
Neuroinflammation in Dementia—Therapeutic Directions in a COVID-19 Pandemic Setting


Abstract
1. Introduction
2. Tumor Necrosis Factor Triggers Dementia Pathology
3. Mechanisms of Chronic Low-Grade Inflammation
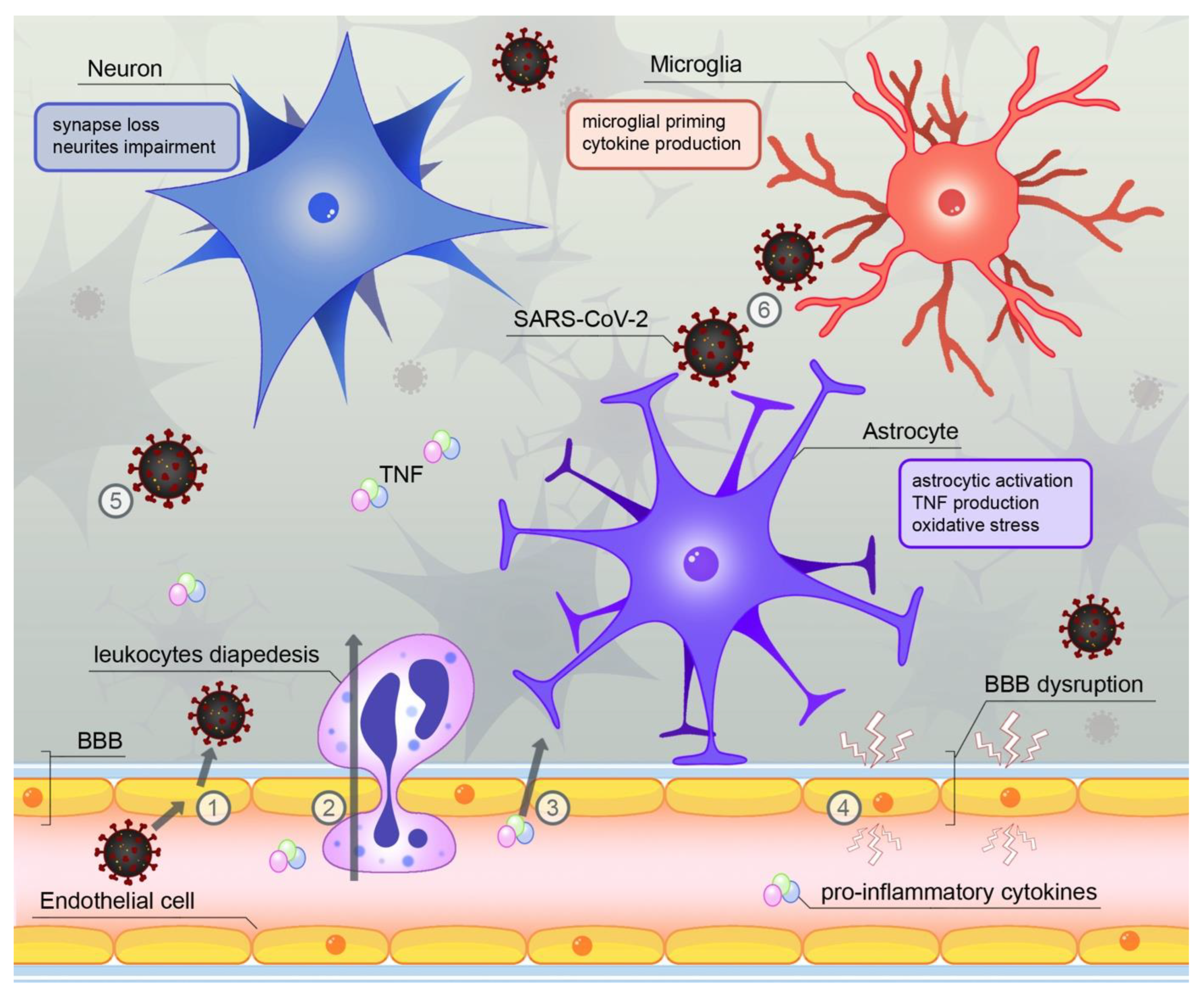
4. SARS-CoV-2—A Novel Source of Neuroinflammation?
5. Glial Involvement in Neuroinflammation
6. Methods for Reduction of Pro-Inflammatory Activation—Critical Appraisal
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ozben, T.; Ozben, S. Neuro-inflammation and anti-inflammatory treatment options for Alzheimer’s disease. Clin. Biochem. 2019, 72, 87–89. [Google Scholar] [CrossRef] [PubMed]
- Irwin, M.R.; Vitiello, M.V. Implications of sleep disturbance and inflammation for Alzheimer’s disease dementia. Lancet Neurol. 2019, 18, 296–306. [Google Scholar] [CrossRef]
- Rosenberg, G.A. Extracellular matrix inflammation in vascular cognitive impairment and dementia. Clin. Sci. 2017, 131, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.X.; Zhang, B.; Xia, R.; Jia, Q.Y. Inflammation, apoptosis and autophagy as critical players in vascular dementia. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 9601–9614. [Google Scholar] [PubMed]
- Zhang, L.Y.; Pan, J.; Mamtilahun, M.; Zhu, Y.; Wang, L.; Venkatesh, A.; Shi, R.; Tu, X.; Jin, K.; Wang, Y.; et al. Microglia exacerbate white matter injury via complement C3/C3aR pathway after hypoperfusion. Theranostics 2020, 10, 74–90. [Google Scholar] [CrossRef] [PubMed]
- Frantellizzi, V.; Pani, A.; Ricci, M.; Locuratolo, N.; Fattapposta, F.; De Vincentis, G. Neuroimaging in Vascular Cognitive Impairment and Dementia: A Systematic Review. J. Alzheimer’s Dis. 2020, 73, 1279–1294. [Google Scholar] [CrossRef]
- Sjogren, M. Increased intrathecal inflammatory activity in frontotemporal dementia: Pathophysiological implications. J. Neurol. Neurosurg. Psychiatry 2004, 75, 1107–1111. [Google Scholar] [CrossRef]
- Miller, Z.A.; Rankin, K.P.; Graff-Radford, N.R.; Takada, L.T.; Sturm, V.E.; Cleveland, C.M.; Criswell, L.A.; Jaeger, P.A.; Stan, T.; Heggeli, K.A.; et al. TDP-43 frontotemporal lobar degeneration and autoimmune disease. J. Neurol. Neurosurg. Psychiatry 2013, 84, 956–962. [Google Scholar] [CrossRef]
- Miller, Z.A.; Sturm, V.E.; Camsari, G.B.; Karydas, A.; Yokoyama, J.S.; Grinberg, L.T.; Boxer, A.L.; Rosen, H.J.; Rankin, K.P.; Gorno-Tempini, M.L.; et al. Increased prevalence of autoimmune disease within C9 and FTD/MND cohorts Completing the picture. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e301. [Google Scholar] [CrossRef]
- McCauley, M.E.; Baloh, R.H. Inflammation in ALS/FTD pathogenesis. Acta Neuropathol. 2019, 137, 715–730. [Google Scholar] [CrossRef]
- Hallett, P.J.; Engelender, S.; Isacson, O. Lipid and immune abnormalities causing age-dependent neurodegeneration and Parkinson’s disease. J. Neuroinflamm. 2019, 16, 153. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Chun, Y.; Lee, J.S.; Lee, S.J. Neuroinflammation in Synucleinopathies. Brain Pathol. 2016, 26, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Ho, D.H.; Suk, J.-E.; You, S.; Michael, S.; Kang, J.; Joong Lee, S.; Masliah, E.; Hwang, D.; Lee, H.-J.; et al. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 2013, 4, 1562. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi Rastegar, D.; Hughes, L.P.; Perera, G.; Keshiya, S.; Zhong, S.; Gao, J.; Halliday, G.M.; Schüle, B.; Dzamko, N. Effect of LRRK2 protein and activity on stimulated cytokines in human monocytes and macrophages. NPJ Parkinson’s Dis. 2022, 8, 34. [Google Scholar] [CrossRef]
- Xu, E.; Boddu, R.; Abdelmotilib, H.A.; Sokratian, A.; Kelly, K.; Liu, Z.; Bryant, N.; Chandra, S.; Carlisle, S.M.; Lefkowitz, E.J.; et al. Pathological α-synuclein recruits LRRK2 expressing pro-inflammatory monocytes to the brain. Mol. Neurodegener. 2022, 17, 7. [Google Scholar] [CrossRef]
- Trudler, D.; Sanz-Blasco, S.; Eisele, Y.S.; Ghatak, S.; Bodhinathan, K.; Akhtar, M.W.; Lynch, W.P.; Piña-Crespo, J.C.; Talantova, M.; Kelly, J.W.; et al. α-Synuclein Oligomers Induce Glutamate Release from Astrocytes and Excessive Extrasynaptic NMDAR Activity in Neurons, Thus Contributing to Synapse Loss. J. Neurosci. 2021, 41, 2264–2273. [Google Scholar] [CrossRef]
- Cyprien, F.; Courtet, P.; Maller, J.; Meslin, C.; Ritchie, K.; Ancelin, M.-L.; Artero, S. Increased serum C-reactive protein and corpus callosum alterations in older adults. Aging Dis. 2019, 10, 463–469. [Google Scholar] [CrossRef]
- Si, S.; Li, J.; Tewara, M.A.; Xue, F. Genetically Determined Chronic Low-Grade Inflammation and Hundreds of Health Outcomes in the UK Biobank and the FinnGen Population: A Phenome-Wide Mendelian Randomization Study. Front. Immunol. 2021, 12, 720876. [Google Scholar] [CrossRef]
- Tao, Q.; Ang, T.F.A.; DeCarli, C.; Auerbach, S.H.; Devine, S.; Stein, T.D.; Zhang, X.; Massaro, J.; Au, R.; Qiu, W.Q. Association of Chronic Low-grade Inflammation with Risk of Alzheimer Disease in ApoE4 Carriers. JAMA Netw. Open 2018, 1, e183597. [Google Scholar] [CrossRef]
- Fard, M.T.; Cribb, L.; Nolidin, K.; Savage, K.; Wesnes, K.; Stough, C. Is there a relationship between low-grade systemic inflammation and cognition in healthy people aged 60–75 years? Behav. Brain Res. 2020, 383, 112502. [Google Scholar] [CrossRef]
- Cervellati, C.; Trentini, A.; Bosi, C.; Valacchi, G.; Morieri, M.L.; Zurlo, A.; Brombo, G.; Passaro, A.; Zuliani, G. Low-grade systemic inflammation is associated with functional disability in elderly people affected by dementia. GeroScience 2018, 40, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Janowitz, D.; Habes, M.; Toledo, J.B.; Hannemann, A.; Frenzel, S.; Terock, J.; Davatzikos, C.; Hoffmann, W.; Grabe, H.J. Inflammatory markers and imaging patterns of advanced brain aging in the general population. Brain Imaging Behav. 2020, 14, 1108–1117. [Google Scholar] [CrossRef] [PubMed]
- Probert, L. TNF and its receptors in the CNS: The essential, the desirable and the deleterious effects. Neuroscience 2015, 302, 2–22. [Google Scholar] [CrossRef] [PubMed]
- Decourt, B.; Lahiri, D.K.; Sabbagh, M.N. Targeting Tumor Necrosis Factor Alpha for Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 14, 412–425. [Google Scholar] [CrossRef]
- Montgomery, S.L.; Bowers, W.J. Tumor necrosis factor-alpha and the roles it plays in homeostatic and degenerative processes within the central nervous system. J. Neuroimmune Pharmacol. 2012, 7, 42–59. [Google Scholar] [CrossRef]
- Tracey, D.; Klareskog, L.; Sasso, E.H.; Salfeld, J.G.; Tak, P.P. Tumor necrosis factor antagonist mechanisms of action: A comprehensive review. Pharmacol. Ther. 2008, 117, 244–279. [Google Scholar] [CrossRef]
- Torres-Acosta, N.; O’Keefe, J.H.; O’Keefe, E.L.; Isaacson, R.; Small, G. The rapeutic Potential of TNF-α Inhibition for Alzheimer’s Disease Prevention. J. Alzheimer’s Dis. 2020, 78, 619–626. [Google Scholar] [CrossRef]
- Brás, J.P.; Bravo, J.; Freitas, J.; Barbosa, M.A.; Santos, S.G.; Summavielle, T.; Almeida, M.I. TNF-alpha-induced microglia activation requires miR-342: Impact on NF-kB signaling and neurotoxicity. Cell Death Dis. 2020, 11, 415. [Google Scholar] [CrossRef]
- Whiten, D.R.; Brownjohn, P.W.; Moore, S.; De, S.; Strano, A.; Zuo, Y.; Haneklaus, M.; Klenerman, D.; Livesey, F.J. Tumour necrosis factor induces increased production of extracellular amyloid-β- and α-synuclein-containing aggregates by human Alzheimer’s disease neurons. Brain Commun. 2020, 2, fcaa146. [Google Scholar] [CrossRef]
- Raffaele, S.; Lombardi, M.; Verderio, C.; Fumagalli, M. TNF Production and Release from Microglia via Extracellular Vesicles: Impact on Brain Functions. Cells 2020, 9, 2145. [Google Scholar] [CrossRef]
- Lindbergh, C.A.; Casaletto, K.B.; Staffaroni, A.M.; Elahi, F.; Walters, S.M.; You, M.; Neuhaus, J.; Contreras, W.R.; Wang, P.; Karydas, A.; et al. Systemic tumor necrosis factor-alpha trajectories relate to brain health in typically aging older adults. J. Gerontol Ser. A Biol. Sci. Med. Sci. 2020, 75, 1558–1565. [Google Scholar] [CrossRef] [PubMed]
- Olsthoorn, L.; Vreeken, D.; Kiliaan, A.J. Gut Microbiome, Inflammation, and Cerebrovascular Function: Link between Obesity and Cognition. Front. Neurosci. 2021, 15, 761456. [Google Scholar] [CrossRef] [PubMed]
- Erion, J.R.; Wosiski-Kuhn, M.; Dey, A.; Hao, S.; Davis, C.L.; Pollock, N.K.; Stranahan, A.M. Obesity elicits interleukin 1-mediated deficits in hippocampal synaptic plasticity. J. Neurosci. 2014, 34, 2618–2631. [Google Scholar] [CrossRef] [PubMed]
- Samara, A.; Murphy, T.; Strain, J.; Rutlin, J.; Sun, P.; Neyman, O.; Sreevalsan, N.; Shimony, J.S.; Ances, B.M.; Song, S.-K.; et al. Neuroinflammation and White Matter Alterations in Obesity Assessed by Diffusion Basis Spectrum Imaging. Front. Hum. Neurosci. 2020, 13, 464. [Google Scholar] [CrossRef]
- Karczewski, J.; Zielińska, A.; Staszewski, R.; Eder, P.; Dobrowolska, A.; Souto, E.B. Obesity and the Brain. Int. J. Mol. Sci. 2022, 23, 6145. [Google Scholar] [CrossRef]
- Litwiniuk, A.; Bik, W.; Kalisz, M.; Baranowska-Bik, A. Inflammasome nlrp3 potentially links obesity-associated low-grade systemic inflammation and insulin resistance with alzheimer’s disease. Int. J. Mol. Sci. 2021, 22, 5603. [Google Scholar] [CrossRef]
- Jeppesen, J. Low-grade chronic inflammation and vascular damage in patients with rheumatoid arthritis: Don’t forget “metabolic inflammation”. J. Rheumatol. 2011, 38, 595–597. [Google Scholar] [CrossRef]
- Calle, M.C.; Fernandez, M.L. Inflammation and type 2 diabetes. Diabetes Metab. 2012, 38, 183–191. [Google Scholar] [CrossRef]
- Duchaine, C.S.; Brisson, C.; Talbot, D.; Gilbert-Ouimet, M.; Trudel, X.; Vézina, M.; Milot, A.; Diorio, C.; Ndjaboué, R.; Giguère, Y.; et al. Psychosocial stressors at work and inflammatory biomarkers: PROspective Quebec Study on Work and Health. Psychoneuroendocrinology 2021, 133, 105400. [Google Scholar] [CrossRef]
- Scheithauer, T.P.M.; Rampanelli, E.; Nieuwdorp, M.; Vallance, B.A.; Verchere, C.B.; van Raalte, D.H.; Herrema, H. Gut Microbiota as a Trigger for Metabolic Inflammation in Obesity and Type 2 Diabetes. Front. Immunol. 2020, 11, 571731. [Google Scholar] [CrossRef]
- Łuc, M.; Misiak, B.; Pawłowski, M.; Stańczykiewicz, B.; Zabłocka, A.; Szcześniak, D.; Pałęga, A.; Rymaszewska, J. Gut microbiota in dementia. Critical review of novel findings and their potential application. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2021, 104, 110039. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Cui, L.; Gao, J.; Zhu, M.; Zhang, Y.; Zhang, H.L. Gut Microbial Metabolites in Parkinson’s Disease: Implications of Mitochondrial Dysfunction in the Pathogenesis and Treatment. Mol. Neurobiol. 2021, 58, 3745–3758. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kwon, S.-H.; Kam, T.-I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal Propagation of Pathologic α-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron 2019, 103, 627–641.e7. [Google Scholar] [CrossRef] [PubMed]
- Giovannini, M.G.; Lana, D.; Traini, C.; Vannucchi, M.G. The microbiota–gut–brain axis and alzheimer disease. From dysbiosis to neurodegeneration: Focus on the central nervous system glial cells. J. Clin. Med. 2021, 10, 2358. [Google Scholar] [CrossRef] [PubMed]
- Al-Obaidi, M.M.J.; Desa, M.N.M. Mechanisms of Blood Brain Barrier Disruption by Different Types of Bacteria, and Bacterial–Host Interactions Facilitate the Bacterial Pathogen Invading the Brain. Cell Mol. Neurobiol. 2018, 38, 1349–1368. [Google Scholar] [CrossRef]
- Morais, L.H.; Schreiber, H.L.; Mazmanian, S.K. The gut microbiota–brain axis in behaviour and brain disorders. Nat. Rev. Microbiol. 2021, 19, 241–255. [Google Scholar] [CrossRef]
- Jang, S.H.; Woo, Y.S.; Lee, S.Y.; Bahk, W.M. The brain–gut–microbiome axis in psychiatry. Int. J. Mol. Sci. 2020, 21, 7122. [Google Scholar] [CrossRef]
- de Erausquin, G.A.; Snyder, H.; Carrillo, M.; Hosseini, A.A.; Brugha, T.S.; Seshadri, S. The chronic neuropsychiatric sequelae of COVID-19: The need for a prospective study of viral impact on brain functioning. Alzheimer’s Dement. 2021, 17, 1056–1065. [Google Scholar] [CrossRef]
- Jacob, F.; Pather, S.R.; Huang, W.K.; Zhang, F.; Wong, S.Z.H.; Zhou, H.; Cubitt, B.; Fan, W.; Chen, C.Z.; Xu, M.; et al. Human Pluripotent Stem Cell-Derived Neural Cells and Brain Organoids Reveal SARS-CoV-2 Neurotropism Predominates in Choroid Plexus Epithelium. Cell Stem Cell 2020, 27, 937–950.e9. [Google Scholar] [CrossRef]
- Hu, J.; Jolkkonen, J.; Zhao, C. Neurotropism of SARS-CoV-2 and its neuropathological alterations: Similarities with other coronaviruses. Neurosci. Biobehav. Rev. 2020, 119, 184–193. [Google Scholar] [CrossRef]
- Buzhdygan, T.P.; DeOre, B.J.; Baldwin-Leclair, A.; Bullock, T.A.; McGary, H.M.; Khan, J.A.; Razmpour, R.; Hale, J.F.; Galie, P.A.; Potula, R.; et al. The SARS-CoV-2 spike protein alters barrier function in 2D static and 3D microfluidic in-vitro models of the human blood–brain barrier. Neurobiol. Dis. 2020, 146, 105131. [Google Scholar] [CrossRef] [PubMed]
- Yachou, Y.; El Idrissi, A.; Belapasov, V.; Ait Benali, S. Neuroinvasion, neurotropic, and neuroinflammatory events of SARS-CoV-2: Understanding the neurological manifestations in COVID-19 patients. Neurol. Sci. 2020, 41, 2657–2669. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhuang, Y.; Kang, L. A Review of Neurological Involvement in Patients with SARS-CoV-2 Infection. Med. Sci. Monit. 2021, 27, e932962. [Google Scholar] [CrossRef]
- Karnik, M.; Beeraka, N.M.; Uthaiah, C.A.; Nataraj, S.M.; Bettadapura, A.D.S.; Aliev, G.; Madhunapantula, S.V. A Review on SARS-CoV-2-Induced Neuroinflammation, Neurodevelopmental Complications, and Recent Updates on the Vaccine Development. Mol. Neurobiol. 2021, 58, 4535–4563. [Google Scholar] [CrossRef] [PubMed]
- Simonetti, B.; Daly, J.L.; Simón-Gracia, L.; Klein, K.; Weeratunga, S.; Antón-Plágaro, C.; Tobi, A.; Hodgson, L.; Lewis, P.A.; Heesom, K.J.; et al. ESCPE-1 mediates retrograde endosomal sorting of the SARS-CoV-2 host factor Neuropilin-1. Proc. Natl. Acad. Sci. USA 2022, 119, e2201980119. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Han, K.; Blair, R.; Kenst, K.; Qin, Z.; Upcin, B.; Wörsdörfer, P.; Midkiff, C.C.; Mudd, J.; Belyaeva, E.; et al. SARS-CoV-2 Infects Endothelial Cells In Vivo and In Vitro. Front. Cell. Infect. Microbiol. 2021, 11, 701278. [Google Scholar] [CrossRef]
- Krasemann, S.; Haferkamp, U.; Pfefferle, S.; Woo, M.S.; Heinrich, F.; Schweizer, M.; Appelt-Menzel, A.; Cubukova, A.; Barenberg, J.; Leu, J.; et al. The blood-brain barrier is dysregulated in COVID-19 and serves as a CNS entry route for SARS-CoV-2. Stem Cell Rep. 2022, 17, 307–320. [Google Scholar] [CrossRef]
- Yang, R.-C.; Huang, K.; Zhang, H.-P.; Li, L.; Zhang, Y.-F.; Tan, C.; Chen, H.-C.; Jin, M.-L.; Wang, X.-R. SARS-CoV-2 productively infects human brain microvascular endothelial cells. J. Neuroinflamm. 2022, 19, 149. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, L.; Bao, L.; Liu, J.; Zhu, H.; Lv, Q.; Liu, R.; Chen, W.; Tong, W.; Wei, Q.; et al. SARS-CoV-2 crosses the blood–brain barrier accompanied with basement membrane disruption without tight junctions alteration. Signal Transduct. Target Ther. 2021, 6, 337. [Google Scholar]
- Huang, J.; Zheng, M.; Tang, X.; Chen, Y.; Tong, A.; Zhou, L. Potential of SARS-CoV-2 to Cause CNS Infection: Biologic Fundamental and Clinical Experience. Front. Neurol. 2020, 11, 659. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, M.; Garcia, G.; Tian, E.; Cui, Q.; Chen, X.; Sun, G.; Wang, J.; Arumugaswami, V.; Shi, Y. ApoE-Isoform-Dependent SARS-CoV-2 Neurotropism and Cellular Response. Cell Stem Cell 2021, 28, 331–342.e5. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Xu, J.; Hou, Y.; Leverenz, J.B.; Kallianpur, A.; Mehra, R.; Liu, Y.; Yu, H.; Pieper, A.A.; Jehi, L.; et al. Network medicine links SARS-CoV-2/COVID-19 infection to brain microvascular injury and neuroinflammation in dementia-like cognitive impairment. Alzheimer’s Res. Ther. 2021, 13, 110. [Google Scholar] [CrossRef] [PubMed]
- Hernández, V.S.; Zetter, M.A.; Guerra, E.C.; Hernández-Araiza, I.; Karuzin, N.; Hernández-Pérez, O.R.; Eiden, L.E.; Zhang, L. ACE2 expression in rat brain: Implications for COVID-19 associated neurological manifestations. Exp. Neurol. 2021, 345, 113837. [Google Scholar] [CrossRef]
- Zhang, Y.; Archie, S.R.; Ghanwatkar, Y.; Sharma, S.; Nozohouri, S.; Burks, E.; Mdzinarishvili, A.; Liu, Z.; Abbruscato, T.J. Potential role of astrocyte angiotensin converting enzyme 2 in the neural transmission of COVID-19 and a neuroinflammatory state induced by smoking and vaping. Fluids Barriers CNS 2022, 19, 46. [Google Scholar] [CrossRef] [PubMed]
- Baig, A.M.; Khaleeq, A.; Ali, U.; Syeda, H. Evidence of the COVID-19 Virus Targeting the CNS: Tissue Distribution, Host-Virus Interaction, and Proposed Neurotropic Mechanisms. ACS Chem. Neurosci. 2020, 11, 995–998. [Google Scholar] [CrossRef] [PubMed]
- Pallanti, S.; Grassi, E.; Makris, N.; Gasic, G.P.; Hollander, E. Neurocovid-19: A clinical neuroscience-based approach to reduce SARS-CoV-2 related mental health sequelae. J. Psychiatr. Res. 2020, 130, 215–217. [Google Scholar] [CrossRef] [PubMed]
- Paterson, R.W.; Brown, R.L.; Benjamin, L.; Nortley, R.; Wiethoff, S.; Bharucha, T.; Jayaseelan, D.L.; Kumar, G.; Raftopoulos, R.E.; Zambreanu, L.; et al. The emerging spectrum of COVID-19 neurology: Clinical, radiological and laboratory findings. Brain 2020, 143, 3104–3120. [Google Scholar]
- Bhaskar, S.; Sinha, A.; Banach, M.; Mittoo, S.; Weissert, R.; Kass, J.S.; Rajagopal, S.; Pai, A.R.; Kutty, S. Cytokine Storm in COVID-19—Immunopathological Mechanisms, Clinical Considerations, and Therapeutic Approaches: The REPROGRAM Consortium Position Paper. Front. Immunol. 2020, 11, 1648. [Google Scholar] [CrossRef]
- Gasmi, A.; Tippairote, T.; Mujawdiya, P.K.; Gasmi Benahmed, A.; Menzel, A.; Dadar, M.; Bjørklund, G. Neurological Involvements of SARS-CoV2 Infection. Mol. Neurobiol. 2021, 58, 944–949. [Google Scholar]
- Jarrahi, A.; Ahluwalia, M.; Khodadadi, H.; Da Silva Lopes Salles, E.; Kolhe, R.; Hess, D.C.; Vale, F.; Kumar, M.; Baban, B.; Vaibhav, K.; et al. Neurological consequences of COVID-19: What have we learned and where do we go from here? J. Neuroinflamm. 2020, 17, 286. [Google Scholar] [CrossRef]
- Tizenberg, B.N.; Brenner, L.A.; Lowry, C.A.; Okusaga, O.O.; Benavides, D.R.; Hoisington, A.J.; Benros, M.E.; Stiller, J.W.; Kessler, R.C.; Postolache, T.T. Biological and Psychological Factors Determining Neuropsychiatric Outcomes in COVID-19. Curr. Psychiatry Rep. 2021, 23, 68. [Google Scholar] [CrossRef] [PubMed]
- Murta, V.; Villarreal, A.; Ramos, A.J. Severe Acute Respiratory Syndrome Coronavirus 2 Impact on the Central Nervous System: Are Astrocytes and Microglia Main Players or Merely Bystanders? ASN Neuro 2020, 12, 1759091420954960. [Google Scholar] [CrossRef] [PubMed]
- Muccioli, L.; Pensato, U.; Cani, I.; Guarino, M.; Cortelli, P.; Bisulli, F. COVID-19–Associated Encephalopathy and Cytokine-Mediated Neuroinflammation. Ann. Neurol. 2020, 88, 860–861. [Google Scholar] [CrossRef]
- Pilotto, A.; Padovani, A. Reply to the Letter “COVID-19-Associated Encephalopathy and Cytokine-Mediated Neuroinflammation”. Ann. Neurol. 2020, 88, 861–862. [Google Scholar] [CrossRef]
- Connors, J.M.; Levy, J.H. COVID-19 and its implications for thrombosis and anticoagulation. Blood 2020, 135, 2033–2040. [Google Scholar] [CrossRef]
- Tirelli, U.; Taibi, R.; Chirumbolo, S. Post COVID syndrome: A new challenge for medicine. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 4422–4425. [Google Scholar] [CrossRef]
- Stevenson, R.; Samokhina, E.; Rossetti, I.; Morley, J.W.; Buskila, Y. Neuromodulation of Glial Function During Neurodegeneration. Front. Cell. Neurosci. 2020, 14, 278. [Google Scholar] [CrossRef]
- Ferrer, I. Oligodendrogliopathy in neurodegenerative diseases with abnormal protein aggregates: The forgotten partner. Prog. Neurobiol. 2018, 169, 24–54. [Google Scholar] [CrossRef]
- Acioglu, C.; Li, L.; Elkabes, S. Contribution of astrocytes to neuropathology of neurodegenerative diseases. Brain Res. 2021, 1758, 147291. [Google Scholar] [CrossRef]
- Gamage, R.; Wagnon, I.; Rossetti, I.; Childs, R.; Niedermayer, G.; Chesworth, R.; Gyengesi, E. Cholinergic Modulation of Glial Function During Aging and Chronic Neuroinflammation. Front. Cell. Neurosci. 2020, 14, 577912. [Google Scholar] [CrossRef]
- Zhou, Y.; Song, W.M.; Andhey, P.S.; Swain, A.; Levy, T.; Miller, K.R.; Poliani, P.L.; Cominelli, M.; Grover, S.; Gilfillan, S.; et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med. 2020, 26, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Court, J.A.; Lloyd, S.; Johnson, M.; Griffiths, M.; Birdsall, N.J.M.; Piggott, M.A.; Oakley, A.; Ince, P.; Perry, E.; Perry, R. Nicotinic and muscarinic cholinergic receptor binding in the human hippocampal formation during development and aging. Dev. Brain Res. 1997, 101, 93–105. [Google Scholar] [CrossRef]
- Sorrentino, Z.A.; Giasson, B.I.; Chakrabarty, P. α-Synuclein and astrocytes: Tracing the pathways from homeostasis to neurodegeneration in Lewy body disease. Acta Neuropathol. 2019, 138, 1–21. [Google Scholar] [CrossRef]
- Radford, R.A.; Morsch, M.; Rayner, S.L.; Cole, N.J.; Pountney, D.L.; Chung, R.S. The established and emerging roles of astrocytes and microglia in amyotrophic lateral sclerosis and frontotemporal dementia. Front. Cell. Neurosci. 2015, 9, 414. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. The neurovascular unit coming of age: A journey through neurovascular coupling in health and disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef]
- Porchet, R.; Probst, A.; Bouras, C.; Dráberová, E.; Dráber, P.; Riederer, B.M. Analysis of gial acidic fibrillary protein in the human entorhinal cortex during aging and in Alzheimer’s disease. Proteomics 2003, 3, 1476–1485. [Google Scholar] [CrossRef]
- Gaikwad, S.; Puangmalai, N.; Bittar, A.; Montalbano, M.; Garcia, S.; McAllen, S.; Bhatt, N.; Sonawane, M.; Sengupta, U.; Kayed, R. Tau oligomer induced HMGB1 release contributes to cellular senescence and neuropathology linked to Alzheimer’s disease and frontotemporal dementia. Cell Rep. 2021, 36, 109419. [Google Scholar] [CrossRef]
- Price, B.R.; Norris, C.M.; Sompol, P.; Wilcock, D.M. An emerging role of astrocytes in vascular contributions to cognitive impairment and dementia. J. Neurochem. 2018, 144, 644–650. [Google Scholar] [CrossRef]
- Kim, J.H.; Ko, P.W.; Lee, H.W.; Jeong, J.Y.; Lee, M.G.; Kim, J.H.; Lee, W.-H.; Yu, R.; Oh, W.-J.; Suk, K. Astrocyte-derived lipocalin-2 mediates hippocampal damage and cognitive deficits in experimental models of vascular dementia. Glia 2017, 65, 1471–1490. [Google Scholar] [CrossRef]
- Llorente, I.L.; Xie, Y.; Mazzitelli, J.A.; Hatanaka, E.A.; Cinkornpumin, J.; Miller, D.R.; Lin, Y.; Lowry, W.E.; Carmichael, S.T. Patient-derived glial enriched progenitors repair functional deficits due to white matter stroke and vascular dementia in rodents. Sci. Transl. Med. 2021, 13, eaaz6747. [Google Scholar] [CrossRef]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139, 136–153. [Google Scholar] [CrossRef] [PubMed]
- López-Valdés, H.E.; Martínez-Coria, H. The Role of Neuroinflammation in Age-Related Dementias. Rev. Investig. Clin. 2016, 68, 40–48. [Google Scholar]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate Mapping Analysis Reveals That Adult Microglia Derive from Primitive Macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Niraula, A.; Sheridan, J.F.; Godbout, J.P. Microglia Priming with Aging and Stress. Neuropsychopharmacology 2017, 42, 318–333. [Google Scholar] [CrossRef] [PubMed]
- Norden, D.M.; Godbout, J.P. Review: Microglia of the aged brain: Primed to be activated and resistant to regulation. Neuropathol. Appl. Neurobiol. 2013, 39, 19–34. [Google Scholar] [CrossRef]
- Wynne, A.M.; Henry, C.J.; Huang, Y.; Cleland, A.; Godbout, J.P. Protracted downregulation of CX3CR1 on microglia of aged mice after lipopolysaccharide challenge. Brain Behav. Immun. 2010, 24, 1190–1201. [Google Scholar] [CrossRef]
- Njie Malick, G.; Boelen, E.; Stassen, F.R.; Steinbusch, H.W.M.; Borchelt, D.R.; Streit, W.J. Ex vivo cultures of microglia from young and aged rodent brain reveal age-related changes in microglial function. Neurobiol. Aging 2012, 33, 195.e1–195.e12. [Google Scholar] [CrossRef]
- Lee, S.; Varvel, N.H.; Konerth, M.E.; Xu, G.; Cardona, A.E.; Ransohoff, R.M.; Lamb, B.T. CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer’s disease mouse models. Am. J. Pathol. 2010, 177, 2549–2562. [Google Scholar] [CrossRef]
- Hampel, H.; Caraci, F.; Cuello, A.C.; Caruso, G.; Nisticò, R.; Corbo, M.; Baldacci, F.; Toschi, N.; Garaci, F.; Chiesa, P.A.; et al. A Path Toward Precision Medicine for Neuroinflammatory Mechanisms in Alzheimer’s Disease. Front. Immunol. 2020, 11, 456. [Google Scholar] [CrossRef]
- Lee, C.Y.D.; Daggett, A.; Gu, X.; Jiang, L.L.; Langfelder, P.; Li, X.; Wang, N.; Zhao, Y.; Park, C.S.; Cooper, Y.; et al. Elevated TREM2 Gene Dosage Reprograms Microglia Responsivity and Ameliorates Pathological Phenotypes in Alzheimer’s Disease Models. Neuron 2018, 97, 1032–1048.e5. [Google Scholar] [CrossRef]
- Yeh, F.L.; Hansen, D.V.; Sheng, M. TREM2, Microglia, and Neurodegenerative Diseases. Trends Mol. Med. 2017, 23, 512–533. [Google Scholar] [CrossRef] [PubMed]
- Deczkowska, A.; Weiner, A.; Amit, I. The Physiology, Pathology, and Potential Therapeutic Applications of the TREM2 Signaling Pathway. Cell 2020, 181, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Ewers, M.; Franzmeier, N.; Suárez-Calvet, M.; Morenas-Rodriguez, E.; Caballero, M.A.A.; Kleinberger, G.; Piccio, L.; Cruchaga, C.; Deming, Y.; Dichgans, M.; et al. Increased soluble TREM2 in cerebrospinal fluid is associated with reduced cognitive and clinical decline in Alzheimer’s disease. Sci. Transl. Med. 2019, 11, eaav6221. [Google Scholar] [CrossRef] [PubMed]
- Jay, T.R.; Hirsch, A.M.; Broihier, M.L.; Miller, C.M.; Neilson, L.E.; Ransohoff, R.M.; Lamb, B.T.; Landreth, G.E. Disease progression-dependent effects of TREM2 deficiency in a mouse model of Alzheimer’s disease. J. Neurosci. 2017, 37, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Leyns, C.E.G.; Ulrich, J.D.; Finn, M.B.; Stewart, F.R.; Koscal, L.J.; Serrano, J.R.; Robinson, G.O.; Anderson, E.; Colonna, M.; Holtzman, D.M. TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc. Natl. Acad. Sci. USA 2017, 114, 11524–11529. [Google Scholar] [CrossRef]
- Parhizkar, S.; Arzberger, T.; Brendel, M.; Kleinberger, G.; Deussing, M.; Focke, C.; Nuscher, B.; Xiong, M.; Ghasemigharagoz, A.; Katzmarski, N.; et al. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat. Neurosci. 2019, 22, 191–204. [Google Scholar] [CrossRef]
- Li, C.; Zhao, B.; Lin, C.; Gong, Z.; An, X. TREM2 inhibits inflammatory responses in mouse microglia by suppressing the PI3K/NF-κB signaling. Cell Biol. Int. 2019, 43, 360–372. [Google Scholar] [CrossRef]
- Carmona-Abellan, M.; Martinez-Valbuena, I.; Marcilla, I.; DiCaudo, C.; Gil, I.; Nuñez, J.; Luquin, M.-R. Microglia is associated with p-Tau aggregates in the olfactory bulb of patients with neurodegenerative diseases. Neurol. Sci. 2021, 42, 1473–1482. [Google Scholar] [CrossRef]
- Stancu, I.C.; Cremers, N.; Vanrusselt, H.; Couturier, J.; Vanoosthuyse, A.; Kessels, S.; Lodder, C.; Brône, B.; Huaux, F.; Octave, J.-N.; et al. Aggregated Tau activates NLRP3–ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded Tau pathology in vivo. Acta Neuropathol. 2019, 137, 599–617. [Google Scholar] [CrossRef]
- Mancuso, R.; Fryatt, G.; Cleal, M.; Obst, J.; Pipi, E.; Monzón-Sandoval, J.; Ribe, E.; Winchester, L.; Webber, C.; Nevado, A.; et al. CSF1R inhibitor JNJ-40346527 attenuates microglial proliferation and neurodegeneration in P301S mice. Brain 2019, 142, 3243–3264. [Google Scholar] [CrossRef]
- Radford, R.; Rcom-H’cheo-Gauthier, A.; Wong, M.B.; Eaton, E.D.; Quilty, M.; Blizzard, C.; Norazit, A.; Meedeniya, A.; Vickers, J.; Gai, W.P.; et al. The degree of astrocyte activation in multiple system atrophy is inversely proportional to the distance to α-synuclein inclusions. Mol. Cell. Neurosci. 2015, 65, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Valdinocci, D.; Radford, R.A.W.; Siow, S.M.; Chung, R.S.; Pountney, D.L. Potential modes of intercellular α-synuclein transmission. Int. J. Mol. Sci. 2017, 18, 469. [Google Scholar] [CrossRef] [PubMed]
- Hartnell, I.J.; Blum, D.; Nicoll, J.A.R.; Dorothee, G.; Boche, D. Glial cells and adaptive immunity in frontotemporal dementia with tau pathology. Brain 2021, 144, 724–745. [Google Scholar] [CrossRef] [PubMed]
- Strohm, L.; Behrends, C. Glia-specific autophagy dysfunction in ALS. Semin. Cell Dev. Biol. 2020, 99, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, A.; Dittlau, K.S.; Corsi, G.I.; Haukedal, H.; Doncheva, N.T.; Ramakrishna, S.; Ambardar, S.; Salcedo, C.; Schmidt, S.I.; Zhang, Y.; et al. Astrocytic reactivity triggered by defective autophagy and metabolic failure causes neurotoxicity in frontotemporal dementia type 3. Stem Cell Rep. 2021, 16, 2736–2751. [Google Scholar] [CrossRef]
- Ghasemi, M.; Keyhanian, K.; Douthwright, C. Glial cell dysfunction in c9orf72-related amyotrophic lateral sclerosis and frontotemporal dementia. Cells 2021, 10, 249. [Google Scholar] [CrossRef]
- Vasefi, M.; Hudson, M.; Ghaboolian-Zare, E. Diet Associated with Inflammation and Alzheimer’s Disease. J. Alzheimer’s Dis. Rep. 2019, 3, 299–309. [Google Scholar] [CrossRef]
- Ozawa, M.; Shipley, M.; Kivimaki, M.; Singh-Manoux, A.; Brunner, E.J. Dietary pattern, inflammation and cognitive decline: The Whitehall II prospective cohort study. Clin. Nutr. 2017, 36, 506–512. [Google Scholar] [CrossRef]
- Shin, D.; Kwon, S.C.; Kim, M.H.; Lee, K.W.; Choi, S.Y.; Shivappa, N.; Hébert, J.R.; Chung, H.-K. Inflammatory potential of diet is associated with cognitive function in an older adult Korean population. Nutrition 2018, 55–56, 56–62. [Google Scholar] [CrossRef]
- Hayden, K.M.; Beavers, D.P.; Steck, S.E.; Hebert, J.R.; Tabung, F.K.; Shivappa, N.; Casanova, R.; Manson, J.E.; Padula, C.B.; Salmoirago-Blotcher, E.; et al. The association between an inflammatory diet and global cognitive function and incident dementia in older women: The Women’s Health Initiative Memory Study. Alzheimer’s Dement. 2017, 13, 1187–1196. [Google Scholar] [CrossRef]
- Frith, E.; Shivappa, N.; Mann, J.R.; Hébert, J.R.; Wirth, M.D.; Loprinzi, P.D. Dietary inflammatory index and memory function: Population-based national sample of elderly Americans. Br. J. Nutr. 2018, 119, 552–558. [Google Scholar] [CrossRef]
- Saita, D.; Ferrarese, R.; Foglieni, C.; Esposito, A.; Canu, T.; Perani, L.; Ceresola, E.R.; Visconti, L.; Burioni, R.; Clementi, M.; et al. Adaptive immunity against gut microbiota enhances apoE-mediated immune regulation and reduces atherosclerosis and western-diet-related inflammation. Sci. Rep. 2016, 6, 29353. [Google Scholar] [CrossRef] [PubMed]
- Milošević, M.; Arsić, A.; Cvetković, Z.; Vučić, V. Memorable Food: Fighting Age-Related Neurodegeneration by Precision Nutrition. Front. Nutr. 2021, 8, 688086. [Google Scholar] [CrossRef] [PubMed]
- Shivappa, N.; Steck, S.E.; Hurley, T.G.; Hussey, J.R.H.J. Dietary Inflammatory Index. Public Health Nutr. 2014, 17, 1689–1696. [Google Scholar] [CrossRef] [PubMed]
- Kesse-Guyot, E.; Assmann, K.E.; Andreeva, V.A.; Touvier, M.; Neufcourt, L.; Shivappa, N.; Hébert, J.R.; Wirth, M.D.; Hercberg, S.; Galan, P.; et al. Long-term association between the dietary inflammatory index and cognitive functioning: Findings from the SU.VI.MAX study. Eur. J. Nutr. 2017, 56, 1647–1655. [Google Scholar] [CrossRef]
- Ostan, R.; Béné, M.C.; Spazzafumo, L.; Pinto, A.; Donini, L.M.; Pryen, F.; Charrouf, Z.; Valentini, L.; Lochs, H.; Bourdel-Marchasson, I.; et al. Impact of diet and nutraceutical supplementation on inflammation in elderly people. Results from the RISTOMED study, an open-label randomized control trial. Clin. Nutr. 2016, 35, 812–818. [Google Scholar] [CrossRef]
- Kim, E.R.; Kim, S.R.; Cho, W.; Lee, S.G.; Kim, S.H.; Kim, J.H.; Choi, E.; Kim, J.-H.; Yu, J.-W.; Lee, B.-W.; et al. Short Term Isocaloric Ketogenic Diet Modulates NLRP3 Inflammasome Via B-hydroxybutyrate and Fibroblast Growth Factor 21. Front. Immunol. 2022, 13, 843520. [Google Scholar] [CrossRef]
- Al-Aubaidy, H.A.; Dayan, A.; Deseo, M.A.; Itsiopoulos, C.; Jamil, D.; Hadi, N.R.; Thomas, C. Twelve-week mediterranean diet intervention increases citrus bioflavonoid levels and reduces inflammation in people with type 2 diabetes mellitus. Nutrients 2021, 13, 1133. [Google Scholar] [CrossRef]
- Georgoulis, M.; Yiannakouris, N.; Tenta, R.; Fragopoulou, E.; Kechribari, I.; Lamprou, K.; Perraki, E.; Vagiakis, E.; Kontogianni, M.D. A weight-loss Mediterranean diet/lifestyle intervention ameliorates inflammation and oxidative stress in patients with obstructive sleep apnea: Results of the “MIMOSA” randomized clinical trial. Eur. J. Nutr. 2021, 60, 3799–3810. [Google Scholar] [CrossRef]
- Ojo, O.; Ojo, O.O.; Zand, N.; Wang, X. The Effect of Dietary Fibre on Gut Microbiota, Lipid Profile, and Inflammatory Markers in Patients with Type 2 Diabetes: A Systematic Review and Meta-Analysis of Randomised Controlled Trials. Nutrients 2021, 13, 1805. [Google Scholar] [CrossRef]
- Shivappa, N.; Hebert, J.R.; Marcos, A.; Diaz, L.E.; Gomez, S.; Nova, E.; Michels, N.; Arouca, A.; González-Gil, E.; Frederic, G.; et al. Association between dietary inflammatory index and inflammatory markers in the HELENA study. Mol. Nutr. Food Res. 2017, 61, 1–23. [Google Scholar] [CrossRef]
- Casas, R.; Urpi-Sardà, M.; Sacanella, E.; Arranz, S.; Corella, D.; Castañer, O.; Lamuela-Raventós, R.-M.; Salas-Salvadó, J.; Lapetra, J.; Portillo, M.P.; et al. Anti-Inflammatory Effects of the Mediterranean Diet in the Early and Late Stages of Atheroma Plaque Development. Mediat. Inflamm. 2017, 2017, 3674390. [Google Scholar] [CrossRef] [PubMed]
- Mazzoli, A.; Spagnuolo, M.S.; Gatto, C.; Nazzaro, M.; Cancelliere, R.; Crescenzo, R.; Iossa, S.; Cigliano, L. Adipose tissue and brain metabolic responses to western diet—is there a similarity between the two? Int. J. Mol. Sci. 2020, 21, 786. [Google Scholar] [CrossRef]
- Jena, P.K.; Sheng, L.; Nguyen, M.; Di Lucente, J.; Hu, Y.; Li, Y.; Maezawa, I.; Jin, L.-W.; Wan, Y.-J.Y. Dysregulated bile acid receptor-mediated signaling and IL-17A induction are implicated in diet-associated hepatic health and cognitive function. Biomark. Res. 2020, 8, 59. [Google Scholar] [CrossRef]
- Godfrey, J.R.; Pincus, M.; Kovacs-Balint, Z.; Feczko, E.; Earl, E.; Miranda-Dominguez, O.; Fair, D.A.; Jones, S.R.; Locke, J.; Sanchez, M.M.; et al. Obesogenic diet-associated C-reactive protein predicts reduced central dopamine and corticostriatal functional connectivity in female rhesus monkeys. Brain Behav. Immun. 2020, 88, 166–173. [Google Scholar] [CrossRef]
- Weng, J.; Zhao, G.; Weng, L.; Guan, J. Aspirin using was associated with slower cognitive decline in patients with Alzheimer’s disease. PLoS ONE 2021, 16, e0252969. [Google Scholar] [CrossRef]
- Sekiyama, K.; Fujita, M.; Sekigawa, A.; Takamatsu, Y.; Waragai, M.; Takenouchi, T.; Sugama, S.; Hashimoto, M. Ibuprofen ameliorates protein aggregation and astrocytic gliosis, but not cognitive dysfunction, in a transgenic mouse expressing dementia with Lewy bodies-linked P123H β-synuclein. Neurosci. Lett. 2012, 515, 97–101. [Google Scholar] [CrossRef]
- Szekely, C.A.; Breitner, J.C.S.; Fitzpatrick, A.L.; Rea, T.D.; Psaty, B.M.; Kuller, L.H.; Zandi, P.P. NSAID use and dementia risk in the Cardiovascular Health Study: Role of APOE and NSAID type. Neurology 2008, 70, 17–24. [Google Scholar] [CrossRef]
- Knopman, D.S.; Petersen, R.C. The quest for dementia prevention does not include an aspirin a day. Neurology 2020, 95, 105–106. [Google Scholar] [CrossRef]
- Tabet, N.; Feldman, H. Ibuprofen for Alzheimer’s disease. Cochrane Database Syst. Rev. 2003, CD004031. [Google Scholar] [CrossRef]
- Ryan, J.; Storey, E.; Murray, A.M.; Woods, R.L.; Wolfe, R.; Reid, C.M.; Nelson, M.R.; Chong, T.T.; Williamson, J.D.; Ward, S.A.; et al. Randomized placebo-controlled trial of the effects of aspirin on dementia and cognitive decline. Neurology 2020, 95, E320–E331. [Google Scholar] [CrossRef]
- Li, H.; Li, W.; Zhang, X.; Ma, X.C.; Zhang, R.W. Aspirin Use on Incident Dementia and Mild Cognitive Decline: A Systematic Review and Meta-Analysis. Front. Aging Neurosci. 2021, 12, 578071. [Google Scholar] [CrossRef]
- Jordan, F.; Quinn, T.J.; McGuinness, B.; Passmore, P.; Kelly, J.P.; Tudur Smith, C.; Murphy, K.; Devane, D. Aspirin and other non-steroidal anti-inflammatory drugs for the prevention of dementia. Cochrane Database Syst. Rev. 2020, CD011459. [Google Scholar] [CrossRef]
- Rands, G.; Orrell, M. Aspirin for vascular dementia. Cochrane Database Syst. Rev. 2000, 2012, CD001296. [Google Scholar] [CrossRef]
- Jaturapatporn, D.; Mgekn, I.; Mccleery, J.; Tabet, N. Aspirin, steroidal and non-steroidal anti-inflammatory drugs for the treatment of Alzheimer’s disease. Cochrane Database Syst. Rev. 2012, 2, CD006378. [Google Scholar] [CrossRef]
- Veronese, N.; Stubbs, B.; Maggi, S.; Thompson, T.; Schofield, P.; Muller, C.; Tseng, P.; Lin, P.; Carvalho, A.F.; Solmi, M. Low-Dose Aspirin Use and Cognitive Function in Older Age: A Systematic Review and Meta-analysis. J. Am. Geriatr. Soc. 2017, 65, 1763–1768. [Google Scholar] [CrossRef]
- Gottlieb, A.B. Tumor necrosis factor blockade: Mechanism of action. J. Investig. Dermatol. Symp. Proc. 2007, 12, 1–4. [Google Scholar] [CrossRef]
- Levin, A.D.; Wildenberg, M.E.; van den Brink, G.R. Mechanism of Action of Anti-TNF Therapy in Inflammatory Bowel Disease. J. Crohn’s Colitis 2016, 10, 989–997. [Google Scholar] [CrossRef]
- Mitoma, H.; Horiuchi, T.; Tsukamoto, H.; Ueda, N. Molecular mechanisms of action of anti-TNF-α agents—Comparison among therapeutic TNF-α antagonists. Cytokine 2018, 101, 56–63. [Google Scholar] [CrossRef]
- Kirman, I.; Whelan, R.L.; Nielsen, O.H. Infliximab: Mechanism of action beyond TNF-α neutralization in inflammatory bowel disease. Eur. J. Gastroenterol. Hepatol. 2004, 16, 639–641. [Google Scholar] [CrossRef]
- Agnholt, J.; Kelsen, J.; Brandsborg, B.; Jakobsen, N.O.; Dahlerup, J.F. Increased production of granulocyte-macrophage colony-stimulating factor in Crohn’s disease—A possible target for infliximab treatment. Eur. J. Gastroenterol. Hepatol. 2004, 16, 649–655. [Google Scholar] [CrossRef]
- Vos, A.C.W.; Wildenberg, M.E.; Duijvestein, M.; Verhaar, A.P.; Van Den Brink, G.R.; Hommes, D.W. AntiTumor necrosis factor-α antibodies induce regulatory macrophages in an Fc region-dependent manner. Gastroenterology 2011, 140, 221–230.e3. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, A.B.; Chamian, F.; Masud, S.; Cardinale, I.; Abello, M.V.; Lowes, M.A.; Chen, F.; Magliocco, M.; Krueger, J.G. TNF Inhibition Rapidly Down-Regulates Multiple Proinflammatory Pathways in Psoriasis Plaques. J. Immunol. 2005, 175, 2721–2729. [Google Scholar] [CrossRef]
- Ou, W.; Yang, J.; Simanauskaite, J.; Choi, M.; Castellanos, D.M.; Chang, R.; Sun, J.; Jagadeesan, N.; Parfitt, K.D.; Cribbs, D.H.; et al. Biologic TNF-α inhibitors reduce microgliosis, neuronal loss, and tau phosphorylation in a transgenic mouse model of tauopathy. J. Neuroinflamm. 2021, 18, 312. [Google Scholar] [CrossRef]
- Chang, R.; Knox, J.; Chang, J.; Derbedrossian, A.; Vasilevko, V.; Cribbs, D.; Boado, R.J.; Pardridge, W.M.; Sumbria, R.K. Blood-Brain Barrier Penetrating Biologic TNF-α Inhibitor for Alzheimer’s Disease. Mol. Pharm. 2017, 14, 2340–2349. [Google Scholar] [CrossRef]
- Abdelhamid, Y.A.; Elyamany, M.F.; Al-Shorbagy, M.Y.; Badary, O.A. Effects of TNF-α antagonist infliximab on fructose-induced metabolic syndrome in rats. Hum. Exp. Toxicol. 2021, 40, 801–811. [Google Scholar] [CrossRef]
- Shi, J.Q.; Shen, W.; Chen, J.; Wang, B.R.; Zhong, L.L.; Zhu, Y.W.; Zhu, H.-Q.; Zhang, Q.-Q.; Zhang, Y.-D.; Xu, J. Anti-TNF-α reduces amyloid plaques and tau phosphorylation and induces CD11c-positive dendritic-like cell in the APP/PS1 transgenic mouse brains. Brain Res. 2011, 1368, 239–247. [Google Scholar] [CrossRef]
- Xu, J.J.; Guo, S.; Xue, R.; Xiao, L.; Kou, J.N.; Liu, Y.Q.; Han, J.-Y.; Fu, J.-J.; Wei, N. Adalimumab ameliorates memory impairments and neuroinflammation in chronic cerebral hypoperfusion rats. Aging 2021, 13, 14001–14014. [Google Scholar] [CrossRef]
- Chou, R.C.; Kane, M.; Ghimire, S.; Gautam, S.; Gui, J. Treatment for Rheumatoid Arthritis and Risk of Alzheimer’s Disease: A Nested Case-Control Analysis. CNS Drugs 2016, 30, 1111–1120. [Google Scholar] [CrossRef]
- Zhou, M.; Xu, R.; Kaelber, D.C.; Gurney, M.E. Tumor Necrosis Factor (TNF) blocking agents are associated with lower risk for Alzheimer’s disease in patients with rheumatoid arthritis and psoriasis. PLoS ONE 2020, 15, e0229819. [Google Scholar] [CrossRef]
- Zhao, J.; Li, T.; Wang, J. Association between psoriasis and dementia: A systematic review. Neurologia 2021. Online ahead of print. [Google Scholar]
- Trzeciak, P.; Herbet, M.; Dudka, J. Common Factors of Alzheimer’s Disease and Rheumatoid Arthritis—Pathomechanism and Treatment. Molecules 2021, 26, 6038. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.M.; Chen, W.S.; Sheu, J.J.; Chen, Y.H.; Chen, J.H.; Chang, C.C. Autoimmune rheumatic diseases increase dementia risk in middle-aged patients: A nationwide cohort study. PLoS ONE 2018, 13, e0186475. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.W.; Lucente, J.D.; Nguyen, H.M.; Singh, V.; Singh, L.; Chavez, M.; Bushong, T.; Wulff, H.; Maezawa, I. Repurposing the KCa3.1 inhibitor senicapoc for Alzheimer’s disease. Ann. Clin. Transl. Neurol. 2019, 6, 723–738. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Study (Type) | Outcomes | Diet/Intervention | Group | Key Findings |
|---|---|---|---|---|
| Ostan et al., 2015 [126] (cohort study) | Inflammatory and metabolic parameters | RISTOMED diet (personalized and balanced) +/− nutraceutics | 125 participants | RISTOMED diet alone or with each nutraceutical supplementation significantly decreased erythrocyte sedimentation rate |
| Kim et al., 2022 [127] (non-randomized intervention study) | Inflammatory parameters Insulin sensitivity | Short-term ketogenic diet (3 days) | 15 participants | Short-term Ketogenic diet resulted in lower IL-1β and TNF secretion; Improved insulin sensitivity |
| Al-Abauidy et al., 2021 [128] (randomized clinical trial) | Oxidative stress and inflammatory parameters | Mediterranean diet (12 weeks) | 19 participants | Mediterranean diet reduced IL-6 levels by 49% and levels of oxidative stress marker, 8-OHdG, by 32.4% |
| Georgoulis et al., 2021 [129] (randomized clinical trial) | Oxidative stress and inflammatory parameters | Mediterranean diet (6 months) | 187 patients with obstructive sleep apnea | Mediterranean diet reduced hs-CRP levels in patients |
| Casas et al., 2017 [132] (randomized clinical trial) | Cytokine levels | Mediterranean diet +/− extra virgin olive oil (5 years) | 66 participants | Mediterranean diet reduced IL-6, IL-8, MCP-1, and MIP-1β levels. Addition of extra virgin olive oil reduced IL-1β, IL-5, IL-7, IL-12p70, IL-18, TNF-α, IFN-γ, GCSF, GMCSF, and ENA78 |
| Omorogieva et al., 2021 [130] (meta-analysis) | Lipid profiles, LPS, BMI, inflammatory markers | Diet rich in fiber | 10 studies included in meta-analysis | Dietary fiber reduces total cholesterol, BMI and CRP, but no significant changes were observed for IL-6 and TNF |
| Shivappa et al., 2016 [131] (cross-sectional study) | Inflammatory markers | - | 532 adolescents | Higher dietary inflammatory index scores were associated with increased levels of various inflammatory markers: TNF-α, IL-1, 2, IFN-γ and VCAM |
| Mazzoli et al., 2020 [133] (animal study) | Inflammatory markers, insulin sensitivity, BDNF | Western diet (4 weeks) | 16 rats | Western diet increased TNF levels in white adipose tissue and hippocampus of rats; brain BDNF and synaptotagmin I were decreased, while PSD-95 was increased. |
| Jena et al., 2020 [134] (animal study) | Interleukin-17, PD-95, BDNF | High sugar and high fat diet (FPC diet) for 3 months, and 5 months +/− inulin supplementation | 12 mice | FPC diet elevated RORγ and IL-17A signaling. Accompanied by microglia activation and reduced hippocampal long-term potentiation, FPC diet intake also reduced postsynaptic density-95 and brain derived neurotrophic factor. |
| Godfrey et al., 2020 [135] (animal study) | CRP levels, CSF dopamine concentrations Functional connectivity | 12 months of obesogenic diet | 34 female rhesus monkeys | CSF dopamine concentrations decreased, and CRP concentrations increased. Resting-state magnetic resonance neuroimaging showed that higher CRP concentrations were associated with decreased functional connectivity. |
| Anti-Inflammatory Agent | Clinicaltrial.gov Indentifier | Clinical Trial Phase | Results |
|---|---|---|---|
| Etanercept (TNF antagonist) | NCT01068353, NCT01716637, NCT00203359, NCT00203320 | 1–2 | Etanercept was well tolerated and showed some trends toward cognitive, functional, and behavioral benefits |
| XPro1595/DN-TNF (TNF antagonist) | NCT03943264, NCT05321498, NCT05522387, NCT05318976 | 1,2 | In phase 1 XPro1595 reduced white matter free water and increased the axonal integrity in adults with mild to moderate Alzheimer’s disease with signs of inflammation. Phase 2 trials are currently active |
| Dapagliflozin (selective sodium-glucose cotransporter 2 inhibitor) | NCT03801642 | 1/2 | Trial ongoing; Alongside beneficial metabolic effects a potential anti-inflammatory effect via reduction in oxidative stress |
| ALZT-OP1/cromolyn + ibuprofen (mast cell stabilizer + NSAID) | NCT04570644, NCT02547818 | 1/2, 3 | The combination of cromolyn and ibuprofen was safe and well tolerated. The concentrations of cromolyn and ibuprofen observed in the CSF are considered sufficient to titrate the estimated daily amyloid production and the associated inflammatory response in patients with AD. Phase 3 results are to be published. |
| Senicapoc (KCa3.1 blocker) | NCT04804241 | 2 | Phase 2 trial is currently active. Previous animal studies show reduced neuroinflammation, decreased cerebral amyloid load, and enhanced hippocampal neuronal plasticity [164]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Łuc, M.; Woźniak, M.; Rymaszewska, J. Neuroinflammation in Dementia—Therapeutic Directions in a COVID-19 Pandemic Setting. Cells 2022, 11, 2959. https://doi.org/10.3390/cells11192959
Łuc M, Woźniak M, Rymaszewska J. Neuroinflammation in Dementia—Therapeutic Directions in a COVID-19 Pandemic Setting. Cells. 2022; 11(19):2959. https://doi.org/10.3390/cells11192959
Chicago/Turabian StyleŁuc, Mateusz, Marta Woźniak, and Joanna Rymaszewska. 2022. "Neuroinflammation in Dementia—Therapeutic Directions in a COVID-19 Pandemic Setting" Cells 11, no. 19: 2959. https://doi.org/10.3390/cells11192959
APA StyleŁuc, M., Woźniak, M., & Rymaszewska, J. (2022). Neuroinflammation in Dementia—Therapeutic Directions in a COVID-19 Pandemic Setting. Cells, 11(19), 2959. https://doi.org/10.3390/cells11192959