
PARP1′s Involvement in RNA Polymerase II Elongation: Pausing and Releasing Regulation through the Integrator and Super Elongation Complex

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. S2 Cell Culture and siRNA Mediated Knockdown
2.2. S2 Cell PARP1 Inhibitor Treatment
2.3. S2 Cell NAD+ Treatment and Heat Shock
2.4. Expression and Purification of Recombinant Proteins
2.5. Recombinant PARP1 PARylation
2.6. Western Blot Analyses
2.7. Analyses and Quantitation of PARP1 Depletion by qRTPCR
2.8. Antibodies
2.9. S2 Cell Lysate Preparation
2.10. Co-Immunoprecipitation and Pull-Down Assays
2.11. Chromatin Immunoprecipitation (ChIP)
2.12. Salt Fractionation of Chromatin
2.13. Genes of Interest
3. Results
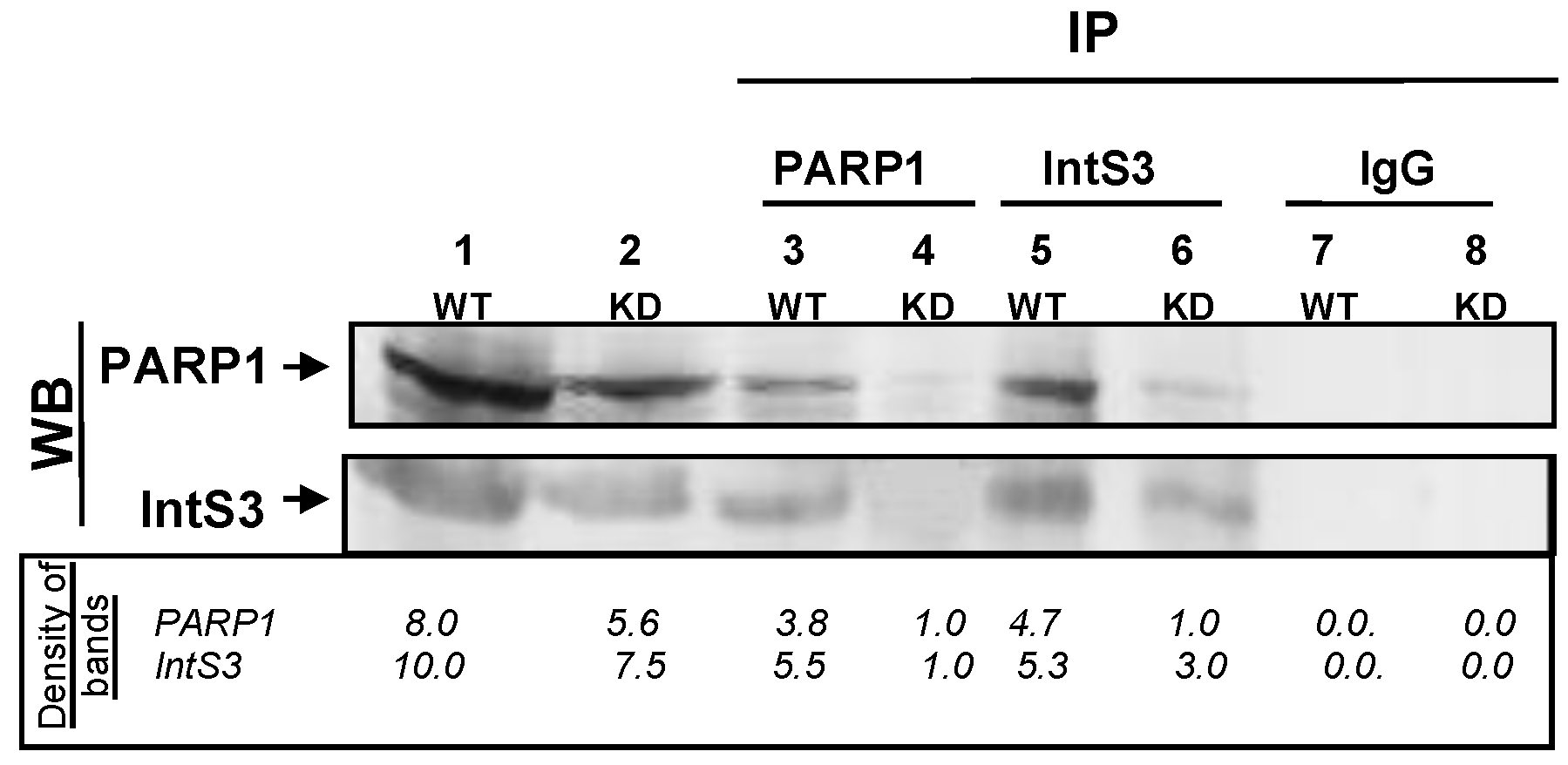
3.1. PARP1 Associates Directly with IntS3 of the Integrator Complex
PARP1 and IntS3 Associate In Vivo
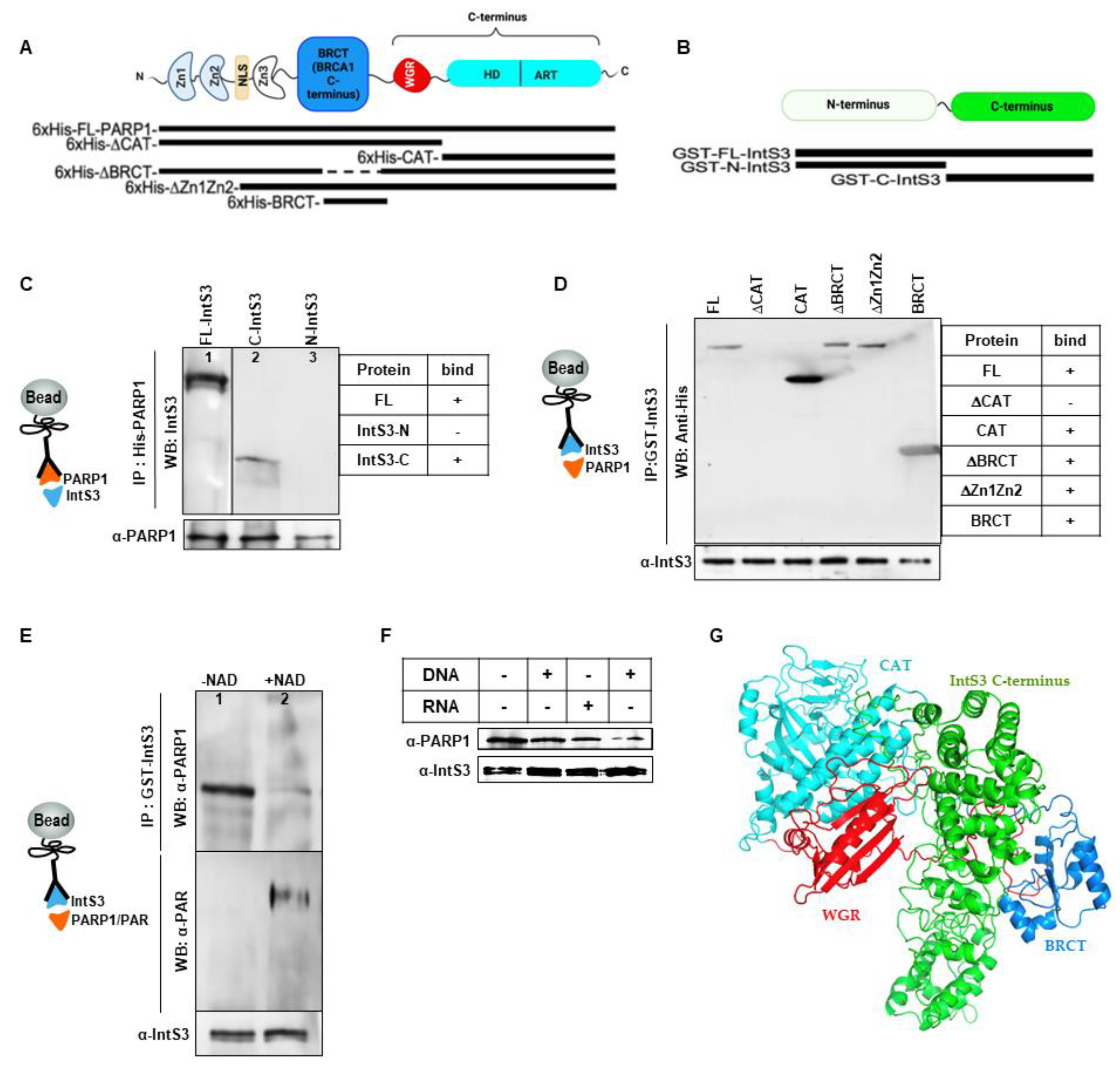
3.2. Critical Domains Necessary for Association of PARP1 and IntS3
PARP1 Binds Directly to C-Terminal Domain of IntS3
3.3. IntS3 Is Not PARylated by PARP1
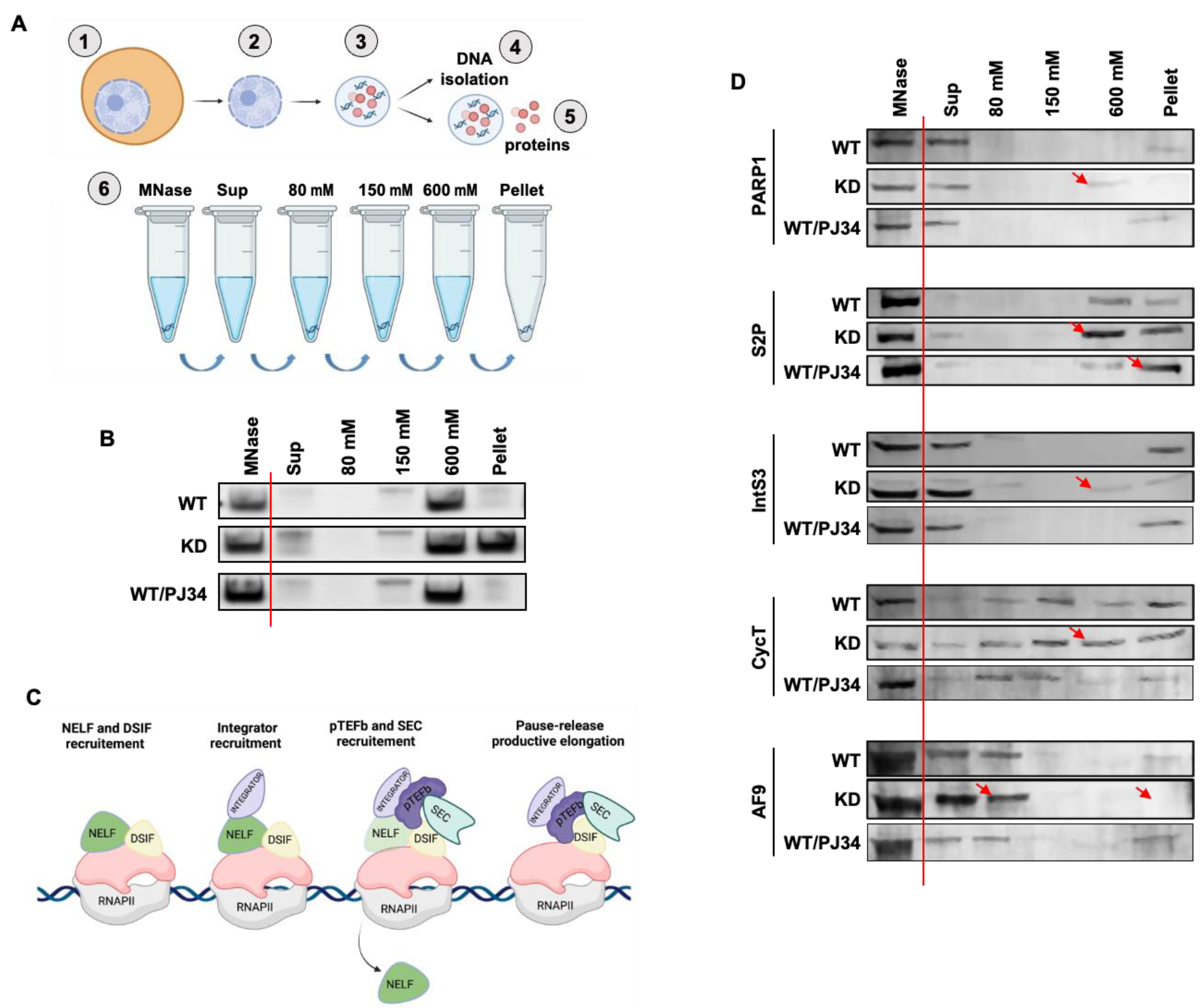
3.4. PARP1 Modulates the Chromatin Association of Elongation Factors
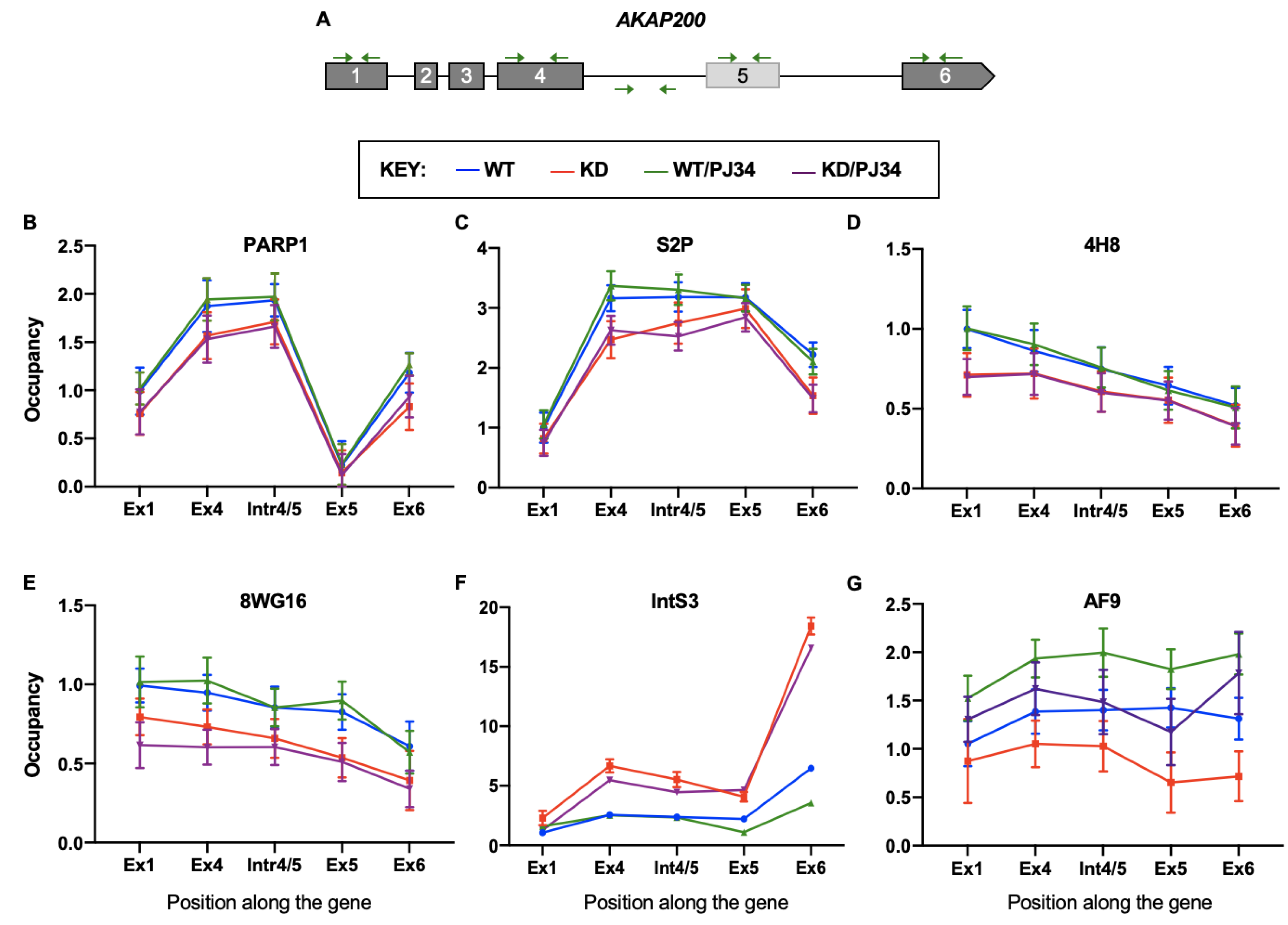
3.5. PARP1 and PARylation Impact Localization and Occupancy of Elongation Factors along the Gene Body
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baralle, F.E.; Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Kelemen, O.; Convertini, P.; Zhang, Z.; Wen, Y.; Shen, M.; Falaleeva, M.; Stamm, S. Function of alternative splicing. Gene 2013, 514, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.S.; Cooper, T.A. Splicing in disease: Disruption of the splicing code and the decoding machinery. Nat. Rev. Genet. 2007, 8, 749–761. [Google Scholar] [CrossRef]
- de la Mata, M.; Alonso, C.R.; Kadener, S.; Fededa, J.P.; Blaustein, M.; Pelisch, F.; Cramer, P.; Bentley, D.; Kornblihtt, A.R. A slow RNA polymerase II affects alternative splicing in vivo. Mol. Cell 2003, 12, 525–532. [Google Scholar] [CrossRef] [PubMed]
- de la Mata, M.; Munoz, M.J.; Allo, M.; Fededa, J.P.; Schor, I.E.; Kornblihtt, A.R. RNA Polymerase II Elongation at the Crossroads of Transcription and Alternative Splicing. Genet. Res. Int. 2011, 2011, 309865. [Google Scholar] [CrossRef] [PubMed]
- Boon, K.; Bailey, N.W.; Yang, J.; Steel, M.P.; Groshong, S.; Kervitsky, D.; Brown, K.K.; Schwarz, M.I.; Schwartz, D.A. Molecular phenotypes distinguish patients with relatively stable from progressive idiopathic pulmonary fibrosis (IPF). PLoS ONE 2009, 4, e5134. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.H.; Silva, J.; Burstein, D.; Pupko, T.; Eyras, E.; Ast, G. Large-scale comparative analysis of splicing signals and their corresponding splicing factors in eukaryotes. Genome Res. 2008, 18, 88–103. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.; Hall, E.; Ast, G. SROOGLE: Webserver for integrative, user-friendly visualization of splicing signals. Nucleic Acids Res. 2009, 37, W189–W192. [Google Scholar] [CrossRef] [PubMed]
- Haque, N.; Oberdoerffer, S. Chromatin and splicing. Methods Mol. Biol. 2014, 1126, 97–113. [Google Scholar] [CrossRef] [PubMed]
- Allemand, E.; Batsche, E.; Muchardt, C. Splicing, transcription, and chromatin: A menage a trois. Curr. Opin. Genet. Dev. 2008, 18, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Lev Maor, G.; Yearim, A.; Ast, G. The alternative role of DNA methylation in splicing regulation. TIG 2015, 31, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Shayevitch, R.; Askayo, D.; Keydar, I.; Ast, G. The importance of DNA methylation of exons on alternative splicing. RNA 2018, 24, 1351–1362. [Google Scholar] [CrossRef]
- Shukla, S.; Kavak, E.; Gregory, M.; Imashimizu, M.; Shutinoski, B.; Kashlev, M.; Oberdoerffer, P.; Sandberg, R.; Oberdoerffer, S. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature 2011, 479, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Schor, I.E.; Fiszbein, A.; Petrillo, E.; Kornblihtt, A.R. Intragenic epigenetic changes modulate NCAM alternative splicing in neuronal differentiation. EMBO J. 2013, 32, 2264–2274. [Google Scholar] [CrossRef]
- Schor, I.E.; Gomez Acuna, L.I.; Kornblihtt, A.R. Coupling between transcription and alternative splicing. Cancer Treat. Res. 2013, 158, 1–24. [Google Scholar] [CrossRef]
- Dujardin, G.; Lafaille, C.; Petrillo, E.; Buggiano, V.; Gomez Acuna, L.I.; Fiszbein, A.; Godoy Herz, M.A.; Nieto Moreno, N.; Munoz, M.J.; Allo, M.; et al. Transcriptional elongation and alternative splicing. Biochim. Biophys. Acta 2013, 1829, 134–140. [Google Scholar] [CrossRef]
- Schwartz, S.; Ast, G. Chromatin density and splicing destiny: On the cross-talk between chromatin structure and splicing. EMBO J. 2010, 29, 1629–1636. [Google Scholar] [CrossRef]
- Andersson, R.; Enroth, S.; Rada-Iglesias, A.; Wadelius, C.; Komorowski, J. Nucleosomes are well positioned in exons and carry characteristic histone modifications. Genome Res. 2009, 19, 1732–1741. [Google Scholar] [CrossRef]
- Schwartz, S.; Meshorer, E.; Ast, G. Chromatin organization marks exon-intron structure. Nat. Struct. Mol. Biol. 2009, 16, 990–995. [Google Scholar] [CrossRef]
- Tilgner, H.; Nikolaou, C.; Althammer, S.; Sammeth, M.; Beato, M.; Valcarcel, J.; Guigo, R. Nucleosome positioning as a determinant of exon recognition. Nat. Struct. Mol. Biol. 2009, 16, 996–1001. [Google Scholar] [CrossRef]
- Parsa, J.Y.; Boudoukha, S.; Burke, J.; Homer, C.; Madhani, H.D. Polymerase pausing induced by sequence-specific RNA-binding protein drives heterochromatin assembly. Genes Dev. 2018, 32, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Aslanzadeh, V.; Huang, Y.; Sanguinetti, G.; Beggs, J.D. Transcription rate strongly affects splicing fidelity and cotranscriptionality in budding yeast. Genome Res. 2018, 28, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Li, W.; Yao, X.; Lin, Q.J.; Yin, J.W.; Liang, Y.; Heiner, M.; Tian, B.; Hui, J.; Wang, G. Mediator complex regulates alternative mRNA processing via the MED23 subunit. Mol. Cell 2012, 45, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Monsalve, M.; Wu, Z.; Adelmant, G.; Puigserver, P.; Fan, M.; Spiegelman, B.M. Direct coupling of transcription and mRNA processing through the thermogenic coactivator PGC-1. Mol. Cell 2000, 6, 307–316. [Google Scholar] [CrossRef]
- Kim, M.Y.; Mauro, S.; Gevry, N.; Lis, J.T.; Kraus, W.L. NAD+-dependent modulation of chromatin structure and transcription by nucleosome binding properties of PARP-1. Cell 2004, 119, 803–814. [Google Scholar] [CrossRef]
- Piao, L.; Fujioka, K.; Nakakido, M.; Hamamoto, R. Regulation of poly(ADP-Ribose) polymerase 1 functions by post-translational modifications. Front. Biosci. (Landmark Ed.) 2018, 23, 13–26. [Google Scholar] [CrossRef]
- Ji, Y.; Tulin, A.V. Poly(ADP-ribosyl)ation of heterogeneous nuclear ribonucleoproteins modulates splicing. Nucleic Acids Res. 2009, 37, 3501–3513. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Tulin, A.V. Post-transcriptional regulation by poly(ADP-ribosyl)ation of the RNA-binding proteins. Int. J. Mol. Sci. 2013, 14, 16168–16183. [Google Scholar] [CrossRef]
- Matveeva, E.; Maiorano, J.; Zhang, Q.; Eteleeb, A.M.; Convertini, P.; Chen, J.; Infantino, V.; Stamm, S.; Wang, J.; Rouchka, E.C.; et al. Involvement of PARP1 in the regulation of alternative splicing. Cell Discov. 2016, 2, 15046. [Google Scholar] [CrossRef]
- Melikishvili, M.; Chariker, J.H.; Rouchka, E.C.; Fondufe-Mittendorf, Y.N. Transcriptome-wide identification of the RNA-binding landscape of the chromatin-associated protein PARP1 reveals functions in RNA biogenesis. Cell Discov. 2017, 3, 17043. [Google Scholar] [CrossRef]
- Matveeva, E.A.; Al-Tinawi, Q.M.H.; Rouchka, E.C.; Fondufe-Mittendorf, Y.N. Coupling of PARP1-mediated chromatin structural changes to transcriptional RNA polymerase II elongation and cotranscriptional splicing. Epigenetics Chromatin 2019, 12, 15. [Google Scholar] [CrossRef] [PubMed]
- Petesch, S.J.; Lis, J.T. Rapid, transcription-independent loss of nucleosomes over a large chromatin domain at Hsp70 loci. Cell 2008, 134, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Kutuzov, M.M.; Belousova, E.A.; Kurgina, T.A.; Ukraintsev, A.A.; Vasil’eva, I.A.; Khodyreva, S.N.; Lavrik, O.I. The contribution of PARP1, PARP2 and poly(ADP-ribosyl)ation to base excision repair in the nucleosomal context. Sci. Rep. 2021, 11, 4849. [Google Scholar] [CrossRef]
- Bartholomeeusen, K.; Xiang, Y.; Fujinaga, K.; Peterlin, B.M. Bromodomain and extra-terminal (BET) bromodomain inhibition activate transcription via transient release of positive transcription elongation factor b (P-TEFb) from 7SK small nuclear ribonucleoprotein. J. Biol. Chem. 2012, 287, 36609–36616. [Google Scholar] [CrossRef]
- Luo, Z.; Lin, C.; Guest, E.; Garrett, A.S.; Mohaghegh, N.; Swanson, S.; Marshall, S.; Florens, L.; Washburn, M.P.; Shilatifard, A. The super elongation complex family of RNA polymerase II elongation factors: Gene target specificity and transcriptional output. Mol. Cell. Biol. 2012, 32, 2608–2617. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Lin, C.; Shilatifard, A. The super elongation complex (SEC) family in transcriptional control. Nat. Rev. Mol. Cell Biol. 2012, 13, 543–547. [Google Scholar] [CrossRef]
- He, N.; Liu, M.; Hsu, J.; Xue, Y.; Chou, S.; Burlingame, A.; Krogan, N.J.; Alber, T.; Zhou, Q. HIV-1 Tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV-1 transcription. Mol. Cell 2010, 38, 428–438. [Google Scholar] [CrossRef]
- Petesch, S.J.; Lis, J.T. Activator-induced spread of poly(ADP-ribose) polymerase promotes nucleosome loss at Hsp70. Mol. Cell 2012, 45, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Thorsell, A.G.; Ekblad, T.; Karlberg, T.; Löw, M.; Pinto, A.F.; Trésaugues, L.; Moche, M.; Cohen, M.S.; Schüler, H. Structural Basis for Potency and Promiscuity in Poly(ADP-ribose) Polymerase (PARP) and Tankyrase Inhibitors. J. Med. Chem. 2017, 60, 1262–1271. [Google Scholar] [CrossRef] [PubMed]
- Langelier, M.F.; Planck, J.L.; Servent, K.M.; Pascal, J.M. Purification of human PARP-1 and PARP-1 domains from Escherichia coli for structural and biochemical analysis. Methods Mol. Biol. 2011, 780, 209–226. [Google Scholar] [CrossRef] [PubMed]
- Vidhyasagar, V.; He, Y.; Guo, M.; Talwar, T.; Singh, R.S.; Yadav, M.; Katselis, G.; Vizeacoumar, F.J.; Lukong, K.E.; Wu, Y. Biochemical characterization of INTS3 and C9ORF80, two subunits of hNABP1/2 heterotrimeric complex in nucleic acid binding. Biochem. J. 2018, 475, 45–60. [Google Scholar] [CrossRef]
- Matveeva, E.A.; Mathbout, L.F.; Fondufe-Mittendorf, Y.N. PARP1 is a versatile factor in the regulation of mRNA stability and decay. Sci. Rep. 2019, 9, 3722. [Google Scholar] [CrossRef] [PubMed]
- Senichkin, V.V.; Prokhorova, E.A.; Zhivotovsky, B.; Kopeina, G.S. Simple and Efficient Protocol for Subcellular Fractionation of Normal and Apoptotic Cells. Cells 2021, 10, 852. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Vivelo, C.A.; Leung, A.K. Proteomics approaches to identify mono-(ADP-ribosyl)ated and poly(ADP-ribosyl)ated proteins. Proteomics 2015, 15, 203–217. [Google Scholar] [CrossRef] [PubMed]
- Ayyappan, V.; Wat, R.; Barber, C.; Vivelo, C.A.; Gauch, K.; Visanpattanasin, P.; Cook, G.; Sazeides, C.; Leung, A.K.L. ADPriboDB 2.0: An updated database of ADP-ribosylated proteins. Nucleic Acids Res. 2021, 49, D261–D265. [Google Scholar] [CrossRef]
- Herrmann, C.; Avgousti, D.C.; Weitzman, M.D. Differential Salt Fractionation of Nuclei to Analyze Chromatin-associated Proteins from Cultured Mammalian Cells. Bio-Protocol 2017, 7, e2175. [Google Scholar] [CrossRef] [PubMed]
- Henikoff, S.; Henikoff, J.G.; Sakai, A.; Loeb, G.B.; Ahmad, K. Genome-wide profiling of salt fractions maps physical properties of chromatin. Genome Res. 2009, 19, 460–469. [Google Scholar] [CrossRef]
- Stadelmayer, B.; Micas, G.; Gamot, A.; Martin, P.; Malirat, N.; Koval, S.; Raffel, R.; Sobhian, B.; Severac, D.; Rialle, S.; et al. Integrator complex regulates NELF-mediated RNA polymerase II pause/release and processivity at coding genes. Nat. Commun. 2014, 5, 5531. [Google Scholar] [CrossRef] [PubMed]
- Skaar, J.R.; Ferris, A.L.; Wu, X.; Saraf, A.; Khanna, K.K.; Florens, L.; Washburn, M.P.; Hughes, S.H.; Pagano, M. The Integrator complex controls the termination of transcription at diverse classes of gene targets. Cell Res. 2015, 25, 288–305. [Google Scholar] [CrossRef] [PubMed]
- Häkkinen, A.; Ribeiro, A.S. Characterizing rate limiting steps in transcription from RNA production times in live cells. Bioinformatics 2016, 32, 1346–1352. [Google Scholar] [CrossRef] [PubMed]
- Saunders, A.; Core, L.J.; Lis, J.T. Breaking barriers to transcription elongation. Nat. Rev. Mol. Cell Biol. 2006, 7, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Gibson, B.A.; Zhang, Y.; Jiang, H.; Hussey, K.M.; Shrimp, J.H.; Lin, H.; Schwede, F.; Yu, Y.; Kraus, W.L. Chemical genetic discovery of PARP targets reveals a role for PARP-1 in transcription elongation. Science 2016, 353, 45–50. [Google Scholar] [CrossRef]
- Petesch, S.J.; Lis, J.T. Overcoming the nucleosome barrier during transcript elongation. Trends Genet. 2012, 28, 285–294. [Google Scholar] [CrossRef]
- Ardehali, M.B.; Lis, J.T. Tracking rates of transcription and splicing in vivo. Nat. Struct. Mol. Biol. 2009, 16, 1123–1124. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Chen, M.; Huang, H.; Zhu, J.; Song, H.; Zhu, J.; Park, J.; Ji, S.J. Dynamic m6A modification regulates local translation of mRNA in axons. Nucleic Acids Res. 2018, 46, 1412–1423. [Google Scholar] [CrossRef]
- Mendoza-Figueroa, M.S.; Tatomer, D.C.; Wilusz, J.E. The Integrator Complex in Transcription and Development. Trends Biochem. Sci. 2020, 45, 923–934. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matveeva, E.A.; Dhahri, H.; Fondufe-Mittendorf, Y. PARP1′s Involvement in RNA Polymerase II Elongation: Pausing and Releasing Regulation through the Integrator and Super Elongation Complex. Cells 2022, 11, 3202. https://doi.org/10.3390/cells11203202
Matveeva EA, Dhahri H, Fondufe-Mittendorf Y. PARP1′s Involvement in RNA Polymerase II Elongation: Pausing and Releasing Regulation through the Integrator and Super Elongation Complex. Cells. 2022; 11(20):3202. https://doi.org/10.3390/cells11203202
Chicago/Turabian StyleMatveeva, Elena A., Hejer Dhahri, and Yvonne Fondufe-Mittendorf. 2022. "PARP1′s Involvement in RNA Polymerase II Elongation: Pausing and Releasing Regulation through the Integrator and Super Elongation Complex" Cells 11, no. 20: 3202. https://doi.org/10.3390/cells11203202
APA StyleMatveeva, E. A., Dhahri, H., & Fondufe-Mittendorf, Y. (2022). PARP1′s Involvement in RNA Polymerase II Elongation: Pausing and Releasing Regulation through the Integrator and Super Elongation Complex. Cells, 11(20), 3202. https://doi.org/10.3390/cells11203202