
ATPase Thorase Deficiency Causes α-Synucleinopathy and Parkinson’s Disease-like Behavior

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mouse Handing
2.2. Mouse Genotyping
2.3. Primary Hippocampal Neuron Culture
2.4. Preparation of Human a-syn-A53T PFFs
2.5. Immunoblot Analysis
2.6. Co-Immunoprecipitation Assay
2.7. Ubiquitination Assay
2.8. Immunohistochemistry
2.9. Immunofluorescence Staining
2.10. Behavioral Measurements
2.10.1. Open Field Activity
2.10.2. Grip Strength
2.10.3. Rotarod Test
2.10.4. Footprint Test
2.11. Antibodies, Reagents, and Plasmids
2.12. Statistical Analysis
3. Results
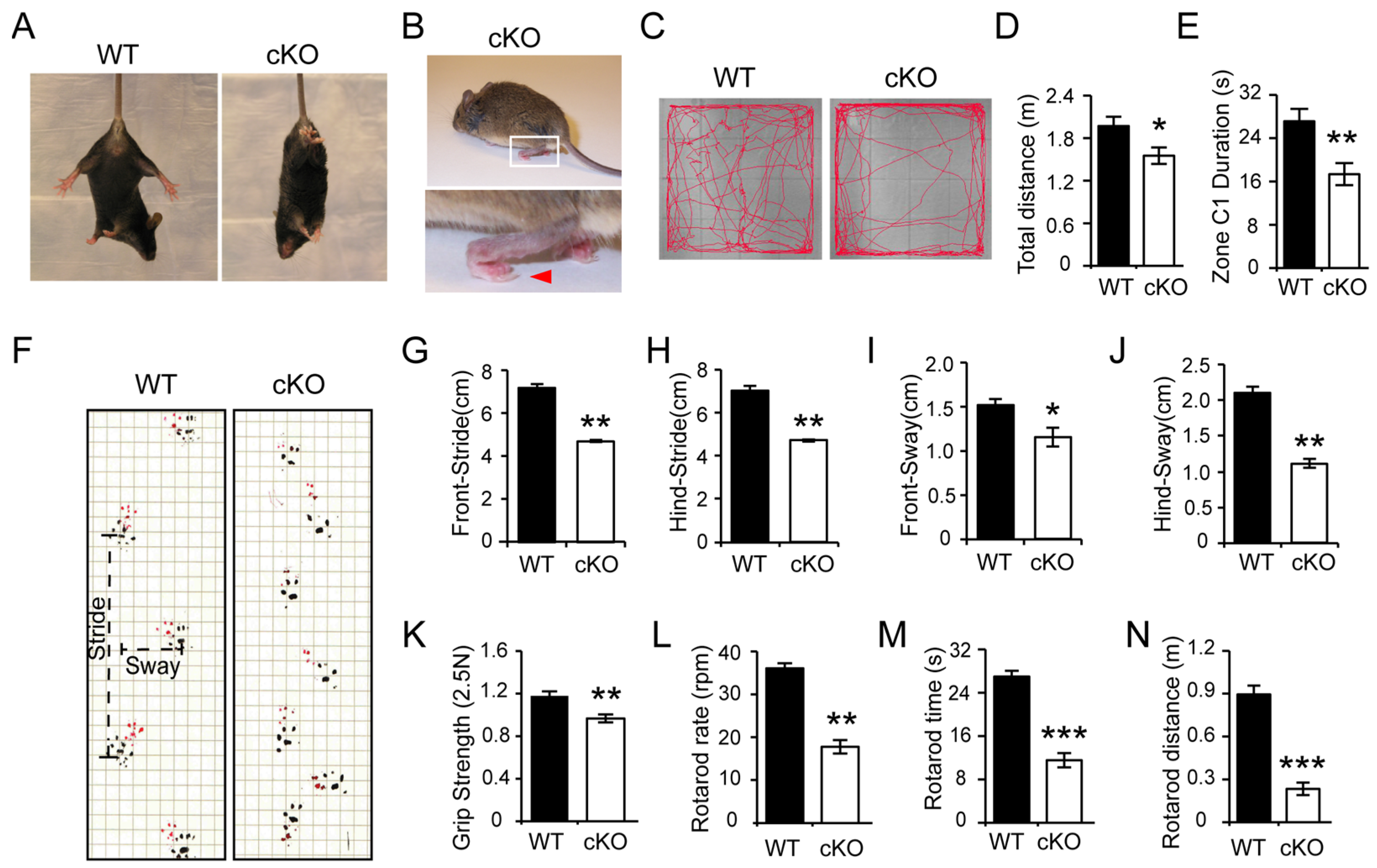
3.1. Thorase Conditional Knockout (cKO) Mice Exhibits Motor Dysfunction Behavior
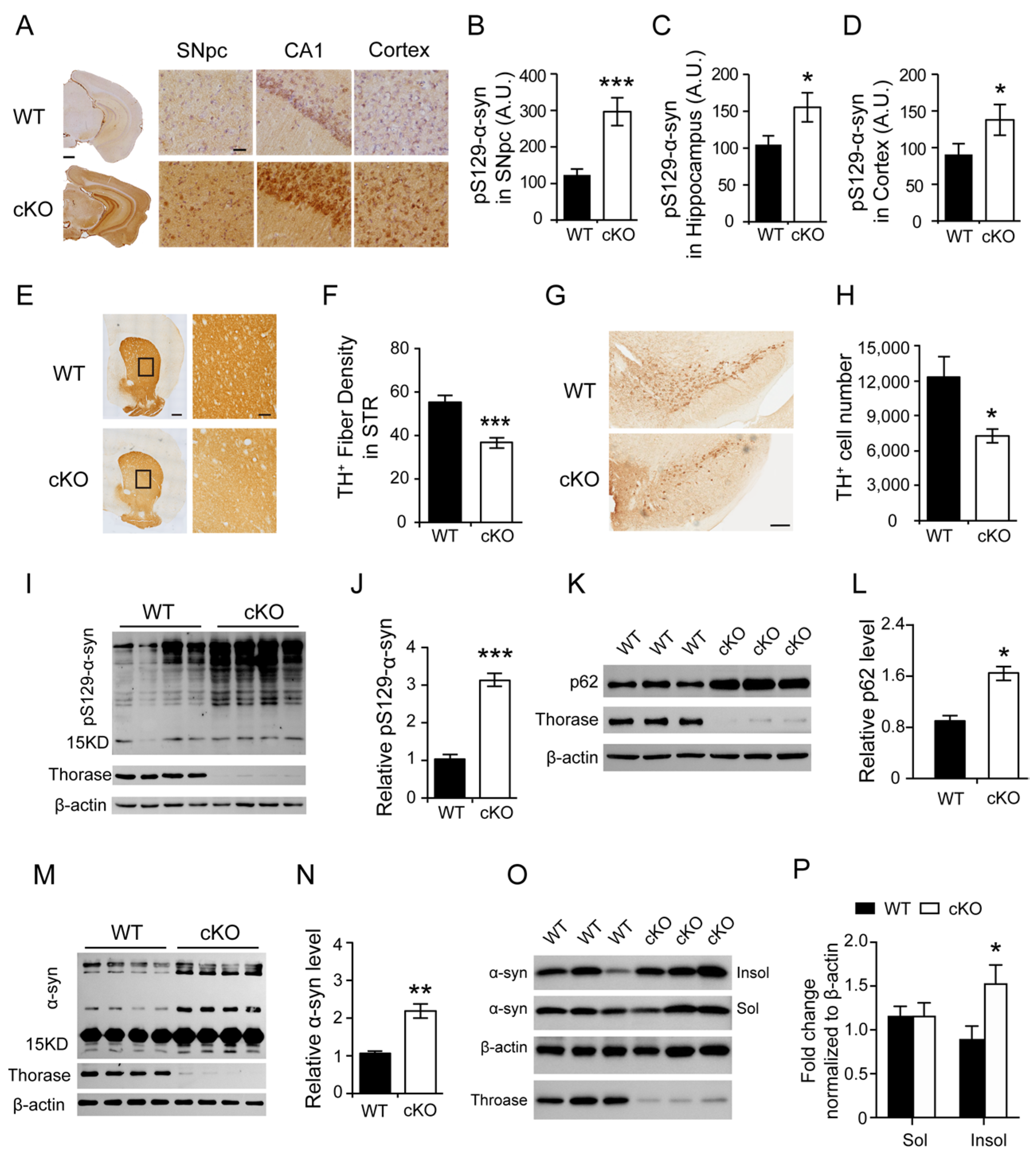
3.2. Thorase Deficiency Results in Extensive α-Synucleinopathy and Reduced TH+ Dopaminergic Neurons
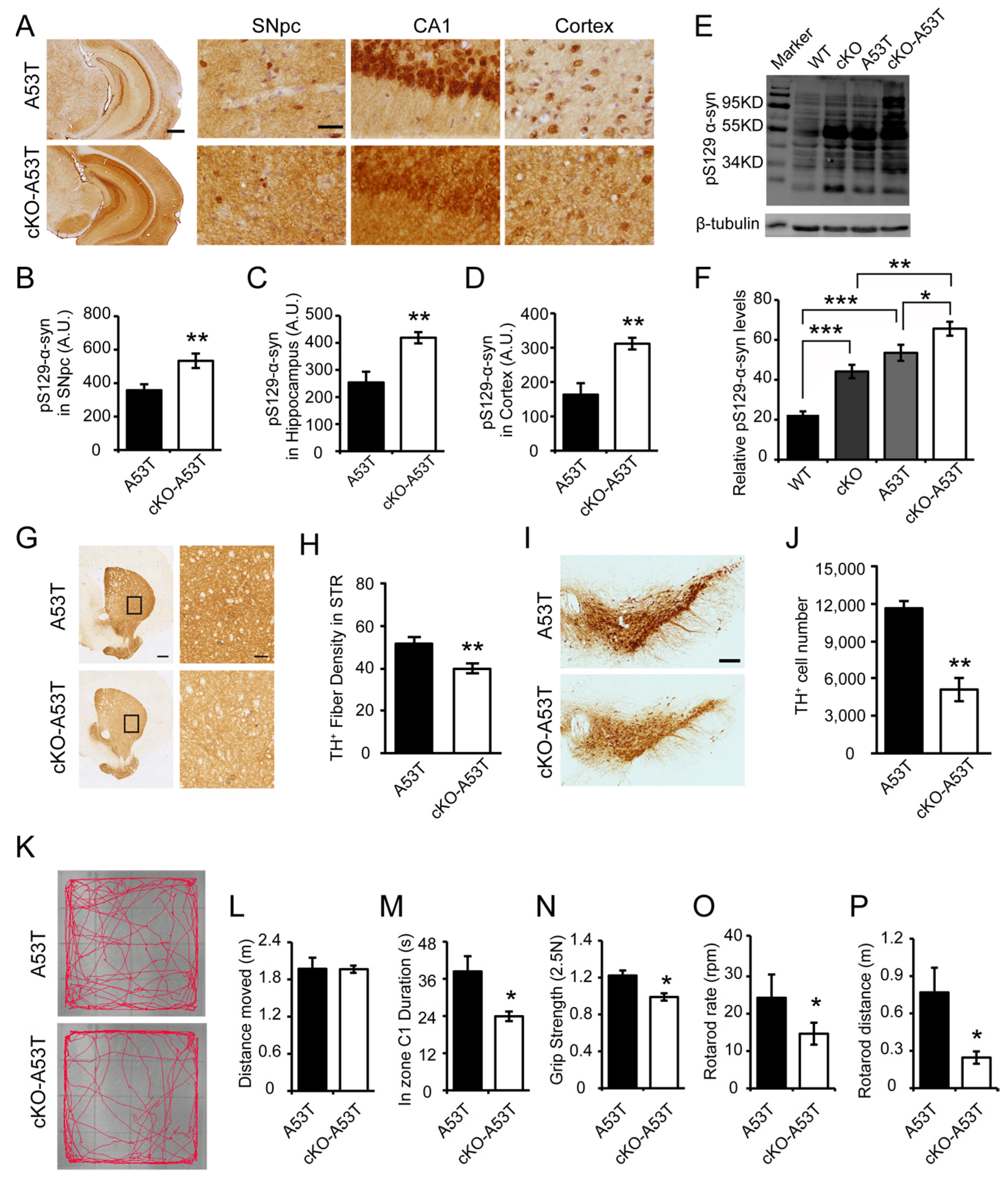
3.3. Thorase Deficiency Accelerates α-Synucleinopathy and Behavioral Impairments in a Familial PD A53T Mouse Model
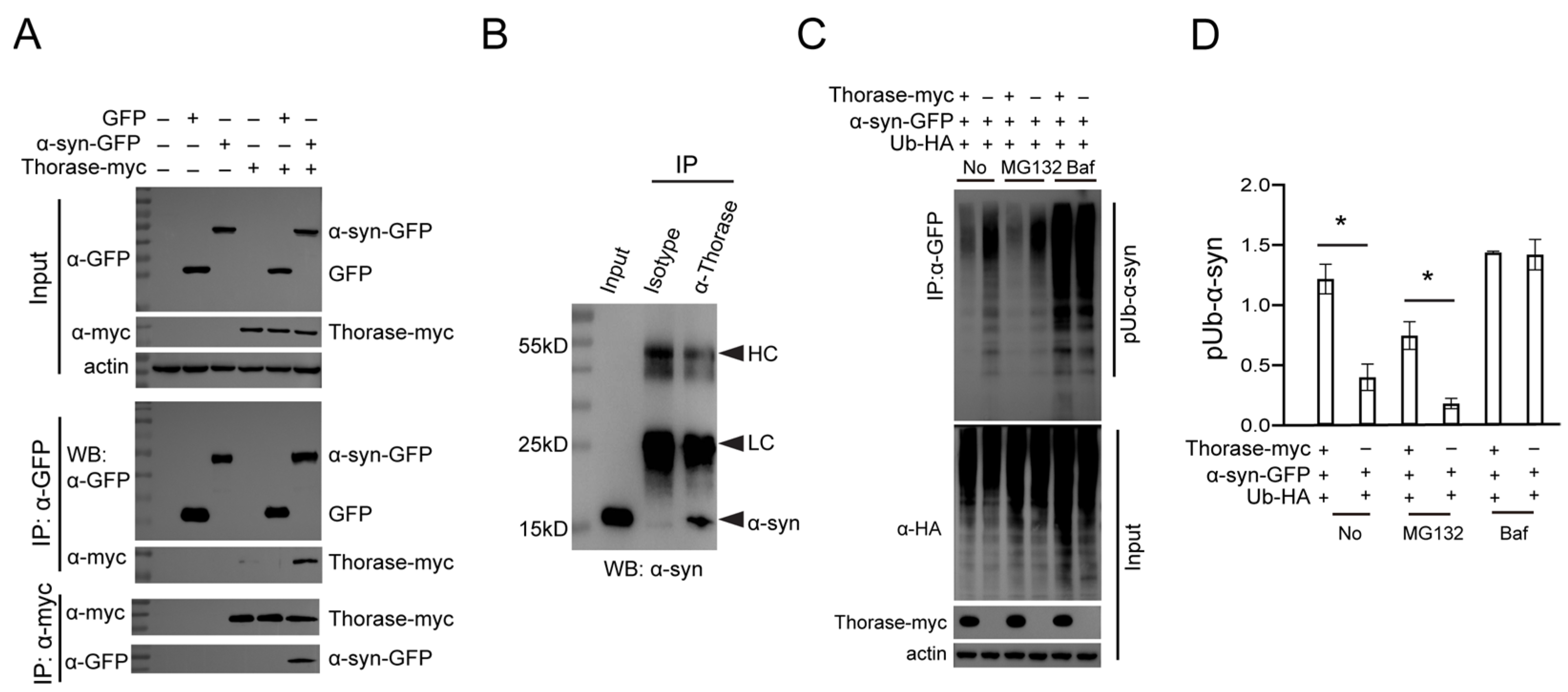
3.4. Thorase Interacts with α-syn and Regulates the Degradation of Ubiquitinated α-syn
3.5. Thorase Deficiency Promotes α-syn Aggregation in Primary Cultured Neurons
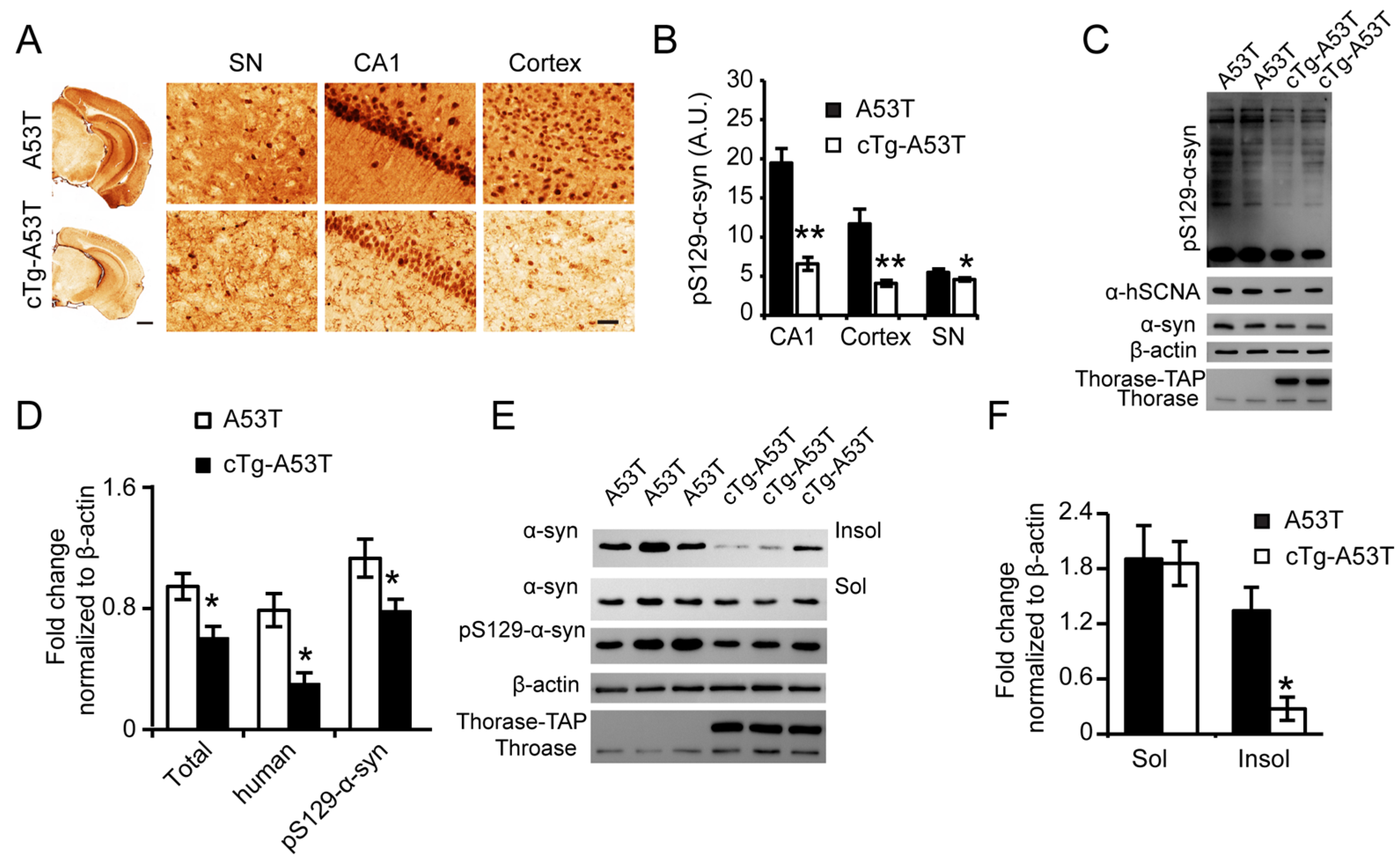
3.6. Thorase Overexpression Prevents α-Synucleinopathy in PD Mouse Model A53T Mice
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ondaro, J.; Hernandez-Eguiazu, H.; Garciandia-Arcelus, M.; Loera-Valencia, R.; Rodriguez-Gómez, L.; Jiménez-Zúñiga, A.; Goikolea, J.; Rodriguez-Rodriguez, P.; Ruiz-Martinez, J.; Moreno, F.; et al. Defects of Nutrient Signaling and Autophagy in Neurodegeneration. Front. Cell Dev. Biol. 2022, 10, 836196. [Google Scholar] [CrossRef] [PubMed]
- Leitão, A.D.G.; Rudolffi-Soto, P.; Chappard, A.; Bhumkar, A.; Lau, D.; Hunter, D.J.B.; Gambin, Y.; Sierecki, E. Selectivity of Lewy Body Protein Interactions along the Aggregation Pathway of α-Synuclein. Commun. Biol. 2021, 4, 1124. [Google Scholar] [CrossRef] [PubMed]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the Alpha-Synuclein Gene Identified in Families with Parkinson’s Disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Selvaraj, S.; Piramanayagam, S. Impact of Gene Mutation in the Development of Parkinson’s Disease. Genes Dis. 2019, 6, 120–128. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Y.; Chi, Z.; Keuss, M.J.; Pai, Y.-M.E.; Kang, H.C.; Shin, J.-H.; Bugayenko, A.; Wang, H.; Xiong, Y.; et al. The AAA+ ATPase Thorase Regulates AMPA Receptor-Dependent Synaptic Plasticity and Behavior. Cell 2011, 145, 284–299. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, J.; Wang, H.; Sherbini, O.; Keuss, M.J.; Umanah, G.K.; Pai, E.L.-L.; Chi, Z.; Paldanius, K.M.; He, W.; et al. The AAA + ATPase Thorase Is Neuroprotective against Ischemic Injury. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2019, 39, 1836–1848. [Google Scholar] [CrossRef]
- Pignatelli, M.; Umanah, G.K.E.; Ribeiro, S.P.; Chen, R.; Karuppagounder, S.S.; Yau, H.-J.; Eacker, S.; Dawson, V.L.; Dawson, T.M.; Bonci, A. Synaptic Plasticity onto Dopamine Neurons Shapes Fear Learning. Neuron 2017, 93, 425–440. [Google Scholar] [CrossRef]
- Umanah, G.K.E.; Pignatelli, M.; Yin, X.; Chen, R.; Crawford, J.; Neifert, S.; Scarffe, L.; Behensky, A.A.; Guiberson, N.; Chang, M.; et al. Thorase Variants Are Associated with Defects in Glutamatergic Neurotransmission That Can Be Rescued by Perampanel. Sci. Transl. Med. 2017, 9, eaah4985. [Google Scholar] [CrossRef]
- Bunod, R.; Doummar, D.; Whalen, S.; Keren, B.; Chantot-Bastaraud, S.; Maincent, K.; Villy, M.-C.; Mayer, M.; Rodriguez, D.; Burglen, L.; et al. Congenital Immobility and Stiffness Related to Biallelic ATAD1 Variants. Neurol. Genet. 2020, 6, e520. [Google Scholar] [CrossRef]
- Piard, J.; Umanah, G.K.E.; Harms, F.L.; Abalde-Atristain, L.; Amram, D.; Chang, M.; Chen, R.; Alawi, M.; Salpietro, V.; Rees, M.I.; et al. A Homozygous ATAD1 Mutation Impairs Postsynaptic AMPA Receptor Trafficking and Causes a Lethal Encephalopathy. Brain J. Neurol. 2018, 141, 651–661. [Google Scholar] [CrossRef]
- Ahrens-Nicklas, R.C.; Umanah, G.K.E.; Sondheimer, N.; Deardorff, M.A.; Wilkens, A.B.; Conlin, L.K.; Santani, A.B.; Nesbitt, A.; Juulsola, J.; Ma, E.; et al. Precision Therapy for a New Disorder of AMPA Receptor Recycling Due to Mutations in ATAD1. Neurol. Genet. 2017, 3, e130. [Google Scholar] [CrossRef] [PubMed]
- Casanova, E.; Fehsenfeld, S.; Mantamadiotis, T.; Lemberger, T.; Greiner, E.; Stewart, A.F.; Schütz, G. A CamKIIalpha ICre BAC Allows Brain-Specific Gene Inactivation. Genes. N. Y. N 2000 2001, 31, 37–42. [Google Scholar] [CrossRef]
- Lee, M.K.; Stirling, W.; Xu, Y.; Xu, X.; Qui, D.; Mandir, A.S.; Dawson, T.M.; Copeland, N.G.; Jenkins, N.A.; Price, D.L. Human Alpha-Synuclein-Harboring Familial Parkinson’s Disease-Linked Ala-53 --> Thr Mutation Causes Neurodegenerative Disease with Alpha-Synuclein Aggregation in Transgenic Mice. Proc. Natl. Acad. Sci. USA 2002, 99, 8968–8973. [Google Scholar] [CrossRef] [PubMed]
- Beaudoin, G.M.J.; Lee, S.-H.; Singh, D.; Yuan, Y.; Ng, Y.-G.; Reichardt, L.F.; Arikkath, J. Culturing Pyramidal Neurons from the Early Postnatal Mouse Hippocampus and Cortex. Nat. Protoc. 2012, 7, 1741–1754. [Google Scholar] [CrossRef]
- Volpicelli-Daley, L.A.; Luk, K.C.; Lee, V.M.-Y. Addition of Exogenous α-Synuclein Preformed Fibrils to Primary Neuronal Cultures to Seed Recruitment of Endogenous α-Synuclein to Lewy Body and Lewy Neurite-like Aggregates. Nat. Protoc. 2014, 9, 2135–2146. [Google Scholar] [CrossRef]
- Ejlerskov, P.; Hultberg, J.G.; Wang, J.; Carlsson, R.; Ambjørn, M.; Kuss, M.; Liu, Y.; Porcu, G.; Kolkova, K.; Friis Rundsten, C.; et al. Lack of Neuronal IFN-β-IFNAR Causes Lewy Body- and Parkinson’s Disease-like Dementia. Cell 2015, 163, 324–339. [Google Scholar] [CrossRef]
- Kraeuter, A.-K.; Guest, P.C.; Sarnyai, Z. The Open Field Test for Measuring Locomotor Activity and Anxiety-Like Behavior. Methods Mol. Biol. Clifton NJ 2019, 1916, 99–103. [Google Scholar] [CrossRef]
- Seibenhener, M.L.; Wooten, M.C. Use of the Open Field Maze to Measure Locomotor and Anxiety-like Behavior in Mice. J. Vis. Exp. JoVE 2015, e52434. [Google Scholar] [CrossRef]
- Shiotsuki, H.; Yoshimi, K.; Shimo, Y.; Funayama, M.; Takamatsu, Y.; Ikeda, K.; Takahashi, R.; Kitazawa, S.; Hattori, N. A Rotarod Test for Evaluation of Motor Skill Learning. J. Neurosci. Methods 2010, 189, 180–185. [Google Scholar] [CrossRef]
- Deacon, R.M.J. Measuring Motor Coordination in Mice. J. Vis. Exp. JoVE 2013, e2609. [Google Scholar] [CrossRef]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. α-Synuclein Is Phosphorylated in Synucleinopathy Lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef]
- Anderson, J.P.; Walker, D.E.; Goldstein, J.M.; de Laat, R.; Banducci, K.; Caccavello, R.J.; Barbour, R.; Huang, J.; Kling, K.; Lee, M.; et al. Phosphorylation of Ser-129 Is the Dominant Pathological Modification of Alpha-Synuclein in Familial and Sporadic Lewy Body Disease. J. Biol. Chem. 2006, 281, 29739–29752. [Google Scholar] [CrossRef] [PubMed]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The Many Faces of α-Synuclein: From Structure and Toxicity to Therapeutic Target. Nat. Rev. Neurosci. 2013, 14, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Kuusisto, E.; Kauppinen, T.; Alafuzoff, I. Use of P62/SQSTM1 Antibodies for Neuropathological Diagnosis. Neuropathol. Appl. Neurobiol. 2008, 34, 169–180. [Google Scholar] [CrossRef]
- Martin, L.J.; Pan, Y.; Price, A.C.; Sterling, W.; Copeland, N.G.; Jenkins, N.A.; Price, D.L.; Lee, M.K. Parkinson’s Disease Alpha-Synuclein Transgenic Mice Develop Neuronal Mitochondrial Degeneration and Cell Death. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.C.; Tresse, E.; Kolaitis, R.-M.; Molliex, A.; Thomas, R.E.; Alami, N.H.; Wang, B.; Joshi, A.; Smith, R.B.; Ritson, G.P.; et al. VCP Is Essential for Mitochondrial Quality Control by PINK1/Parkin and This Function Is Impaired by VCP Mutations. Neuron 2013, 78, 65–80. [Google Scholar] [CrossRef]
- Volpicelli-Daley, L.A.; Luk, K.C.; Patel, T.P.; Tanik, S.A.; Riddle, D.M.; Stieber, A.; Meaney, D.F.; Trojanowski, J.Q.; Lee, V.M.-Y. Exogenous α-Synuclein Fibrils Induce Lewy Body Pathology Leading to Synaptic Dysfunction and Neuron Death. Neuron 2011, 72, 57–71. [Google Scholar] [CrossRef]
- Aarsland, D.; Batzu, L.; Halliday, G.M.; Geurtsen, G.J.; Ballard, C.; Ray Chaudhuri, K.; Weintraub, D. Parkinson Disease-Associated Cognitive Impairment. Nat. Rev. Dis. Primer 2021, 7, 47. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-Synuclein in Lewy Bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- White, S.R.; Lauring, B. AAA+ ATPases: Achieving Diversity of Function with Conserved Machinery. Traffic Cph. Den. 2007, 8, 1657–1667. [Google Scholar] [CrossRef]
- Wohlever, M.L.; Mateja, A.; McGilvray, P.T.; Day, K.J.; Keenan, R.J. Msp1 Is a Membrane Protein Dislocase for Tail-Anchored Proteins. Mol. Cell 2017, 67, 194–202.e6. [Google Scholar] [CrossRef] [PubMed]
- Weidberg, H.; Amon, A. MitoCPR-A Surveillance Pathway That Protects Mitochondria in Response to Protein Import Stress. Science 2018, 360, eaan4146. [Google Scholar] [CrossRef]
- Sharma, C.; Kim, S.; Nam, Y.; Jung, U.J.; Kim, S.R. Mitochondrial Dysfunction as a Driver of Cognitive Impairment in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 4850. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Rockenstein, E.; Crews, L.; Masliah, E. Role of Protein Aggregation in Mitochondrial Dysfunction and Neurodegeneration in Alzheimer’s and Parkinson’s Diseases. Neuromolecular Med. 2003, 4, 21–36. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, F.; Zhang, H.; Yang, J.; Cai, M.; Yang, Q.; Wang, H.; Xu, Y.; Chen, H.; Hu, Y.; He, W.; et al. ATPase Thorase Deficiency Causes α-Synucleinopathy and Parkinson’s Disease-like Behavior. Cells 2022, 11, 2990. https://doi.org/10.3390/cells11192990
Gao F, Zhang H, Yang J, Cai M, Yang Q, Wang H, Xu Y, Chen H, Hu Y, He W, et al. ATPase Thorase Deficiency Causes α-Synucleinopathy and Parkinson’s Disease-like Behavior. Cells. 2022; 11(19):2990. https://doi.org/10.3390/cells11192990
Chicago/Turabian StyleGao, Fei, Han Zhang, Jia Yang, Menghua Cai, Qi Yang, Huaishan Wang, Yi Xu, Hui Chen, Yu Hu, Wei He, and et al. 2022. "ATPase Thorase Deficiency Causes α-Synucleinopathy and Parkinson’s Disease-like Behavior" Cells 11, no. 19: 2990. https://doi.org/10.3390/cells11192990
APA StyleGao, F., Zhang, H., Yang, J., Cai, M., Yang, Q., Wang, H., Xu, Y., Chen, H., Hu, Y., He, W., & Zhang, J. (2022). ATPase Thorase Deficiency Causes α-Synucleinopathy and Parkinson’s Disease-like Behavior. Cells, 11(19), 2990. https://doi.org/10.3390/cells11192990