti, Jayati, Ritu Pandey, Jared M. Churko, Jennifer Eschbacher, Saptarshi Mallick, Yuliang Chen, Beth Hermes, Palash Mallick, Ben N. Stansfield, Kelvin W. Pond, and et al. 2022. "Development of Human Pituitary Neuroendocrine Tumor Organoids to Facilitate Effective Targeted Treatments of Cushing’s Disease" Cells 11, no. 21: 3344. https://doi.org/10.3390/cells11213344
APA StyleChakrabarti, J., Pandey, R., Churko, J. M., Eschbacher, J., Mallick, S., Chen, Y., Hermes, B., Mallick, P., Stansfield, B. N., Pond, K. W., Thorne, C. A., Yuen, K. C. J., Little, A. S., & Zavros, Y. (2022). Development of Human Pituitary Neuroendocrine Tumor Organoids to Facilitate Effective Targeted Treatments of Cushing’s Disease. Cells, 11(21), 3344. https://doi.org/10.3390/cells11213344