
Immunological Insights into Cigarette Smoking-Induced Cardiovascular Disease Risk

 , ,
, , {kind=link}
Abstract
1. Introduction
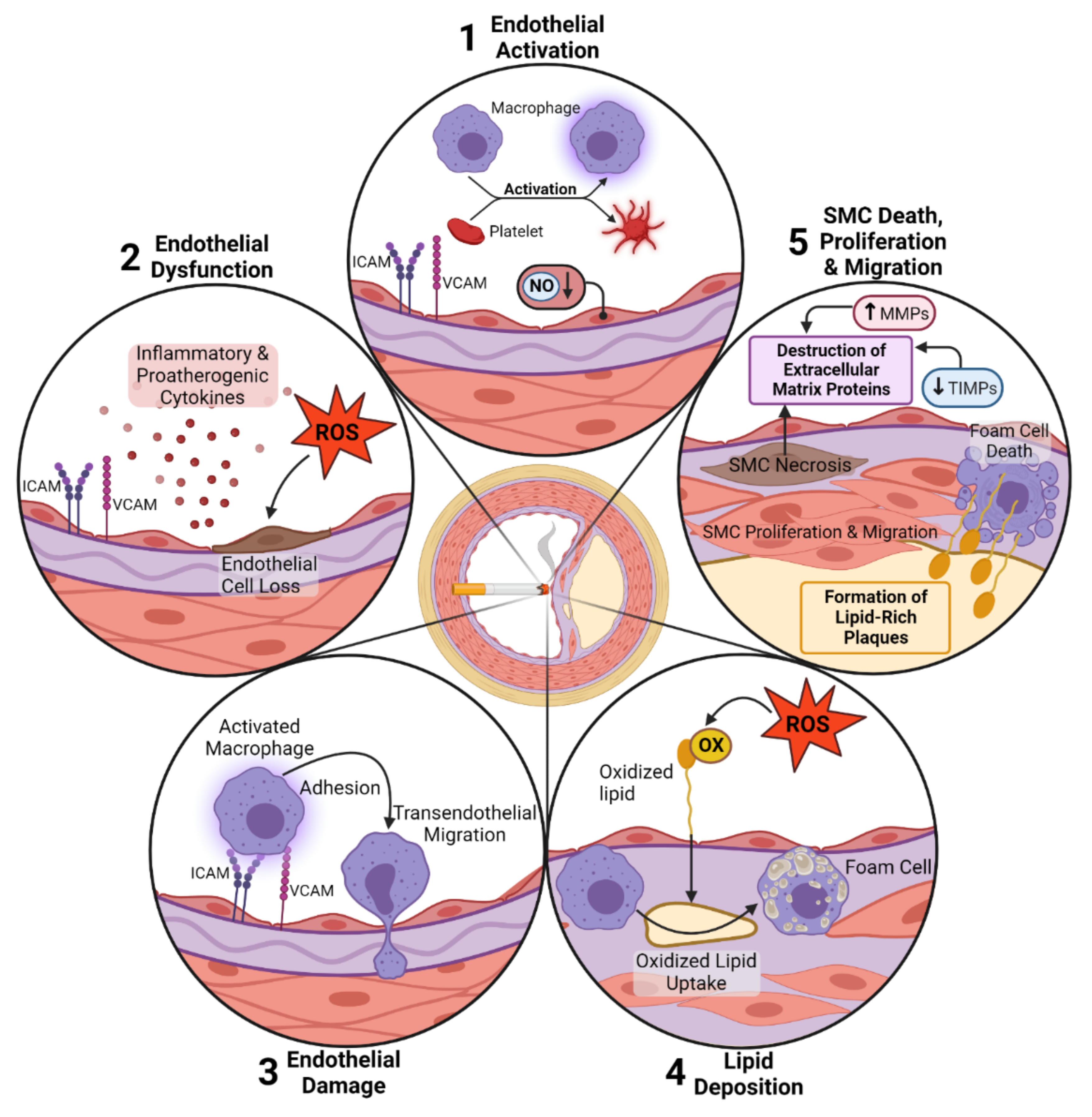
2. Smoking-Induced Pulmonary and Cardiovascular Effects
3. Conventional vs. Electronic Cigarettes
4. Effect of Smoking on Inflammation and Immune System
5. Effect of Cigarette Smoking on the Adaptive Immune System
6. Effect of Cigarette Smoking on Macrophages
7. Effect of Cigarette Smoking on Neutrophils
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- National Center for Chronic Disease Prevention and Health Promotion (NCCDPHP). Reports of the Surgeon General. In The Health Consequences of Smoking—50 Years of Progress: A Report of the Surgeon General; Centers for Disease Control and Prevention (US): Atlanta, GA, USA, 2014. [Google Scholar]
- CDC. Achievements in Public Health, 1900–1999: Tobacco Use—United States. MMWR 1999, 48, 986–993. [Google Scholar]
- Hackshaw, A.; Morris, J.K.; Boniface, S.; Tang, J.L.; Milenkovic, D. Low cigarette consumption and risk of coronary heart disease and stroke: Meta-analysis of 141 cohort studies in 55 study reports. BMJ 2018, 360, j5855. [Google Scholar] [CrossRef] [PubMed]
- National Center for Chronic Disease Prevention and Health Promotion (NCCDPHP). Publications and Reports of the Surgeon General. In How Tobacco Smoke Causes Disease: The Biology and Behavioral Basis for Smoking-Attributable Disease: A Report of the Surgeon General; Surgeon General, Centers for Disease Control and Prevention (US): Atlanta, GA, USA, 2010. [Google Scholar]
- Services UDoHaH. How Tobacco Smoke Causes Disease. The Biology and Behavioral Basis for Smoking—Attributable Disease; CDC: Atlanta, GA, USA, 2010.
- Sasco, A.J.; Secretan, M.B.; Straif, K. Tobacco smoking and cancer: A brief review of recent epidemiological evidence. Lung Cancer 2004, 45, S3–S9. [Google Scholar] [CrossRef] [PubMed]
- Valesini, G.; Gerardi, M.C.; Iannuccelli, C.; Pacucci, V.A.; Pendolino, M.; Shoenfeld, Y. Citrullination and autoimmunity. Autoimmun. Rev. 2015, 14, 490–497. [Google Scholar] [CrossRef]
- Dwivedi, N.; Radic, M. Citrullination of autoantigens implicates NETosis in the induction of autoimmunity. Ann. Rheum. Dis. 2014, 73, 483–491. [Google Scholar] [CrossRef]
- Papadopoulos, N.G.; Alamanos, Y.; Voulgari, P.V.; Epagelis, E.K.; Tsifetaki, N.; Drosos, A.A. Does cigarette smoking influence disease expression, activity and severity in early rheumatoid arthritis patients? Clin. Exp. Rheumatol. 2005, 23, 861–866. [Google Scholar]
- Kallberg, H.; Ding, B.; Padyukov, L.; Bengtsson, C.; Ronnelid, J.; Klareskog, L.; Alfredsson, L.; Group, E.S. Smoking is a major preventable risk factor for rheumatoid arthritis: Estimations of risks after various exposures to cigarette smoke. Ann. Rheum. Dis. 2011, 70, 508–511. [Google Scholar] [CrossRef]
- Vincent, C.; Nogueira, L.; Sebbag, M.; Chapuy-Regaud, S.; Arnaud, M.; Letourneur, O.; Rolland, D.; Fournie, B.; Cantagrel, A.; Jolivet, M.; et al. Detection of antibodies to deiminated recombinant rat filaggrin by enzyme-linked immunosorbent assay: A highly effective test for the diagnosis of rheumatoid arthritis. Arthritis Rheum. 2002, 46, 2051–2058. [Google Scholar] [CrossRef]
- Nishimura, K.; Sugiyama, D.; Kogata, Y.; Tsuji, G.; Nakazawa, T.; Kawano, S.; Saigo, K.; Morinobu, A.; Koshiba, M.; Kuntz, K.M.; et al. Meta-analysis: Diagnostic accuracy of anti-cyclic citrullinated peptide antibody and rheumatoid factor for rheumatoid arthritis. Ann. Intern. Med. 2007, 146, 797–808. [Google Scholar] [CrossRef]
- Makrygiannakis, D.; af Klint, E.; Lundberg, I.E.; Lofberg, R.; Ulfgren, A.K.; Klareskog, L.; Catrina, A.I. Citrullination is an inflammation-dependent process. Ann. Rheum. Dis. 2006, 65, 1219–1222. [Google Scholar] [CrossRef]
- Makrygiannakis, D.; Hermansson, M.; Ulfgren, A.K.; Nicholas, A.P.; Zendman, A.J.; Eklund, A.; Grunewald, J.; Skold, C.M.; Klareskog, L.; Catrina, A.I. Smoking increases peptidylarginine deiminase 2 enzyme expression in human lungs and increases citrullination in BAL cells. Ann. Rheum. Dis. 2008, 67, 1488–1492. [Google Scholar] [CrossRef]
- Sopori, M. Effects of cigarette smoke on the immune system. Nat. Rev. Immunol. 2002, 2, 372–377. [Google Scholar] [CrossRef]
- Battaglia, M. Experiments by nature: Lessons on type 1 diabetes. Tissue Antigens 2014, 83, 1–9. [Google Scholar] [CrossRef]
- Roep, B.O.; Tree, T.I. Immune modulation in humans: Implications for type 1 diabetes mellitus. Nat. Rev. Endocrinol. 2014, 10, 229–242. [Google Scholar] [CrossRef]
- Hedstrom, A.K.; Baarnhielm, M.; Olsson, T.; Alfredsson, L. Tobacco smoking, but not Swedish snuff use, increases the risk of multiple sclerosis. Neurology 2009, 73, 696–701. [Google Scholar] [CrossRef]
- Odoardi, F.; Sie, C.; Streyl, K.; Ulaganathan, V.K.; Schlager, C.; Lodygin, D.; Heckelsmiller, K.; Nietfeld, W.; Ellwart, J.; Klinkert, W.E.; et al. T cells become licensed in the lung to enter the central nervous system. Nature 2012, 488, 675–679. [Google Scholar] [CrossRef]
- Hernan, M.A.; Jick, S.S.; Logroscino, G.; Olek, M.J.; Ascherio, A.; Jick, H. Cigarette smoking and the progression of multiple sclerosis. Brain A J. Neurol. 2005, 128, 1461–1465. [Google Scholar] [CrossRef]
- Pittas, F.; Ponsonby, A.L.; van der Mei, I.A.; Taylor, B.V.; Blizzard, L.; Groom, P.; Ukoumunne, O.C.; Dwyer, T. Smoking is associated with progressive disease course and increased progression in clinical disability in a prospective cohort of people with multiple sclerosis. J. Neurol. 2009, 256, 577–585. [Google Scholar] [CrossRef]
- Healy, B.C.; Ali, E.N.; Guttmann, C.R.; Chitnis, T.; Glanz, B.I.; Buckle, G.; Houtchens, M.; Stazzone, L.; Moodie, J.; Berger, A.M.; et al. Smoking and disease progression in multiple sclerosis. Arch. Neurol. 2009, 66, 858–864. [Google Scholar] [CrossRef]
- Gao, Z.; Nissen, J.C.; Ji, K.; Tsirka, S.E. The experimental autoimmune encephalomyelitis disease course is modulated by nicotine and other cigarette smoke components. PLoS ONE 2014, 9, e107979. [Google Scholar] [CrossRef]
- Khanna, A.; Guo, M.; Mehra, M.; Royal, W., 3rd. Inflammation and oxidative stress induced by cigarette smoke in Lewis rat brains. J. Neuroimmunol. 2013, 254, 69–75. [Google Scholar] [CrossRef]
- Correale, J.; Farez, M.F. Smoking worsens multiple sclerosis prognosis: Two different pathways are involved. J. Neuroimmunol. 2015, 281, 23–34. [Google Scholar] [CrossRef]
- Perricone, C.; Versini, M.; Ben-Ami, D.; Gertel, S.; Watad, A.; Segel, M.J.; Ceccarelli, F.; Conti, F.; Cantarini, L.; Bogdanos, D.P.; et al. Smoke and autoimmunity: The fire behind the disease. Autoimmun. Rev. 2016, 15, 354–374. [Google Scholar] [CrossRef]
- Katanoda, K.; Yako-Suketomo, H. Mortality attributable to tobacco by selected countries based on the WHO Global Report. Jpn. J. Clin. Oncol. 2012, 42, 561–562. [Google Scholar] [CrossRef]
- Celermajer, D.S.; Sorensen, K.E.; Gooch, V.M.; Spiegelhalter, D.J.; Miller, O.I.; Sullivan, I.D.; Lloyd, J.K.; Deanfield, J.E. Non-invasive detection of endothelial dysfunction in children and adults at risk of atherosclerosis. Lancet 1992, 340, 1111–1115. [Google Scholar] [CrossRef]
- Johnson, H.M.; Gossett, L.K.; Piper, M.E.; Aeschlimann, S.E.; Korcarz, C.E.; Baker, T.B.; Fiore, M.C.; Stein, J.H. Effects of smoking and smoking cessation on endothelial function: 1-year outcomes from a randomized clinical trial. J. Am. Coll. Cardiol. 2010, 55, 1988–1995. [Google Scholar] [CrossRef]
- Craig, W.Y.; Palomaki, G.E.; Haddow, J.E. Cigarette smoking and serum lipid and lipoprotein concentrations: An analysis of published data. BMJ 1989, 298, 784–788. [Google Scholar] [CrossRef]
- Alyan, O.; Kacmaz, F.; Ozdemir, O.; Maden, O.; Topaloglu, S.; Ozbakir, C.; Metin, F.; Karadede, A.; Ilkay, E. Effects of cigarette smoking on heart rate variability and plasma N-terminal pro-B-type natriuretic peptide in healthy subjects: Is there the relationship between both markers? Ann. Noninvasive Electrocardiol. 2008, 13, 137–144. [Google Scholar] [CrossRef]
- McGrath, M.F.; de Bold, M.L.K.; Adolfo, J. The endocrine function of the heart. Trends Endocrinol. Metab. 2005, 16, 469–477. [Google Scholar] [CrossRef]
- Newton-Cheh, C.; Larson, M.G.; Vasan, R.S.; Levy, D.; Bloch, K.D.; Surti, A.; Guiducci, C.; Kathiresan, S.; Benjamin, E.J.; Struck, J.; et al. Association of common variants in NPPA and NPPB with circulating natriuretic peptides and blood pressure. Nat. Genet. 2009, 41, 348–353. [Google Scholar] [CrossRef]
- Ventura, H.O.; Silver, M.A. Natriuretic peptides as markers of cardiovascular risk: The story continues. Mayo Clin. Proc. 2011, 86, 1143–1145. [Google Scholar] [CrossRef] [PubMed]
- Nadruz, W., Jr.; Gonçalves, A.; Claggett, B.; Roca, G.Q.; Shah, A.M.; Cheng, S.; Heiss, G.; Ballantyne, C.M.; Solomon, S.D. Influence of cigarette smoking on cardiac biomarkers: The Atherosclerosis Risk in Communities (ARIC) Study. Eur. J. Heart Fail. 2016, 18, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Khaled, S.A.; El-amir, A.M. Effect of exposure to second-hand smoke on serum levels of N-terminal pro-brain natriuretic peptide. Kasr Al Ainy Med. J. 2015, 21, 22. [Google Scholar] [CrossRef]
- Otsuka, T.; Kawada, T.; Seino, Y.; Ibuki, C.; Katsumata, M.; Kodani, E. Relation of smoking status to serum levels of N-terminal pro–brain natriuretic peptide in middle-aged men without overt cardiovascular disease. Am. J. Cardiol. 2010, 106, 1456–1460. [Google Scholar] [CrossRef] [PubMed]
- Lyngbakken, M.N.; Skranes, J.B.; de Lemos, J.A.; Nygård, S.; Dalen, H.; Hveem, K.; Røsjø, H.; Omland, T. Impact of smoking on circulating cardiac troponin I concentrations and cardiovascular events in the general population: The HUNT Study (Nord-Trøndelag Health Study). Circulation 2016, 134, 1962–1972. [Google Scholar] [CrossRef] [PubMed]
- Bhatnagar, A. Cardiovascular pathophysiology of environmental pollutants. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H479–H485. [Google Scholar] [CrossRef] [PubMed]
- Schane, R.E.; Ling, P.M.; Glantz, S.A. Health effects of light and intermittent smoking: A review. Circulation 2010, 121, 1518–1522. [Google Scholar] [CrossRef]
- Benowitz, N.L.; Jacob, P.; Jones, R.T.; Rosenberg, J. Interindividual variability in the metabolism and cardiovascular effects of nicotine in man. J. Pharmacol. Exp. Ther. 1982, 221, 368–372. [Google Scholar]
- De Couck, M.; Cserjesi, R.; Caers, R.; Zijlstra, W.; Widjaja, D.; Wolf, N.; Luminet, O.; Ellrich, J.; Gidron, Y. Effects of short and prolonged transcutaneous vagus nerve stimulation on heart rate variability in healthy subjects. Auton. Neurosci. 2017, 203, 88–96. [Google Scholar] [CrossRef]
- Yun, M.; Li, S.; Sun, D.; Ge, S.; Lai, C.C.; Fernandez, C.; Chen, W.; Srinivasan, S.R.; Berenson, G.S. Tobacco smoking strengthens the association of elevated blood pressure with arterial stiffness: The Bogalusa Heart Study. J. Hypertens 2015, 33, 266–274. [Google Scholar] [CrossRef]
- Messner, B.; Bernhard, D. Smoking and cardiovascular disease: Mechanisms of endothelial dysfunction and early atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 509–515. [Google Scholar] [CrossRef]
- Freedman, D.S.; Srinivasan, S.R.; Shear, C.L.; Hunter, S.M.; Croft, J.B.; Webber, L.S.; Berenson, G.S. Cigarette smoking initiation and longitudinal changes in serum lipids and lipoproteins in early adulthood: The Bogalusa Heart Study. Am. J. Epidemiol. 1986, 124, 207–219. [Google Scholar] [CrossRef]
- Neufeld, E.J.; Mietus-Snyder, M.; Beiser, A.S.; Baker, A.L.; Newburger, J.W. Passive cigarette smoking and reduced HDL cholesterol levels in children with high-risk lipid profiles. Circulation 1997, 96, 1403–1407. [Google Scholar] [CrossRef]
- Gepner, A.D.; Piper, M.E.; Johnson, H.M.; Fiore, M.C.; Baker, T.B.; Stein, J.H. Effects of smoking and smoking cessation on lipids and lipoproteins: Outcomes from a randomized clinical trial. Am. Heart J. 2011, 161, 145–151. [Google Scholar] [CrossRef]
- Morrow, J.D.; Frei, B.; Longmire, A.W.; Gaziano, J.M.; Lynch, S.M.; Shyr, Y.; Strauss, W.E.; Oates, J.A.; Roberts, L.J., 2nd. Increase in circulating products of lipid peroxidation (F2-isoprostanes) in smokers. Smoking as a cause of oxidative damage. N. Engl. J. Med. 1995, 332, 1198–1203. [Google Scholar] [CrossRef]
- Pilz, H.; Oguogho, A.; Chehne, F.; Lupattelli, G.; Palumbo, B.; Sinzinger, H. Quitting cigarette smoking results in a fast improvement of in vivo oxidation injury (determined via plasma, serum and urinary isoprostane). Thromb. Res. 2000, 99, 209–221. [Google Scholar] [CrossRef]
- Reilly, M.; Delanty, N.; Lawson, J.A.; FitzGerald, G.A. Modulation of oxidant stress in vivo in chronic cigarette smokers. Circulation 1996, 94, 19–25. [Google Scholar] [CrossRef]
- Solak, Z.A.; Kabaroglu, C.; Cok, G.; Parildar, Z.; Bayindir, U.; Ozmen, D.; Bayindir, O. Effect of different levels of cigarette smoking on lipid peroxidation, glutathione enzymes and paraoxonase 1 activity in healthy people. Clin. Exp. Med. 2005, 5, 99–105. [Google Scholar] [CrossRef]
- Frostegård, J. Immunity, atherosclerosis and cardiovascular disease. BMC Med. 2013, 11, 117. [Google Scholar] [CrossRef]
- Battistoni, A.; Rubattu, S.; Volpe, M. Circulating biomarkers with preventive, diagnostic and prognostic implications in cardiovascular diseases. Int. J. Cardiol. 2012, 157, 160–168. [Google Scholar] [CrossRef]
- Bazzano, L.A.; He, J.; Muntner, P.; Vupputuri, S.; Whelton, P.K. Relationship between cigarette smoking and novel risk factors for cardiovascular disease in the United States. Ann. Intern. Med. 2003, 138, 891–897. [Google Scholar] [CrossRef]
- Bermudez, E.A.; Rifai, N.; Buring, J.E.; Manson, J.E.; Ridker, P.M. Relation between markers of systemic vascular inflammation and smoking in women. Am. J. Cardiol. 2002, 89, 1117–1119. [Google Scholar] [CrossRef]
- Frost-Pineda, K.; Liang, Q.; Liu, J.; Rimmer, L.; Jin, Y.; Feng, S.; Kapur, S.; Mendes, P.; Roethig, H.; Sarkar, M. Biomarkers of potential harm among adult smokers and nonsmokers in the total exposure study. Nicotine Tob. Res. 2011, 13, 182–193. [Google Scholar] [CrossRef]
- Levitzky, Y.S.; Guo, C.-Y.; Rong, J.; Larson, M.G.; Walter, R.E.; Keaney, J.F., Jr.; Sutherland, P.A.; Vasan, A.; Lipinska, I.; Evans, J.C. Relation of smoking status to a panel of inflammatory markers: The framingham offspring. Atherosclerosis 2008, 201, 217–224. [Google Scholar] [CrossRef]
- McEvoy, J.W.; Nasir, K.; DeFilippis, A.P.; Lima, J.A.; Bluemke, D.A.; Hundley, W.G.; Barr, R.G.; Budoff, M.J.; Szklo, M.; Navas-Acien, A.; et al. Relationship of cigarette smoking with inflammation and subclinical vascular disease: The Multi-Ethnic Study of Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1002–1010. [Google Scholar] [CrossRef]
- Wannamethee, S.G.; Lowe, G.D.; Shaper, A.G.; Rumley, A.; Lennon, L.; Whincup, P.H. Associations between cigarette smoking, pipe/cigar smoking, and smoking cessation, and haemostatic and inflammatory markers for cardiovascular disease. Eur. Heart J. 2005, 26, 1765–1773. [Google Scholar] [CrossRef]
- Al Rifai, M.; DeFilippis, A.P.; McEvoy, J.W.; Hall, M.E.; Acien, A.N.; Jones, M.R.; Keith, R.; Magid, H.S.; Rodriguez, C.J.; Barr, G.R. The relationship between smoking intensity and subclinical cardiovascular injury: The Multi-Ethnic Study of Atherosclerosis (MESA). Atherosclerosis 2017, 258, 119–130. [Google Scholar] [CrossRef]
- Barbieri, S.S.; Zacchi, E.; Amadio, P.; Gianellini, S.; Mussoni, L.; Weksler, B.B.; Tremoli, E. Cytokines present in smokers’ serum interact with smoke components to enhance endothelial dysfunction. Cardiovasc. Res. 2011, 90, 475–483. [Google Scholar] [CrossRef] [PubMed]
- César-Neto, J.B.; Duarte, P.M.; De Oliveira, M.C.G.; Casati, M.Z.; Tambeli, C.H.; Parada, C.A.; Sallum, E.A.; Nociti, F.H., Jr. Smoking modulates interferon-γ expression in the gingival tissue of patients with chronic periodontitis. Eur. J. Oral Sci. 2006, 114, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Friedrichs, B.; Neumann, U.; Schüller, J.; Peck, M.J. Cigarette-smoke-induced priming of neutrophils from smokers and non-smokers for increased oxidative burst response is mediated by TNF-α. Toxicol. Vitr. 2014, 28, 1249–1258. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, R.; Coletta, R.; Silvério, K.; Benevides, L.; Casati, M.; Da Silva, J.; Nociti, F. Impact of smoking on inflammation: Overview of molecular mechanisms. Inflamm. Res. 2011, 60, 409–424. [Google Scholar] [CrossRef]
- Hagiwara, E.; Takahashi, K.-I.; Okubo, T.; Ohno, S.; Ueda, A.; Aoki, A.; Odagiri, S.; Ishigatsubo, Y. Cigarette smoking depletes cells spontaneously secreting th1cytokines in the human airway. Cytokine 2001, 14, 121–126. [Google Scholar] [CrossRef]
- Meuronen, A.; Majuri, M.-L.; Alenius, H.; Mäntylä, T.; Wolff, H.; Piirilä, P.; Laitinen, A. Decreased cytokine and chemokine mRNA expression in bronchoalveolar lavage in asymptomatic smoking subjects. Respiration 2008, 75, 450–458. [Google Scholar] [CrossRef]
- Ridker, P.M.; Rifai, N.; Stampfer, M.J.; Hennekens, C.H. Plasma concentration of interleukin-6 and the risk of future myocardial infarction among apparently healthy men. Circulation 2000, 101, 1767–1772. [Google Scholar] [CrossRef]
- Stoner, L.; Lucero, A.A.; Palmer, B.R.; Jones, L.M.; Young, J.M.; Faulkner, J. Inflammatory biomarkers for predicting cardiovascular disease. Clin. Biochem. 2013, 46, 1353–1371. [Google Scholar] [CrossRef]
- De Lemos, J.A.; Morrow, D.A.; Sabatine, M.S.; Murphy, S.A.; Gibson, C.M.; Antman, E.M.; McCabe, C.H.; Cannon, C.P.; Braunwald, E. Association between plasma levels of monocyte chemoattractant protein-1 and long-term clinical outcomes in patients with acute coronary syndromes. Circulation 2003, 107, 690–695. [Google Scholar] [CrossRef]
- Deo, R.; Khera, A.; McGuire, D.K.; Murphy, S.A.; Neto, J.D.P.M.; Morrow, D.A.; de Lemos, J.A. Association among plasma levels of monocyte chemoattractant protein-1, traditional cardiovascular risk factors, and subclinical atherosclerosis. J. Am. Coll. Cardiol. 2004, 44, 1812–1818. [Google Scholar] [CrossRef]
- Hoogeveen, R.C.; Morrison, A.; Boerwinkle, E.; Miles, J.S.; Rhodes, C.E.; Sharrett, A.R.; Ballantyne, C.M. Plasma MCP-1 level and risk for peripheral arterial disease and incident coronary heart disease: Atherosclerosis Risk in Communities study. Atherosclerosis 2005, 183, 301–307. [Google Scholar] [CrossRef]
- McDermott, D.H.; Yang, Q.; Kathiresan, S.; Cupples, L.A.; Massaro, J.M.; Keaney, J.F., Jr.; Larson, M.G.; Vasan, R.S.; Hirschhorn, J.N.; O’Donnell, C.J. CCL2 polymorphisms are associated with serum monocyte chemoattractant protein-1 levels and myocardial infarction in the Framingham Heart Study. Circulation 2005, 112, 1113–1120. [Google Scholar] [CrossRef]
- Lavi, S.; Prasad, A.; Yang, E.H.; Mathew, V.; Simari, R.D.; Rihal, C.S.; Lerman, L.O.; Lerman, A. Smoking is associated with epicardial coronary endothelial dysfunction and elevated white blood cell count in patients with chest pain and early coronary artery disease. Circulation 2007, 115, 2621–2627. [Google Scholar] [CrossRef]
- Newby, D.E.; Wright, R.A.; Labinjoh, C.; Ludlam, C.A.; Fox, K.A.; Boon, N.A.; Webb, D.J. Endothelial dysfunction, impaired endogenous fibrinolysis, and cigarette smoking: A mechanism for arterial thrombosis and myocardial infarction. Circulation 1999, 99, 1411–1415. [Google Scholar] [CrossRef]
- Martin, J.L.; Wilson, J.R.; Ferraro, N.; Laskey, W.K.; Kleaveland, J.P.; Hirshfeld, J.W., Jr. Acute coronary vasoconstrictive effects of cigarette smoking in coronary heart disease. Am. J. Cardiol. 1984, 54, 56–60. [Google Scholar] [CrossRef]
- Cavusoglu, Y.; Timuralp, B.; Us, T.; Akgun, Y.; Kudaiberdieva, G.; Gorenek, B.; Unalir, A.; Goktekin, O.; Ata, N. Cigarette smoking increases plasma concentrations of vascular cell adhesion molecule-1 in patients with coronary artery disease. Angiology 2004, 55, 397–402. [Google Scholar] [CrossRef]
- Brunner, H.; Cockcroft, J.R.; Deanfield, J.; Donald, A.; Ferrannini, E.; Halcox, J.; Kiowski, W.; Luscher, T.F.; Mancia, G.; Natali, A.; et al. Endothelial function and dysfunction. Part II: Association with cardiovascular risk factors and diseases. A statement by the Working Group on Endothelins and Endothelial Factors of the European Society of Hypertension. J. Hypertens 2005, 23, 233–246. [Google Scholar] [CrossRef]
- Deanfield, J.E.; Selwyn, A.P. Character and causes of transient myocardial ischemia during daily life. Implications for treatment of patients with coronary disease. Am. J. Med. 1986, 80, 18–24. [Google Scholar] [CrossRef]
- McGarvey, L.P.; John, M.; Anderson, J.A.; Zvarich, M.; Wise, R.A. Ascertainment of cause-specific mortality in COPD: Operations of the TORCH Clinical Endpoint Committee. Thorax 2007, 62, 411–415. [Google Scholar] [CrossRef]
- Saetta, M.; Di Stefano, A.; Turato, G.; Facchini, F.M.; Corbino, L.; Mapp, C.E.; Maestrelli, P.; Ciaccia, A.; Fabbri, L.M. CD8+ T-lymphocytes in peripheral airways of smokers with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1998, 157, 822–826. [Google Scholar] [CrossRef]
- Yu, M.Q.; Liu, X.S.; Wang, J.M.; Xu, Y.J. CD8(+) Tc-lymphocytes immunodeviation in peripheral blood and airway from patients of chronic obstructive pulmonary disease and changes after short-term smoking cessation. Chin. Med. J. 2013, 126, 3608–3615. [Google Scholar]
- Alwis, K.U.; deCastro, B.R.; Morrow, J.C.; Blount, B.C. Acrolein Exposure in U.S. Tobacco Smokers and Non-Tobacco Users: NHANES 2005–2006. Environ. Health Perspect. 2015, 123, 1302–1308. [Google Scholar] [CrossRef]
- Koch, A.; Gaczkowski, M.; Sturton, G.; Staib, P.; Schinkothe, T.; Klein, E.; Rubbert, A.; Bacon, K.; Wassermann, K.; Erdmann, E. Modification of surface antigens in blood CD8+ T-lymphocytes in COPD: Effects of smoking. Eur. Respir. J. 2007, 29, 42–50. [Google Scholar] [CrossRef]
- Brassington, K.; Selemidis, S.; Bozinovski, S.; Vlahos, R. New frontiers in the treatment of comorbid cardiovascular disease in chronic obstructive pulmonary disease. Clin. Sci. 2019, 133, 885–904. [Google Scholar] [CrossRef] [PubMed]
- Vlahos, R.; Bozinovski, S. Recent advances in pre-clinical mouse models of COPD. Clin. Sci. 2014, 126, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Doiron, D.; de Hoogh, K.; Probst-Hensch, N.; Fortier, I.; Cai, Y.; De Matteis, S.; Hansell, A.L. Air pollution, lung function and COPD: Results from the population-based UK Biobank study. Eur. Respir. J. 2019, 54, 1802140. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.; Marron, R.; Voelker, H.; Albert, R.; Connett, J.; Bailey, W.; Casaburi, R.; Cooper, J.A.; Curtis, J.L.; Dransfield, M. Effect of beta-blockers on exacerbation rate and lung function in chronic obstructive pulmonary disease (COPD). Respir. Res. 2017, 18, 1–7. [Google Scholar] [CrossRef]
- Kluchová, Z.; Petrášová, D.; Joppa, P.; Dorková, Z.; Tkáčová, R. The association between oxidative stress and obstructive lung impairment in patients with COPD. Physiol. Res. 2007, 56, 51–56. [Google Scholar] [CrossRef]
- Barnes, P.J. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2016, 138, 16–27. [Google Scholar] [CrossRef]
- Pelgrim, C.E.; Peterson, J.D.; Gosker, H.R.; Schols, A.; van Helvoort, A.; Garssen, J.; Folkerts, G.; Kraneveld, A.D. Psychological co-morbidities in COPD: Targeting systemic inflammation, a benefit for both? Eur. J. Pharmacol. 2019, 842, 99–110. [Google Scholar] [CrossRef]
- Szucs, B.; Szucs, C.; Petrekanits, M.; Varga, J.T. Molecular characteristics and treatment of endothelial dysfunction in patients with COPD: A review article. Int. J. Mol. Sci. 2019, 20, 4329. [Google Scholar] [CrossRef]
- Barcelo, B.; Pons, J.; Ferrer, J.M.; Sauleda, J.; Fuster, A.; Agusti, A.G. Phenotypic characterisation of T-lymphocytes in COPD: Abnormal CD4+CD25+ regulatory T-lymphocyte response to tobacco smoking. Eur. Respir. J. 2008, 31, 555–562. [Google Scholar] [CrossRef]
- Chiappori, A.; Folli, C.; Balbi, F.; Caci, E.; Riccio, A.M.; De Ferrari, L.; Melioli, G.; Braido, F.; Canonica, G.W. CD4(+)CD25(high)CD127(-) regulatory T-cells in COPD: Smoke and drugs effect. World Allergy Organ. J. 2016, 9, 5. [Google Scholar] [CrossRef]
- Brandsma, C.A.; Hylkema, M.N.; Geerlings, M.; van Geffen, W.H.; Postma, D.S.; Timens, W.; Kerstjens, H.A. Increased levels of (class switched) memory B cells in peripheral blood of current smokers. Respir. Res. 2009, 10, 108. [Google Scholar] [CrossRef]
- Brandsma, C.A.; Kerstjens, H.A.; van Geffen, W.H.; Geerlings, M.; Postma, D.S.; Hylkema, M.N.; Timens, W. Differential switching to IgG and IgA in active smoking COPD patients and healthy controls. Eur. Respir. J. 2012, 40, 313–321. [Google Scholar] [CrossRef]
- Arnson, Y.; Shoenfeld, Y.; Amital, H. Effects of tobacco smoke on immunity, inflammation and autoimmunity. J. Autoimmun. 2010, 34, J258–J265. [Google Scholar] [CrossRef]
- Hoenderdos, K.; Condliffe, A. The neutrophil in chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2013, 48, 531–539. [Google Scholar] [CrossRef]
- O’Donnell, R.; Breen, D.; Wilson, S.; Djukanovic, R. Inflammatory cells in the airways in COPD. Thorax 2006, 61, 448–454. [Google Scholar] [CrossRef]
- Mills, K.H.; Dunne, A. Immune modulation: IL-1, master mediator or initiator of inflammation. Nat. Med. 2009, 15, 1363–1364. [Google Scholar] [CrossRef]
- Striz, I.; Brabcova, E.; Kolesar, L.; Sekerkova, A. Cytokine networking of innate immunity cells: A potential target of therapy. Clin. Sci. 2014, 126, 593–612. [Google Scholar] [CrossRef]
- Eltom, S.; Belvisi, M.G.; Stevenson, C.S.; Maher, S.A.; Dubuis, E.; Fitzgerald, K.A.; Birrell, M.A. Role of the inflammasome-caspase1/11-IL-1/18 axis in cigarette smoke driven airway inflammation: An insight into the pathogenesis of COPD. PLoS ONE 2014, 9, e112829. [Google Scholar] [CrossRef]
- Barnes, P.J. The cytokine network in chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2009, 41, 631–638. [Google Scholar] [CrossRef]
- Wang, H.; Peng, W.; Weng, Y.; Ying, H.; Li, H.; Xia, D.; Yu, W. Imbalance of Th17/Treg cells in mice with chronic cigarette smoke exposure. Int. Immunopharmacol. 2012, 14, 504–512. [Google Scholar] [CrossRef]
- Nadigel, J.; Prefontaine, D.; Baglole, C.J.; Maltais, F.; Bourbeau, J.; Eidelman, D.H.; Hamid, Q. Cigarette smoke increases TLR4 and TLR9 expression and induces cytokine production from CD8(+) T cells in chronic obstructive pulmonary disease. Respir. Res. 2011, 12, 149. [Google Scholar] [CrossRef]
- Zhang, M.Q.; Wan, Y.; Jin, Y.; Xin, J.B.; Zhang, J.C.; Xiong, X.Z.; Chen, L.; Chen, G. Cigarette smoking promotes inflammation in patients with COPD by affecting the polarization and survival of Th/Tregs through up-regulation of muscarinic receptor 3 and 5 expression. PLoS ONE 2014, 9, e112350. [Google Scholar] [CrossRef]
- Almirall, J.; Gonzalez, C.A.; Balanzo, X.; Bolibar, I. Proportion of community-acquired pneumonia cases attributable to tobacco smoking. Chest 1999, 116, 375–379. [Google Scholar] [CrossRef]
- Erhardt, L. Cigarette smoking: An undertreated risk factor for cardiovascular disease. Atherosclerosis 2009, 205, 23–32. [Google Scholar] [CrossRef]
- Vargas-Rojas, M.I.; Ramirez-Venegas, A.; Limon-Camacho, L.; Ochoa, L.; Hernandez-Zenteno, R.; Sansores, R.H. Increase of Th17 cells in peripheral blood of patients with chronic obstructive pulmonary disease. Respir. Med. 2011, 105, 1648–1654. [Google Scholar] [CrossRef]
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef]
- Mantovani, A.; Biswas, S.K.; Galdiero, M.R.; Sica, A.; Locati, M. Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 2013, 229, 176–185. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Allen, J.; Hankey-Giblin, P.A. Ontogeny and polarization of macrophages in inflammation: Blood monocytes versus tissue macrophages. Front. Immunol. 2014, 5, 683. [Google Scholar] [CrossRef] [PubMed]
- Cornwell, W.D.; Kim, V.; Fan, X.; Vega, M.E.; Ramsey, F.V.; Criner, G.J.; Rogers, T.J. Activation and polarization of circulating monocytes in severe chronic obstructive pulmonary disease. BMC Pulm. Med. 2018, 18, 101. [Google Scholar] [CrossRef] [PubMed]
- Dragoljevic, D.; Kraakman, M.J.; Nagareddy, P.R.; Ngo, D.; Shihata, W.; Kammoun, H.L.; Whillas, A.; Lee, M.K.S.; Al-Sharea, A.; Pernes, G.; et al. Defective cholesterol metabolism in haematopoietic stem cells promotes monocyte-driven atherosclerosis in rheumatoid arthritis. Eur. Heart J. 2018, 39, 2158–2167. [Google Scholar] [CrossRef]
- Fadini, G.P.; Simoni, F.; Cappellari, R.; Vitturi, N.; Galasso, S.; de Kreutzenberg, S.V.; Previato, L.; Avogaro, A. Pro-inflammatory monocyte-macrophage polarization imbalance in human hypercholesterolemia and atherosclerosis. Atherosclerosis 2014, 237, 805–808. [Google Scholar] [CrossRef]
- Ziegler-Heitbrock, L. Blood Monocytes and Their Subsets: Established Features and Open Questions. Front. Immunol. 2015, 6, 423. [Google Scholar] [CrossRef]
- Laniado-Laborín, R. Smoking and chronic obstructive pulmonary disease (COPD). Parallel epidemics of the 21 century. Int. J. Environ. Res. Public Health 2009, 6, 209–224. [Google Scholar] [CrossRef]
- Samara, K.D.; Margaritopoulos, G.; Wells, A.U.; Siafakas, N.M.; Antoniou, K.M. Smoking and pulmonary fibrosis: Novel insights. Pulm. Med. 2011, 2011, 461439. [Google Scholar] [CrossRef]
- Niewoehner, D.E.; Kleinerman, J.; Rice, D.B. Pathologic changes in the peripheral airways of young cigarette smokers. N. Engl. J. Med. 1974, 291, 755–758. [Google Scholar] [CrossRef]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef]
- Dewhurst, J.A.; Lea, S.; Hardaker, E.; Dungwa, J.V.; Ravi, A.K.; Singh, D. Characterisation of lung macrophage subpopulations in COPD patients and controls. Sci. Rep. 2017, 7, 7143. [Google Scholar] [CrossRef]
- Eapen, M.S.; Hansbro, P.M.; McAlinden, K.; Kim, R.Y.; Ward, C.; Hackett, T.L.; Walters, E.H.; Sohal, S.S. Abnormal M1/M2 macrophage phenotype profiles in the small airway wall and lumen in smokers and chronic obstructive pulmonary disease (COPD). Sci. Rep. 2017, 7, 13392. [Google Scholar] [CrossRef]
- Phaybouth, V.; Wang, S.Z.; Hutt, J.A.; McDonald, J.D.; Harrod, K.S.; Barrett, E.G. Cigarette smoke suppresses Th1 cytokine production and increases RSV expression in a neonatal model. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L222–L231. [Google Scholar] [CrossRef]
- Shaykhiev, R.; Krause, A.; Salit, J.; Strulovici-Barel, Y.; Harvey, B.G.; O’Connor, T.P.; Crystal, R.G. Smoking-dependent reprogramming of alveolar macrophage polarization: Implication for pathogenesis of chronic obstructive pulmonary disease. J. Immunol. 2009, 183, 2867–2883. [Google Scholar] [CrossRef]
- Van Zyl-Smit, R.N.; Binder, A.; Meldau, R.; Semple, P.L.; Evans, A.; Smith, P.; Bateman, E.D.; Dheda, K. Cigarette smoke impairs cytokine responses and BCG containment in alveolar macrophages. Thorax 2014, 69, 363–370. [Google Scholar] [CrossRef]
- Park, E.J.; Lee, H.S.; Lee, S.J.; Park, Y.J.; Park, S.I.; Chang, J.; Lee, K. Cigarette smoke condensate may disturb immune function with apoptotic cell death by impairing function of organelles in alveolar macrophages. Toxicol. Vitr. 2018, 52, 351–364. [Google Scholar] [CrossRef]
- Karimi, K.; Sarir, H.; Mortaz, E.; Smit, J.J.; Hosseini, H.; De Kimpe, S.J.; Nijkamp, F.P.; Folkerts, G. Toll-like receptor-4 mediates cigarette smoke-induced cytokine production by human macrophages. Respir. Res. 2006, 7, 66. [Google Scholar] [CrossRef]
- Yang, S.R.; Wright, J.; Bauter, M.; Seweryniak, K.; Kode, A.; Rahman, I. Sirtuin regulates cigarette smoke-induced proinflammatory mediator release via RelA/p65 NF-kappaB in macrophages in vitro and in rat lungs in vivo: Implications for chronic inflammation and aging. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L567–L576. [Google Scholar] [CrossRef]
- Rubins, J.B. Alveolar macrophages: Wielding the double-edged sword of inflammation. Am. J. Respir. Crit. Care Med. 2003, 167, 103–104. [Google Scholar] [CrossRef]
- Schagat, T.L.; Wofford, J.A.; Wright, J.R. Surfactant protein A enhances alveolar macrophage phagocytosis of apoptotic neutrophils. J. Immunol. 2001, 166, 2727–2733. [Google Scholar] [CrossRef] [PubMed]
- The BAL Cooperative Group Steering Committee. Bronchoalveolar lavage constituents in healthy individuals, idiopathic pulmonary fibrosis, and selected comparison groups. Am. Rev. Respir. Dis. 1990, 141, S169–S202. [Google Scholar] [CrossRef]
- Karimi, R.; Tornling, G.; Grunewald, J.; Eklund, A.; Sköld, C.M. Cell recovery in bronchoalveolar lavage fluid in smokers is dependent on cumulative smoking history. PLoS ONE 2012, 7, e34232. [Google Scholar] [CrossRef]
- Koch, A.; Giembycz, M.; Stirling, R.G.; Lim, S.; Adcock, I.; Wassermann, K.; Erdmann, E.; Chung, K.F. Effect of smoking on MAP kinase-induced modulation of IL-8 in human alveolar macrophages. Eur. Respir. J. 2004, 23, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Mio, T.; Romberger, D.J.; Thompson, A.B.; Robbins, R.A.; Heires, A.; Rennard, S.I. Cigarette smoke induces interleukin-8 release from human bronchial epithelial cells. Am. J. Respir. Crit. Care Med. 1997, 155, 1770–1776. [Google Scholar] [CrossRef]
- Nishikawa, M.; Kakemizu, N.; Ito, T.; Kudo, M.; Kaneko, T.; Suzuki, M.; Udaka, N.; Ikeda, H.; Okubo, T. Superoxide mediates cigarette smoke-induced infiltration of neutrophils into the airways through nuclear factor-kappaB activation and IL-8 mRNA expression in guinea pigs in vivo. Am. J. Respir. Cell Mol. Biol. 1999, 20, 189–198. [Google Scholar] [CrossRef]
- Barnes, P.J. Alveolar macrophages in chronic obstructive pulmonary disease (COPD). Cell. Mol. Biol. 2004, 50, OL627-37. [Google Scholar]
- Reynolds, H.Y. Bronchoalveolar lavage. Am. Rev. Respir. Dis. 1987, 135, 250–263. [Google Scholar] [CrossRef]
- Martin, R.R. Cigarette smoking and human pulmonary macrophages. Hosp. Pract. 1977, 12, 97–104. [Google Scholar] [CrossRef]
- Skold, C.M.; Lundahl, J.; Hallden, G.; Hallgren, M.; Eklund, A. Chronic smoke exposure alters the phenotype pattern and the metabolic response in human alveolar macrophages. Clin. Exp. Immunol. 1996, 106, 108–113. [Google Scholar] [CrossRef]
- Kirkham, P.A.; Spooner, G.; Rahman, I.; Rossi, A.G. Macrophage phagocytosis of apoptotic neutrophils is compromised by matrix proteins modified by cigarette smoke and lipid peroxidation products. Biochem. Biophys. Res. Commun. 2004, 318, 32–37. [Google Scholar] [CrossRef]
- Green, C.R.; Rodgman, A. The Tobacco Chemists’ Research Conference: A half century forum for advances in analytical methodology of tobacco and its products. Recent Adv. Tob. Sci. 1996, 22, 131–304. [Google Scholar]
- Smith, C.J.; Fischer, T.H. Particulate and vapor phase constituents of cigarette mainstream smoke and risk of myocardial infarction. Atherosclerosis 2001, 158, 257–267. [Google Scholar] [CrossRef]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans; World Health Organization; International Agency for Research on Cancer. Tobacco Smoke and Involuntary Smoking; Iarc: Lyon, France, 2004; Volume 83. [Google Scholar]
- Matsunaga, K.; Klein, T.W.; Friedman, H.; Yamamoto, Y. Involvement of nicotinic acetylcholine receptors in suppression of antimicrobial activity and cytokine responses of alveolar macrophages to Legionella pneumophila infection by nicotine. J. Immunol. 2001, 167, 6518–6524. [Google Scholar] [CrossRef]
- Harris, J.O.; Swenson, E.W.; Johnson, J.E., 3rd. Human alveolar macrophages: Comparison of phagocytic ability, glucose utilization, and ultrastructure in smokers and nonsmokers. J. Clin. Investig. 1970, 49, 2086–2096. [Google Scholar] [CrossRef] [PubMed]
- Pratt, S.A.; Smith, M.H.; Ladman, A.J.; Finley, T.N. The ultrastructure of alveolar macrophages from human cigarette smokers and nonsmokers. Lab. Investig. J. Tech. Methods Pathol. 1971, 24, 331–338. [Google Scholar]
- Skold, C.M.; Eklund, A.; Hed, J.; Hernbrand, R. Endocytosis of cigarette-smoke condensate by rabbit alveolar macrophages in vitro measured as fluorescence intensity. Eur. Respir. J. 1992, 5, 53–58. [Google Scholar]
- Dougherty, G.J.; Hogg, N. The role of monocyte lymphocyte function-associated antigen 1 (LFA-1) in accessory cell function. Eur. J. Immunol. 1987, 17, 943–947. [Google Scholar] [CrossRef]
- Ortega, E.; Hueso, F.; Collazos, M.E.; Pedrera, M.I.; Barriga, C.; Rodriguez, A.B. Phagocytosis of latex beads by alveolar macrophages from mice exposed to cigarette smoke. Comp. Immunol. Microbiol. Infect. Dis. 1992, 15, 137–142. [Google Scholar] [CrossRef]
- Green, G.M. Mechanisms of tobacco smoke toxicity on pulmonary macrophage cells. Eur. J. Respir. Dis. Suppl. 1985, 139, 82–85. [Google Scholar] [PubMed]
- King, T.E., Jr.; Savici, D.; Campbell, P.A. Phagocytosis and killing of Listeria monocytogenes by alveolar macrophages: Smokers versus nonsmokers. J. Infect. Dis. 1988, 158, 1309–1316. [Google Scholar] [CrossRef]
- Harris, J.O.; Gonzalez-Rothi, R.J. Abnormal phagolysosome fusion in pulmonary alveolar macrophages of rats exposed chronically to cigarette smoke. Am. Rev. Respir. Dis. 1984, 130, 467–471. [Google Scholar] [CrossRef]
- Heguy, A.; O’Connor, T.P.; Luettich, K.; Worgall, S.; Cieciuch, A.; Harvey, B.G.; Hackett, N.R.; Crystal, R.G. Gene expression profiling of human alveolar macrophages of phenotypically normal smokers and nonsmokers reveals a previously unrecognized subset of genes modulated by cigarette smoking. J. Mol. Med. 2006, 84, 318–328. [Google Scholar] [CrossRef]
- Silvestre-Roig, C.; Fridlender, Z.G.; Glogauer, M.; Scapini, P. Neutrophil Diversity in Health and Disease. Trends Immunol. 2019, 40, 565–583. [Google Scholar] [CrossRef]
- Ley, K.; Hoffman, H.M.; Kubes, P.; Cassatella, M.A.; Zychlinsky, A.; Hedrick, C.C.; Catz, S.D. Neutrophils: New insights and open questions. Sci. Immunol. 2018, 3, eaat4579. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.G.; Ostuni, R.; Hidalgo, A. Heterogeneity of neutrophils. Nat. Rev. Immunol. 2019, 19, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Hampton, H.R.; Bailey, J.; Tomura, M.; Brink, R.; Chtanova, T. Microbe-dependent lymphatic migration of neutrophils modulates lymphocyte proliferation in lymph nodes. Nat. Commun. 2015, 6, 7139. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Zychlinsky, A. Beneficial suicide: Why neutrophils die to make NETs. Nat. Rev. Microbiol. 2007, 5, 577–582. [Google Scholar] [CrossRef]
- Carmona-Rivera, C.; Kaplan, M.J. Low-density granulocytes: A distinct class of neutrophils in systemic autoimmunity. Semin. Immunopathol. 2013, 35, 455–463. [Google Scholar] [CrossRef]
- Bagaitkar, J.; Demuth, D.R.; Scott, D.A. Tobacco use increases susceptibility to bacterial infection. Tob. Induc. Dis. 2008, 4, 12. [Google Scholar] [CrossRef]
- O’Donnell, R.A.; Peebles, C.; Ward, J.A.; Daraker, A.; Angco, G.; Broberg, P.; Pierrou, S.; Lund, J.; Holgate, S.T.; Davies, D.E.; et al. Relationship between peripheral airway dysfunction, airway obstruction, and neutrophilic inflammation in COPD. Thorax 2004, 59, 837–842. [Google Scholar] [CrossRef]
- Zhang, Y.; Geng, S.; Prasad, G.L.; Li, L. Suppression of Neutrophil Antimicrobial Functions by Total Particulate Matter from Cigarette Smoke. Front. Immunol. 2018, 9, 2274. [Google Scholar] [CrossRef]
- Petrovski, G.; Zahuczky, G.; Katona, K.; Vereb, G.; Martinet, W.; Nemes, Z.; Bursch, W.; Fesus, L. Clearance of dying autophagic cells of different origin by professional and non-professional phagocytes. Cell Death Differ. 2007, 14, 1117–1128. [Google Scholar] [CrossRef]
- Guzik, K.; Skret, J.; Smagur, J.; Bzowska, M.; Gajkowska, B.; Scott, D.A.; Potempa, J.S. Cigarette smoke-exposed neutrophils die unconventionally but are rapidly phagocytosed by macrophages. Cell Death Dis. 2011, 2, e131. [Google Scholar] [CrossRef]
- Palmer, R.M.; Wilson, R.F.; Hasan, A.S.; Scott, D.A. Mechanisms of action of environmental factors—Obacco smoking. J. Clin. Periodontol. 2005, 32, 180–195. [Google Scholar] [CrossRef]
- Ryder, M.I.; Fujitaki, R.; Johnson, G.; Hyun, W. Alterations of neutrophil oxidative burst by in vitro smoke exposure: Implications for oral and systemic diseases. Ann. Periodontol. 1998, 3, 76–87. [Google Scholar] [CrossRef]
- Sorensen, L.T.; Nielsen, H.B.; Kharazmi, A.; Gottrup, F. Effect of smoking and abstention on oxidative burst and reactivity of neutrophils and monocytes. Surgery 2004, 136, 1047–1053. [Google Scholar] [CrossRef]
- Fredriksson, M.I.; Figueredo, C.M.; Gustafsson, A.; Bergstrom, K.G.; Asman, B.E. Effect of periodontitis and smoking on blood leukocytes and acute-phase proteins. J. Periodontol. 1999, 70, 1355–1360. [Google Scholar] [CrossRef]
- Gustafsson, A.; Asman, B.; Bergstrom, K. Cigarette smoking as an aggravating factor in inflammatory tissue-destructive diseases. Increase in tumor necrosis Factor-alpha priming of peripheral neutrophils measured as generation of oxygen radicals. Int. J. Clin. Lab. Res. 2000, 30, 187–190. [Google Scholar] [CrossRef]
- Steinberg, B.E.; Grinstein, S. Unconventional roles of the NADPH oxidase: Signaling, ion homeostasis, and cell death. Sci. STKE Signal Transduct. Knowl. Environ. 2007, 2007, pe11. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Kaplan, M.J.; Radic, M. Neutrophil extracellular traps: Double-edged swords of innate immunity. J. Immunol. 2012, 189, 2689–2695. [Google Scholar] [CrossRef]
- White, P.C.; Hirschfeld, J.; Milward, M.R.; Cooper, P.R.; Wright, H.J.; Matthews, J.B.; Chapple, I.L.C. Cigarette smoke modifies neutrophil chemotaxis, neutrophil extracellular trap formation and inflammatory response-related gene expression. J. Periodontal Res. 2018, 53, 525–535. [Google Scholar] [CrossRef]
- Qiu, S.L.; Zhang, H.; Tang, Q.Y.; Bai, J.; He, Z.Y.; Zhang, J.Q.; Li, M.H.; Deng, J.M.; Liu, G.N.; Zhong, X.N. Neutrophil extracellular traps induced by cigarette smoke activate plasmacytoid dendritic cells. Thorax 2017, 72, 1084–1093. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, H.; Qi, W.; Zhang, Y.; Li, J.; Li, Z.; Lin, Y.; Bai, X.; Liu, X.; Chen, X.; et al. Nicotine promotes atherosclerosis via ROS-NLRP3-mediated endothelial cell pyroptosis. Cell Death Dis. 2018, 9, 171. [Google Scholar] [CrossRef]
- Avanzas, P.; Arroyo-Espliguero, R.; Cosín-Sales, J.; Quiles, J.; Zouridakis, E.; Kaski, J.C. Multiple complex stenoses, high neutrophil count and C-reactive protein levels in patients with chronic stable angina. Atherosclerosis 2004, 175, 151–157. [Google Scholar] [CrossRef]
- Dinerman, J.L.; Mehta, J.L.; Saldeen, T.G.; Emerson, S.; Wallin, R.; Davda, R.; Davidson, A. Increased neutrophil elastase release in unstable angina pectoris and acute myocardial infarction. J. Am. Coll. Cardiol. 1990, 15, 1559–1563. [Google Scholar] [CrossRef]
- Li, D.; Yu, J.; Zeng, R.; Zhao, L.; Wan, Z.; Zeng, Z.; Cao, Y. Neutrophil Count Is Associated With Risks of Cardiovascular Diseases. J. Am. Coll. Cardiol. 2017, 70, 911–912. [Google Scholar] [CrossRef]
- Shah, A.D.; Denaxas, S.; Nicholas, O.; Hingorani, A.D.; Hemingway, H. Neutrophil Counts and Initial Presentation of 12 Cardiovascular Diseases: A CALIBER Cohort Study. J. Am. Coll. Cardiol. 2017, 69, 1160–1169. [Google Scholar] [CrossRef]
- Yalcin, M.; Aparci, M.; Uz, O.; Isilak, Z.; Balta, S.; Dogan, M.; Kardesoglu, E.; Uzun, M. Neutrophil-lymphocyte ratio may predict left atrial thrombus in patients with nonvalvular atrial fibrillation. Clin. Appl. Thromb. Hemost. 2015, 21, 166–171. [Google Scholar] [CrossRef]
- Koza, Y. What is the clinical benefit of neutrophil-lymphocyte ratio in cardiovascular patients? J. Cardiovasc. Thorac. Res. 2014, 6, 131–132. [Google Scholar] [CrossRef] [PubMed]
- Kraakman, M.J.; Lee, M.K.; Al-Sharea, A.; Dragoljevic, D.; Barrett, T.J.; Montenont, E.; Basu, D.; Heywood, S.; Kammoun, H.L.; Flynn, M.; et al. Neutrophil-derived S100 calcium-binding proteins A8/A9 promote reticulated thrombocytosis and atherogenesis in diabetes. J. Clin. Investig. 2017, 127, 2133–2147. [Google Scholar] [CrossRef] [PubMed]
- Papanicolaou, D.A.; Wilder, R.L.; Manolagas, S.C.; Chrousos, G.P. The pathophysiologic roles of interleukin-6 in human disease. Ann. Intern. Med. 1998, 128, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Fanola, C.L.; Morrow, D.A.; Cannon, C.P.; Jarolim, P.; Lukas, M.A.; Bode, C.; Hochman, J.S.; Goodrich, E.L.; Braunwald, E.; O’Donoghue, M.L. Interleukin-6 and the Risk of Adverse Outcomes in Patients After an Acute Coronary Syndrome: Observations From the SOLID-TIMI 52 (Stabilization of Plaque Using Darapladib-Thrombolysis in Myocardial Infarction 52) Trial. J. Am. Heart Assoc. 2017, 6, e005637. [Google Scholar] [CrossRef]
- Harris, T.B.; Ferrucci, L.; Tracy, R.P.; Corti, M.C.; Wacholder, S.; Ettinger, W.H., Jr.; Heimovitz, H.; Cohen, H.J.; Wallace, R. Associations of elevated interleukin-6 and C-reactive protein levels with mortality in the elderly. Am. J. Med. 1999, 106, 506–512. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D., Jr.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef]
- Stakos, D.A.; Kambas, K.; Konstantinidis, T.; Mitroulis, I.; Apostolidou, E.; Arelaki, S.; Tsironidou, V.; Giatromanolaki, A.; Skendros, P.; Konstantinides, S.; et al. Expression of functional tissue factor by neutrophil extracellular traps in culprit artery of acute myocardial infarction. Eur. Heart J. 2015, 36, 1405–1414. [Google Scholar] [CrossRef]
- Helseth, R.; Knudsen, E.C.; Eritsland, J.; Opstad, T.B.; Arnesen, H.; Andersen, G.; Seljeflot, I. Glucose associated NETosis in patients with ST-elevation myocardial infarction: An observational study. BMC Cardiovasc. Disord. 2019, 19, 221. [Google Scholar] [CrossRef]
- Liberale, L.; Holy, E.W.; Akhmedov, A.; Bonetti, N.R.; Nietlispach, F.; Matter, C.M.; Mach, F.; Montecucco, F.; Beer, J.H.; Paneni, F.; et al. Interleukin-1β Mediates Arterial Thrombus Formation via NET-Associated Tissue Factor. J. Clin. Med. 2019, 8, 72. [Google Scholar] [CrossRef]
- Liu, J.; Yang, D.; Wang, X.; Zhu, Z.; Wang, T.; Ma, A.; Liu, P. Neutrophil extracellular traps and dsDNA predict outcomes among patients with ST-elevation myocardial infarction. Sci. Rep. 2019, 9, 11599. [Google Scholar] [CrossRef]
- Mangold, A.; Hofbauer, T.M.; Ondracek, A.S.; Artner, T.; Scherz, T.; Speidl, W.S.; Krychtiuk, K.A.; Sadushi-Kolici, R.; Jakowitsch, J.; Lang, I.M. Neutrophil extracellular traps and monocyte subsets at the culprit lesion site of myocardial infarction patients. Sci. Rep. 2019, 9, 16304. [Google Scholar] [CrossRef]
- Fernández-Ruiz, I. Neutrophil-driven SMC death destabilizes atherosclerotic plaques. Nat. Rev. Cardiol. 2019, 16, 455. [Google Scholar] [CrossRef]
- Gomez, I.; Ward, B.; Souilhol, C.; Recarti, C.; Ariaans, M.; Johnston, J.; Burnett, A.; Mahmoud, M.; Luong, L.A.; West, L.; et al. Neutrophil microvesicles drive atherosclerosis by delivering miR-155 to atheroprone endothelium. Nat. Commun. 2020, 11, 214. [Google Scholar] [CrossRef]
- Ionita, M.G.; van den Borne, P.; Catanzariti, L.M.; Moll, F.L.; de Vries, J.P.; Pasterkamp, G.; Vink, A.; de Kleijn, D.P. High neutrophil numbers in human carotid atherosclerotic plaques are associated with characteristics of rupture-prone lesions. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1842–1848. [Google Scholar] [CrossRef]
- Libby, P.; Pasterkamp, G.; Crea, F.; Jang, I.K. Reassessing the Mechanisms of Acute Coronary Syndromes. Circ. Res. 2019, 124, 150–160. [Google Scholar] [CrossRef]
- Silvestre-Roig, C.; Braster, Q.; Wichapong, K.; Lee, E.Y.; Teulon, J.M.; Berrebeh, N.; Winter, J.; Adrover, J.M.; Santos, G.S.; Froese, A.; et al. Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature 2019, 569, 236–240. [Google Scholar] [CrossRef]
- Drechsler, M.; Megens, R.T.; van Zandvoort, M.; Weber, C.; Soehnlein, O. Hyperlipidemia-triggered neutrophilia promotes early atherosclerosis. Circulation 2010, 122, 1837–1845. [Google Scholar] [CrossRef]
- Silvestre-Roig, C.; Braster, Q.; Ortega-Gomez, A.; Soehnlein, O. Neutrophils as regulators of cardiovascular inflammation. Nat. Rev. Cardiol. 2020, 17, 327–340. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dahdah, A.; Jaggers, R.M.; Sreejit, G.; Johnson, J.; Kanuri, B.; Murphy, A.J.; Nagareddy, P.R. Immunological Insights into Cigarette Smoking-Induced Cardiovascular Disease Risk. Cells 2022, 11, 3190. https://doi.org/10.3390/cells11203190
Dahdah A, Jaggers RM, Sreejit G, Johnson J, Kanuri B, Murphy AJ, Nagareddy PR. Immunological Insights into Cigarette Smoking-Induced Cardiovascular Disease Risk. Cells. 2022; 11(20):3190. https://doi.org/10.3390/cells11203190
Chicago/Turabian StyleDahdah, Albert, Robert M. Jaggers, Gopalkrishna Sreejit, Jillian Johnson, Babunageswararao Kanuri, Andrew J. Murphy, and Prabhakara R. Nagareddy. 2022. "Immunological Insights into Cigarette Smoking-Induced Cardiovascular Disease Risk" Cells 11, no. 20: 3190. https://doi.org/10.3390/cells11203190
APA StyleDahdah, A., Jaggers, R. M., Sreejit, G., Johnson, J., Kanuri, B., Murphy, A. J., & Nagareddy, P. R. (2022). Immunological Insights into Cigarette Smoking-Induced Cardiovascular Disease Risk. Cells, 11(20), 3190. https://doi.org/10.3390/cells11203190