
Neuroinflammation and Parkinson’s Disease—From Neurodegeneration to Therapeutic Opportunities

 , , ,
, , , 
Abstract
1. Introduction
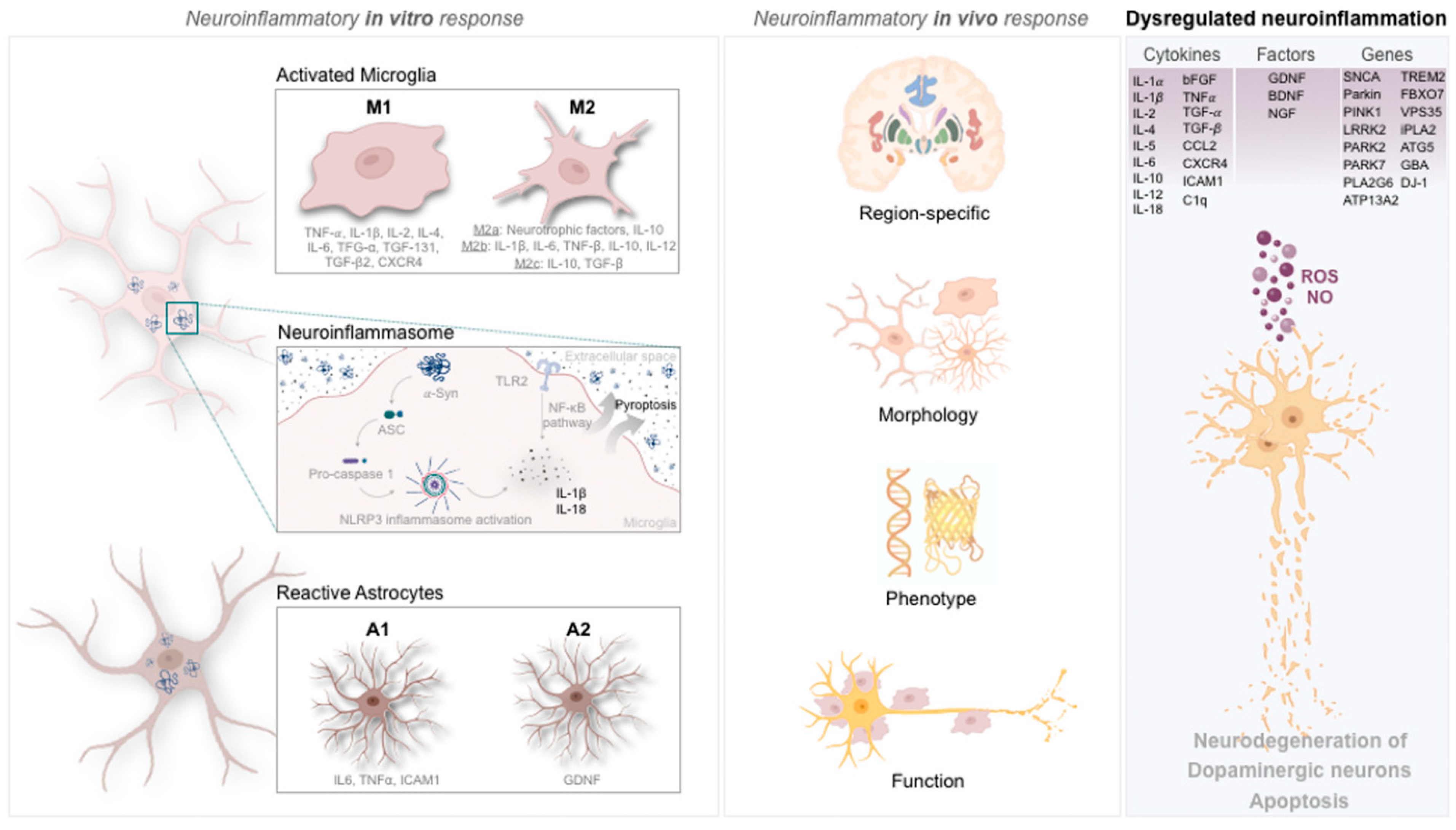
2. Microglia in Parkinson’s Disease
2.1. Cytokine and Genetic Signature of Microglia in Parkinson’s Disease
2.2. Microglia Phenotypic Portrayal in Parkinson’s Disease
3. Astrocytes in Parkinson’s Disease
3.1. Cytokine and Genetic Signature of Astrocytes in Parkinson’s Disease
3.2. Astrocytes’ Phenotypic Portrayal in Parkinson’s Disease
4. Inflammasome
5. Acute Versus Chronic Inflammation
5.1. What Came First: Neuroinflammation or Dopaminergic Neurodegeneration?
5.2. Microglia Activation Preceding the Neurodegenerative Processes: From α-Syn Accumulation to Aging
5.3. Astrogliosis Manifestation in Parkinson’s Disease
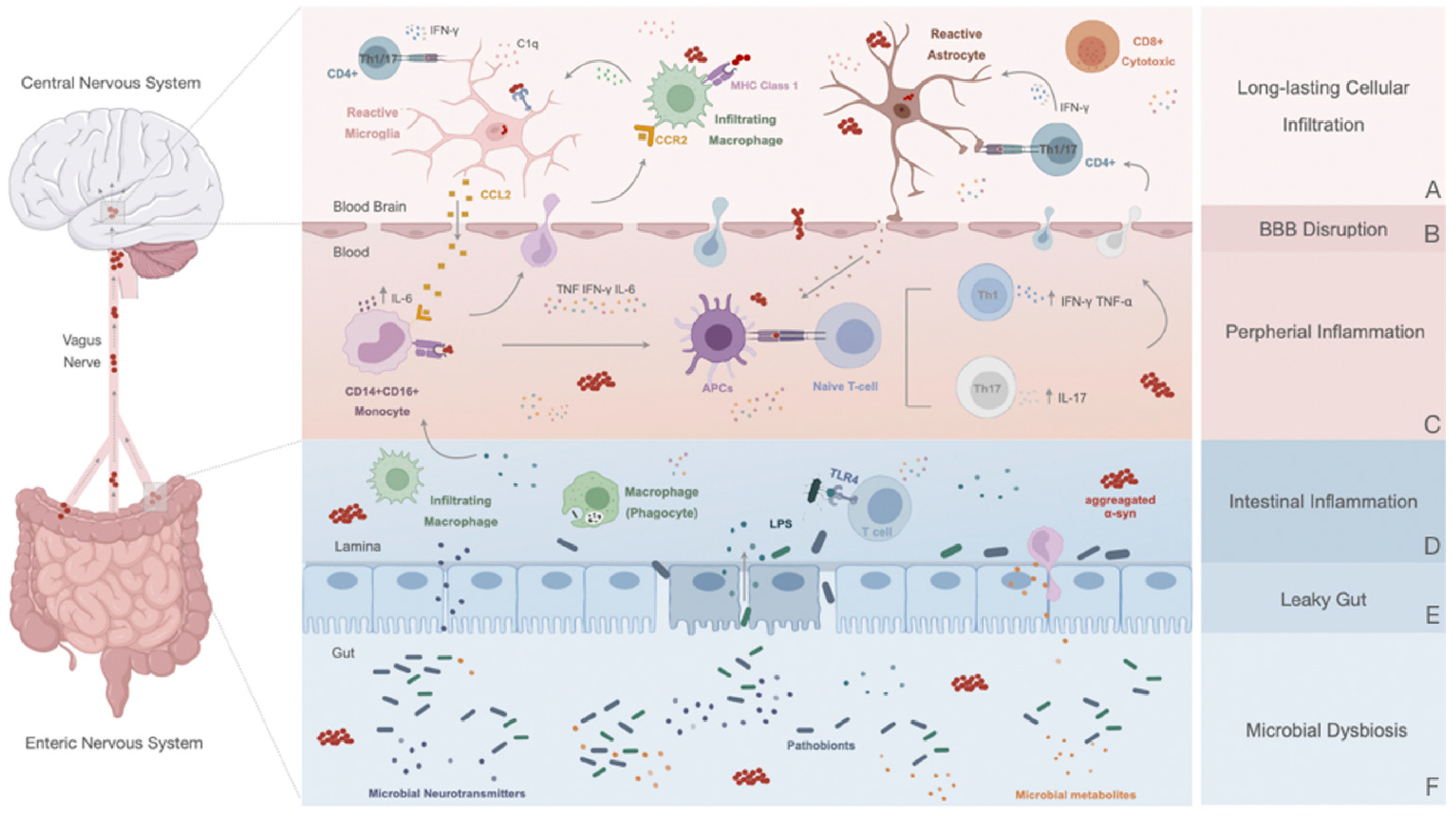
6. Peripheral Response in Parkinson’s Disease
6.1. Disruption of the Blood–Brain Barrier: A Contributor to PD Immune Dysfunction
6.2. Innate Immune Response
6.3. Adaptive Immune Response: The Role of T and B Cells in PD
7. The Peripheral Enteric Nervous System and the Gut–Brain Axis
7.1. Altered Gut in Parkinson’s Disease: A Promotor or a Consequence of the Inflammatory State?
7.2. Gut–Brain Communication: α-Syn Spreading and Novel Initial Sites of Inflammation
8. Therapeutic Approaches to Target Neuroinflammation in PD
8.1. Immunotherapy for Alpha-Synuclein (α-Syn) Aggregation
8.2. Vagotomy and Appendectomy
8.3. Nonsteroidal Anti-Inflammatory Drugs
8.4. Food-Based Therapies and Physical Exercise
9. Gaps in the Literature and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Kölliker-Frers, R.; Udovin, L.; Otero-Losada, M.; Kobiec, T.; Herrera, M.I.; Palacios, J.; Razzitte, G.; Capani, F. Neuroinflammation: An Integrating Overview of Reactive-Neuroimmune Cell Interactions in Health and Disease. Mediat. Inflamm. 2021, 2021, 9999146. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J.; Tan, E.K. Parkinson’s disease: Etiopathogenesis and treatment. J. Neurol. Neurosurg. Psychiatry 2020, 91, 795–808. [Google Scholar] [CrossRef] [PubMed]
- Seppi, K.; Ray Chaudhuri, K.; Coelho, M.; Fox, S.H.; Katzenschlager, R.; Perez Lloret, S.; Weintraub, D.; Sampaio, C.; Chahine, L.; Hametner, E.M.; et al. Update on treatments for nonmotor symptoms of Parkinson’s disease—An evidence-based medicine review. Mov. Disord. 2019, 34, 180–198. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Trapp, B.D. Microglia and neuroprotection. J. Neurochem. 2016, 136, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Surmeier, D.J.; Obeso, J.A.; Halliday, G.M. Selective neuronal vulnerability in Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Hijaz, B.A.; Volpicelli-Daley, L.A. Initiation and propagation of alpha-synuclein aggregation in the nervous system. Mol. Neurodegener. 2020, 15, 19. [Google Scholar] [CrossRef]
- El-Agnaf, O.M.; Salem, S.A.; Paleologou, K.E.; Cooper, L.J.; Fullwood, N.J.; Gibson, M.J.; Curran, M.D.; Court, J.A.; Mann, D.M.; Ikeda, S.; et al. Alpha-synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. FASEB J. 2003, 17, 1945–1947. [Google Scholar] [CrossRef]
- Li, Y.Y.; Zhou, T.T.; Zhang, Y.; Chen, N.H.; Yuan, Y.H. Distribution of alpha-Synuclein Aggregation in the Peripheral Tissues. Neurochem. Res. 2022; Online ahead of print. [Google Scholar] [CrossRef]
- Mollenhauer, B.; Locascio, J.J.; Schulz-Schaeffer, W.; Sixel-Döring, F.; Trenkwalder, C.; Schlossmacher, M.G. α-Synuclein and tau concentrations in cerebrospinal fluid of patients presenting with parkinsonism: A cohort study. Lancet Neurol. 2011, 10, 230–240. [Google Scholar] [CrossRef]
- Braak, H.; Sastre, M.; Del Tredici, K. Development of alpha-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson’s disease. Acta Neuropathol. 2007, 114, 231–241. [Google Scholar] [CrossRef]
- Rostami, J.; Holmqvist, S.; Lindstrom, V.; Sigvardson, J.; Westermark, G.T.; Ingelsson, M.; Bergstrom, J.; Roybon, L.; Erlandsson, A. Human Astrocytes Transfer Aggregated Alpha-Synuclein via Tunneling Nanotubes. J. Neurosci. 2017, 37, 11835–11853. [Google Scholar] [CrossRef] [PubMed]
- Scheiblich, H.; Dansokho, C.; Mercan, D.; Schmidt, S.V.; Bousset, L.; Wischhof, L.; Eikens, F.; Odainic, A.; Spitzer, J.; Griep, A.; et al. Microglia jointly degrade fibrillar alpha-synuclein cargo by distribution through tunneling nanotubes. Cell 2021, 184, 5089–5106.e5021. [Google Scholar] [CrossRef] [PubMed]
- George, S.; Rey, N.L.; Tyson, T.; Esquibel, C.; Meyerdirk, L.; Schulz, E.; Pierce, S.; Burmeister, A.R.; Madaj, Z.; Steiner, J.A.; et al. Microglia affect alpha-synuclein cell-to-cell transfer in a mouse model of Parkinson’s disease. Mol. Neurodegener. 2019, 14, 34. [Google Scholar] [CrossRef] [PubMed]
- Hanisch, U.K. Microglia as a source and target of cytokines. Glia 2002, 40, 140–155. [Google Scholar] [CrossRef] [PubMed]
- Kam, T.I.; Hinkle, J.T.; Dawson, T.M.; Dawson, V.L. Microglia and astrocyte dysfunction in parkinson’s disease. Neurobiol. Dis. 2020, 144, 105028. [Google Scholar] [CrossRef]
- Salter, M.W.; Stevens, B. Microglia emerge as central players in brain disease. Nat. Med. 2017, 23, 1018–1027. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef]
- Glezer, I.; Simard, A.R.; Rivest, S. Neuroprotective role of the innate immune system by microglia. Neuroscience 2007, 147, 867–883. [Google Scholar] [CrossRef]
- Le, W.; Wu, J.; Tang, Y. Protective microglia and their regulation in Parkinson’s disease. Front. Mol. Neurosci. 2016, 9, 89. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Riederer, P.; Narabayashi, H.; Fujita, K.; Nagatsu, T. Tumor necrosis factor-α (TNF-α) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci. Lett. 1994, 165, 208–210. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Kondo, T.; Riederer, P.; Inagaki, H.; Minami, M.; Nagatsu, T. Interleukin-1β, interleukin-6, epidermal growth factor and transforming growth factor-α are elevated in the brain from parkinsonian patients. Neurosci. Lett. 1994, 180, 147–150. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Kondo, T.; Riederer, P.; Nagatsu, T. Interleukin-2 but not basic fibroblast growth factor is elevated in parkinsonian brain. J. Neural Transm. 1996, 103, 1077–1081. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Kondo, T.; Narabayashi, H.; Riederer, P.; Nagatsu, T. Transforming growth factor-β1 levels are elevated in the striatum and in ventricular cerebrospinal fluid in Parkinson’s disease. Neurosci. Lett. 1995, 193, 129–132. [Google Scholar] [CrossRef]
- Vawter, M.P.; Dillon-Carter, O.; Tourtellotte, W.W.; Carvey, P.; Freed, W.J. TGFβ1 and TGFβ2 concentrations are elevated in Parkinson’s disease in ventricular cerebrospinal fluid. Exp. Neurol. 1996, 142, 313–322. [Google Scholar] [CrossRef]
- Scalzo, P.; Kümmer, A.; Cardoso, F.; Teixeira, A.L. Increased serum levels of soluble tumor necrosis factor-α receptor-1 in patients with Parkinson’s disease. J. Neuroimmunol. 2009, 216, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Harada, M.; Narabayashi, H.; Inagaki, H.; Minami, M.; Nagatsu, T. Interleukin (IL)-1β, IL-2, IL-4, IL-6 and transforming growth factor-α levels are elevated in ventricular cerebrospinal fluid in juvenile parkinsonism and Parkinson’s disease. Neurosci. Lett. 1996, 211, 13–16. [Google Scholar] [CrossRef]
- Sawada, M.; Suzumura, A.; Marunouchi, T. Cytokine network in the central nervous system and its roles in growth and differentiation of glial and neuronal cells. Int. J. Dev. Neurosci. 1995, 13, 253–264. [Google Scholar] [CrossRef]
- Sawada, M.; Suzumura, A.; Hosoya, H.; Marunouchi, T.; Nagatsu, T. Interleukin-10 inhibits both production of cytokines and expression of cytokine receptors in microglia. J. Neurochem. 1999, 72, 1466–1471. [Google Scholar] [CrossRef]
- Dobbs, R.J.; Charlett, A.; Purkiss, A.G.; Dobbs, S.M.; Weller, C.; Peterson, D.W. Association of circulating TNF-alpha and IL-6 with ageing and parkinsonism. Acta Neurol. Scand. 1999, 100, 34–41. [Google Scholar] [CrossRef]
- Gerhard, A.; Pavese, N.; Hotton, G.; Turkheimer, F.; Es, M.; Hammers, A.; Eggert, K.; Oertel, W.; Banati, R.B.; Brooks, D.J. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol. Dis. 2006, 21, 404–412. [Google Scholar] [CrossRef]
- Ouchi, Y.; Yoshikawa, E.; Sekine, Y.; Futatsubashi, M.; Kanno, T.; Ogusu, T.; Torizuka, T. Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann. Neurol. 2005, 57, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Stokholm, M.G.; Iranzo, A.; Østergaard, K.; Serradell, M.; Otto, M.; Svendsen, K.B.; Garrido, A.; Vilas, D.; Borghammer, P.; Santamaria, J.; et al. Assessment of neuroinflammation in patients with idiopathic rapid-eye-movement sleep behaviour disorder: A case-control study. Lancet Neurol. 2017, 16, 789–796. [Google Scholar] [CrossRef]
- Chen, D.; Zhu, C.; Wang, X.; Feng, X.; Pang, S.; Huang, W.; Hawley, R.G.; Yan, B. A novel and functional variant within the ATG5 gene promoter in sporadic Parkinson’s disease. Neurosci. Lett. 2013, 538, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Wu, J.; Koc, S.; Lu, G. Genetic Imaging of Neuroinflammation in Parkinson’s Disease: Recent Advancements. Front. Cell Dev. Biol. 2021, 9, 655819. [Google Scholar] [CrossRef]
- Gosselin, D.; Skola, D.; Coufal, N.G.; Holtman, I.R.; Schlachetzki, J.C.M.; Sajti, E.; Jaeger, B.N.; O’Connor, C.; Fitzpatrick, C.; Pasillas, M.P.; et al. An environment-dependent transcriptional network specifies human microglia identity. Science 2017, 356, 1248–1259. [Google Scholar] [CrossRef]
- Belloli, S.; Morari, M.; Murtaj, V.; Valtorta, S.; Moresco, R.M.; Gilardi, M.C. Translation Imaging in Parkinson’s Disease: Focus on Neuroinflammation. Front. Aging Neurosci. 2020, 12, 152. [Google Scholar] [CrossRef]
- Booth, H.D.E.; Hirst, W.D.; Wade-Martins, R. The Role of Astrocyte Dysfunction in Parkinson’s Disease Pathogenesis. Trends Neurosci. 2017, 40, 358–370. [Google Scholar] [CrossRef]
- Ferreira, S.A.; Romero-Ramos, M. Microglia response during Parkinson’s disease: Alpha-synuclein intervention. Front. Cell. Neurosci. 2018, 12, 247. [Google Scholar] [CrossRef]
- Mullin, S.; Stokholm, M.G.; Hughes, D.; Mehta, A.; Parbo, P.; Hinz, R.; Pavese, N.; Brooks, D.J.; Schapira, A.H.V. Brain Microglial Activation Increased in Glucocerebrosidase (GBA) Mutation Carriers without Parkinson’s disease. Mov. Disord. 2021, 36, 774–779. [Google Scholar] [CrossRef]
- Brunialti, E.; Villa, A.; Mekhaeil, M.; Mornata, F.; Vegeto, E.; Maggi, A.; Di Monte, D.A.; Ciana, P. Inhibition of microglial β-glucocerebrosidase hampers the microglia-mediated antioxidant and protective response in neurons. J. Neuroinflamm. 2021, 18, 220. [Google Scholar] [CrossRef]
- Keatinge, M.; Bui, H.; Menke, A.; Chen, Y.C.; Sokol, A.M.; Bai, Q.; Ellett, F.; Da Costa, M.; Burke, D.; Gegg, M.; et al. Glucocerebrosidase 1 deficient Danio rerio mirror key pathological aspects of human Gaucher disease and provide evidence of early microglial activation preceding alpha-synuclein-independent neuronal cell death. Hum. Mol. Genet. 2015, 24, 6640–6652. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Dammer, E.B.; Malovic, E.; Olsen, A.L.; Raza, S.A.; Gao, T.; Xiao, H.; Oliver, D.L.; Duong, D.; Joers, V.; et al. Molecular Signatures of Neuroinflammation Induced by αSynuclein Aggregates in Microglial Cells. Front. Immunol. 2020, 11, 33. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Cho, E.D.; Lee, K.W.; Kim, J.H.; Cho, S.G.; Lee, S.J. Autophagic failure promotes the exocytosis and intercellular transfer of α-synuclein. Exp. Mol. Med. 2013, 45, e22. [Google Scholar] [CrossRef] [PubMed]
- Tu, H.Y.; Yuan, B.S.; Hou, X.O.; Zhang, X.J.; Pei, C.S.; Ma, Y.T.; Yang, Y.P.; Fan, Y.; Qin, Z.H.; Liu, C.F.; et al. α-synuclein suppresses microglial autophagy and promotes neurodegeneration in a mouse model of Parkinson’s disease. Aging Cell 2021, 20, e13522. [Google Scholar] [CrossRef]
- Butler, C.A.; Popescu, A.S.; Kitchener, E.J.A.; Allendorf, D.H.; Puigdellívol, M.; Brown, G.C. Microglial phagocytosis of neurons in neurodegeneration, and its regulation. J. Neurochem. 2021, 158, 621–639. [Google Scholar] [CrossRef]
- Janda, E.; Boi, L.; Carta, A.R. Microglial phagocytosis and its regulation: A therapeutic target in parkinson’s disease? Front. Mol. Neurosci. 2018, 11. [Google Scholar] [CrossRef]
- Miki, Y.; Shimoyama, S.; Kon, T.; Ueno, T.; Hayakari, R.; Tanji, K.; Matsumiya, T.; Tsushima, E.; Mori, F.; Wakabayashi, K.; et al. Alteration of autophagy-related proteins in peripheral blood mononuclear cells of patients with Parkinson’s disease. Neurobiol. Aging 2018, 63, 33–43. [Google Scholar] [CrossRef]
- Subramaniam, S.R.; Federoff, H.J. Targeting microglial activation states as a Therapeutic Avenue in Parkinson’s disease. Front. Aging Neurosci. 2017, 9, 176. [Google Scholar] [CrossRef]
- Sousa, C.; Golebiewska, A.; Poovathingal, S.K.; Kaoma, T.; Pires-Afonso, Y.; Martina, S.; Coowar, D.; Azuaje, F.; Skupin, A.; Balling, R.; et al. Single-cell transcriptomics reveals distinct inflammation-induced microglia signatures. EMBO Rep. 2018, 19, e46171. [Google Scholar] [CrossRef]
- He, M.; Dong, H.; Huang, Y.; Lu, S.; Zhang, S.; Qian, Y.; Jin, W. Astrocyte-derived CCL2 is associated with m1 activation and recruitment of cultured microglial cells. Cell. Physiol. Biochem. 2016, 38, 859–870. [Google Scholar] [CrossRef]
- Pajares, M.; Rojo, A.I.; Manda, G.; Boscá, L.; Cuadrado, A. Inflammation in Parkinson’s Disease: Mechanisms and Therapeutic Implications. Cells 2020, 9, 1687. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Togari, A.; Kondo, T.; Mizuno, Y.; Komure, O.; Kuno, S.; Ichinose, H.; Nagatsu, T. Caspase Activities and Tumor Necrosis Factor Receptor R1 (p55) Level Are Elevated in the Substantia Nigra from Parkinsonian Brain. J. Neural Transm. 2000, 107, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Shih, R.H.; Wang, C.Y.; Yang, C.M. NF-kappaB signaling pathways in neurological inflammation: A mini review. Front. Mol. Neurosci. 2015, 8, 77. [Google Scholar] [CrossRef]
- Shimoji, M.; Pagan, F.; Healton, E.B.; Mocchetti, I. CXCR4 and CXCL12 expression is increased in the nigro-striatal system of Parkinson’s disease. Neurotox. Res. 2009, 16, 318–328. [Google Scholar] [CrossRef]
- Parrella, E.; Bellucci, A.; Porrini, V.; Benarese, M.; Lanzillotta, A.; Faustini, G.; Longhena, F.; Abate, G.; Uberti, D.; Pizzi, M. NF-κB/c-Rel deficiency causes Parkinson’s disease-like prodromal symptoms and progressive pathology in mice. Transl. Neurodegener. 2019, 8, 16. [Google Scholar] [CrossRef]
- Chhor, V.; Le Charpentier, T.; Lebon, S.; Oré, M.V.; Celador, I.L.; Josserand, J.; Degos, V.; Jacotot, E.; Hagberg, H.; Sävman, K.; et al. Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia In vitro. Brain Behav. Immun. 2013, 32, 70–85. [Google Scholar] [CrossRef]
- Tan, Y.L.; Yuan, Y.; Tian, L. Microglial regional heterogeneity and its role in the brain. Mol. Psychiatry 2020, 25, 351–367. [Google Scholar] [CrossRef]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 2019, 50, 253–271.e256. [Google Scholar] [CrossRef]
- Crotti, A.; Ransohoff, R.M. Microglial Physiology and Pathophysiology: Insights from Genome-wide Transcriptional Profiling. Immunity 2016, 44, 505–515. [Google Scholar] [CrossRef]
- De Biase, L.M.; Schuebel, K.E.; Fusfeld, Z.H.; Jair, K.; Hawes, I.A.; Cimbro, R.; Zhang, H.Y.; Liu, Q.R.; Shen, H.; Xi, Z.X.; et al. Local Cues Establish and Maintain Region-Specific Phenotypes of Basal Ganglia Microglia. Neuron 2017, 95, 341–356.e346. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Cheng, Z.; Zhou, L.; Darmanis, S.; Neff, N.F.; Okamoto, J.; Gulati, G.; Bennett, M.L.; Sun, L.O.; Clarke, L.E.; et al. Developmental Heterogeneity of Microglia and Brain Myeloid Cells Revealed by Deep Single-Cell RNA Sequencing. Neuron 2019, 101, 207–223.e210. [Google Scholar] [CrossRef] [PubMed]
- Smajic, S.; Prada-Medina, C.A.; Landoulsi, Z.; Ghelfi, J.; Delcambre, S.; Dietrich, C.; Jarazo, J.; Henck, J.; Balachandran, S.; Pachchek, S.; et al. Single-cell sequencing of human midbrain reveals glial activation and a Parkinson-specific neuronal state. Brain 2022, 145, 964–978. [Google Scholar] [CrossRef] [PubMed]
- Uriarte Huarte, O.; Kyriakis, D.; Heurtaux, T.; Pires-Afonso, Y.; Grzyb, K.; Halder, R.; Buttini, M.; Skupin, A.; Mittelbronn, M.; Michelucci, A. Single-Cell Transcriptomics and In Situ Morphological Analyses Reveal Microglia Heterogeneity Across the Nigrostriatal Pathway. Front. Immunol. 2021, 12, 639613. [Google Scholar] [CrossRef] [PubMed]
- Björklund, A.; Dunnett, S.B. Dopamine neuron systems in the brain: An update. Trends Neurosci. 2007, 30, 194–202. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist. Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef]
- Vasile, F.; Dossi, E.; Rouach, N. Human astrocytes: Structure and functions in the healthy brain. Brain Struct. Funct. 2017, 222, 2017–2029. [Google Scholar] [CrossRef]
- Abbott, N.J. Astrocyte-endothelial interactions and blood-brain barrier permeability. J. Anat. 2002, 200, 629–638. [Google Scholar] [CrossRef]
- Mena, M.A.; De Bernardo, S.; Casarejos, M.J.; Canals, S.; Rodríguez-Martín, E. The role of astroglia on the survival of dopamine neurons. Mol. Neurobiol. 2002, 25, 245–263. [Google Scholar] [CrossRef]
- Lee, H.-J.; Kim, C.; Lee, S.-J. Alpha-synuclein stimulation of astrocytes: Potential role for neuroinflammation and neuroprotection. Oxidative Med. Cell. Longev. 2010, 3, 283–287. [Google Scholar] [CrossRef]
- Gu, X.L.; Long, C.X.; Sun, L.; Xie, C.; Lin, X.; Cai, H. Astrocytic expression of Parkinson’s disease-related A53T -synuclein causes neurodegeneration in mice. Mol. Brain 2010, 3, 12. [Google Scholar] [CrossRef] [PubMed]
- Klegeris, A.; Giasson, B.I.; Zhang, H.; Maguire, J.; Pelech, S.; McGeer, P.L. Alpha-synuclein and its disease-causing mutants induce ICAM-1 and IL-6 in human astrocytes and astrocytoma cells. FASEB J. 2006, 20, 2000–2008. [Google Scholar] [CrossRef]
- Diniz, L.P.; Araujo, A.P.B.; Matias, I.; Garcia, M.N.; Barros-Aragão, F.G.Q.; de Melo Reis, R.A.; Foguel, D.; Braga, C.; Figueiredo, C.P.; Romão, L.; et al. Astrocyte glutamate transporters are increased in an early sporadic model of synucleinopathy. Neurochem. Int. 2020, 138, 104758. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, S.; Gharagozloo, M.; Simard, C.; Gris, D. Astrocytes Maintain Glutamate Homeostasis in the CNS by Controlling the Balance between Glutamate Uptake and Release. Cells 2019, 8, 184. [Google Scholar] [CrossRef]
- Argaw, A.T.; Zhang, Y.; Snyder, B.J.; Zhao, M.-L.; Kopp, N.; Lee, S.C.; Raine, C.S.; Brosnan, C.F.; John, G.R. IL-1β Regulates Blood-Brain Barrier Permeability via Reactivation of the Hypoxia-Angiogenesis Program. J. Immunol. 2006, 177, 5574–5584. [Google Scholar] [CrossRef] [PubMed]
- Argaw, A.T.; Gurfein, B.T.; Zhang, Y.; Zameer, A.; John, G.R. VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc. Natl. Acad. Sci. USA 2009, 106, 1977–1982. [Google Scholar] [CrossRef]
- Rite, I.; Machado, A.; Cano, J.; Venero, J.L. Blood-brain barrier disruption induces in vivo degeneration of nigral dopaminergic neurons. J. Neurochem. 2007, 101, 1567–1582. [Google Scholar] [CrossRef]
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020, 19, 170–178. [Google Scholar] [CrossRef]
- Sonninen, T.-M.; Hämäläinen, R.H.; Koskuvi, M.; Oksanen, M.; Shakirzyanova, A.; Wojciechowski, S.; Puttonen, K.; Naumenko, N.; Goldsteins, G.; Laham-Karam, N.; et al. Metabolic alterations in Parkinson’s disease astrocytes. Sci. Rep. 2020, 10, 14474. [Google Scholar] [CrossRef]
- Lee, S.; Jha, M.K.; Suk, K. Lipocalin-2 in the inflammatory activation of brain astrocytes. Crit. Rev. Immunol. 2015, 35, 77–84. [Google Scholar] [CrossRef] [PubMed]
- MacMahon Copas, A.N.; McComish, S.F.; Fletcher, J.M.; Caldwell, M.A. The Pathogenesis of Parkinson’s Disease: A Complex Interplay Between Astrocytes, Microglia, and T Lymphocytes? Front. Neurol. 2021, 12, 666737. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.L.; Zhao, L.; Zhao, Y.; Sun, D.; Zhu, X.J.; Mei, L.; Xiong, W.C. Coupling of terminal differentiation deficit with neurodegenerative pathology in Vps35-deficient pyramidal neurons. Cell Death Differ. 2020, 27, 2099–2116. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, Y.H.; Berman, D.E.; Marsh, S.E.; Klein, R.L.; Patel, V.M.; Simoes, S.; Kannan, S.; Petsko, G.A.; Stevens, B.; Small, S.A. The neuronal retromer can regulate both neuronal and microglial phenotypes of Alzheimer’s disease. Cell Rep. 2022, 38, 110262. [Google Scholar] [CrossRef] [PubMed]
- Qiao, C.; Yin, N.; Gu, H.Y.; Zhu, J.L.; Ding, J.H.; Lu, M.; Hu, G. Atp13a2 Deficiency Aggravates Astrocyte-Mediated Neuroinflammation via NLRP3 Inflammasome Activation. CNS Neurosci. Ther. 2016, 22, 451–460. [Google Scholar] [CrossRef]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic analysis of reactive astrogliosis. J. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The Inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Walsh, J.G.; Muruve, D.A.; Power, C. Inflammasomes in the CNS. Nat. Rev. Neurosci. 2014, 15, 84–97. [Google Scholar] [CrossRef]
- Fan, Z.; Pan, Y.T.; Zhang, Z.Y.; Yang, H.; Yu, S.Y.; Zheng, Y.; Ma, J.H.; Wang, X.M. Systemic activation of NLRP3 inflammasome and plasma α-synuclein levels are correlated with motor severity and progression in Parkinson’s disease. J. Neuroinflamm. 2020, 17, 11. [Google Scholar] [CrossRef]
- Gustot, A.; Gallea, J.I.; Sarroukh, R.; Celej, M.S.; Ruysschaert, J.M.; Raussens, V. Amyloid fibrils are the molecular trigger of inflammation in Parkinson’s disease. Biochem. J. 2015, 471, 323–333. [Google Scholar] [CrossRef]
- Heneka, M.T.; McManus, R.M.; Latz, E. Inflammasome signalling in brain function and neurodegenerative disease. Nat. Rev. Neurosci. 2018, 19, 610–621. [Google Scholar] [CrossRef]
- Codolo, G.; Plotegher, N.; Pozzobon, T.; Brucale, M.; Tessari, I.; Bubacco, L.; de Bernard, M. Triggering of Inflammasome by Aggregated α-Synuclein, an Inflammatory Response in Synucleinopathies. PLoS ONE 2013, 8, e55375. [Google Scholar] [CrossRef]
- Koprich, J.B.; Reske-Nielsen, C.; Mithal, P.; Isacson, O. Neuroinflammation mediated by IL-1β increases susceptibility of dopamine neurons to degeneration in an animal model of Parkinson’s disease. J. Neuroinflamm. 2008, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef]
- Zhou, Y.; Lu, M.; Du, R.H.; Qiao, C.; Jiang, C.Y.; Zhang, K.Z.; Ding, J.H.; Hu, G. MicroRNA-7 targets Nod-like receptor protein 3 inflammasome to modulate neuroinflammation in the pathogenesis of Parkinson’s disease. Mol. Neurodegener. 2016, 11, 28. [Google Scholar] [CrossRef] [PubMed]
- Mejias, N.H.; Martinez, C.C.; Stephens, M.E.; De Rivero Vaccari, J.P. Contribution of the inflammasome to inflammaging. J. Inflamm. 2018, 15, 23. [Google Scholar] [CrossRef]
- Sarkar, S.; Malovic, E.; Harishchandra, D.S.; Ghaisas, S.; Panicker, N.; Charli, A.; Palanisamy, B.N.; Rokad, D.; Jin, H.; Anantharam, V.; et al. Mitochondrial impairment in microglia amplifies NLRP3 inflammasome proinflammatory signaling in cell culture and animal models of Parkinson’s disease. NPJ Parkinson’s Disease 2017, 3, 30. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Ma, H.; Yv, Q.; Ye, F.; He, Z.; Chen, S.; Keram, A.; Li, W.; Zhu, M. Alpha-synuclein/MPP+ mediated activation of NLRP3 inflammasome through microtubule-driven mitochondrial perinuclear transport. Biochem. Biophys. Res. Commun. 2022, 594, 161–167. [Google Scholar] [CrossRef]
- Coll, R.C.; Hill, J.R.; Day, C.J.; Zamoshnikova, A.; Boucher, D.; Massey, N.L.; Chitty, J.L.; Fraser, J.A.; Jennings, M.P.; Robertson, A.A.B.; et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat. Chem. Biol. 2019, 15, 556–559. [Google Scholar] [CrossRef]
- Gordon, R.; Albornoz, E.A.; Christie, D.C.; Langley, M.R.; Kumar, V.; Mantovani, S.; Robertson, A.A.B.; Butler, M.S.; Rowe, D.B.; O’Neill, L.A.; et al. Inflammasome inhibition prevents -synuclein pathology and dopaminergic neurodegeneration in mice. Sci. Transl. Med. 2018, 10, 1–13. [Google Scholar] [CrossRef]
- Huang, S.; Chen, Z.; Fan, B.; Chen, Y.; Zhou, L.; Jiang, B.; Long, H.; Zhong, W.; Li, X.; Li, Y. A selective NLRP3 inflammasome inhibitor attenuates behavioral deficits and neuroinflammation in a mouse model of Parkinson’s disease. J. Neuroimmunol. 2021, 354, 577543. [Google Scholar] [CrossRef] [PubMed]
- Gelders, G.; Baekelandt, V.; Van der Perren, A. Linking neuroinflammation and neurodegeneration in parkinson’s disease. J. Immunol. Res. 2018, 2018, 4784268. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Federoff, H.J.; Maguire-Zeiss, K.A. Mutant alpha-synuclein overexpression mediates early proinflammatory activity. Neurotox. Res. 2009, 16, 238–254. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Maguire-Zeiss, K.A.; Giuliano, R.; Prifti, L.; Venkatesh, K.; Federoff, H.J. Synuclein activates microglia in a model of Parkinson’s disease. Neurobiol. Aging 2008, 29, 1690–1701. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.M.; Kiser, G.L.; Kaysser-Kranich, T.; Casaceli, C.; Colla, E.; Lee, M.K.; Palaniappan, C.; Federoff, H.J. Wild-type and mutant alpha-synuclein induce a multi-component gene expression profile consistent with shared pathophysiology in different transgenic mouse models of PD. Exp. Neurol. 2007, 204, 421–432. [Google Scholar] [CrossRef]
- Rodriguez-Pallares, J.; Parga, J.A.; Muñoz, A.; Rey, P.; Guerra, M.J.; Labandeira-Garcia, J.L. Mechanism of 6-hydroxydopamine neurotoxicity: The role of NADPH oxidase and microglial activation in 6-hydroxydopamine-induced degeneration of dopaminergic neurons. J. Neurochem. 2007, 103, 145–156. [Google Scholar] [CrossRef]
- Sherer, T.B.; Betarbet, R.; Kim, J.H.; Greenamyre, J.T. Selective microglial activation in the rat rotenone model of Parkinson’s disease. Neurosci. Lett. 2003, 341, 87–90. [Google Scholar] [CrossRef]
- Castaño, A.; Herrera, A.J.; Cano, J.; Machado, A. Lipopolysaccharide Intranigral Injection Induces Inflammatory Reaction and Damage in Nigrostriatal Dopaminergic System. J. Neurochem. 1998, 70, 1584–1592. [Google Scholar] [CrossRef] [PubMed]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef]
- Harms, A.S.; Ferreira, S.A.; Romero-Ramos, M. Periphery and brain, innate and adaptive immunity in Parkinson’s disease. Acta Neuropathol. 2021, 141, 527–545. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhang, W.; Zhou, Y.; Hong, J.-S.; et al. Aggregated α-synuclein activates microglia: A process leading to disease progression in Parkinson’s disease. FASEB J. 2005, 19, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chu, C.H.; Stewart, T.; Ginghina, C.; Wang, Y.; Nie, H.; Guo, M.; Wilson, B.; Hong, J.S.; Zhang, J. α-Synuclein, a chemoattractant, directs microglial migration via H2O2-dependent Lyn phosphorylation. Proc. Natl. Acad. Sci. USA 2015, 112, E1926–E1935. [Google Scholar] [CrossRef] [PubMed]
- Croisier, E.; Moran, L.B.; Dexter, D.T.; Pearce, R.K.B.; Graeber, M.B. Microglial inflammation in the parkinsonian substantia nigra: Relationship to alpha-synuclein deposition. J. Neuroinflamm. 2005, 2, 14. [Google Scholar] [CrossRef]
- Couch, Y.; Alvarez-Erviti, L.; Sibson, N.R.; Wood, M.J.A.; Anthony, D.C. The acute inflammatory response to intranigral α-synuclein differs significantly from intranigral lipopolysaccharide and is exacerbated by peripheral inflammation. J. Neuroinflamm. 2011, 8, 166. [Google Scholar] [CrossRef]
- Drobny, A.; Ngo, P.A.; Neurath, M.F.; Zunke, F.; López-Posadas, R. Molecular Communication Between Neuronal Networks and Intestinal Epithelial Cells in Gut Inflammation and Parkinson’s Disease. Front. Med. 2021, 8, 1–24. [Google Scholar] [CrossRef]
- Lee, H.-J.; Suk, J.-E.; Bae, E.-J.; Lee, S.-J. Clearance and deposition of extracellular alpha-synuclein aggregates in microglia. Biochem. Biophys. Res. Commun. 2008, 372, 423–428. [Google Scholar] [CrossRef]
- Bliederhaeuser, C.; Grozdanov, V.; Speidel, A.; Zondler, L.; Ruf, W.P.; Bayer, H.; Kiechle, M.; Feiler, M.S.; Freischmidt, A.; Brenner, D.; et al. Age-dependent defects of alpha-synuclein oligomer uptake in microglia and monocytes. Acta Neuropathol. 2016, 131, 379–391. [Google Scholar] [CrossRef]
- Fuzzati-Armentero, M.T.; Cerri, S.; Blandini, F. Peripheral-central neuroimmune crosstalk in parkinson’s disease: What do patients and animal models tell us? Front. Neurol. 2019, 10, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Zecca, L.; Hong, J.-S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef]
- Niranjan, R. Recent advances in the mechanisms of neuroinflammation and their roles in neurodegeneration. Neurochem. Int. 2018, 120, 13–20. [Google Scholar] [CrossRef]
- Troncoso-Escudero, P.; Parra, A.; Nassif, M.; Vidal, R.L. Outside in: Unraveling the role of neuroinflammation in the progression of Parkinson’s disease. Front. Neurol. 2018, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, J.; Morales, L.; Barreto, G.E. Metabolic and Inflammatory Adaptation of Reactive Astrocytes: Role of PPARs. Mol. Neurobiol. 2017, 54, 2518–2538. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Hu, Z.; Han, X.; Wang, D.; Jiang, Q.; Ding, J.; Xiao, M.; Wang, C.; Lu, M.; Hu, G. Dopamine D2 receptor restricts astrocytic NLRP3 inflammasome activation via enhancing the interaction of β-arrestin2 and NLRP3. Cell Death Differ. 2018, 25, 2037–2049. [Google Scholar] [CrossRef] [PubMed]
- Chinta, S.J.; Woods, G.; Demaria, M.; Rane, A.; Zou, Y.; McQuade, A.; Rajagopalan, S.; Limbad, C.; Madden, D.T.; Campisi, J.; et al. Cellular Senescence Is Induced by the Environmental Neurotoxin Paraquat and Contributes to Neuropathology Linked to Parkinson’s Disease. Cell Rep. 2018, 22, 930–940. [Google Scholar] [CrossRef]
- Salminen, A.; Ojala, J.; Kaarniranta, K.; Haapasalo, A.; Hiltunen, M.; Soininen, H. Astrocytes in the aging brain express characteristics of senescence-associated secretory phenotype. Eur. J. Neurosci. 2011, 34, 3–11. [Google Scholar] [CrossRef]
- Lee, H.-J.; Suk, J.-E.; Patrick, C.; Bae, E.-J.; Cho, J.-H.; Rho, S.; Hwang, D.; Masliah, E.; Lee, S.-J. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J. Biol. Chem. 2010, 285, 9262–9272. [Google Scholar] [CrossRef]
- Ambrosi, G.; Kustrimovic, N.; Siani, F.; Rasini, E.; Cerri, S.; Ghezzi, C.; Dicorato, G.; Caputo, S.; Marino, F.; Cosentino, M.; et al. Complex Changes in the Innate and Adaptive Immunity Accompany Progressive Degeneration of the Nigrostriatal Pathway Induced by Intrastriatal Injection of 6-Hydroxydopamine in the Rat. Neurotox. Res. 2017, 32, 71–81. [Google Scholar] [CrossRef]
- Saijo, K.; Winner, B.; Carson, C.T.; Collier, J.G.; Boyer, L.; Rosenfeld, M.G.; Gage, F.H.; Glass, C.K. A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell 2009, 137, 47–59. [Google Scholar] [CrossRef]
- Joshi, A.U.; Minhas, P.S.; Liddelow, S.A.; Haileselassie, B.; Andreasson, K.I.; Dorn, G.W., 2nd; Mochly-Rosen, D. Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat. Neurosci. 2019, 22, 1635–1648. [Google Scholar] [CrossRef]
- Nakajima, K.; Tohyama, Y.; Maeda, S.; Kohsaka, S.; Kurihara, T. Neuronal regulation by which microglia enhance the production of neurotrophic factors for GABAergic, catecholaminergic, and cholinergic neurons. Neurochem. Int. 2007, 50, 807–820. [Google Scholar] [CrossRef]
- Schonhoff, A.M.; Williams, G.P.; Wallen, Z.D.; Standaert, D.G.; Harms, A.S. Innate and adaptive immune responses in Parkinson’s disease. Prog. Brain Res. 2020, 252, 169–216. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.T.; Woulfe, J.M. Striatal blood-brain barrier permeability in Parkinson’s disease. J. Cereb. Blood Flow Metab. 2015, 35, 747–750. [Google Scholar] [CrossRef]
- Al-Bachari, S.; Vidyasagar, R.; Emsley, H.C.A.; Parkes, L.M. Structural and physiological neurovascular changes in idiopathic Parkinson’s disease and its clinical phenotypes. J. Cereb. Blood Flow Metab. 2017, 37, 3409–3421. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, M.; Yamashita, T.; Ohta, Y.; Tadokoro, K.; Omote, Y.; Morihara, R.; Abe, K. Cerebral Microbleeds in Patients with Parkinson’s Disease and Dementia with Lewy Bodies: Comparison Using Magnetic Resonance Imaging and 99 mTc-ECD SPECT Subtraction Imaging. J. Alzheimers Dis. JAD 2021, 80, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Kortekaas, R.; Leenders, K.L.; Van Oostrom, J.C.H.; Vaalburg, W.; Bart, J.; Willemsen, A.T.M.; Hendrikse, N.H. Blood-brain barrier dysfunction in Parkinsonian midbrain in vivo. Ann. Neurol. 2005, 57, 176–179. [Google Scholar] [CrossRef]
- Bradaric, B.D.; Patel, A.; Schneider, J.A.; Carvey, P.M.; Hendey, B. Evidence for angiogenesis in Parkinson’s disease, incidental Lewy body disease, and progressive supranuclear palsy. J. Neural Transm. 2012, 119, 59–71. [Google Scholar] [CrossRef]
- Barcia, C.; Bautista, V.; Sánchez-Bahillo, Á.; Fernández-Villalba, E.; Faucheux, B.; Poza Y Poza, M.; Fernandez Barreiro, A.; Hirsch, E.C.; Herrero, M.T. Changes in vascularization in substantia nigra pars compacta of monkeys rendered parkinsonian. J. Neural Transm. 2005, 112, 1237–1248. [Google Scholar] [CrossRef]
- Yasuda, T.; Fukuda-Tani, M.; Nihira, T.; Wada, K.; Hattori, N.; Mizuno, Y.; Mochizuki, H. Correlation between levels of pigment epithelium-derived factor and vascular endothelial growth factor in the striatum of patients with Parkinson’s disease. Exp. Neurol. 2007, 206, 308–317. [Google Scholar] [CrossRef]
- Carvey, P.M.; Zhao, C.H.; Hendey, B.; Lum, H.; Trachtenberg, J.; Desai, B.S.; Snyder, J.; Zhu, Y.G.; Ling, Z.D. 6-Hydroxydopamine-induced alterations in blood-brain barrier permeability. Eur. J. Neurosci. 2005, 22, 1158–1168. [Google Scholar] [CrossRef]
- Chen, X.; Lan, X.; Roche, I.; Liu, R.; Geiger, J.D. Caffeine protects against MPTP-induced blood-brain barrier dysfunction in mouse striatum. J. Neurochem. 2008, 107, 1147–1157. [Google Scholar] [CrossRef]
- Lynch, N.J.; Willis, C.L.; Nolan, C.C.; Roscher, S.; Fowler, M.J.; Weihe, E.; Ray, D.E.; Schwaeble, W.J. Microglial activation and increased synthesis of complement component C1q precedes blood-brain barrier dysfunction in rats. Mol. Immunol. 2004, 40, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Stoll, G.; Jander, S.; Schroeter, M. Detrimental and beneficial effects of injury-induced inflammation and cytokine expression in the nervous system. Adv. Exp. Med. Biol. 2002, 513, 87–113. [Google Scholar] [CrossRef] [PubMed]
- Mark, K.S.; Miller, D.W. Increased permeability of primary cultured brain microvessel endothelial cell monolayers following TNF-α exposure. Life Sci. 1999, 64, 1941–1953. [Google Scholar] [CrossRef]
- Tsao, N.; Hsu, H.P.; Wu, C.M.; Liu, C.C.; Lei, H.Y. Tumour necrosis factor-α causes an increase in blood-brain barrier permeability during sepsis. J. Med. Microbiol. 2001, 50, 812–821. [Google Scholar] [CrossRef] [PubMed]
- Didier, N.; Romero, I.A.; Créminon, C.; Wijkhuisen, A.; Grassi, J.; Mabondzo, A. Secretion of interleukin-1β by astrocytes mediates endothelin-1 and tumour necrosis factor-α effects on human brain microvascular endothelial cell permeability. J. Neurochem. 2003, 86, 246–254. [Google Scholar] [CrossRef]
- Rostami, J.; Fotaki, G.; Sirois, J.; Mzezewa, R.; Bergström, J.; Essand, M.; Healy, L.; Erlandsson, A. Astrocytes have the capacity to act as antigen-presenting cells in the Parkinson’s disease brain. J. Neuroinflamm. 2020, 17, 119. [Google Scholar] [CrossRef]
- Argaw, A.T.; Asp, L.; Zhang, J.; Navrazhina, K.; Pham, T.; Mariani, J.N.; Mahase, S.; Dutta, D.J.; Seto, J.; Kramer, E.G.; et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J. Clin. Investig. 2012, 122, 2454–2468. [Google Scholar] [CrossRef]
- Dohgu, S.; Takata, F.; Matsumoto, J.; Kimura, I.; Yamauchi, A.; Kataoka, Y. Monomeric α-synuclein induces blood–brain barrier dysfunction through activated brain pericytes releasing inflammatory mediators in vitro. Microvasc. Res. 2019, 124, 61–66. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, Y.; Jiao, T.; Shi, D.; Zhu, X.; Zhang, M.; Shi, M.; Zhou, H. CXCR2 is essential for cerebral endothelial activation and leukocyte recruitment during neuroinflammation. J. Neuroinflamm. 2015, 12, 98. [Google Scholar] [CrossRef]
- Chui, R.; Dorovini-Zis, K. Regulation of CCL2 and CCL3 expression in human brain endothelial cells by cytokines and lipopolysaccharide. J. Neuroinflamm. 2010, 7, 1. [Google Scholar] [CrossRef]
- Fu, Q.; Song, R.; Yang, Z.; Shan, Q.; Chen, W. 6-Hydroxydopamine induces brain vascular endothelial inflammation. IUBMB Life 2017, 69, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Kuan, W.L.; Bennett, N.; He, X.; Skepper, J.N.; Martynyuk, N.; Wijeyekoon, R.; Moghe, P.V.; Williams-Gray, C.H.; Barker, R.A. α-Synuclein pre-formed fibrils impair tight junction protein expression without affecting cerebral endothelial cell function. Exp. Neurol. 2016, 285, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Machida, T.; Takata, F.; Matsumoto, J.; Miyamura, T.; Hirata, R.; Kimura, I.; Kataoka, Y.; Dohgu, S.; Yamauchi, A. Contribution of thrombin-reactive brain pericytes to blood-brain barrier dysfunction in an in vivo mouse model of obesity-associated diabetes and an in vitro rat model. PLoS ONE 2017, 12, e0177447. [Google Scholar] [CrossRef] [PubMed]
- Wijeyekoon, R.S.; Kronenberg-Versteeg, D.; Scott, K.M.; Hayat, S.; Jones, J.L.; Clatworthy, M.R.; Floto, R.A.; Barker, R.A.; Williams-Gray, C.H. Monocyte Function in Parkinson’s Disease and the Impact of Autologous Serum on Phagocytosis. Front. Neurol. 2018, 9, 870. [Google Scholar] [CrossRef]
- Grozdanov, V.; Bliederhaeuser, C.; Ruf, W.P.; Roth, V.; Fundel-Clemens, K.; Zondler, L.; Brenner, D.; Martin-Villalba, A.; Hengerer, B.; Kassubek, J.; et al. Inflammatory dysregulation of blood monocytes in Parkinson’s disease patients. Acta Neuropathol. 2014, 128, 651–663. [Google Scholar] [CrossRef]
- Witoelar, A.; Jansen, I.E.; Wang, Y.; Desikan, R.S.; Gibbs, J.R.; Blauwendraat, C.; Thompson, W.K.; Hernandez, D.G.; Djurovic, S.; Schork, A.J.; et al. Genome-wide Pleiotropy Between Parkinson Disease and Autoimmune Diseases. JAMA Neurol. 2017, 74, 780–792. [Google Scholar] [CrossRef]
- Serbina, N.V.; Pamer, E.G. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 2006, 7, 311–317. [Google Scholar] [CrossRef]
- D’Mello, C.; Le, T.; Swain, M.G. Cerebral microglia recruit monocytes into the brain in response to tumor necrosis factoralpha signaling during peripheral organ inflammation. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 2089–2102. [Google Scholar] [CrossRef]
- Satoh, J.i.; Kino, Y.; Asahina, N.; Takitani, M.; Miyoshi, J.; Ishida, T.; Saito, Y. TMEM119 marks a subset of microglia in the human brain. Neuropathology 2016, 36, 39–49. [Google Scholar] [CrossRef]
- Prinz, M.; Priller, J. Tickets to the brain: Role of CCR2 and CX3CR1 in myeloid cell entry in the CNS. J. Neuroimmunol. 2010, 224, 80–84. [Google Scholar] [CrossRef]
- Harms, A.S.; Thome, A.D.; Yan, Z.; Schonhoff, A.M.; Williams, G.P.; Li, X.; Liu, Y.; Qin, H.; Benveniste, E.N.; Standaert, D.G. Peripheral monocyte entry is required for alpha-Synuclein induced inflammation and Neurodegeneration in a model of Parkinson disease. Exp. Neurol. 2018, 300, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Parillaud, V.R.; Lornet, G.; Monnet, Y.; Privat, A.L.; Haddad, A.T.; Brochard, V.; Bekaert, A.; de Chanville, C.B.; Hirsch, E.C.; Combadière, C.; et al. Analysis of monocyte infiltration in MPTP mice reveals that microglial CX3CR1 protects against neurotoxic over-induction of monocyte-attracting CCL2 by astrocytes. J. Neuroinflamm. 2017, 14, 60. [Google Scholar] [CrossRef] [PubMed]
- Gardai, S.J.; Mao, W.; Schüle, B.; Babcock, M.; Schoebel, S.; Lorenzana, C.; Alexander, J.; Kim, S.; Glick, H.; Hilton, K.; et al. Elevated Alpha-Synuclein Impairs Innate Immune Cell Function and Provides a Potential Peripheral Biomarker for Parkinson’s Disease. PLoS ONE 2013, 8, e71634. [Google Scholar] [CrossRef] [PubMed]
- Caggiu, E.; Arru, G.; Hosseini, S.; Niegowska, M.; Sechi, G.P.; Zarbo, I.R.; Sechi, L.A. Inflammation, infectious triggers, and Parkinson’s disease. Front. Neurol. 2019, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.K.; Chao, Y.X.; West, A.; Chan, L.L.; Poewe, W.; Jankovic, J. Parkinson disease and the immune system—Associations, mechanisms and therapeutics. Nat. Rev. Neurol. 2020, 16, 303–318. [Google Scholar] [CrossRef]
- Bas, J.; Calopa, M.; Mestre, M.; Molleví, D.G.; Cutillas, B.; Ambrosio, S.; Buendia, E. Lymphocyte populations in Parkinson’s disease and in rat models of parkinsonism. J. Neuroimmunol. 2001, 113, 146–152. [Google Scholar] [CrossRef]
- Stevens, C.H.; Rowe, D.; Morel-Kopp, M.-C.; Orr, C.; Russell, T.; Ranola, M.; Ward, C.; Halliday, G.M. Reduced T helper and B lymphocytes in Parkinson’s disease. J. Neuroimmunol. 2012, 252, 95–99. [Google Scholar] [CrossRef]
- Baba, Y.; Kuroiwa, A.; Uitti, R.J.; Wszolek, Z.K.; Yamada, T. Alterations of T-lymphocyte populations in Parkinson disease. Parkinsonism Relat. Disord. 2005, 11, 493–498. [Google Scholar] [CrossRef]
- Niwa, F.; Kuriyama, N.; Nakagawa, M.; Imanishi, J. Effects of peripheral lymphocyte subpopulations and the clinical correlation with Parkinson’s disease. Geriatr. Gerontol. Int. 2012, 12, 102–107. [Google Scholar] [CrossRef]
- Fiszer, U.; Mix, E.; Fredrikson, S.; Kostulas, V.; Link, H. Parkinson’s disease and immunological abnormalities: Increase of HLA-DR expression on monocytes in cerebrospinal fluid and of CD45RO+ T cells in peripheral blood. Acta Neurol. Scand. 1994, 90, 160–166. [Google Scholar] [CrossRef]
- Schröder, J.B.; Pawlowski, M.; zu Hörste, G.; Gross, C.C.; Wiendl, H.; Meuth, S.G.; Ruck, T.; Warnecke, T. Immune Cell Activation in the Cerebrospinal Fluid of Patients With Parkinson’s Disease. Front. Neurol. 2018, 9, 1081. [Google Scholar] [CrossRef] [PubMed]
- Brochard, V.; Combadière, B.; Prigent, A.; Laouar, Y.; Perrin, A.; Beray-Berthat, V.; Bonduelle, O.; Alvarez-Fischer, D.; Callebert, J.; Launay, J.-M.; et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J. Clin. Investig. 2009, 119, 182–192. [Google Scholar] [CrossRef]
- Kustrimovic, N.; Comi, C.; Magistrelli, L.; Rasini, E.; Legnaro, M.; Bombelli, R.; Aleksic, I.; Blandini, F.; Minafra, B.; Riboldazzi, G.; et al. Parkinson’s disease patients have a complex phenotypic and functional Th1 bias: Cross-sectional studies of CD4+ Th1/Th2/T17 and Treg in drug-naïve and drug-treated patients. J. Neuroinflamm. 2018, 15, 205. [Google Scholar] [CrossRef] [PubMed]
- Rosenkranz, D.; Weyer, S.; Tolosa, E.; Gaenslen, A.; Berg, D.; Leyhe, T.; Gasser, T.; Stoltze, L. Higher frequency of regulatory T cells in the elderly and increased suppressive activity in neurodegeneration. J. Neuroimmunol. 2007, 188, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Saunders, J.A.H.; Estes, K.A.; Kosloski, L.M.; Allen, H.E.; Dempsey, K.M.; Torres-Russotto, D.R.; Meza, J.L.; Santamaria, P.M.; Bertoni, J.M.; Murman, D.L.; et al. CD4+ regulatory and effector/memory T cell subsets profile motor dysfunction in Parkinson’s disease. J. Neuroimmune Pharmacol. 2012, 7, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, A.D.; Banerjee, R.; Liu, J.; Gendelman, H.E.; Mosley, R.L. Neuroprotective activities of CD4+CD25+ regulatory T cells in an animal model of Parkinson’s disease. J. Leukoc. Biol. 2007, 82, 1083–1094. [Google Scholar] [CrossRef]
- Sulzer, D.; Alcalay, R.N.; Garretti, F.; Cote, L.; Kanter, E.; Agin-Liebes, J.; Liong, C.; McMurtrey, C.; Hildebrand, W.H.; Mao, X.; et al. T cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature 2017, 546, 656–661. [Google Scholar] [CrossRef]
- Blandini, F.; Mangiagalli, A.; Cosentino, M.; Marino, F.; Samuele, A.; Rasini, E.; Fancellu, R.; Martignoni, E.; Riboldazzi, G.; Calandrella, D.; et al. Peripheral Markers of Apoptosis in Parkinson’s Disease: The Effect of Dopaminergic Drugs. Ann. N. Y. Acad. Sci. 2003, 1010, 675–678. [Google Scholar] [CrossRef]
- Migliore, L.; Petrozzi, L.; Lucetti, C.; Gambaccini, G.; Bernardini, S.; Scarpato, R.; Trippi, F.; Barale, R.; Frenzilli, G.; Rodilla, V.; et al. Oxidative damage and cytogenetic analysis in leukocytes of Parkinson’s disease patients. Neurology 2002, 58, 1809–1815. [Google Scholar] [CrossRef]
- Benner, E.J.; Banerjee, R.; Reynolds, A.D.; Sherman, S.; Pisarev, V.M.; Tsiperson, V.; Nemachek, C.; Ciborowski, P.; Przedborski, S.; Mosley, R.L.; et al. Nitrated α–Synuclein Immunity Accelerates Degeneration of Nigral Dopaminergic Neurons. PLoS ONE 2008, 3, e1376. [Google Scholar] [CrossRef]
- Wang, L.; Xie, Y.; Zhu, L.-J.; Chang, T.-T.; Mao, Y.-Q.; Li, J. An association between immunosenescence and CD4(+)CD25(+) regulatory T cells: A systematic review. Biomed. Environ. Sci. BES 2010, 23, 327–332. [Google Scholar] [CrossRef]
- Reynolds, A.D.; Stone, D.K.; Hutter, J.A.L.; Benner, E.J.; Mosley, R.L.; Gendelman, H.E. Regulatory T Cells Attenuate Th17 Cell-Mediated Nigrostriatal Dopaminergic Neurodegeneration in a Model of Parkinson’s Disease. J. Immunol. 2010, 184, 2261–2271. [Google Scholar] [CrossRef] [PubMed]
- Theodore, S.; Cao, S.; McLean, P.J.; Standaert, D.G. Targeted Overexpression of Human α-Synuclein Triggers Microglial Activation and an Adaptive Immune Response in a Mouse Model of Parkinson Disease. J. Neuropathol. Exp. Neurol. 2008, 67, 1149–1158. [Google Scholar] [CrossRef] [PubMed]
- Harm, A.S.; Cao, S.; Rowse, A.L.; Thome, A.D.; Li, X.; Mangieri, L.R.; Cron, R.Q.; Shacka, J.J.; Raman, C.; Standaert, D.G. MHCII is required for α-Synuclein-induced activation of microglia, CD4 T cell proliferation, and dopaminergic neurodegeneration. J. Neurosci. 2013, 33, 9592–9600. [Google Scholar] [CrossRef]
- Imamura, K.; Hishikawa, N.; Sawada, M.; Nagatsu, T.; Yoshida, M.; Hashizume, Y. Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol. 2003, 106, 518–526. [Google Scholar] [CrossRef]
- Armentero, M.-T.; Levandis, G.; Nappi, G.; Bazzini, E.; Blandini, F. Peripheral inflammation and neuroprotection: Systemic pretreatment with complete Freund’s adjuvant reduces 6-hydroxydopamine toxicity in a rodent model of Parkinson’s disease. Neurobiol. Dis. 2006, 24, 492–505. [Google Scholar] [CrossRef]
- Wheeler, C.J.; Seksenyan, A.; Koronyo, Y.; Rentsendorj, A.; Sarayba, D.; Wu, H.; Gragg, A.; Siegel, E.; Thomas, D.; Espinosa, A.; et al. T-Lymphocyte Deficiency Exacerbates Behavioral Deficits in the 6-OHDA Unilateral Lesion Rat Model for Parkinson’s Disease. J. Neurol. Neurophysiol. 2014, 5, 209. [Google Scholar] [CrossRef]
- Sommer, A.; Marxreiter, F.; Krach, F.; Fadler, T.; Grosch, J.; Maroni, M.; Graef, D.; Eberhardt, E.; Riemenschneider, M.J.; Yeo, G.W.; et al. Th17 Lymphocytes Induce Neuronal Cell Death in a Human iPSC-Based Model of Parkinson’s Disease. Cell Stem Cell 2018, 23, 123–131.e126. [Google Scholar] [CrossRef]
- Cebrián, C.; Zucca, F.A.; Mauri, P.; Steinbeck, J.A.; Studer, L.; Scherzer, C.R.; Kanter, E.; Budhu, S.; Mandelbaum, J.; Vonsattel, J.P.; et al. MHC-I expression renders catecholaminergic neurons susceptible to T-cell-mediated degeneration. Nat. Commun. 2014, 5, 3633. [Google Scholar] [CrossRef]
- Matheoud, D.; Sugiura, A.; Bellemare-Pelletier, A.; Laplante, A.; Rondeau, C.; Chemali, M.; Fazel, A.; Bergeron, J.J.; Trudeau, L.E.; Burelle, Y.; et al. Parkinson’s Disease-Related Proteins PINK1 and Parkin Repress Mitochondrial Antigen Presentation. Cell 2016, 166, 314–327. [Google Scholar] [CrossRef]
- Houser, M.C.; Tansey, M.G. The gut-brain axis: Is intestinal inflammation a silent driver of Parkinson’s disease pathogenesis? NPJ Parkinsons Dis. 2017, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Chapelet, G.; Leclair-Visonneau, L.; Clairembault, T.; Neunlist, M.; Derkinderen, P. Can the gut be the missing piece in uncovering PD pathogenesis? Parkinsonism Relat. Disord. 2019, 59, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Selkrig, J.; Wong, P.; Zhang, X.; Pettersson, S. Metabolic tinkering by the gut microbiome. Gut Microbes 2014, 5, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Menozzi, E.; Macnaughtan, J.; Schapira, A.H.V. The gut-brain axis and Parkinson disease: Clinical and pathogenetic relevance. Ann. Med. 2021, 53, 611–625. [Google Scholar] [CrossRef]
- Chen, H.; Zhao, E.J.; Zhang, W.; Lu, Y.; Liu, R.; Huang, X.; Ciesielski-Jones, A.J.; Justice, M.A.; Cousins, D.S.; Peddada, S. Meta-analyses on prevalence of selected Parkinson’s nonmotor symptoms before and after diagnosis. Transl. Neurodegener. 2015, 4, 1. [Google Scholar] [CrossRef]
- Siddiqui, M.F.; Rast, S.; Lynn, M.J.; Auchus, A.P.; Pfeiffer, R.F. Autonomic dysfunction in Parkinson’s disease: A comprehensive symptom survey. Parkinsonism Relat. Disord. 2002, 8, 277–284. [Google Scholar] [CrossRef]
- Ueki, A.; Otsuka, M. Life style risks of Parkinson’s disease: Association between decreased water intake and constipation. J. Neurol. 2004, 251, vII18-23. [Google Scholar] [CrossRef]
- Devos, D.; Lebouvier, T.; Lardeux, B.; Biraud, M.; Rouaud, T.; Pouclet, H.; Coron, E.; Bruley des Varannes, S.; Naveilhan, P.; Nguyen, J.-M.; et al. Colonic inflammation in Parkinson’s disease. Neurobiol. Dis. 2013, 50, 42–48. [Google Scholar] [CrossRef]
- Hui, K.Y.; Heriberto, F.-H.; Jianzhong, H.; Adam, S.; Nathan, P.; Nai-Yun, H.; Ling-Shiang, C.; Shai, C.; Nicole, V.; Xianting, L.; et al. Functional variants in the LRRK2 gene confer shared effects on risk for Crohn’s disease and Parkinson’s disease. Sci. Transl. Med. 2018, 10, eaai7795. [Google Scholar] [CrossRef]
- Bialecka, M.; Kurzawski, M.; Klodowska-Duda, G.; Opala, G.; Juzwiak, S.; Kurzawski, G.; Tan, E.-K.; Drozdzik, M. CARD15 variants in patients with sporadic Parkinson’s disease. Neurosci. Res. 2007, 57, 473–476. [Google Scholar] [CrossRef]
- Lee, H.S.; Lobbestael, E.; Vermeire, S.; Sabino, J.; Cleynen, I. Inflammatory bowel disease and Parkinson’s disease: Common pathophysiological links. Gut 2021, 70, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Erny, D.; Hrabě de Angelis, A.L.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Rooks, M.G.; Garrett, W.S. Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 2016, 16, 341–352. [Google Scholar] [CrossRef]
- Bercik, P.; Denou, E.; Collins, J.; Jackson, W.; Lu, J.; Jury, J.; Deng, Y.; Blennerhassett, P.; Macri, J.; McCoy, K.D.; et al. The Intestinal Microbiota Affect Central Levels of Brain-Derived Neurotropic Factor and Behavior in Mice. Gastroenterology 2011, 141, 599–609.e593. [Google Scholar] [CrossRef] [PubMed]
- Heijtz, R.D.; Wang, S.; Anuar, F.; Qian, Y.; Björkholm, B.; Samuelsson, A.; Hibberd, M.L.; Forssberg, H.; Pettersson, S. Normal gut microbiota modulates brain development and behavior. Proc. Natl. Acad. Sci. USA 2011, 108, 3047–3052. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-H.; Chen, C.-C.; Chiang, H.-L.; Liou, J.-M.; Chang, C.-M.; Lu, T.-P.; Chuang, E.Y.; Tai, Y.-C.; Cheng, C.; Lin, H.-Y.; et al. Altered gut microbiota and inflammatory cytokine responses in patients with Parkinson’s disease. J. Neuroinflamm. 2019, 16, 129. [Google Scholar] [CrossRef]
- Heintz-Buschart, A.; Pandey, U.; Wicke, T.; Sixel-Döring, F.; Janzen, A.; Sittig-Wiegand, E.; Trenkwalder, C.; Oertel, W.H.; Mollenhauer, B.; Wilmes, P. The nasal and gut microbiome in Parkinson’s disease and idiopathic rapid eye movement sleep behavior disorder. Mov. Disord. 2018, 33, 88–98. [Google Scholar] [CrossRef]
- Keshavarzian, A.; Green, S.J.; Engen, P.A.; Voigt, R.M.; Naqib, A.; Forsyth, C.B.; Mutlu, E.; Shannon, K.M. Colonic bacterial composition in Parkinson’s disease. Mov. Disord. 2015, 30, 1351–1360. [Google Scholar] [CrossRef]
- Petrov, V.A.; Saltykova, I.V.; Zhukova, I.A.; Alifirova, V.M.; Zhukova, N.G.; Dorofeeva, Y.B.; Tyakht, A.V.; Kovarsky, B.A.; Alekseev, D.G.; Kostryukova, E.S.; et al. Analysis of Gut Microbiota in Patients with Parkinson’s Disease. Bull. Exp. Biol. Med. 2017, 162, 734–737. [Google Scholar] [CrossRef]
- Wallen, Z.D.; Appah, M.; Dean, M.N.; Sesler, C.L.; Factor, S.A.; Molho, E.; Zabetian, C.P.; Standaert, D.G.; Payami, H. Characterizing dysbiosis of gut microbiome in PD: Evidence for overabundance of opportunistic pathogens. NPJ Parkinsons Dis. 2020, 6, 11. [Google Scholar] [CrossRef]
- Takeda, K.; Akira, S. Toll-like receptors in innate immunity. Int. Immunol. 2005, 17, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Perez-Pardo, P.; Dodiya, H.B.; Engen, P.A.; Forsyth, C.B.; Huschens, A.M.; Shaikh, M.; Voigt, R.M.; Naqib, A.; Green, S.J.; Kordower, J.H.; et al. Role of TLR4 in the gut-brain axis in Parkinson’s disease: A translational study from men to mice. Gut 2019, 68, 829–843. [Google Scholar] [CrossRef] [PubMed]
- Rani, L.; Mondal, A.C. Unravelling the role of gut microbiota in Parkinson’s disease progression: Pathogenic and therapeutic implications. Neurosci. Res. 2021, 168, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Gil, P.; Rodriguez-Perez, A.I.; Dominguez-Meijide, A.; Guerra, M.J.; Labandeira-Garcia, J.L. Bidirectional Neural Interaction Between Central Dopaminergic and Gut Lesions in Parkinson’s Disease Models. Mol. Neurobiol. 2018, 55, 7297–7316. [Google Scholar] [CrossRef]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 2016, 167, 1469–1480.e1412. [Google Scholar] [CrossRef]
- Pull, S.L.; Doherty, J.M.; Mills, J.C.; Gordon, J.I.; Stappenbeck, T.S. Activated macrophages are an adaptive element of the colonic epithelial progenitor niche necessary for regenerative responses to injury. Proc. Natl. Acad. Sci. USA 2005, 102, 99–104. [Google Scholar] [CrossRef]
- Shannon, K.M.; Keshavarzian, A.; Mutlu, E.; Dodiya, H.B.; Daian, D.; Jaglin, J.A.; Kordower, J.H. Alpha-synuclein in colonic submucosa in early untreated Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2012, 27, 709–715. [Google Scholar] [CrossRef]
- Böttner, M.; Zorenkov, D.; Hellwig, I.; Barrenschee, M.; Harde, J.; Fricke, T.; Deuschl, G.; Egberts, J.-H.; Becker, T.; Fritscher-Ravens, A.; et al. Expression pattern and localization of alpha-synuclein in the human enteric nervous system. Neurobiol. Dis. 2012, 48, 474–480. [Google Scholar] [CrossRef]
- Gold, A.; Turkalp, Z.T.; Munoz, D.G. Enteric alpha-synuclein expression is increased in Parkinson’s disease but not Alzheimer’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2013, 28, 237–240. [Google Scholar] [CrossRef]
- Braak, H.; Rüb, U.; Gai, W.P.; Del Tredici, K. Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural Transm. 2003, 110, 517–536. [Google Scholar] [CrossRef]
- Kim, S.; Kwon, S.H.; Kam, T.I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal Propagation of Pathologic α-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron 2019, 103, 627–641.e627. [Google Scholar] [CrossRef] [PubMed]
- Uemura, N.; Yagi, H.; Uemura, M.T.; Hatanaka, Y.; Yamakado, H.; Takahashi, R. Inoculation of α-synuclein preformed fibrils into the mouse gastrointestinal tract induces Lewy body-like aggregates in the brainstem via the vagus nerve. Mol. Neurodegener. 2018, 13, 21. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Heppner, F.L.; Heikenwalder, M.; Prinz, M.; Mertz, K.; Seeger, H.; Glatzel, M. Immune system and peripheral nerves in propagation of prions to CNS. Br. Med. Bull. 2003, 66, 141–159. [Google Scholar] [CrossRef] [PubMed]
- Phillips, R.J.; Walter, G.C.; Wilder, S.L.; Baronowsky, E.A.; Powley, T.L. Alpha-synuclein-immunopositive myenteric neurons and vagal preganglionic terminals: Autonomic pathway implicated in Parkinson’s disease? Neuroscience 2008, 153, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Holmqvist, S.; Chutna, O.; Bousset, L.; Aldrin-Kirk, P.; Li, W.; Björklund, T.; Wang, Z.Y.; Roybon, L.; Melki, R.; Li, J.Y. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 2014, 128, 805–820. [Google Scholar] [CrossRef]
- Pan-Montojo, F.; Anichtchik, O.; Dening, Y.; Knels, L.; Pursche, S.; Jung, R.; Jackson, S.; Gille, G.; Spillantini, M.G.; Reichmann, H.; et al. Progression of Parkinson’s disease pathology is reproduced by intragastric administration of rotenone in mice. PLoS ONE 2010, 5, e8762. [Google Scholar] [CrossRef]
- Choi, J.G.; Kim, N.; Ju, I.G.; Eo, H.; Lim, S.-M.; Jang, S.-E.; Kim, D.-H.; Oh, M.S. Oral administration of Proteus mirabilis damages dopaminergic neurons and motor functions in mice. Sci. Rep. 2018, 8, 1275. [Google Scholar] [CrossRef]
- O’Donovan, S.M.; Crowley, E.K.; Brown, J.R.M.; O’Sullivan, O.; O’Leary, O.F.; Timmons, S.; Nolan, Y.M.; Clarke, D.J.; Hyland, N.P.; Joyce, S.A.; et al. Nigral overexpression of α-synuclein in a rat Parkinson’s disease model indicates alterations in the enteric nervous system and the gut microbiome. Neurogastroenterol. Motil. 2020, 32, e13726. [Google Scholar] [CrossRef]
- Hui-Ming, G.; Feng, Z.; Hui, Z.; Wayneho, K.; Belinda, W.; Jau-Shyong, H. Neuroinflammation and α-Synuclein Dysfunction Potentiate Each Other, Driving Chronic Progression of Neurodegeneration in a Mouse Model of Parkinson’s Disease. Environ. Health Perspect. 2011, 119, 807–814. [Google Scholar] [CrossRef]
- Zhang, W.; Gao, J.-h.; Yan, Z.-f.; Huang, X.-y.; Guo, P.; Sun, L.; Liu, Z.; Hu, Y.; Zuo, L.-j.; Yu, S.-y.; et al. Minimally Toxic Dose of Lipopolysaccharide and α-Synuclein Oligomer Elicit Synergistic Dopaminergic Neurodegeneration: Role and Mechanism of Microglial NOX2 Activation. Mol. Neurobiol. 2018, 55, 619–632. [Google Scholar] [CrossRef]
- Peralta Ramos, J.M.; Iribarren, P.; Bousset, L.; Melki, R.; Baekelandt, V.; Van der Perren, A. Peripheral Inflammation Regulates CNS Immune Surveillance Through the Recruitment of Inflammatory Monocytes Upon Systemic α-Synuclein Administration. Front. Immunol. 2019, 10, 80. [Google Scholar] [CrossRef] [PubMed]
- Beach, T.G.; White, C.L.; Hladik, C.L.; Sabbagh, M.N.; Connor, D.J.; Shill, H.A.; Sue, L.I.; Sasse, J.; Bachalakuri, J.; Henry-Watson, J.; et al. Olfactory bulb α-synucleinopathy has high specificity and sensitivity for Lewy body disorders. Acta Neuropathol. 2008, 117, 169. [Google Scholar] [CrossRef] [PubMed]
- Doty, R.L. Olfaction in Parkinson’s disease and related disorders. Neurobiol. Dis. 2012, 46, 527–552. [Google Scholar] [CrossRef] [PubMed]
- Zapiec, B.; Dieriks, B.V.; Tan, S.; Faull, R.L.M.; Mombaerts, P.; Curtis, M.A. A ventral glomerular deficit in Parkinson’s disease revealed by whole olfactory bulb reconstruction. Brain A J. Neurol. 2017, 140, 2722–2736. [Google Scholar] [CrossRef] [PubMed]
- Flores-Cuadrado, A.; Saiz-Sanchez, D.; Mohedano-Moriano, A.; Lamas-Cenjor, E.; Leon-Olmo, V.; Martinez-Marcos, A.; Ubeda-Bañon, I. Astrogliosis and sexually dimorphic neurodegeneration and microgliosis in the olfactory bulb in Parkinson’s disease. NPJ Parkinsons Dis. 2021, 7, 11. [Google Scholar] [CrossRef]
- Doursout, M.-F.; Schurdell, M.S.; Young, L.M.; Osuagwu, U.; Hook, D.M.; Poindexter, B.J.; Schiess, M.C.; Bick, D.L.M.; Bick, R.J. Inflammatory cells and cytokines in the olfactory bulb of a rat model of neuroinflammation; insights into neurodegeneration? J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 2013, 33, 376–383. [Google Scholar] [CrossRef]
- Stefanis, L.; Emmanouilidou, E.; Pantazopoulou, M.; Kirik, D.; Vekrellis, K.; Tofaris, G.K. How is alpha-synuclein cleared from the cell? J. Neurochem. 2019, 150, 577–590. [Google Scholar] [CrossRef]
- Bergstrom, A.L.; Kallunki, P.; Fog, K. Development of Passive Immunotherapies for Synucleinopathies. Mov. Disord. 2016, 31, 203–213. [Google Scholar] [CrossRef]
- Schneeberger, A.; Tierney, L.; Mandler, M. Active immunization therapies for Parkinson’s disease and multiple system atrophy. Mov. Disord. 2016, 31, 214–224. [Google Scholar] [CrossRef]
- Zella, S.M.A.; Metzdorf, J.; Ciftci, E.; Ostendorf, F.; Muhlack, S.; Gold, R.; Tonges, L. Emerging Immunotherapies for Parkinson Disease. Neurol. Ther. 2019, 8, 29–44. [Google Scholar] [CrossRef]
- Volc, D.; Poewe, W.; Kutzelnigg, A.; Lührs, P.; Thun-Hohenstein, C.; Schneeberger, A.; Galabova, G.; Majbour, N.; Vaikath, N.; El-Agnaf, O.; et al. Safety and immunogenicity of the α-synuclein active immunotherapeutic PD01A in patients with Parkinson’s disease: A randomised, single-blinded, phase 1 trial. Lancet Neurol. 2020, 19, 591–600. [Google Scholar] [CrossRef]
- Masliah, E.; Rockenstein, E.; Adame, A.; Alford, M.; Crews, L.; Hashimoto, M.; Seubert, P.; Lee, M.; Goldstein, J.; Chilcote, T.; et al. Effects of α-synuclein immunization in a mouse model of Parkinson’s disease. Neuron 2005, 46, 857–868. [Google Scholar] [CrossRef]
- Ugen, K.E.; Lin, X.; Bai, G.; Liang, Z.; Cai, J.; Li, K.; Song, S.; Cao, C.; Sanchez-Ramos, J. Evaluation of an α synuclein sensitized dendritic cell based vaccine in a transgenic mouse model of Parkinson disease. Hum. Vaccines Immunother. 2015, 11, 922–930. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Yang, Y.; Yang, X.; Zhou, C.; Li, F.; Lei, P.; Zhong, L.; Jin, X.; Peng, G. Immune effects of optimized DNA vaccine and protective effects in a MPTP model of Parkinson’s disease. Neurol. Sci. 2013, 34, 1559–1570. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Rockenstein, E.; Mante, M.; Crews, L.; Spencer, B.; Adame, A.; Patrick, C.; Trejo, M.; Ubhi, K.; Rohn, T.T.; et al. Passive immunization reduces behavioral and neuropathological deficits in an alpha-synuclein transgenic model of lewy body disease. PLoS ONE 2011, 6, e19338. [Google Scholar] [CrossRef]
- Bae, E.J.; Lee, H.J.; Rockenstein, E.; Ho, D.H.; Park, E.B.; Yang, N.Y.; Desplats, P.; Masliah, E.; Lee, S.J. Antibody-aided clearance of extracellular α-synuclein prevents cell-to-cell aggregate transmission. J. Neurosci. 2012, 32, 13454–13469. [Google Scholar] [CrossRef]
- Games, D.; Valera, E.; Spencer, B.; Rockenstein, E.; Mante, M.; Adame, A.; Patrick, C.; Ubhi, K.; Nuber, S.; Sacayon, P.; et al. Reducing C-terminal-truncated alpha-synuclein by immunotherapy attenuates neurodegeneration and propagation in Parkinson’s disease-like models. J. Neurosci. 2014, 34, 9441–9454. [Google Scholar] [CrossRef]
- Lindström, V.; Fagerqvist, T.; Nordström, E.; Eriksson, F.; Lord, A.; Tucker, S.; Andersson, J.; Johannesson, M.; Schell, H.; Kahle, P.J.; et al. Immunotherapy targeting α-synuclein protofibrils reduced pathology in (Thy-1)-h[A30P] α-synuclein mice. Neurobiol. Dis. 2014, 69, 134–143. [Google Scholar] [CrossRef]
- Tran, H.T.; Chung, C.H.Y.; Iba, M.; Zhang, B.; Trojanowski, J.Q.; Luk, K.C.; Lee, V.M.Y. α-Synuclein Immunotherapy Blocks Uptake and Templated Propagation of Misfolded α-Synuclein and Neurodegeneration. Cell Rep. 2014, 7, 2054–2065. [Google Scholar] [CrossRef]
- Shahaduzzaman, M.; Nash, K.; Hudson, C.; Sharif, M.; Grimmig, B.; Lin, X.; Bai, G.; Liu, H.; Ugen, K.E.; Cao, C.; et al. Anti-human α-synuclein N-terminal peptide antibody protects against dopaminergic cell death and ameliorates behavioral deficits in an AAV-α-synuclein rat model of Parkinson’s disease. PLoS ONE 2015, 10, e0116841. [Google Scholar] [CrossRef]
- Mandler, M.; Valera, E.; Rockenstein, E.; Weninger, H.; Patrick, C.; Adame, A.; Santic, R.; Meindl, S.; Vigl, B.; Smrzka, O.; et al. Next-generation active immunization approach for synucleinopathies: Implications for Parkinson’s disease clinical trials. Acta Neuropathol. 2014, 127, 861–879. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Fang, F.; Pedersen, N.L.; Tillander, A.; Ludvigsson, J.F.; Ekbom, A.; Svenningsson, P.; Chen, H.; Karin, W. Vagotomy and Parkinson disease A Swedish register-based matched-cohort study). Neurology 2017, 88, 1996–2002. [Google Scholar] [CrossRef] [PubMed]
- Tysnes, O.B.; Kenborg, L.; Herlofson, K.; Steding-Jessen, M.; Horn, A.; Olsen, J.H.; Reichmann, H. Does vagotomy reduce the risk of Parkinson’s disease? Ann. Neurol. 2015, 78, 1011–1012. [Google Scholar] [CrossRef]
- Svensson, E.; Horváth-Puhó, E.; Thomsen, R.W.; Djurhuus, J.C.; Pedersen, L.; Borghammer, P.; Sørensen, H.T. Vagotomy and subsequent risk of Parkinson’s disease. Ann. Neurol. 2015, 78, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Killinger, B.A.; Madaj, Z.; Sikora, J.W.; Rey, N.; Haas, A.J.; Vepa, Y.; Lindqvist, D.; Chen, H.; Thomas, P.M.; Brundin, P.; et al. The vermiform appendix impacts the risk of developing Parkinson’s disease. Sci. Transl. Med. 2018, 10, eaar5280. [Google Scholar] [CrossRef] [PubMed]
- Teismann, P.; Tieu, K.; Choi, D.K.; Wu, D.C.; Naini, A.; Hunot, S.; Vila, M.; Jackson-Lewis, V.; Przedborski, S. Cyclooxygenase-2 is instrumental in Parkinson’s disease neurodegeneration. Proc. Natl. Acad. Sci. USA 2003, 100, 5473–5478. [Google Scholar] [CrossRef]
- Wilkinson, B.L.; Cramer, P.E.; Varvel, N.H.; Reed-Geaghan, E.; Jiang, Q.; Szabo, A.; Herrup, K.; Lamb, B.T.; Landreth, G.E. Ibuprofen attenuates oxidative damage through NOX2 inhibition in Alzheimer’s disease. Neurobiol. Aging 2012, 33, 197.e21–197.e32. [Google Scholar] [CrossRef]
- Zaminelli, T.; Gradowski, R.W.; Bassani, T.B.; Barbiero, J.K.; Santiago, R.M.; Maria-Ferreira, D.; Baggio, C.H.; Vital, M.A. Antidepressant and antioxidative effect of Ibuprofen in the rotenone model of Parkinson’s disease. Neurotox. Res. 2014, 26, 351–362. [Google Scholar] [CrossRef]
- Rees, K.; Stowe, R.; Patel, S.; Ives, N.; Breen, K.; Clarke, C.E.; Ben-Shlomo, Y. Non-steroidal anti-inflammatory drugs as disease-modifying agents for Parkinson’s disease: Evidence from observational studies. Cochrane Database Syst. Rev. 2011, 11, CD008454. [Google Scholar] [CrossRef]
- Joshi, N.; Singh, S. Updates on immunity and inflammation in Parkinson disease pathology. J. Neurosci. Res. 2018, 96, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Kata, D.; Földesi, I.; Feher, L.Z.; Hackler, L.; Puskas, L.G.; Gulya, K. A novel pleiotropic effect of aspirin: Beneficial regulation of pro- and anti-inflammatory mechanisms in microglial cells. Brain Res. Bull. 2017, 132, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Liu, X.; Ye, Y.; Yan, X.; Cheng, Y.; Zhao, L.; Chen, F.; Ling, Z. Gut Microbiota: A Novel Therapeutic Target for Parkinson’s Disease. Front. Immunol. 2022, 13, 937555. [Google Scholar] [CrossRef] [PubMed]
- Perez-Pardo, P.; Kliest, T.; Dodiya, H.B.; Broersen, L.M.; Garssen, J.; Keshavarzian, A.; Kraneveld, A.D. The gut-brain axis in Parkinson’s disease: Possibilities for food-based therapies. Eur. J. Pharmacol. 2017, 817, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.; Forsyth, C.B.; Shaikh, M.; Voigt, R.M.; Engen, P.A.; Ramirez, V.; Keshavarzian, A. Diet in Parkinson’s Disease: Critical Role for the Microbiome. Front. Neurol. 2019, 10, 1245. [Google Scholar] [CrossRef] [PubMed]
- Bousquet, M.; Saint-Pierre, M.; Julien, C.; Salem, N., Jr.; Cicchetti, F.; Calon, F. Beneficial effects of dietary omega-3 polyunsaturated fatty acid on toxin-induced neuronal degeneration in an animal model of Parkinson’s disease. FASEB J. 2008, 22, 1213–1225. [Google Scholar] [CrossRef]
- Bousquet, M.; Calon, F.; Cicchetti, F. Impact of omega-3 fatty acids in Parkinson’s disease. Ageing Res. Rev. 2011, 10, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Cassidy, A.; Schwarzschild, M.A.; Rimm, E.B.; Ascherio, A. Habitual intake of dietary flavonoids and risk of Parkinson disease. Neurology 2012, 78, 1138–1145. [Google Scholar] [CrossRef]
- Ross, G.W.; Abbott, R.D.; Petrovitch, H.; Morens, D.M.; Grandinetti, A.; Tung, K.H.; Tanner, C.M.; Masaki, K.H.; Blanchette, P.L.; Curb, J.D.; et al. Association of coffee and caffeine intake with the risk of Parkinson disease. JAMA 2000, 283, 2674–2679. [Google Scholar] [CrossRef]
- Murray, D.K.; Sacheli, M.A.; Eng, J.J.; Stoessl, A.J. The effects of exercise on cognition in Parkinson’s disease: A systematic review. Transl. Neurodegener. 2014, 3, 5. [Google Scholar] [CrossRef]
- Ahlskog, J.E. Does vigorous exercise have a neuroprotective effect in Parkinson disease? Neurology 2011, 77, 288–294. [Google Scholar] [CrossRef]
- Monteiro-Junior, R.S.; Cevada, T.; Oliveira, B.R.R.; Lattari, E.; Portugal, E.M.M.; Carvalho, A.; Deslandes, A.C. We need to move more: Neurobiological hypotheses of physical exercise as a treatment for Parkinson’s disease. Med. Hypotheses 2015, 85, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Zoladz, J.A.; Majerczak, J.; Zeligowska, E.; Mencel, J.; Jaskolski, A.; Jaskolska, A.; Marusiak, J. Moderate-intensity interval training increases serum brain-derived neurotrophic factor level and decreases inflammation in parkinson’s disease patients. J. Physiol. Pharmacol. 2014, 65, 441–448. [Google Scholar] [PubMed]
- Schenk, D.B.; Koller, M.; Ness, D.K.; Griffith, S.G.; Grundman, M.; Zago, W.; Soto, J.; Atiee, G.; Ostrowitzki, S.; Kinney, G.G. First-in-human assessment of PRX002, an anti–α-synuclein monoclonal antibody, in healthy volunteers. Mov. Disord. 2017, 32, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Weihofen, A.; Liu, Y.T.; Arndt, J.W.; Huy, C.; Quan, C.; Smith, B.A.; Baeriswyl, J.L.; Cavegn, N.; Senn, L.; Su, L.; et al. Development of an aggregate-selective, human-derived α-synuclein antibody BIIB054 that ameliorates disease phenotypes in Parkinson’s disease models. Neurobiol. Dis. 2019, 124, 276–288. [Google Scholar] [CrossRef]
- Schofield, D.J.; Irving, L.; Calo, L.; Bogstedt, A.; Rees, G.; Nuccitelli, A.; Narwal, R.; Petrone, M.; Roberts, J.; Brown, L.; et al. Preclinical development of a high affinity α-synuclein antibody, MEDI1341, that can enter the brain, sequester extracellular α-synuclein and attenuate α-synuclein spreading in vivo. Neurobiol. Dis. 2019, 132, 104582. [Google Scholar] [CrossRef]
- Nimmo, J.T.; Smith, H.; Wang, C.Y.; Teeling, J.L.; Nicoll, J.A.R.; Verma, A.; Dodart, J.C.; Liu, Z.; Lin, F.; Carare, R.O. Immunisation with UB-312 in the Thy1SNCA mouse prevents motor performance deficits and oligomeric α-synuclein accumulation in the brain and gut. Acta Neuropathol. 2022, 143, 55–73. [Google Scholar] [CrossRef]
- Mullin, S.; Smith, L.; Lee, K.; D’Souza, G.; Woodgate, P.; Elflein, J.; Hällqvist, J.; Toffoli, M.; Streeter, A.; Hosking, J.; et al. Ambroxol for the Treatment of Patients with Parkinson Disease with and Without Glucocerebrosidase Gene Mutations: A Nonrandomized, Noncontrolled Trial. JAMA Neurol. 2020, 77, 427–434. [Google Scholar] [CrossRef]
- Simuni, T.; Fiske, B.; Merchant, K.; Coffey, C.S.; Klingner, E.; Caspell-Garcia, C.; Lafontant, D.-E.; Matthews, H.; Wyse, R.K.; Brundin, P.; et al. Efficacy of Nilotinib in Patients With Moderately Advanced Parkinson Disease: A Randomized Clinical Trial. JAMA Neurol. 2021, 78, 312–320. [Google Scholar] [CrossRef]
- Savitt, D.; Jankovic, J. Targeting α-Synuclein in Parkinson’s Disease: Progress Towards the Development of Disease-Modifying Therapeutics. Drugs 2019, 79, 797–810. [Google Scholar] [CrossRef]
- Gendelman, H.E.; Zhang, Y.; Santamaria, P.; Olson, K.E.; Schutt, C.R.; Bhatti, D.; Shetty, B.L.D.; Lu, Y.; Estes, K.A.; Standaert, D.G.; et al. Evaluation of the safety and immunomodulatory effects of sargramostim in a randomized, double-blind phase 1 clinical Parkinson’s disease trial. NPJ Parkinsons Dis. 2017, 3, 10. [Google Scholar] [CrossRef]
- Singh, A.; Tripathi, P.; Singh, S. Neuroinflammatory responses in Parkinson’s disease: Relevance of Ibuprofen in therapeutics. Inflammopharmacology 2021, 29, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Borah, A.; Mohanakumar, K.P. Salicylic acid protects against chronic l-DOPA-induced 6-OHDA generation in experimental model of parkinsonism. Brain Res. 2010, 1344, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Racette, B.A.; Gross, A.; Vouri, S.M.; Camacho-Soto, A.; Willis, A.W.; Searles Nielsen, S. Immunosuppressants and risk of Parkinson disease. Ann. Clin. Transl. Neurol. 2018, 5, 870–875. [Google Scholar] [CrossRef]
- Braak, H.; De Vos, R.A.I.; Bohl, J.; Del Tredici, K. Gastric α-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci. Lett. 2006, 396, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Henry, C.J.; Huang, Y.; Wynne, A.; Hanke, M.; Himler, J.; Bailey, M.T.; Sheridan, J.F.; Godbout, J.P. Minocycline attenuates lipopolysaccharide (LPS)-induced neuroinflammation, sickness behavior, and anhedonia. J. Neuroinflamm. 2008, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Cankaya, S.; Cankaya, B.; Kilic, U.; Kilic, E.; Yulug, B. The therapeutic role of minocycline in Parkinson’s disease. Drugs Context 2019, 8, 212553. [Google Scholar] [CrossRef]
- Kurkowska-Jastrzȩbska, I.; Litwin, T.; Joniec, I.; Ciesielska, A.; Przybyłkowski, A.; Członkowski, A.; Członkowska, A. Dexamethasone protects against dopaminergic neurons damage in a mouse model of Parkinson’s disease. Int. Immunopharmacol. 2004, 4, 1307–1318. [Google Scholar] [CrossRef]
- Jin, H.; Zhang, J.; Hu, Q.; Ping, J.; Jiang, T.; Du, B.; Duan, X. Naloxone Alleviate the Severity of Delirium in Hospitalized Patients With Parkinsonism: Three Case Reports. Front. Psychiatry 2021, 12, 1–6. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Treatment | Target | Species Tested | Results | Reference |
|---|---|---|---|---|
| Animal Models-Active Immunization | ||||
| Vaccination of human aSyn | α-Syn | Transgenic mice human α-Syn |
| [242] |
| PSDC (peptide-sensitized dendritic cells) | Vaccine based on dendritic cells sensitized with α-Syn | Transgenic mice that expressed the human disease-associated A53T mutation of α-Syn with bone marrow-derived dendritic cells |
| [243] |
| DNA vaccination | Induced overexpression of growth factors | C57BL/6 mice |
| [244] |
| Animal models—passive immunization | ||||
| 9E.4 | C-terminus of α-Syn | Transgenic mice PDGF-hu-wt-α-Syn |
| [245] |
| AB274 | C-terminus of α-Syn | Transgenic mice PDGF-hu-wt-α-Syn |
| [246] |
| 1H7, 5C1 | C-terminus of α-Syn | Transgenic mice PDGF-hu-wt-α-Syn |
| [247] |
| 5D12 | C-terminus of α-Syn | Transgenic mice PDGF-hu-wt-α-Syn |
| [247] |
| Ab47 | Protofibrils of α-Syn | Transgenic mice expressing the human pathologic A30P variant of α-syn under a Thy1 promoter (Thy-1)-h[A30P] α-Syn transgenic mice) |
| [248] |
| Syn303 | N-terminal of α-Syn | C57BL6 mice |
| [249] |
| AB1 | N-terminal of α-Syn | Harlan rats injected with AAV-α-Syn-AB1 |
| [250] |
| AB2 | Central region of α-Syn | Harlan rats injected with AAV-α-Syn-AB2 |
| [250] |
| AFF1 | C-terminus of α-Syn | Transgenic mice PDGF-hu-wt- α-Syn Or Transgenic mice mThy1-α-Syn |
| [251] |
| Treatment | Target | Criteria | Phase | ClinicalTrials.gov Identifier | Description | Company/Class | Reference |
|---|---|---|---|---|---|---|---|
| Clinical trials for reducing extracellular α-Syn | |||||||
| PD01A | Oligomeric α-Syn | 45 and 65 years old with early-stage idiopathic Parkinson’s disease on stable medication | II (To begin) | NCT 01568099 | Active vaccine to α-Syn composed by amino acid peptide | AFFirRIS | [241] |
| PRX002 | C-terminus of α-Syn | Patients with early PD who are untreated or treated with MAO-B | II (Ongoing) | NCT03100149 | Monoclonal antibody | Prothena | [273] |
| BIIB054 | N-terminal of α-Syn | 40–80 years | II (Ongoing) | NCT03318523 | Human-derived α-Syn antibody | Biogen | [274] |
| MEDI1341 | α-Syn | Healthy volunteers aged 18 to 65 years | II (Completed) | NCT03272165 | Monoclonal antibody | AstraZeneca | [275] |
| 805 BAN | Oligomeric/protofibrillar α-Syn | Patients with idiopathic, mild to moderate Parkinson’s | I (Ongoing) | NCT04127695 | Humanized monoclonal antibody targeting α-Syn | Abbvie/Bioarctic | |
| UB-312 | C-terminus of α-Syn | Healthy participants and participants with PD | I (Ongoing) | NCT04075318 | UBITh-enhanced synthetic peptide-based vaccine | United neuroscience | [276] |
| Clinical trial for GCASE stimulation | |||||||
| Ambroxol | β-glucocerebrosidase pathway/expression | 40–80 years | II (Completed) | NCT02941822 | Mucolytic compound that acts as a molecular chaperone for the lysosomal enzyme glucocerebrosidase (GCase) | Cure PD | [277] |
| Clinical trial for inhibiting αSyn aggregation | |||||||
| NPT200-11 | α-Syn | 18–55 years | I (Completed) | NCT02606682 | Small-molecule inhibitor of α-Syn misfolding and aggregation | Celerion/NeuroPore | |
| Clinical trials for enhancing autophagy | |||||||
| Nilotinib | Abl tyrosine kinase inhibitor | 40–79 years with idiopathic PD | II (Completed) | NCT03205488 | Abl tyrosine kinase inhibitor | Michael J. Fox Foundation for Parkinson’s Research | [278] |
| K0706 | Abl tyrosine kinase inhibitor | More than 50 years early PD not receiving dopaminergic therapy | II (Ongoing) | NCT03655236 | Suppressor of an enzyme called Abl tyrosine kinase | Sun Pharma Advanced Research Company Limited | [279] |
| Other recent clinical trials | |||||||
| GE180 | Mitochondrial translocator protein (TSPO) | 55–90 years | Ongoing | NCT03702816 | PET ligand | The Cleveland Clinic | |
| [18F]DPA-714 | Mitochondrial translocator protein (TSPO) | 30 years and older | Ongoing | NCT03457493 | PET tracer | University of Alabama at Birmingham | |
| Leukine | Granulocyte-macrophage colony stimulating factor receptor | 35–85 years | I (Ongoing) | NCT03790670 | Human recombinant granulocyte macrophage colony-stimulating factor expressed in yeast | University of Nebraska | [280] |
| Treatment | Target | Description | Class | Reference |
|---|---|---|---|---|
| Potential Therapies for PD | ||||
| Ibuprofen | Inhibitor of COX |
| NSAIDs | [259,281] |
| Aspirin | Inhibitor of COX |
| NSAIDs | [261] |
| Salicyclic acid | Metabolite of aspirin |
| NSAIDs | [282] |
| MCC950 | Walker B motif within the NLRP3 NACHT domain |
| Inflammasome inhibitor | [99,100,101] |
| Inosine monophosphate dehydrogenase (IMDH) inhibitors | Intracellular signaling pathways |
| Immunosuppressors | [283,284] |
| Minocycline |
| Antibiotic | [285,286] | |
| Dexamethasone | Cytoplasmic glucocorticoid receptor |
| Glucocorticoid | [287] |
| Naloxone | μ-Opioid receptors |
| Opioid | [288] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Araújo, B.; Caridade-Silva, R.; Soares-Guedes, C.; Martins-Macedo, J.; Gomes, E.D.; Monteiro, S.; Teixeira, F.G. Neuroinflammation and Parkinson’s Disease—From Neurodegeneration to Therapeutic Opportunities. Cells 2022, 11, 2908. https://doi.org/10.3390/cells11182908
Araújo B, Caridade-Silva R, Soares-Guedes C, Martins-Macedo J, Gomes ED, Monteiro S, Teixeira FG. Neuroinflammation and Parkinson’s Disease—From Neurodegeneration to Therapeutic Opportunities. Cells. 2022; 11(18):2908. https://doi.org/10.3390/cells11182908
Chicago/Turabian StyleAraújo, Bruna, Rita Caridade-Silva, Carla Soares-Guedes, Joana Martins-Macedo, Eduardo D. Gomes, Susana Monteiro, and Fábio G. Teixeira. 2022. "Neuroinflammation and Parkinson’s Disease—From Neurodegeneration to Therapeutic Opportunities" Cells 11, no. 18: 2908. https://doi.org/10.3390/cells11182908
APA StyleAraújo, B., Caridade-Silva, R., Soares-Guedes, C., Martins-Macedo, J., Gomes, E. D., Monteiro, S., & Teixeira, F. G. (2022). Neuroinflammation and Parkinson’s Disease—From Neurodegeneration to Therapeutic Opportunities. Cells, 11(18), 2908. https://doi.org/10.3390/cells11182908