1. Introduction
HAX1 (HCLS1-associated protein X-1) is known as an antiapoptotic protein with a role in the regulation of cell migration, cell adhesion, and calcium homeostasis [
1]. HAX1 deficiency, due to mutations in the
HAX1 gene, results in the autosomal recessive severe congenital neutropenia (SCN) called Kostmann disease [
2]. This myelopoietic disorder is caused by the arrest of granulocyte maturation and the resulting paucity of mature neutrophils, leading to life-threatening infections. This effect has been attributed to excessive apoptosis caused by HAX1 deficiency, but the exact molecular mechanism was not demonstrated. On the other hand,
HAX1 overexpression was documented in several neoplasms [
3,
4,
5,
6]. Here, we propose that these effects may be linked to ribosome dysfunction.
To date, HAX1’s RNA-binding propensity has been reported in two particular cases: for the vimentin transcript [
7] and the DNA polymerase beta transcript [
8]. Both instances pertained to strong hairpin structures at the 3′UTR of these transcripts, although the comparison of the hairpin motifs in vimentin and Pol β mRNAs did not reveal any significant similarities, apart from the presence of U-rich single-stranded regions [
8]. In our recent report [
9], in which we described the HAX1 protein interactome, we also suggested the possibility of RNA-binding by HAX1 deduced from neighboring proteins (first observed by Brannan et al. [
10]).
The role of HAX1 RNA binding in cellular processes has not been clarified so far, except for some suggestions of its involvement in the regulation of specific mRNAs [
11,
12].
The HAX1 protein has no homologues or known domains, except for a PEST sequence, a specific signal which was linked to a shorter half-life of the protein and its proteasomal degradation [
13]. The presence of BCL2-like domains was disproved [
14]. Consequently, HAX1 does not possess any known RNA-binding domain, and a large proportion of the protein is predicted to be disordered [
15,
16], so RNA binding probably occurs in a nonconventional manner.
The current study provides for the first time a comprehensive analysis of the HAX1–RNA interactome, with two independent approaches for the isolation of its RNA targets. Subsequent analysis of the impact of HAX1 on the cell transcriptome and analysis of expression in several cancer databases produced coherent results that indicate the unanticipated role of HAX1 in ribosome biogenesis and translation. Comparison of the experimental data obtained for HAX1 RNA binding and transcriptome profiling indicates that HAX1 may regulate the stability of the bound transcripts. These results were corroborated by the observation that HAX1 KO affects the ribosomal profile, especially with respect to the ratio of the small ribosomal subunit to the monosome. The involvement of HAX1 in ribosome biogenesis and translation emerging from this work may help to elucidate its many-sided effects on cellular processes and HAX1-associated diseases.
3. Discussion
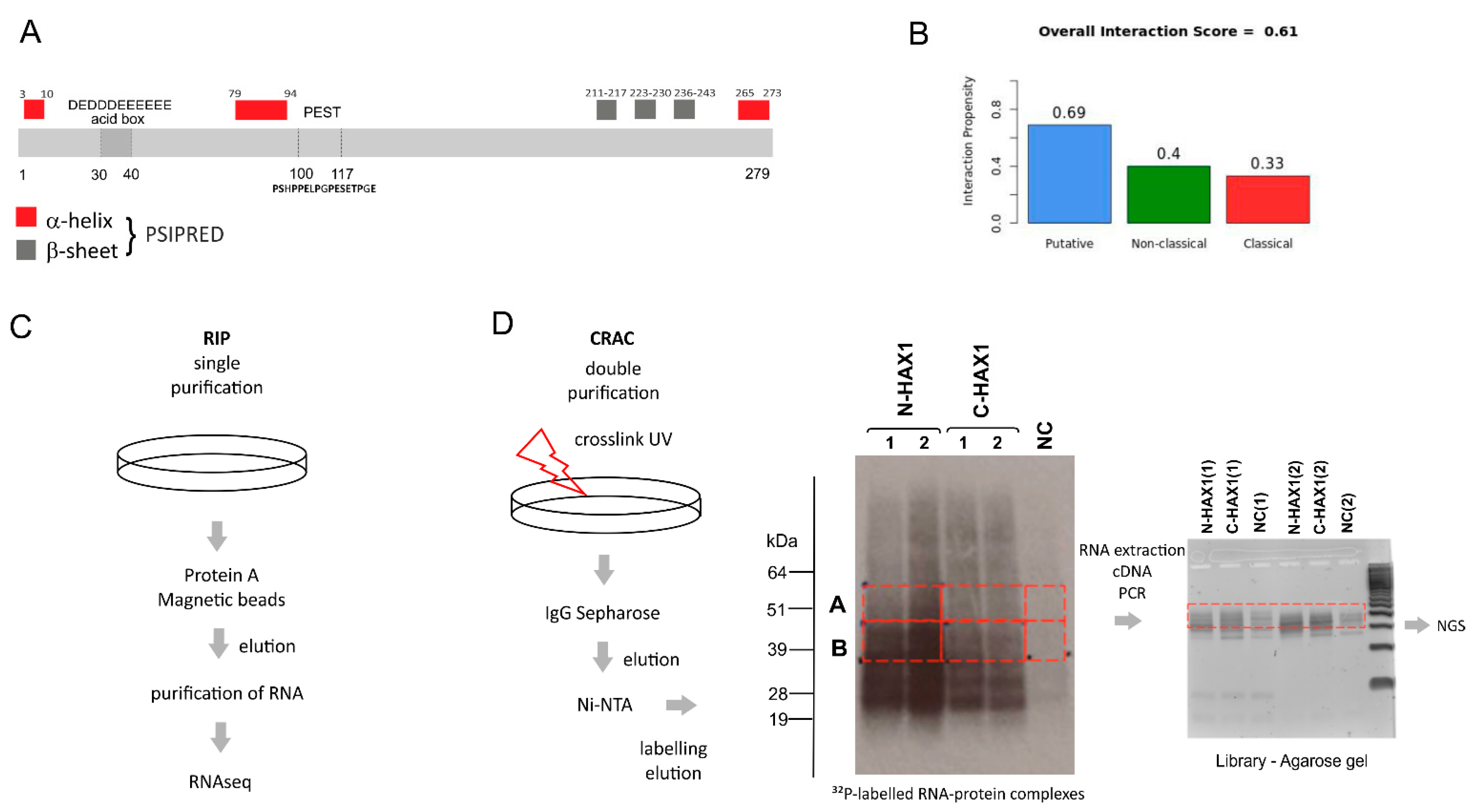
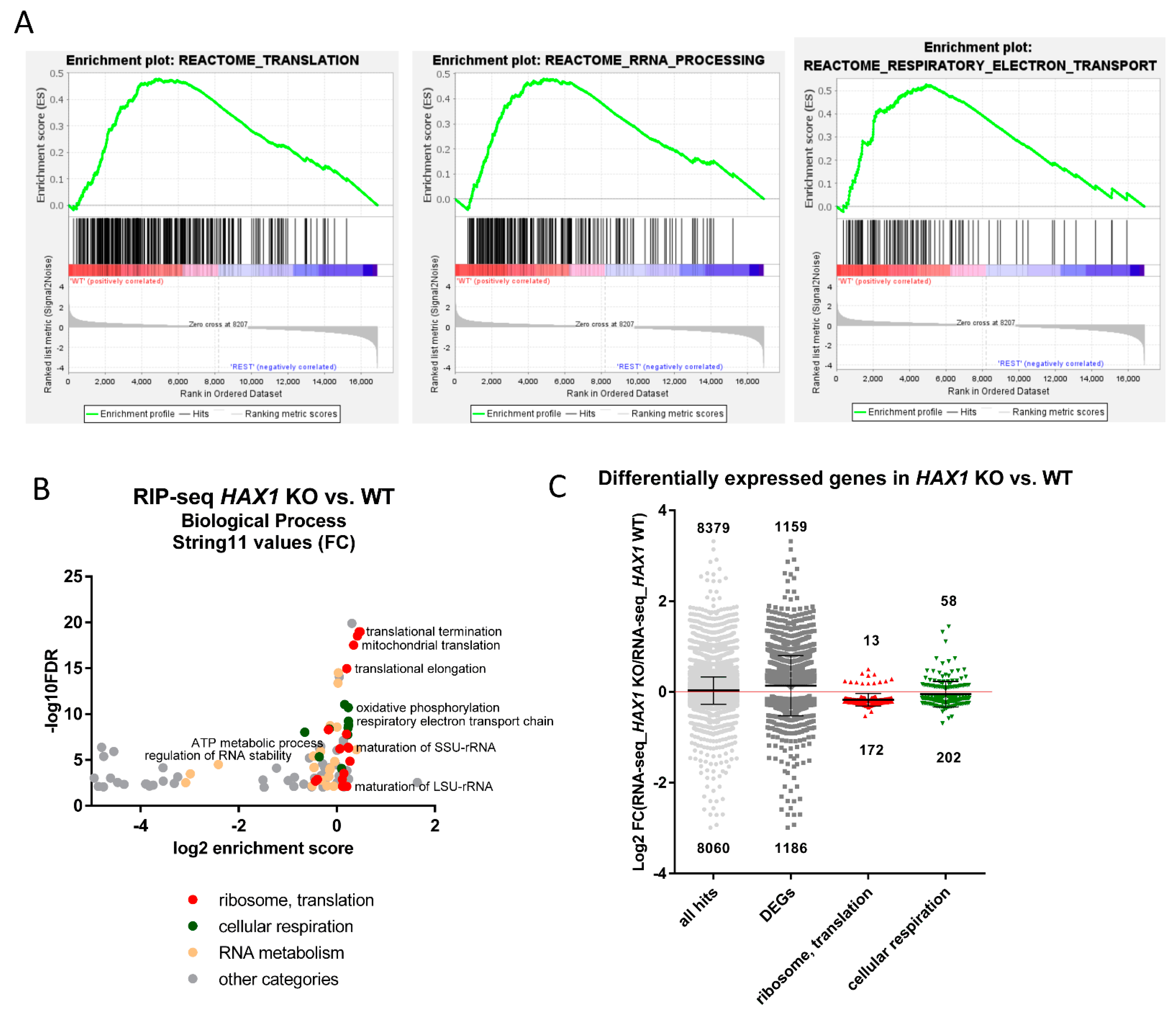
In this manuscript we report a comprehensive analysis of the HAX1–RNA interactome by two independent, high-throughput methods, which both suggest HAX1 involvement in the regulation of transcripts controlling translation, rRNA processing, and ribosome biogenesis. The two methods differ in complexity and specificity (
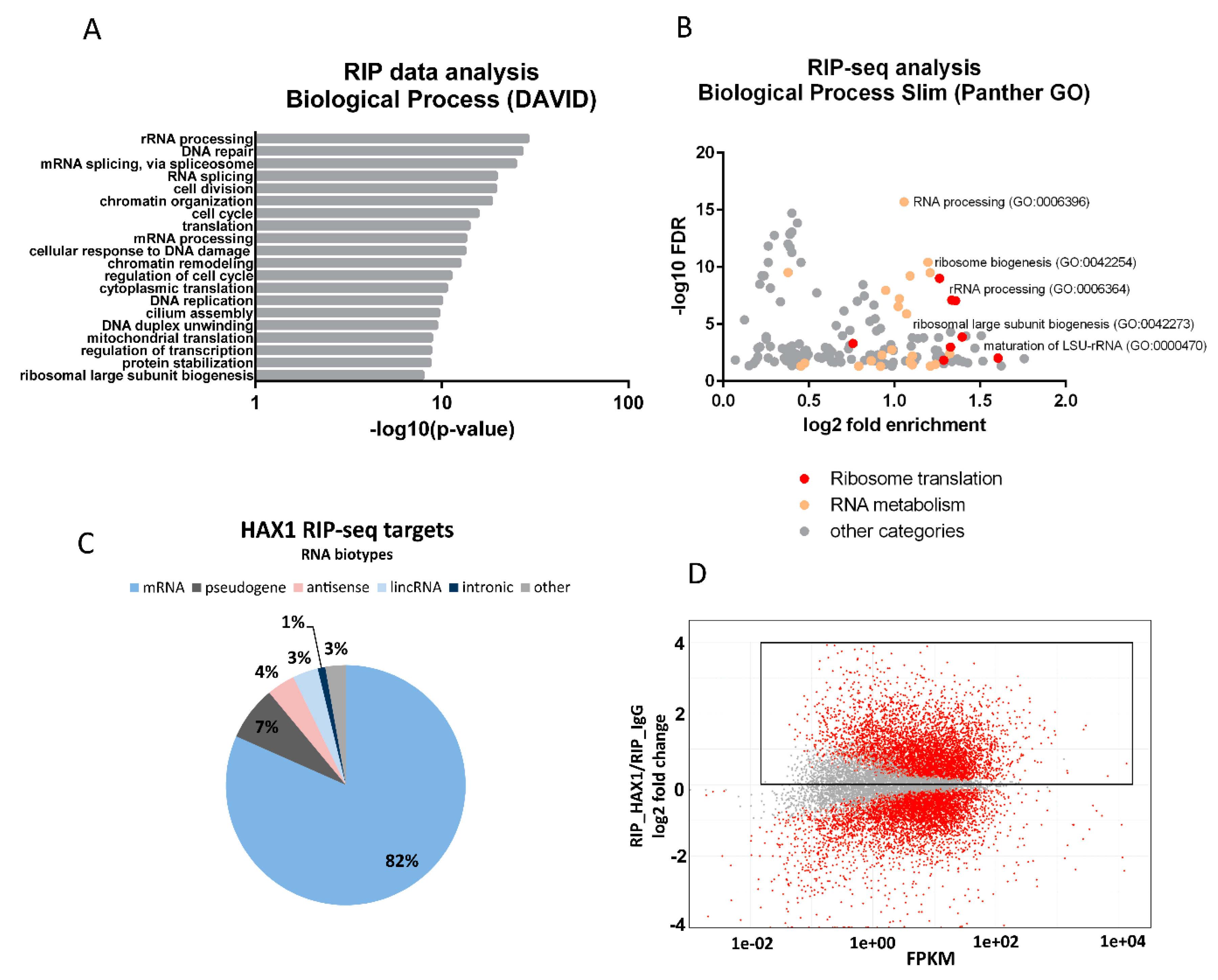
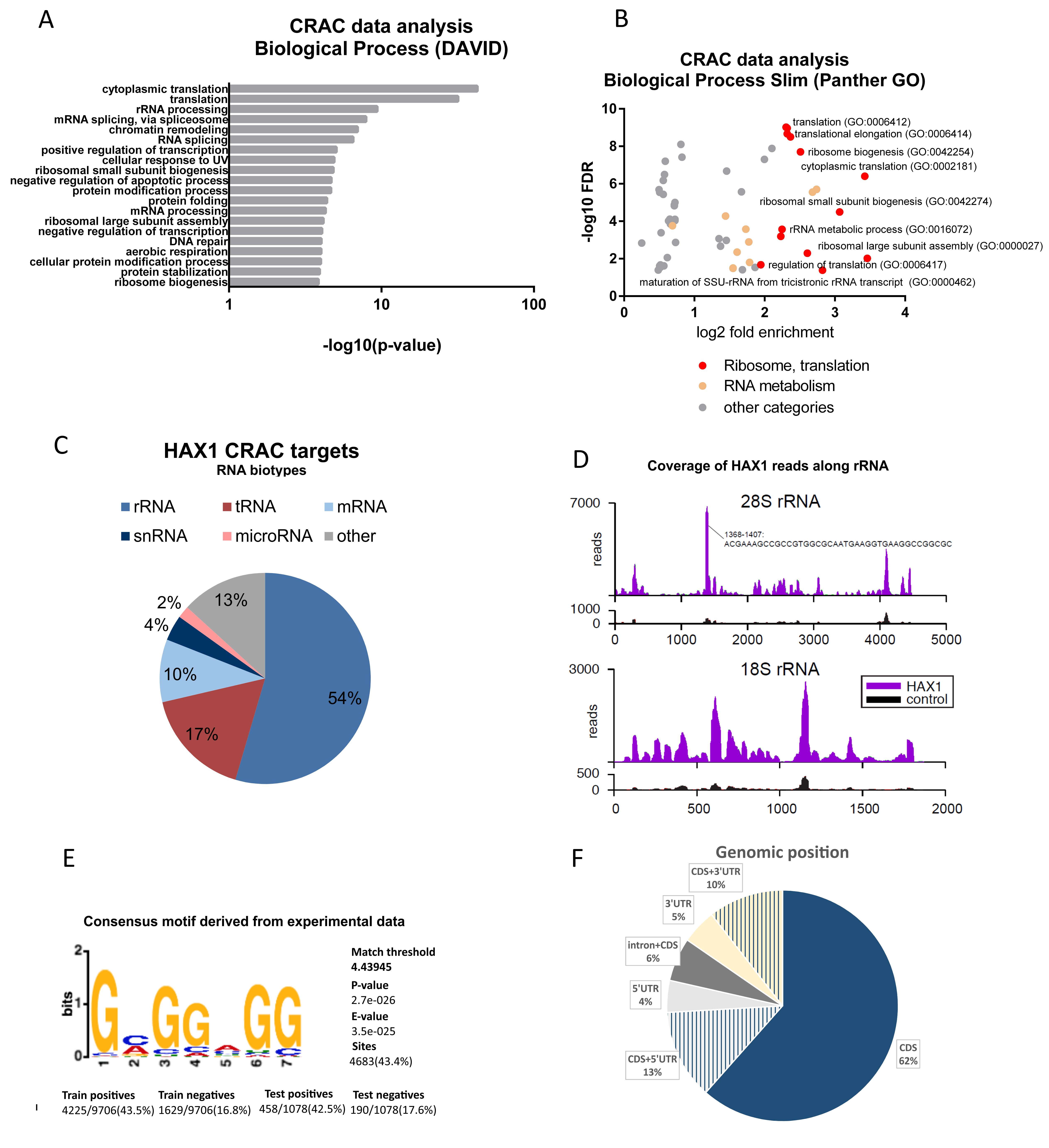
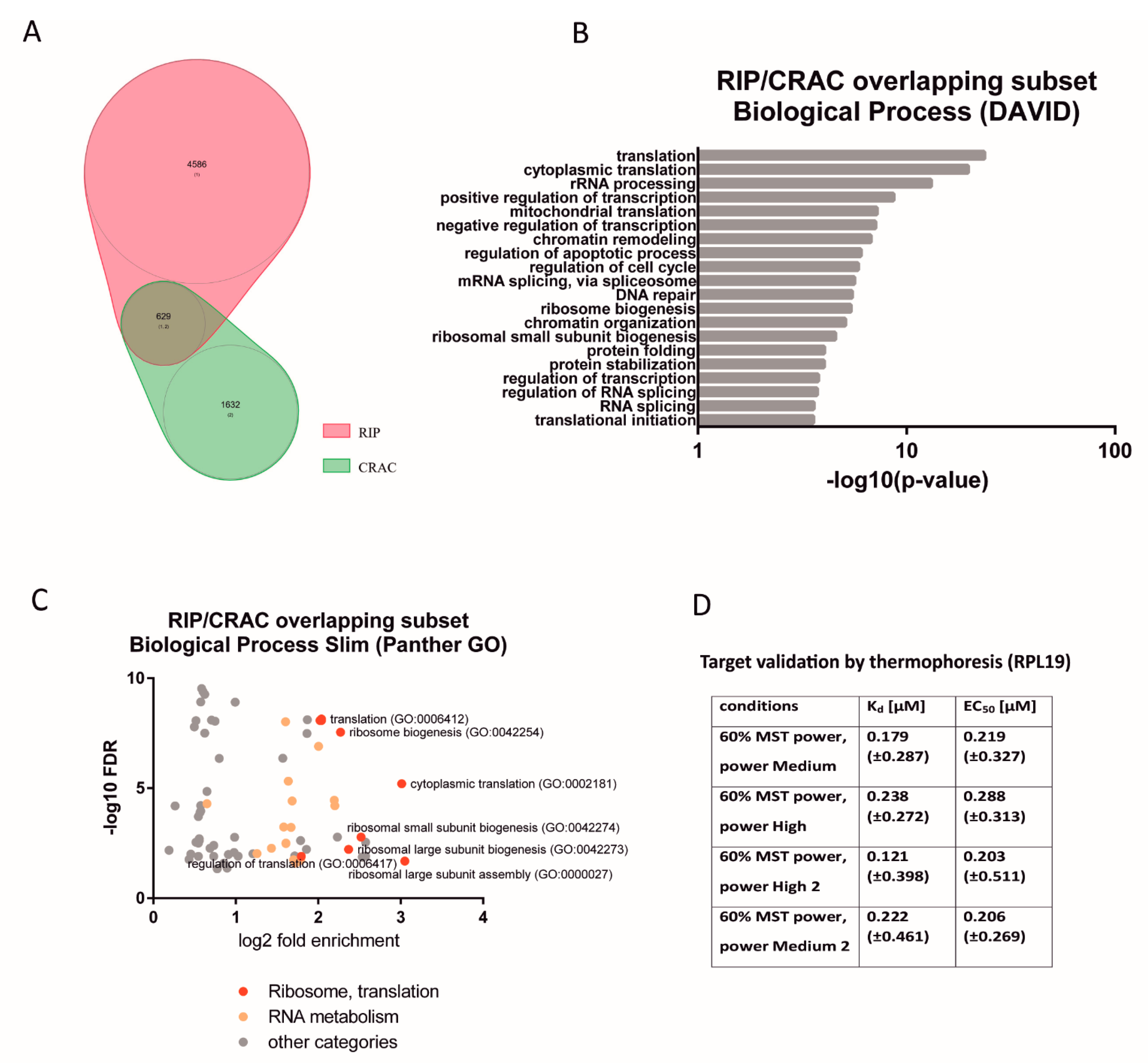
Figure 1A), and the experiments were carried out in two different cell lines (HL-60 promyelocytic cell line, selected due to the strong effect of HAX1 inactivation in these cells, and HEK293 embryonic kidney cells with adrenal endocrine characteristics, selected for technical reasons). Nevertheless, both approaches yielded enrichment in similar GO terms in the Biological Process category (translation, rRNA processing, and ribosome biogenesis). The pool of overlapping targets obtained simultaneously by both methods also shows involvement in translation, rRNA processing, and ribosome biogenesis.
The distribution of RNA biotypes detected in both methods was different, with RIP targets predominantly consisting of mRNA and CRAC targets with a high proportion of rRNA and tRNA. These differences may stem from the fact that in CRAC: (1) the protein of interest was overexpressed, which should increase the abundance of nonmitochondrial protein able to interact with rRNA compared to endogenous RIP; (2) RNA targets were UV-crosslinked to it, which may shift the distribution towards less stable, transient interactions eliminated in RIP; (3) the background (negative control sample) is cleaner than in RIP-seq, and thus it may be easier to detect significant fold changes for very abundant transcripts (rRNAs, tRNAs, and snRNAs).
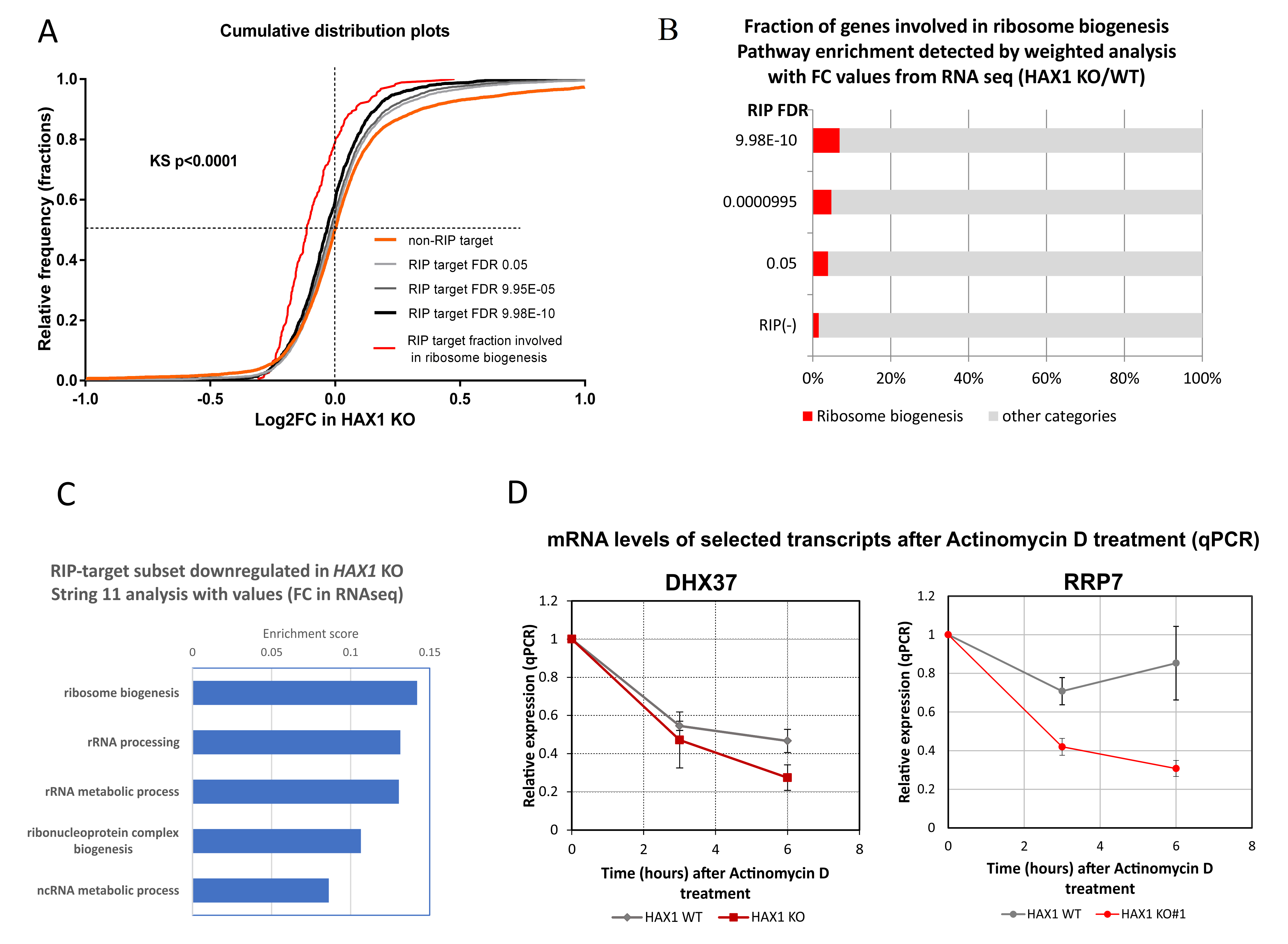
The next important question was if the expression of HAX1 RNA targets has changed after HAX1 knockout. This comparison was made for HL-60 cells, since this cell line was used in both RIP and RNA-seq expression profiling for HAX1 KO vs. WT. The results indicate a partial overlap between HAX1 RNA targets and mRNAs downregulated in HAX1 KO and the overlap is related to transcripts involved in ribosome biogenesis and translation, and, only to a very small extent, to the transcripts involved in energy generation in mitochondria, which are also significantly downregulated in HAX1 KO. This result suggests that only the subset involved in ribosome biogenesis and translation is regulated through direct HAX1 binding and the subsequent stabilization of the mRNA. Therefore, the other detected changes (especially the downregulation of a very important subset of transcripts involved in energy generation) must be therefore regulated by a different mechanism.
Further support for the hypothesis of the HAX1 transcript-stabilizing role is provided by the quantitative assessment of DHX37 and RRP7A mRNA degradation. The DHX37 RNA helicase is involved in ribosome biogenesis, including the formation of the central pseudoknot structure of the small ribosomal subunit [
20]. RRP7A is also predicted to be involved in rRNA processing and assembly of the small ribosomal subunit. Both transcripts represent top RIP/CRAC targets, which are downregulated in
HAX1 KO cells. Quantification of the degradation of the DHX37 and RRP7A transcripts revealed more dynamic degradation in
HAX1 KO cells, suggesting stabilization by HAX1.
Analysis of the genomic position of the RNA targets obtained using the CRAC method revealed the prevalence of coding sequence (CDS) regions, which is not typical for the regulatory RNA sequence and is not consistent with the genomic position of previously characterized HAX1 binding regions (3′UTR). However, new high-throughput analyses demonstrated that binding to CDS is not as uncommon as previously thought and may play a role in the regulation of mRNA stability [
21]. Interestingly, this stabilization should refer to the situation when mRNA is not actively translated, and thus it is not covered and protected by ribosomes and susceptible to endonuclease attack, as in the case of protein CRD-BP, which binds to c-myc mRNA, thus protecting it [
22]. Furthermore, CDS binding was also observed for the FMRP protein and was related to the recruitment of the APP mRNA to processing bodies (P-bodies), which was proposed to restrict translation [
23]. In line with this observation, we previously reported that HAX1 was observed to colocalize with the P-body marker, Dcp1 [
12], pointing to its possible role in transcript stabilization during storage.
Interaction with one of the targets within CDS reported by both methods (RPL19) was confirmed by microscale thermophoresis, with the dissociation constants indicating relatively weak binding (K
d in a range of 0.1–0.2 μM, comparing, for example, to the strong FMRP interaction with N19 RNA with a K
d of 1 nM [
24]). These values suggest a transient, regulatory interaction. Interestingly, the K
d values previously reported for HAX1 binding to 3′UTR are lower [
8], indicating a different strength of interaction and, possibly, a different mode of binding for the CDS and 3′UTR regions. A similar phenomenon was also observed for the GLD-1 and FMRP proteins, involved in translation regulation [
21].
Analysis of the possibility of indirect regulation mediated by transcription factors indicated that such regulation is improbable, since the expression of TFs itself is not changed in HAX1 KO. However, this hypothesis cannot be totally dismissed, since TF-encoding transcripts may be differentially translated, or their protein product may be degraded in HAX1 KO cells, resulting in differences in TFs at the protein level and subsequent changes in specific groups of transcripts regulated by those TFs.
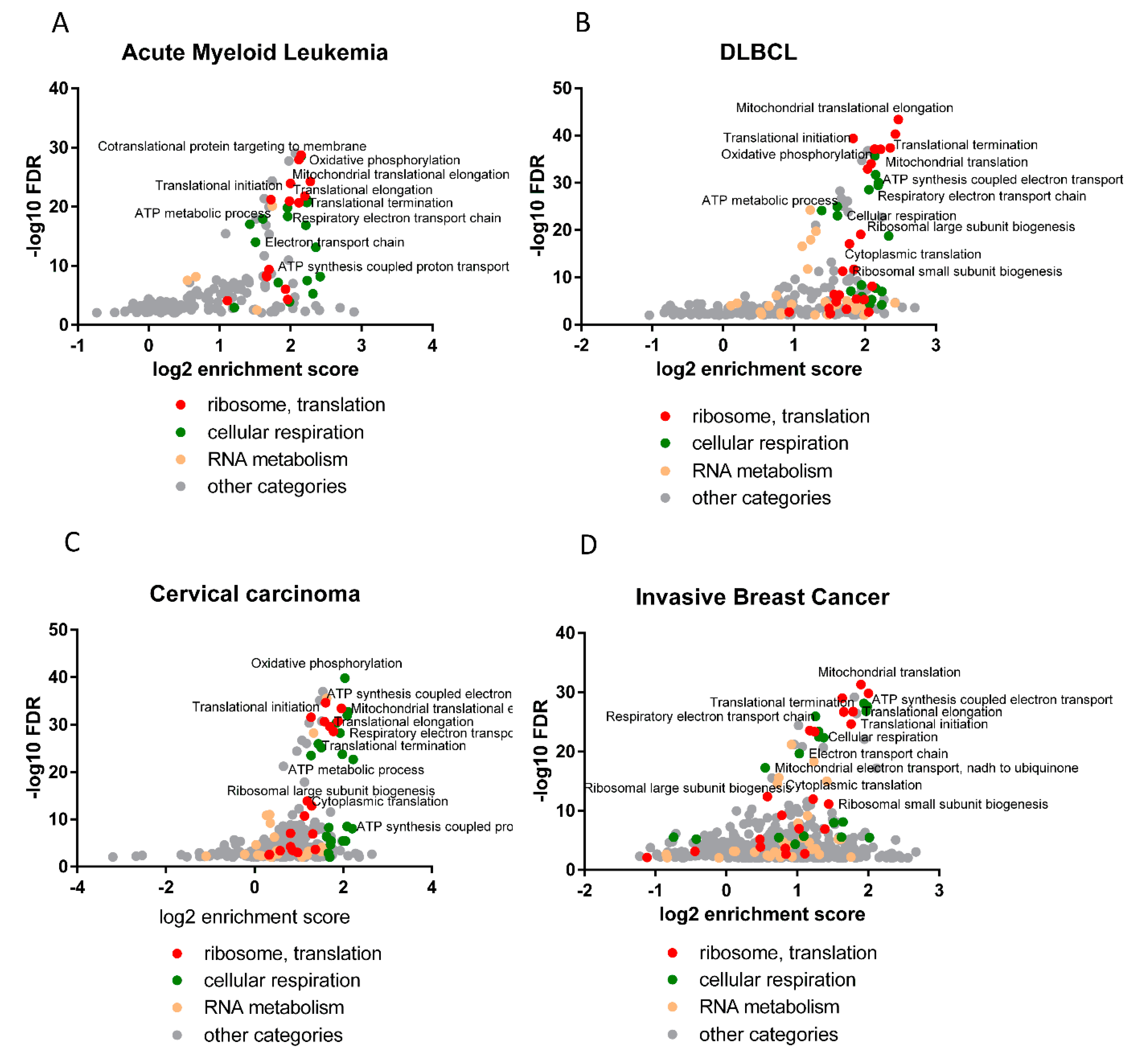
Interestingly, the analysis of the correlation of expression with HAX1, performed for the TCGA database in four neoplasms (AML, DLBCL, breast cancer, cervical cancer), identified enrichment in the same biological processes as detected for HAX1 KO vs. WT in the HL-60 cell line, indicating that these results are not cell line or neoplasm-specific, and thus further corroborating these findings.
The observed changes in expression related to ribosome assembly and translation are not huge, but reproducible, and refer to a relatively large group of transcripts and do not appear to be cell-type or neoplasm-specific. Relatively weak binding and small but reliable changes in expression suggest regulation via small, additive effects. It is an open question whether these effects can manifest more robustly in nonquiescent cells subjected to some kind of stress. This conjecture is supported by the reported changes in the location of HAX1 after stress, including nucleocytoplasmic shuttling [
11], which could be linked to ribosome biogenesis and RNA binding in the nucleus/nucleolus. Furthermore, the abundance of rRNA observed as a potential target in the CRAC analysis suggests the possibility of a more direct involvement in ribosome biogenesis. The suggested binding site (
Figure S3) maps within rRNA expansion segments, for which the function in ribosome biogenesis was proposed [
25]. Thus, the involvement of HAX1 in ribosome assembly could encompass not only the regulation of the stability of mRNAs that encode ribosomal proteins and assembly factors, but also a direct interaction with ribosomal RNA. Interestingly, the possibility of the simultaneous regulation of translation by direct ribosome binding and controlling mRNA stability was described for the FMRP protein [
26], already mentioned here for similar mode of binding and possible recruitment of transcripts to P-bodies.
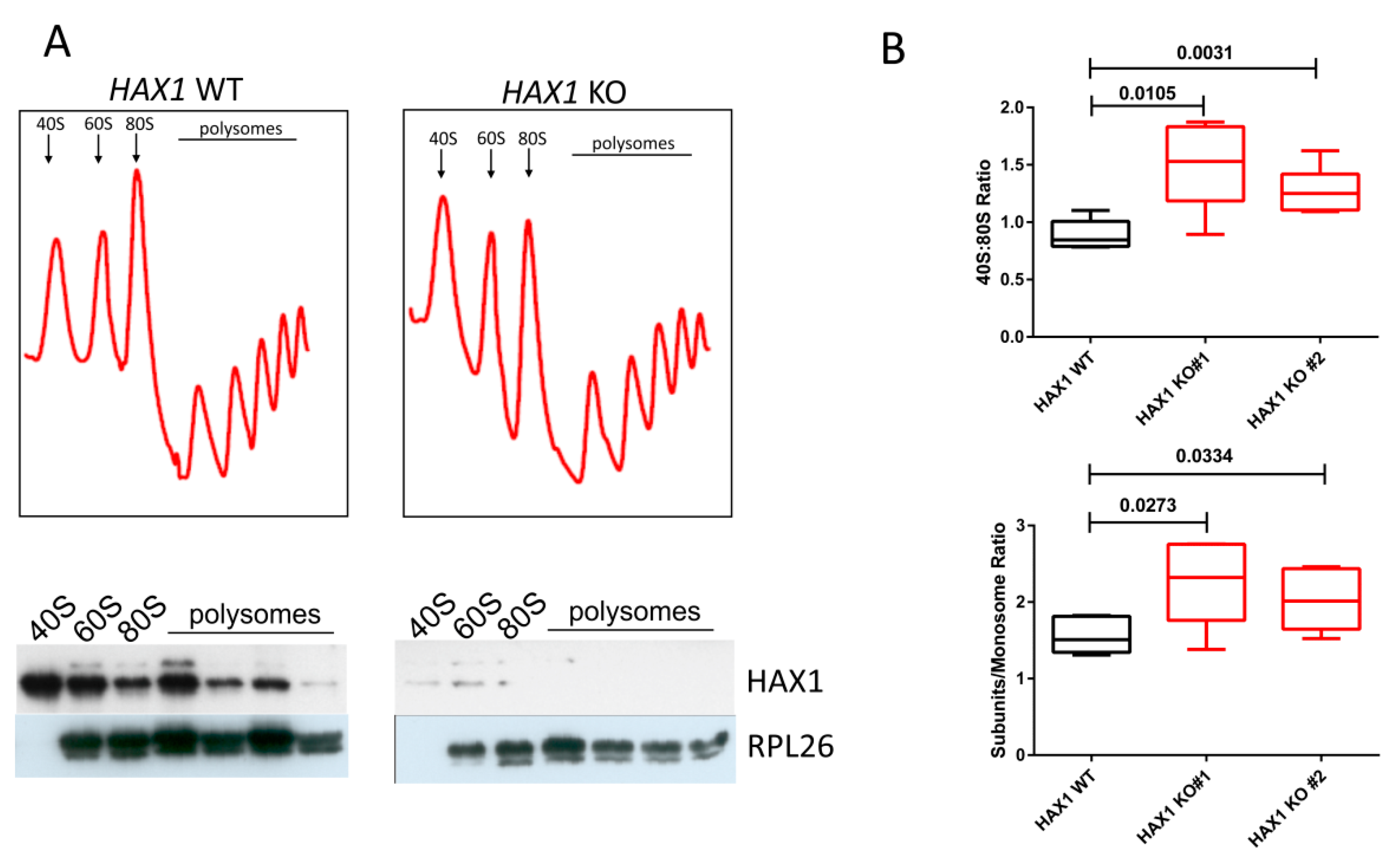
To test the physiological consequences of HAX1 KO on ribosome status, we performed ribosome sedimentation profiling, demonstrating a difference in the 40S:monosome ratio for HAX1-deficient cells. The shift towards free subunits in HAX1 KO may indicate less efficient monosome assembly, resulting from lower expression of ribosomal assembly factors and ribosomal components.
In conclusion, we provide evidence for the involvement of HAX1 in ribosome assembly and translation, which represents a new finding. Previously, it was reported that HAX1 has an interaction with PELO, a protein involved in ribosomal rescue during ribosome stalling, but no mechanism for HAX1 involvement was proposed and no physiological effect was observed [
27]. Recently, You et al. [
28] demonstrated, among other things, that HAX1 levels correlate with ribosome formation and that HAX1 promotes the translation of the transcript encoding integrin subunit beta 6 in endothelial cells, partially matching the findings presented here.
Thus, the results presented suggest the possibility that HAX1 binds to the CDS of the nontranslated transcripts, protecting them from degradation, and that the main mRNA targets subjected to this regulation include transcripts involved in ribosome biogenesis.
Changes in ribosomal status and translation efficiency affect proliferation, which in turn can contribute (in opposite directions) to neutropenia and/or cancer. Indeed, the status of HAX1 has already been shown to affect proliferation [
29], and we also observed this effect in our cell lines (data not shown). Further research should elucidate the exact role of HAX1 in maintaining translation efficiency, but the results presented here provide a starting point to explore these new and unanticipated possibilities.
4. Materials and Methods
4.1. Generation of Cell Lines
4.1.1. HEK293FlpInTRex with Induced Overexpression of HAX1
Plasmid design and molecular subcloning: HAX1 CDS was obtained by PCR and cloned into prepared vector (pcDNA5FRT TO) with a special gene coding tag (Protein A fragment, TEV protease cleavage site, and His-tag) in two orientations (tag at the 3′ or 5′ end of the HAX1 coding sequence).
Cell line generation: HEK293-FlpIn cells (ThermoFisher Scientific) were transfected with an empty vector (negative control, NC) or plasmid with the HAX1 cDNA with a tag at the 3′ end or 5′ end of the gene, and cotransfected with pOG44 vector, containing Flp recombinase, for Flp–FRT recombination. Transfection was carried out according to the manufacturers’ instructions (LipofectamineTM2000, ThermoFisher Scientific, Waltham, MA, USA). Cells were detached and seeded on 100 mm plates in a concentration enabling obtaining single colonies (selection: Blasticidin 15 μg/mL and Higromycin B 100 μg/mL) for 2 weeks. Single colonies were passaged to 24-well plates and tested by Western blot and qPCR with and without doxycycline induction (18–48 h).
4.1.2. HL-60 HAX1 CRISPR Knockout
Plasmid design and molecular subcloning: two pairs (4 oligonucleotides) of small guide RNA (sgRNA) complementary to the
HAX1 gene were designed using the online bioinformatic tool (
https://CRISPR.mit.edu, accessed on 15 August 2022). Each pair of sgRNA was introduced to the AIO-GFP plasmid that encodes the Cas9 nickase tagged with EGFP (D10A), as described in [
30]. Briefly, each of the four oligonucleotides contained 5’ overhangs (forward: ACCG, reverse: AAAC) compatible with BbsI and BsaI restriction enzymes. The BbsI site present in AIO-GFP was utilized to introduce antisense (LC—left CRISPER) oligonucleotides and therefore generate AIO-GFP HAX1 LC1 and AIO-GFP HAX1 LC2 plasmids. To each respective plasmid, the second (sense, RC—right CRISPER) sgRNA of a given pair was introduced utilizing the BsaI site. As a result, two different constructs were generated, AIO-GFP HAX1 LCRC1 and AIO-GFP HAX1 LCRC2.
Cell line generation: HL-60 cell line was grown in RPMI1640 medium with L-glutamine (Biowest) and 10% Fetal Bovine Serum (Gibco) at 37 °C in a 5% CO
2. Electroporation of 5 × 106 HL-60 cells was carried out with AIO-GFP HAX1 LCRC1 and AIO-GFP HAX1 LCRC2 using the CLB-Transfection™ Kit (Lonza, Austria) and the CLB-Transfection™ System (Lonza, Austria) with the default program 9 setting. Single transfected cells were sorted into separate wells of 96-well culture plate using BD FACSAria™ III (Becton Dickinson, Franklin Lakes, NJ, USA). Cells were cultured in RPMI for 14–21 days, colonies were propagated, and successful KO was validated by Western blot in four cell lines, two of which were used in experiments (
Figure S1).
4.2. RIP-Seq
The HL-60 promyelocytic cell line (DSMZ, Braunschweig, Germany) was grown in RPMI1640 medium with L-glutamine (Biowest) and 10% Fetal Bovine Serum (Gibco) at 37 °C in a 5% CO
2. Sample preparation: The experiment was carried out in five replicates. The EZ-Magna RIP RNA-Binding Protein Immunoprecipitation Kit (Millipore, 17–700 Sigma-Aldrich) was used according to the manufacturer’s protocol. A single freeze–thaw was employed to gently lyse the cells, as described by Keene et al. [
31]. An amount of 30 × 106 cells per sample were collected by centrifugation at 966×
g, 5 min at 4 °C, and washed two times with PBS containing protease inhibitors, resuspended in 200 μL of RIP lysis buffer (Millipore) containing protease and RNase inhibitors, incubated for 5 min on ice, snap-frozen in liquid nitrogen, and stored at −80 °C. Magnetic Beads Protein A/G (from Magna RIP Kit) were incubated overnight (4 °C) with 10 µg of anti-HAX1 rabbit polyclonal antibody (Thermo Fisher Scientific, Waltham, MA, USA; cat. PA5-27592) and Rabbit IgG (Millipore) as a negative control. The lysates from the previous step were quickly thawed, centrifuged at 14,000 rpm for 10 min at 4 °C, and 150 μL of supernatants were added to each antibody complex in the RIP Immunoprecipitation Buffer. The final volume of the immunoprecipitation reaction was 1.5 mL. A total of 10% of the sample was taken and stored as a total input. The lysate was incubated with antibody-coated beads for 4 h at 4 °C. After immunoprecipitation, the beads were washed 5 times with 1 mL of cold RIP Wash Buffer. The last, sixth wash was performed with 0.5 mL of wash buffer and 50 μL out of 500 μL of each beads’ suspension was taken to test the efficiency of immunoprecipitation by Western blotting. The remaining 450 μL of each suspension was collected with magnetic separator, immune complexes and input were eluted and treated with proteinase K (55 °C for 30 min). RNA was purified by extraction of phenol/chloroform followed by ethanol precipitation. The concentration of the precipitated RNA samples was checked using QuantiFluor RNA System (Promega), and the samples were used for library preparation and RIP-seq.
4.3. RNA-Seq
Sample preparation: HL-60 cell lines (WT and HAX1 KO#1 and #2) were grown to 3.5 × 106 cells in each culture (each cell line in four replicates). RNA was isolated using RNA PureLink Mini (Thermofisher, Waltham, MA, USA). Genomic DNA was removed from the samples using the TURBO DNA-free kit (Thermofisher, Waltham, MA, USA). RNA integrity was evaluated using Agilent RNA 6000 Nano Kit (Agilent Technologies, Waltham, MA, USA). RNA samples with a RIN score ≥ 9 were used for the preparation of cDNA libraries.
4.4. NGS Library Preparation and Sequencing (RIP-Seq, RNA-Seq)
The cDNA libraries were prepared using TruSeq™ Stranded Total RNA Library Prep Gold (Illumina, San Diego, CA, USA) according to the manufacturer’s procedure. The average size of the libraries was determined using the Agilent 2100 Bioanalyzer and High Sensitivity DNA Kit (Agilent Technologies, USA), while the concentration was assessed using the Qubit Fluorometer and dsDNA HS Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA). Uniquely indexed libraries were pooled, mixed with Illumina PhiX Control v3 Library (1% of the total amount), and sequenced on HiSeq 1500 (Illumina) in Rapid Run Mode. Single-read sequencing (1 × 50 bp) and paired-end sequencing (2 × 100 bp) were performed for RIP-seq and RNA-seq, respectively.
4.5. RIP-Seq and RNA-Seq Data Analysis
4.5.1. RIP-Seq
All experiments were performed in five replicates. Raw sequences were trimmed according to quality using Trimmomatic [
32] (version 0.39) using default parameters, except MINLEN, which was set to 50. Trimmed sequences were mapped to the human reference genome provided by ENSEMBL (version grch38_snp_tran) using Hisat2 [
33] with default parameters. Optical duplicates were removed using the MarkDuplicates tool from GATK [
34] package (version 4.1.2.0) with default parameters, except OPTICAL_DUPLICATE_PIXEL_DISTANCE, which was set to 12000. The mapped reads were associated with transcripts from the grch38 database [
35] (Ensembl, version 96) using HTSeq-count [
36] (version 0.9.1) with default parameters, except for the stranded, which set to “reverse”. The variation was assessed by visual inspection of the first two components from the principal component analysis (PCA), which revealed correct clustering. FPKM was calculated with the fpkm function of the deseq2 package. Differentially expressed genes were selected using the DESeq2 package [
37] (version 1.16.1). The fold change was corrected using apeglm. PCA, FPKM, and Deseq2 calculations were performed in the R environment (version 3.6).
4.5.2. RNA-Seq
All experiments were carried out in four replicates. The DEGs selection was performed as described above.
4.6. CRAC (Crosslinking and Analyses of cDNAs)
4.6.1. Sample Preparation
Stable established cell lines with HAX1 overexpression (coding protein tagged at the C or N terminal end, two replicates for each construct) were induced with 1 μg/mL doxycycline and incubated 18–48 h. After, the incubation cells were UV-crosslinked in Stratalinker 1800 (E = 400 mJ/cm2) at a wavelength 254 nm. Cells were lysed for 10 min on ice in lysis buffer (50 mM Tris-HCl pH 7.5, 300 mM NaCl, 1% NP-40, 5 mM EDTA, 10% glycerol, 5 mM β-mercaptoethanol). Lysates were spun at 4600 RPM, 4 °C for 5 min, and then filtered using syringe filter of 0.45 µm with PES membrane. IgG Sepharose was added to the lysates and incubated overnight at 4 °C with rotation. The beads were washed with IgG wash buffer (50 mM Tris-HCl pH 7.5, 800 mM NaCl, 0.5% NP-40, 5 mM MgCl2) and PNK wash buffer (25 mM Tris-HCl pH 7.5, 50 mM NaCl, 0.1% NP-40, 1 mM MgCl2). RNAs were trimmed on beads using 1 unit of RNAce-IT in 0.4 of PNK buffer for 7 min at 37 °C. To stop the reaction, the supernatant with RNaceIT was removed and the beads were resuspended in the room temperature denaturing elution buffer Ni-WBI (50 mM Tris-HCl pH 7.5, 300 mM NaCl, 1.5 mM MgCl2, 10 mM Imidazole pH 8.0, 0.1% NP-40, and 6 M guanidine hydrochloride). The elution was repeated one more time, both fractions were combined, and the Ni-NTA beads were added for overnight incubation at 4 °C. The NI-NTA beads were washed and transferred to Pierce columns. RNA was dephosphorylated with 8 U of Thermosensitive Alkaline Phosphatase (Promega) in supplied MultiCore buffer with 80 U RNAsin for 30 min at 37 °C. Beads were washed with Ni-WBI and PNK wash buffer. The 3′ linker ligation was performed overnight at 16 °C with 1 μM 3′ linker, 800 U of truncated T4 RNA ligase 2 K227Q (New England Biolabs) in supplied PNK buffer with RNAsin (Promega), and 10% (PEG8000). The beads were washed with WBI and PNK wash buffer. The RNA–protein complexes were radioactively labeled with 32P-γ-ATP (20 μCi) using 40 U T4 Polynucleotide Kinase (New England Biolabs) in the supplied PNK buffer for 30 min at 37 °C. The 5′ linker ligation was performed in the same reaction mixture by addition of the 5′ linker to the final 2.5 mM, nonradioactive ATP to final 1.25 mM, and 40 U T4 RNA ligase 1 for 8 h at 16 °C. After washing the beads with the Ni-WBI and PNK wash buffer, the RNA–protein complexes were eluted with elution buffer (50 mM Tris-Hcl pH 7.8, 300 mM NaCl, 150 mM Imidazole pH 8.0, 0.1% NP-40, 5 mM 2-mercaptoethanol) at room temperature for 5 min. Protein–RNA complexes were precipitated with 80% acetone in the presence of GlycoBlue at −20 °C overnight and spun for 20 min with max speed at 4 °C. Pellets were resuspended in LDS sample buffer (ThermoFisher, Waltham, MA, USA), DTT, and EDTA and denatured for 3 min at 90 °C.
4.6.2. Autoradiography
Samples were resolved on 4–12% Bis-Tris NuPAGE gel at constant voltage (120 V) using NuPAGE MOPS SDS running buffer (Thermo Fisher Scientific, Waltham, MA, USA) and transferred to nitrocellulose membrane in NuPAGE transfer buffer using BioRad Protean wet transfer system at constant voltage (100 V) for 1 h. Exposition was performed at −80 °C overnight.
4.6.3. RNA Isolation from Membrane
Bands corresponding to RNA crosslinked to the HAX1 protein (
Figure 1D) were cut out and incubated with 450 μL Proteinase K buffer (50 mM Tris-HCl pH 7.8, 50 mM NaCl, 10 mM imidazole pH 8.0, 0.1% NP-40, 1% SDS, 5 mM EDTA, 5 mM 2-mercaptoethanol) and 200 μg Proteinase K for 2 h at 55 °C. The 3M sodium acetate pH 5.2 was added to final 10% and the RNA was extracted with 500 µL phenol:chloroform:isoamyl alcohol. After 5 min spin with a maximum speed at 4 °C, the aqueous phase was collected to a new tube and the RNA was precipitated with 3 volumes of ethanol in the presence of GlucoBlue.
4.6.4. cDNA Library
Isolated RNA was incubated with SSIV reverse transcriptase (ThermoFisher) and a primer binding to 3′ linker. The cDNA was amplified using LA Takara Taq polymerase. PCR products were resolved on 3% Metaphor agarose gel (Lonza), and DNA fragments of sizes approximately 150–200 nt were isolated from the gel using Qiagen’s Gel Extraction Kit (
Figure 1D). The cDNA library was sequenced on the Illumina MiSeq platform in Edinburgh Genomics (the University of Edinburgh).
4.7. CRAC Data Analysis
For CRAC experiments, all experiments (for N- and C-tagged protein) were performed in two replicates each. Fold change for each gene was calculated as a Log2 from the experiment/negative control ratio, normalized for total number of hits, and means from two replicates. NGS results were analyzed using algorithms: flexbar (preprocessing), tophat (genome mapping), and bedtools (analysis of genomic annotations). To identify HAX1 binding motifs, we extracted the coordinates of all HAX1 binding sites in mRNAs and created a control dataset by randomly placing the coordinates of these binding sites on the same mRNAs using shuffleBed. We then used the STREME sequence motif discovery algorithm (minimum motif length 4 nt, maximum length 8 nt) to identify enriched motifs [
38] (
https://meme-suite.org/meme/doc/streme.html, accessed on 15 August 2022).
Genomic position of HAX1 binding identification was done using UCSC Genome Browser on Human Feb. 2009 (GRCh37/hg19) Assembly.
4.8. Gene Ontology and Correlation Analysis
Enrichment plots for RNA-seq data were generated by GSEA (gene set enrichment analysis, [
39,
40]) software. Table of transcripts, identified during the RNA-seq experiment, with an associated number of counts per transcript (data obtained with HTSeq-count), was used as input data. Number of permutations was set to 1000 and permutation type was set to “gene_set”. Gene set database (version 7.2) used for analysis is included in the respective enrichment plots titles. The other gene ontology enrichment analyses were performed using packages: Gene Ontology resource (
http://geneontology.org, accessed on 15 August 2022); ([
41] Gene Ontology 2021), DAVID Functional Annotation Tool, DAVID Bioinformatics Resources, NIAID/NIH (
https://david.ncifcrf.gov/home.jsp, accessed on 15 August 2022) [
42,
43], and String 11 Functional Enrichment Analysis for proteins with values/ranks (
https://string-db.org, accessed on 15 August 2022) [
44]. Transcription factors were analyzed using Enrichr ENCODE and ChEA Consensus TFs from ChIP-X (
https://maayanlab.cloud/Enrichr/, accessed on 15 August 2022) [
45,
46].
Correlation analysis with high-throughput data accumulated in TCGA (The Cancer Genome Atlas) database was performed using cBioPortal for Cancer Genomics, a platform for exploring multidimensional cancer genomic data [
47,
48] (
https://www.cbioportal.org/, accessed on 15 August 2022 ).
4.9. Transcription In Vitro
Transcription in vitro was performed for 140 nt RPL19 fragment of CDS from exon 3 (primers: FW 5′-GGTGCATTATGCTTTCCCAGGTCAG-3′, REV 5′-CTATGCCCATGTGCCTGCCCTTC-3′) cloned into pGEM-T Easy vector in sense and antisense orientation. M13 fwd and rev primers were used in the PCR reaction for template generation. MEGAscript T7 transcription Kit (ThermoFisher Scientific) was used for in vitro transcription with T7 RNA polymerase, according to the manufacturers’ protocol. Transcripts were purified using MEGAclear Kit (ThermoFisher Scientific, Waltham, MA, USA).
4.10. Microscale Thermophoresis (MST)
MST experiments were performed using Monolith NT.115 (NanoTemper Technologies GmbH, Munich, Germany). Purified HAX1 protein (Proteintech, Ag27244, fused with His-tag) was labeled with RED-NHS 2nd Generation dye according to the supplied labeling protocol Monolith NT™ Protein Labeling Kit. A series of dilutions of ligand RNA (sense and antisense transcript) were prepared using buffer solution containing PBS with 0.2% Tween-20. The solution of labeled protein was mixed 1:1 with different concentrations of RNA strand, yielding a final concentration of 50 nM of the protein and the ligand in a range of final concentrations between 10.8 µM and 0.000328 µM. After 5 min of incubation, the NT.115 premium capillaries (NanoTemper Technologies, Munich, Germany) were filled with the RNA/protein solution and thermophoresis was measured at an LED power of 100% and an MST power of 60% at RT. Each operation was controlled using the MO control software. The Kd was determined by nonlinear fitting of the thermophoresis responses and EC50 was determined by Hill fitting model using the MO Affinity Analysis v2.3 for both types of the calculations.
4.11. Western Blot
Protein extracts were heat-denaturated (95 °C) in Laemmli buffer (50 mM Tris/HCl, 0.01% Bromophenol Blue, 1.75% 2-mercaptoethanol, 11% glycerol, 2% SDS) and separated by 10–12% SDS/PAGE electrophoresis. Proteins were transferred to Immobilon-P PVDF membrane (Merck Millipore, Burlington, MA, USA). The membranes were incubated for 1h using 5% low-fat milk solution in 1X TBS (50 mM Tris-Cl, pH 7.5, 150 mM NaCl) as a blocking buffer, and then overnight at 4 °C in the same blocking solution containing one of the following antibodies: anti-HAX1 (rabbit, Proteintech 11266-1-AP) or anti-RPL26 (rabbit, 1:5000, Abcam, ab59567). After washing (3 × 10 min in TBS), membranes were incubated for 2 h at room temperature with the adequate HRP-conjugated secondary antibody: goat anti-rabbit IgG (1:5000, Abcam, GB; cat. 97051) or goat anti-mouse IgG (1:10,000, Abcam, GB; cat. ab97023). Membranes were developed using HRP detection kit WesternBright Quantum (Advansta, San Jose, CA, USA; cat. K-12042).
4.12. qPCR
Quantitative PCR was performed as described [
11]. Briefly, stable cell lines with HAX1 WT and HAX1 KO were subjected to Actinomycin D treatment (10 μg/mL). Cells were harvested in a designated time points and used for total RNA preparation (PureLink RNA mini kit; Invitrogen), followed by the treatment with recombinant DNase I (Roche). An amount of 1 μg of the obtained RNA was used for cDNA synthesis using Superscript III (Invitrogen). The cDNA was quantified by quantitative PCR on an ABI Prism 7500 real-time PCR system using Power SYBR Green PCR Master Mix (Applied Biosystems, Life Technologies, Carlsbad, CA, USA) and primers amplifying a fragment of DHX37 transcript (forward 5′- CCCGATATCGAGAAAGCCTGG-3′; reverse 5′-CGTCCAGCACGTGAGATGAA-3′), RRP7 transcript: (forward 5′- TTCTCGTCACAAGGCACAGG-3′; reverse 5′-GAAGGGCCACACCTAAGTCC-3′) and, as a reference, ACTB transcript (forward 5′-AGCCTCGCCTTTGCCGA-3′; reverse 5′-GCGCGGCGATATCATCATC-3′). The ΔΔ CT method was used for calculating mRNA expression levels.
4.13. Sucrose Gradient Centrifugation
HL-60 cell lines (WT and HAX1 KO#1 and #2) were grown and subcultured until achieving 6 T-75 or 3 T-175 flasks with cells at a density of 1 × 106/mL. Cells were treated with cycloheximide (100 μg/mL) at 37 °C for 10 min, harvested by centrifugation for 5 min at 500× g, 4 °C, and washed 3 times with ice-cold PBS supplemented with 100 μg/mL cycloheximide. After final wash and complete removal of PBS, the cell pellet was resuspended in 0.75 mL of lysis buffer A (10 mM Tris-HCl pH 7.4, 12.5 mM MgCl2, 100 mM KCl, 0.5% Triton X-100 reduced, 2 mM DTT, 100 µg/mL CHX, 200 units SUPERaseIn™ RNase Inhibitor (20 U/μL; ThermoScientific), and cOmplete EDTA-free protease inhibitor (Roche)). Cells were lysed by thorough pipetting and incubation for 15 min at 4 °C on a rotating wheel. Lysates were aspirated into 1 mL syringe, passed through a 26G needle seven times, and then centrifuged for 10 min at 16,000× g, 4 °C. RNA concentration in clarified cytoplasmic extracts thus obtained was measured using Nanodrop 2000c (ThermoScientific). A total of 14–20 OD260 units of cytoplasmic extract in 500 μL of lysis buffer was layered on top of 10–50% linear sucrose gradients, prepared using ÅKTA Purifier FPLC system and 0.22 µm-filtered sucrose solutions in polysome buffer (20 mM Hepes-KOH pH 7.4, 12.5 mM MgCl2, 100 mM KCl; 2 mM DTT, 100 µg/mL CHX, and cOmplete EDTA-free protease inhibitor), and ultracentrifuged for 3 h 15′ at 36,000 rpm, 4 °C in SW-41Ti rotor (Beckman Coulter). Subsequently, 0.5 mL fractions were collected from gradient by pumping 60% sucrose solution in polysome buffer to the bottom of tubes, and OD254 was monitored on an ÅKTA Purifier.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







