

Augmented Antitumor Effect of Unripe Rubus coreanus Miquel Combined with Oxaliplatin in a Humanized PD-1/PD-L1 Knock-In Colorectal Cancer Mouse Model



Abstract
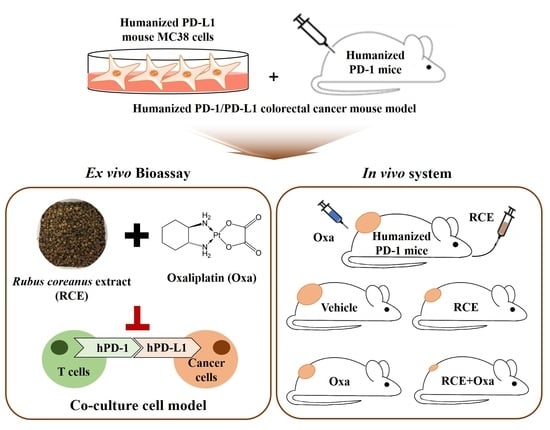
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Preparation of RCE
2.2. Materials
2.3. Humanized PD-L1 MC38 Cell Lines
2.4. Humanized PD-1 Mice
2.5. Tumor Allograft Mice Model
2.6. Isolation and Activation of Tumor-Infiltrating T Cells
2.7. Cell Counting Kit-8 (CCK) Assay
2.8. The Co-Culture System with Tumor-Infiltrating T Cells and MC38 Cells
2.9. IL-2 Measurement
2.10. Granzyme B Measurement
2.11. In Vivo RCE and Oxa Treatment
2.12. Blood Biochemistry
2.13. Immunohistochemistry
2.14. Statistical Analysis
3. Results
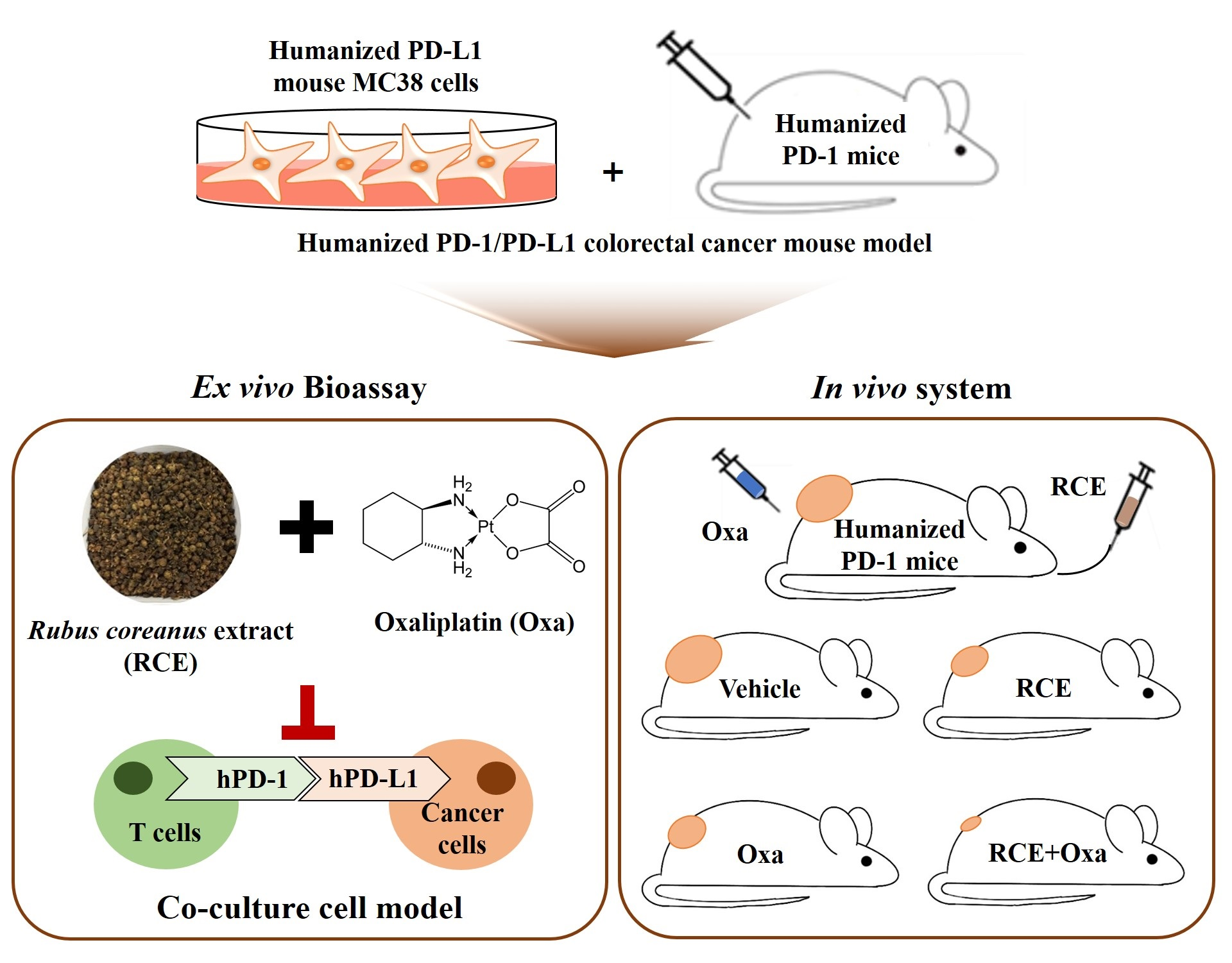
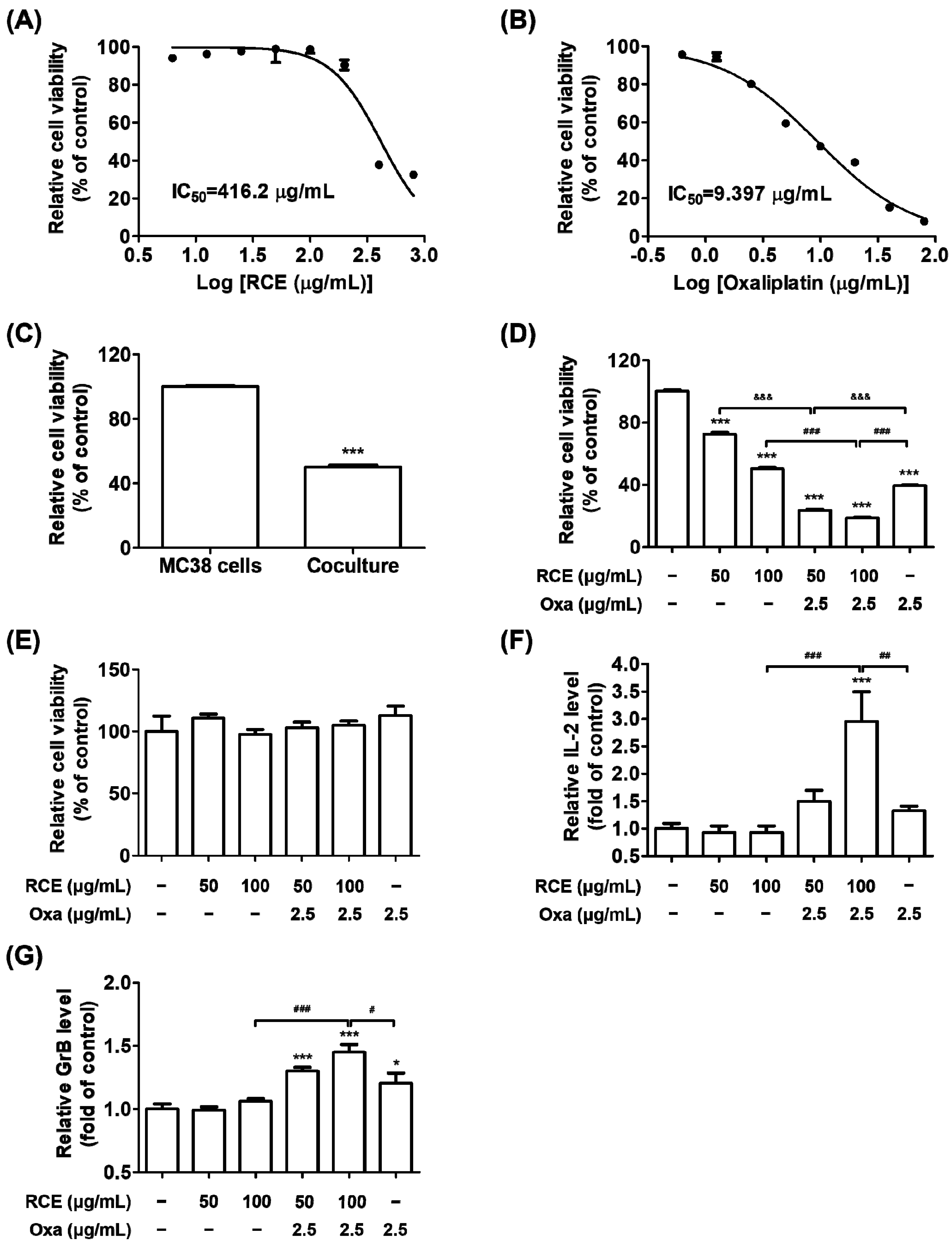
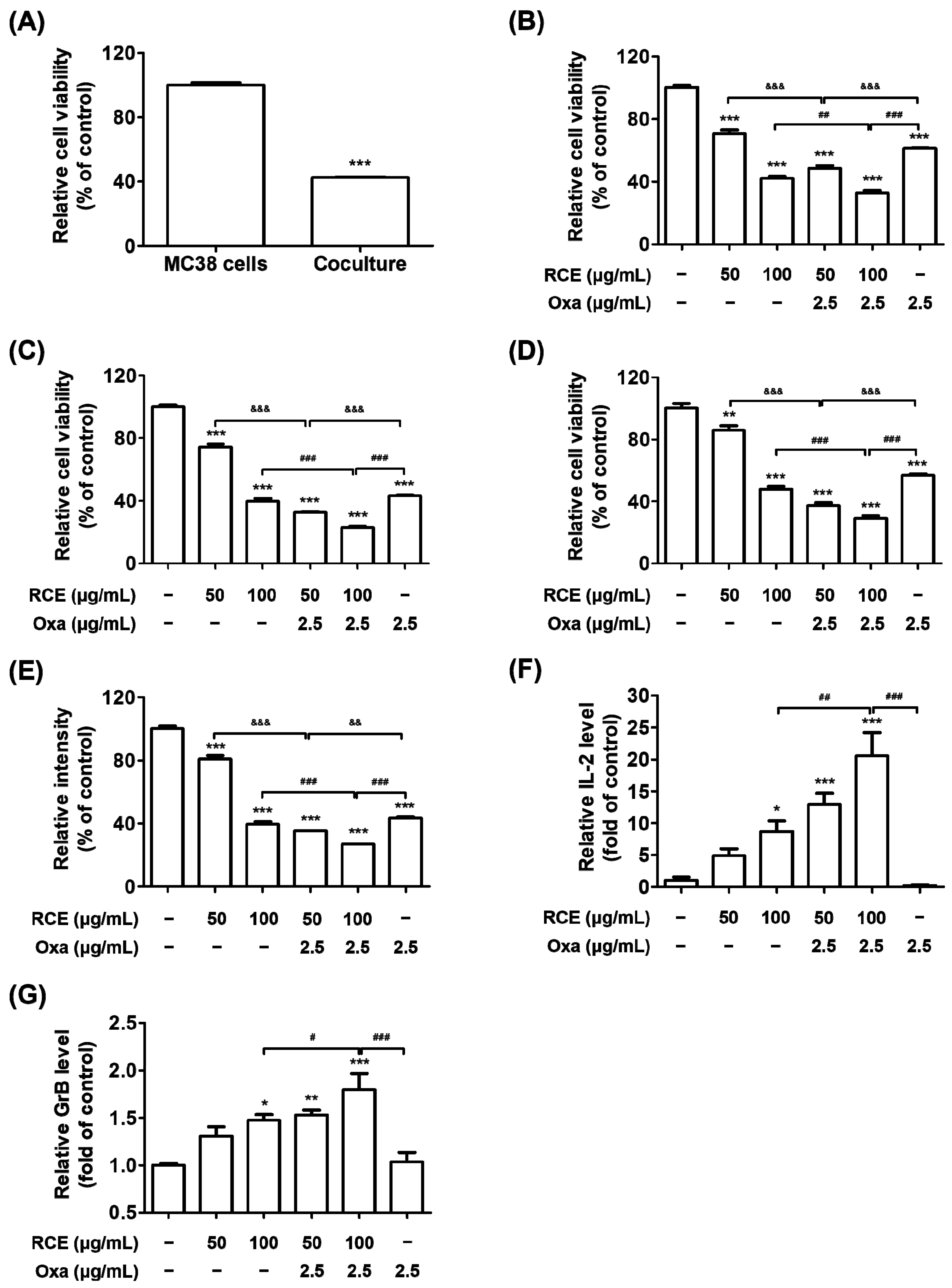
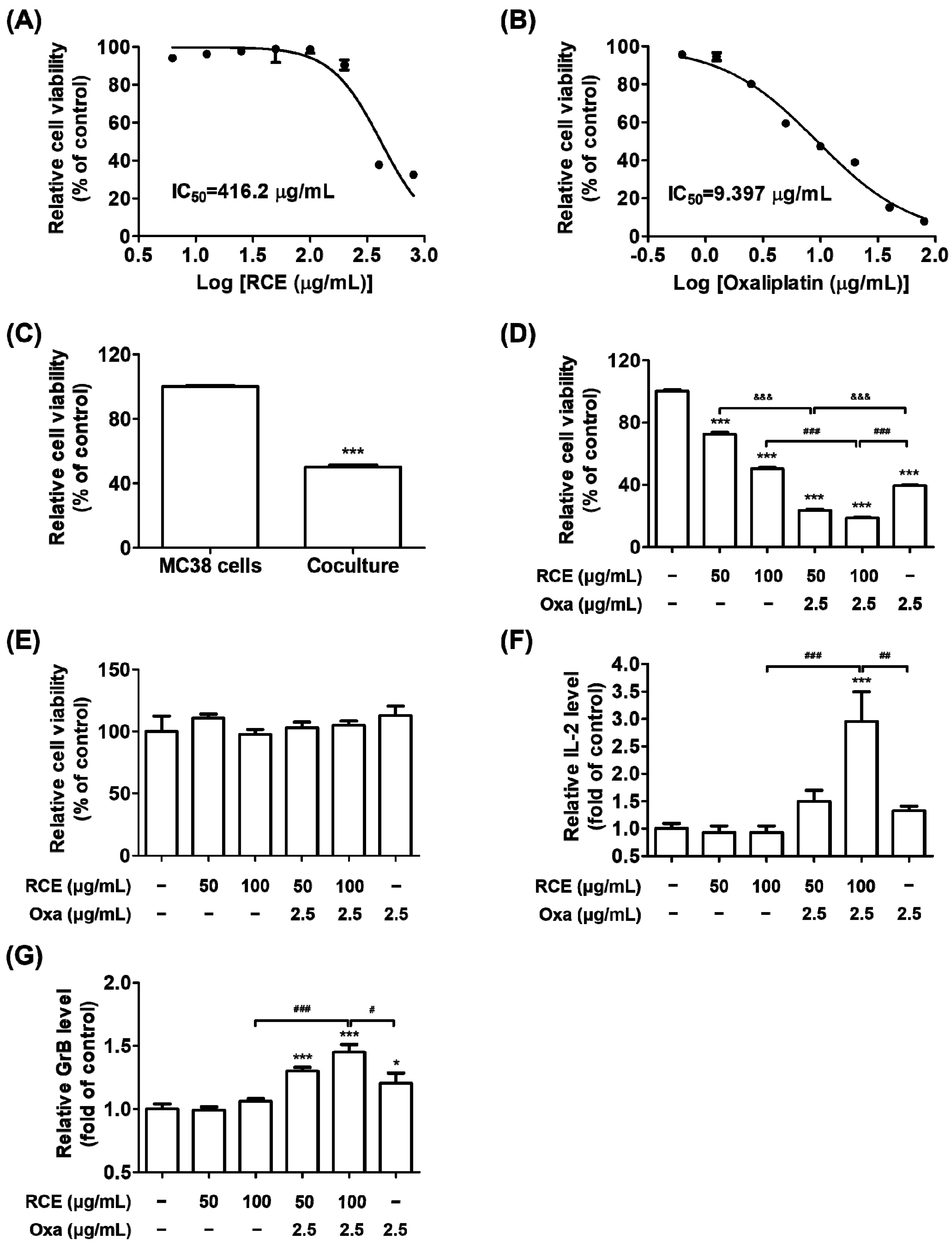
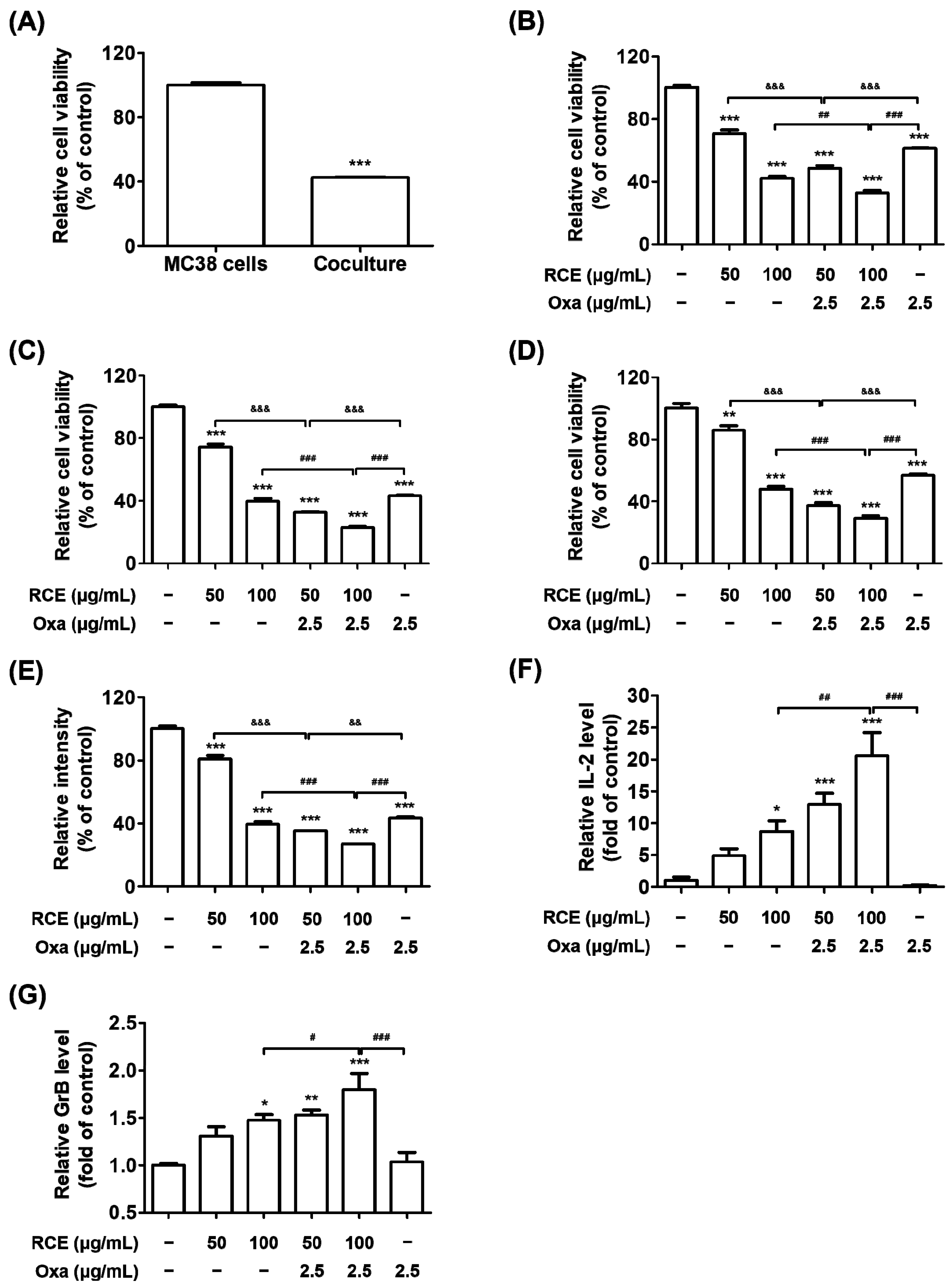
3.1. Augmented Antitumor Effect of Tumor-Infiltrating CD3+ T Cell-Mediated CRC Cell Killing by RCE plus Oxa Combination Therapy
3.2. RCE plus Oxa Enhances the Activation of Tumor-Infiltrating CD3+CD8+ T Cells Yielding an Augmented Antitumor Effect
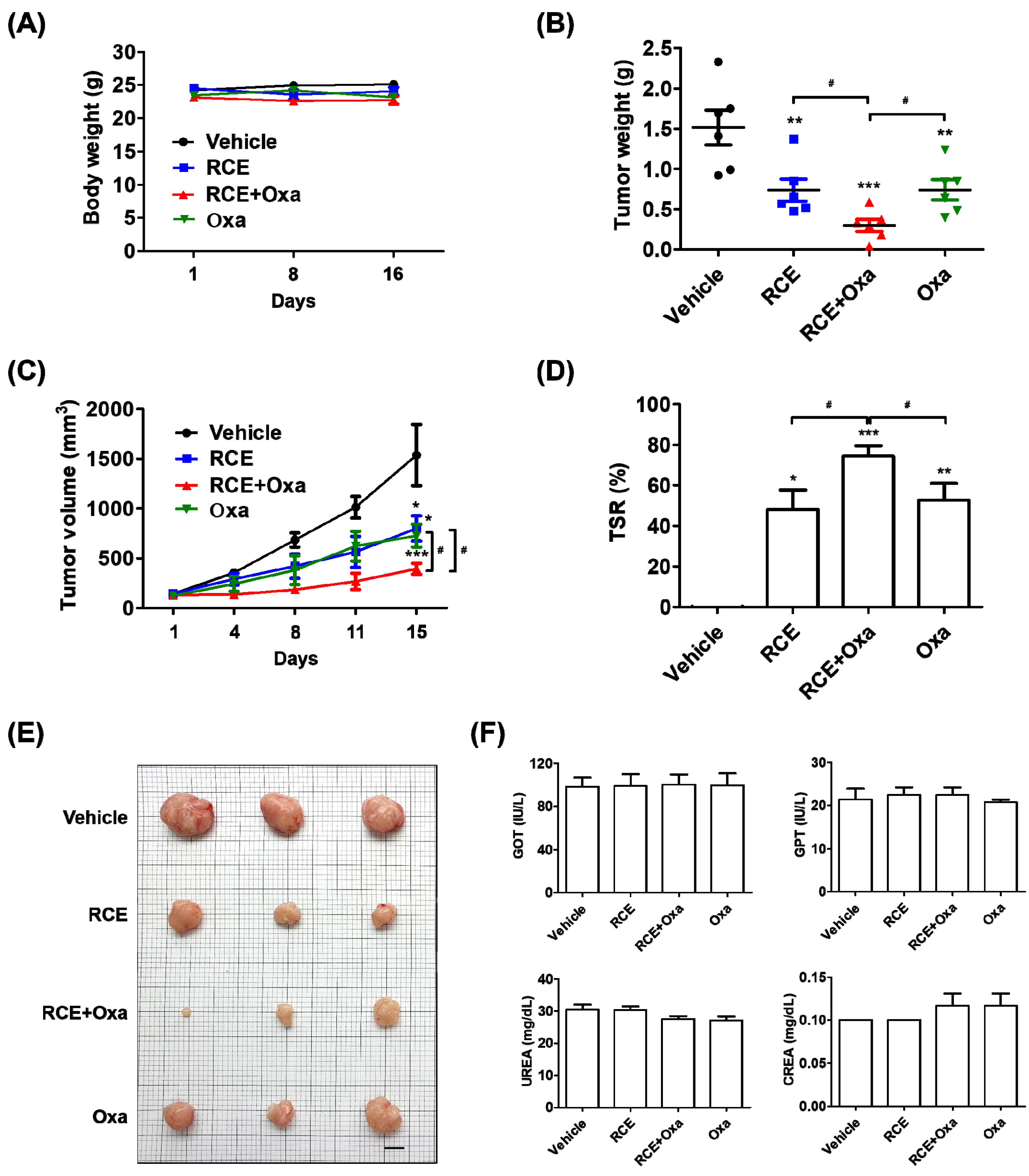
3.3. Augmented Antitumor Effect of RCE plus Oxa in Humanized PD-1/PD-L1 MC38 Tumor Mouse Models
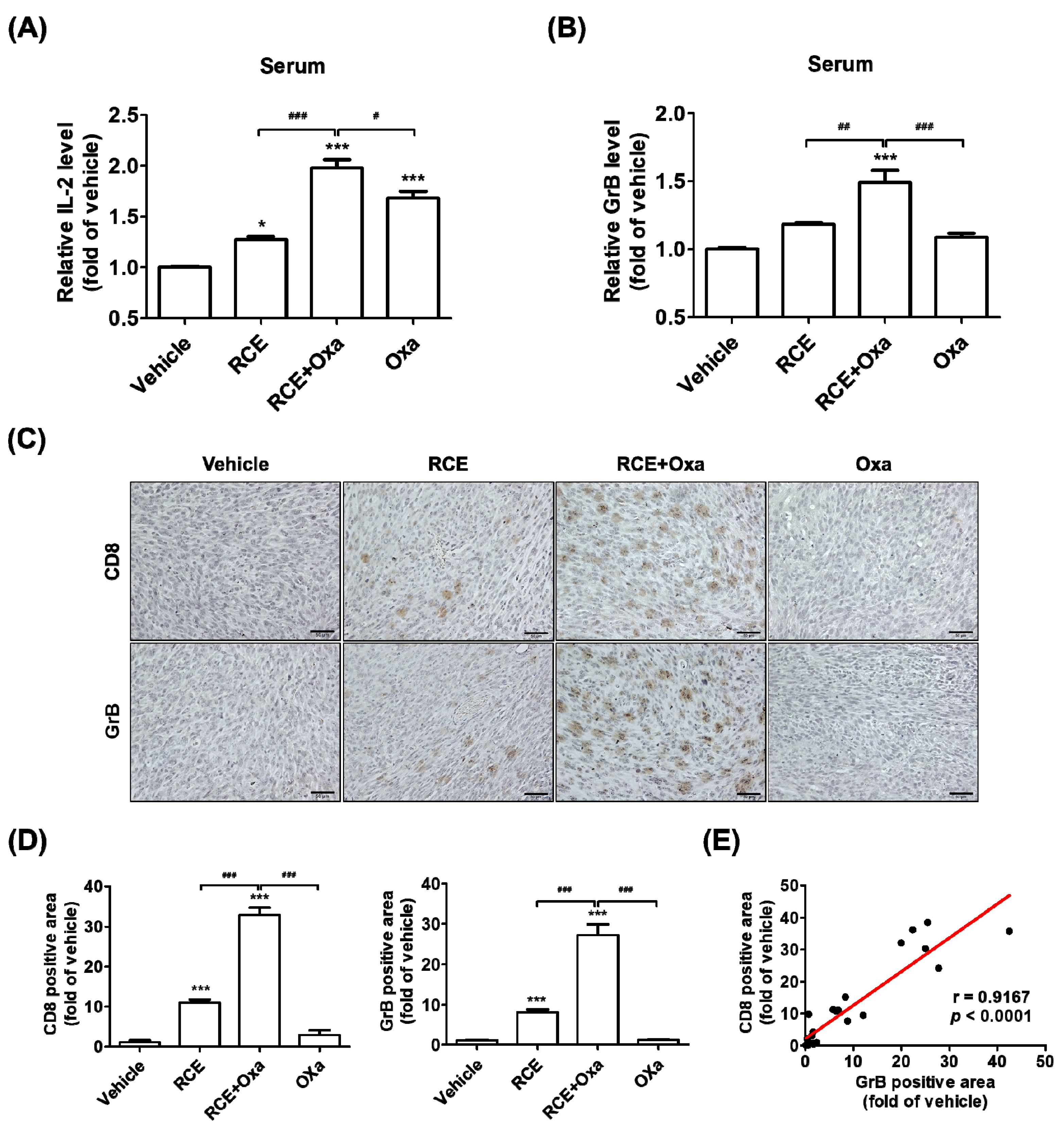
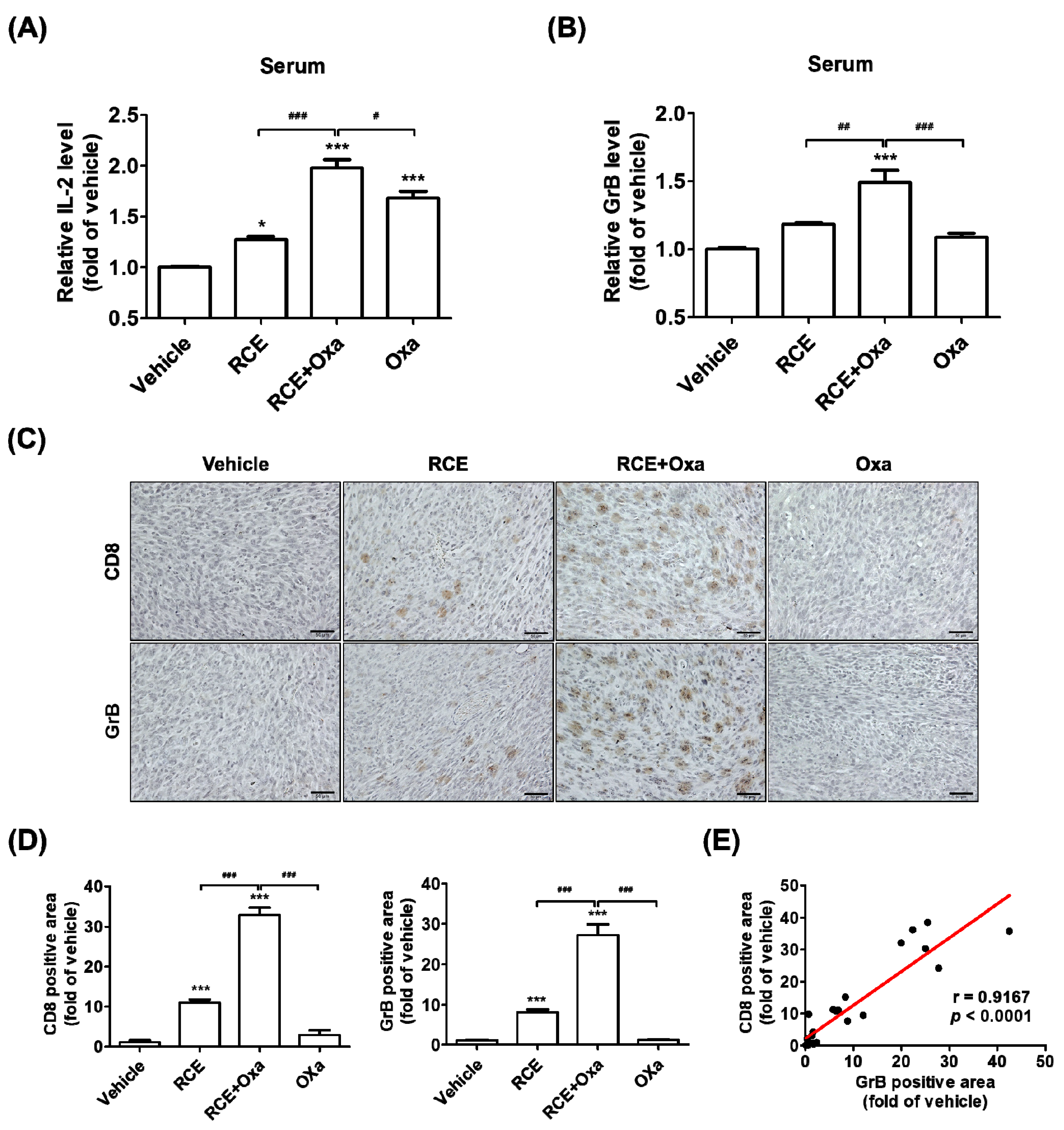
3.4. RCE plus Oxa Increased CD8+ T Cell Infiltration in Humanized PD-1/PD-L1 MC38 Tumor Tissues
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Xi, Y.; Xu, P. Global colorectal cancer burden in 2020 and projections to 2040. Transl. Oncol. 2021, 14, 101174. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA A Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Xie, Y.-H.; Chen, Y.-X.; Fang, J.-Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther. 2020, 5, 22. [Google Scholar] [CrossRef]
- Moutabian, H.; Majdaeen, M.; Ghahramani-Asl, R.; Yadollahi, M.; Gharepapagh, E.; Ataei, G.; Falahatpour, Z.; Bagheri, H.; Farhood, B. A systematic review of the therapeutic effects of resveratrol in combination with 5-fluorouracil during colorectal cancer treatment: With a special focus on the oxidant, apoptotic, and anti-inflammatory activities. Cancer Cell Int. 2022, 22, 142. [Google Scholar] [CrossRef]
- Ren, D.; Hua, Y.; Yu, B.; Ye, X.; He, Z.; Li, C.; Wang, J.; Mo, Y.; Wei, X.; Chen, Y.; et al. Predictive biomarkers and mechanisms underlying resistance to PD1/PD-L1 blockade cancer immunotherapy. Mol. Cancer 2020, 19, 19. [Google Scholar] [CrossRef]
- Spranger, S.; Gajewski, T.F. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat. Cancer 2018, 18, 139–147. [Google Scholar] [CrossRef]
- Hamanishi, J.; Mandai, M.; Matsumura, N.; Abiko, K.; Baba, T.; Konishi, I. PD-1/PD-L1 blockade in cancer treatment: Perspectives and issues. Int. J. Clin. Oncol. 2016, 21, 462–473. [Google Scholar] [CrossRef]
- Twomey, J.D.; Zhang, B. Cancer Immunotherapy Update: FDA-Approved Checkpoint Inhibitors and Companion Diagnostics. AAPS J. 2021, 23, 39. [Google Scholar] [CrossRef]
- Picard, E.; Verschoor, C.P.; Ma, G.W.; Pawelec, G. Relationships Between Immune Landscapes, Genetic Subtypes and Responses to Immunotherapy in Colorectal Cancer. Front. Immunol. 2020, 11, 369. [Google Scholar] [CrossRef]
- Yarchoan, M.; Johnson, B.A., III; Lutz, E.R.; Laheru, D.A.; Jaffee, E.M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 2017, 17, 209–222. [Google Scholar] [CrossRef]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef]
- Martinez-Lostao, L.; Anel, A.; Pardo, J. How Do Cytotoxic Lymphocytes Kill Cancer Cells? Clin. Cancer Res. 2015, 21, 5047–5056. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol. Cancer 2019, 18, 10. [Google Scholar] [CrossRef]
- Kim, J.-H.; Lee, K.-J.L.; Lee, S.-W. Cancer immunotherapy with T-cell targeting cytokines: IL-2 and IL-7. BMB Rep. 2021, 54, 21–30. [Google Scholar] [CrossRef]
- Zhan, M.-M.; Hu, X.-Q.; Liu, X.-X.; Ruan, B.-F.; Xu, J.; Liao, C. From monoclonal antibodies to small molecules: The development of inhibitors targeting the PD-1/PD-L1 pathway. Drug Discov. Today 2016, 21, 1027–1036. [Google Scholar] [CrossRef]
- Lumish, M.A.; Cercek, A. Immunotherapy for the treatment of colorectal cancer. J. Surg. Oncol. 2021, 123, 760–774. [Google Scholar] [CrossRef]
- Zitvogel, L.; Apetoh, L.; Ghiringhelli, F.; Kroemer, G. Immunological aspects of cancer chemotherapy. Nat. Rev. Immunol. 2008, 8, 59–73. [Google Scholar] [CrossRef]
- Galluzzi, L.; Senovilla, L.; Zitvogel, L.; Kroemer, G. The secret ally: Immunostimulation by anticancer drugs. Nat. Rev. Drug Discov. 2012, 11, 215–233. [Google Scholar] [CrossRef]
- Dosset, M.; Vargas, T.R.; Lagrange, A.; Boidot, R.; Vegran, F.; Roussey, A.; Chalmin, F.; Dondaine, L.; Paul, C.; Marie-Joseph, E.L.; et al. PD-1/PD-L1 pathway: An adaptive immune resistance mechanism to immunogenic chemotherapy in colorectal cancer. OncoImmunology 2018, 7, e1433981. [Google Scholar] [CrossRef]
- Calvo, E.; Cortes, J.; González-Cao, M.; Rodríguez, J.; Aramendía, J.M.; Fernández-Hidalgo, O.; Martín-Algarra, S.; Salgado, J.E.; Martínez-Monge, R.; de Irala, J.; et al. Combined Irinotecan, Oxaliplatin and 5-Fluorouracil in Patients with Advanced Colorectal Cancer. Oncology 2002, 63, 254–265. [Google Scholar] [CrossRef]
- Golshani, G.; Zhang, Y. Advances in immunotherapy for colorectal cancer: A review. Ther. Adv. Gastroenterol. 2020, 13. [Google Scholar] [CrossRef]
- Jung, K.-A.; Han, D.; Kwon, E.-K.; Lee, C.-H.; Kim, Y.-E. Antifatigue Effect of Rubus coreanus Miquel Extract in Mice. J. Med. Food 2007, 10, 689–693. [Google Scholar] [CrossRef]
- Kim, Y.; Lee, S.M.; Kim, J.-H. Unripe Rubus coreanus Miquel suppresses migration and invasion of human prostate cancer cells by reducing matrix metalloproteinase expression. Biosci. Biotechnol. Biochem. 2014, 78, 1402–1411. [Google Scholar] [CrossRef]
- Choi, C.; Lee, H.; Lim, H.; Park, S.; Lee, J.; Do, S. Effect of Rubus coreanus extracts on diabetic osteoporosis by simultaneous regulation of osteoblasts and osteoclasts. Menopause 2012, 19, 1043–1051. [Google Scholar] [CrossRef]
- Kim, S.K.; Kim, H.; Kim, S.A.; Park, H.K.; Kim, W. Anti-Inflammatory and Anti-Superbacterial Activity of Polyphenols Isolated from Black Raspberry. Korean J. Physiol. Pharmacol. 2013, 17, 73–79. [Google Scholar] [CrossRef]
- Lee, J.E.; Park, E.; Lee, J.E.; Auh, J.H.; Choi, H.-K.; Lee, J.; Cho, S.; Kim, J.-H. Effects of aRubus coreanusMiquel supplement on plasma antioxidant capacity in healthy Korean men. Nutr. Res. Pract. 2011, 5, 429–434. [Google Scholar] [CrossRef]
- Wang, L.-S.; Kuo, C.-T.; Cho, S.-J.; Seguin, C.; Siddiqui, J.; Stoner, K.; Weng, Y.-I.; Huang, T.H.-M.; Tichelaar, J.; Yearsley, M.; et al. Black Raspberry-Derived Anthocyanins Demethylate Tumor Suppressor Genes Through the Inhibition of DNMT1 and DNMT3B in Colon Cancer Cells. Nutr. Cancer 2013, 65, 118–125. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, Y.S.; Kim, T.I.; Li, W.; Mun, J.-G.; Jeon, H.D.; Kee, J.-Y.; Choi, J.-G.; Chung, H.-S. Unripe Black Raspberry (Rubus coreanus Miquel) Extract and Its Constitute, Ellagic Acid Induces T Cell Activation and Antitumor Immunity by Blocking PD-1/PD-L1 Interaction. Foods 2020, 9, 1590. [Google Scholar] [CrossRef]
- Liu, C.; Seeram, N.P.; Ma, H. Small molecule inhibitors against PD-1/PD-L1 immune checkpoints and current methodologies for their development: A review. Cancer Cell Int. 2021, 21, 239. [Google Scholar] [CrossRef]
- Gomez-Cadena, A.; Barreto, A.; Fioretino, S.; Jandus, C. Immune system activation by natural products and complex fractions: A network pharmacology approach in cancer treatment. Cell Stress 2020, 4, 154–166. [Google Scholar] [CrossRef]
- Li, W.; Kim, T.I.; Kim, J.H.; Chung, H.-S. Immune Checkpoint PD-1/PD-L1 CTLA-4/CD80 are Blocked by Rhus verniciflua Stokes and its Active Compounds. Molecules 2019, 24, 4062. [Google Scholar] [CrossRef]
- Lee, E.-J.; Kim, J.H.; Kim, T.I.; Kim, Y.-J.; Pak, M.E.; Jeon, C.H.; Park, Y.J.; Li, W.; Kim, Y.S.; Choi, J.-G.; et al. Sanguisorbae Radix Suppresses Colorectal Tumor Growth Through PD-1/PD-L1 Blockade and Synergistic Effect with Pembrolizumab in a Humanized PD-L1-Expressing Colorectal Cancer Mouse Model. Front. Immunol. 2021, 12, 737076. [Google Scholar] [CrossRef]
- Choi, J.-G.; Kim, Y.S.; Kim, J.H.; Kim, T.I.; Li, W.; Oh, T.W.; Jeon, C.H.; Kim, S.J.; Chung, H.-S. Anticancer Effect of Salvia plebeia and Its Active Compound by Improving T-Cell Activity via Blockade of PD-1/PD-L1 Interaction in Humanized PD-1 Mouse Model. Front. Immunol. 2020, 11, 598556. [Google Scholar] [CrossRef]
- Opzoomer, J.; Sosnowska, D.; Anstee, J.E.; Spicer, J.; Arnold, J. Cytotoxic Chemotherapy as an Immune Stimulus: A Molecular Perspective on Turning Up the Immunological Heat on Cancer. Front. Immunol. 2019, 10, 1654. [Google Scholar] [CrossRef]
- Almquist, D.R.; Ahn, D.H.; Bekaii-Saab, T.S. The Role of Immune Checkpoint Inhibitors in Colorectal Adenocarcinoma. BioDrugs 2020, 34, 349–362. [Google Scholar] [CrossRef]
- Zhao, P.; Li, L.; Jiang, X.; Li, Q. Mismatch repair deficiency/microsatellite instability-high as a predictor for anti-PD-1/PD-L1 immunotherapy efficacy. J. Hematol. Oncol. 2019, 12, 54. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.-J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Eng, C.; Kim, T.W.; Bendell, J.; Argilés, G.; Tebbutt, N.C.; Di Bartolomeo, M.; Falcone, A.; Fakih, M.; Kozloff, M.; Segal, N.H.; et al. Atezolizumab with or without cobimetinib versus regorafenib in previously treated metastatic colorectal cancer (IMblaze370): A multicentre, open-label, phase 3, randomised, controlled trial. Lancet Oncol. 2019, 20, 849–861. [Google Scholar] [CrossRef]
- Ri, M.H.; Ma, J.; Jin, X. Development of natural products for anti-PD-1/PD-L1 immunotherapy against cancer. J. Ethnopharmacol. 2021, 281, 114370. [Google Scholar] [CrossRef]
- Liu, L.; Mayes, P.A.; Eastman, S.; Shi, H.; Yadavilli, S.; Zhang, T.; Yang, J.; Seestaller-Wehr, L.; Zhang, S.-Y.; Hopson, C.; et al. The BRAF and MEK Inhibitors Dabrafenib and Trametinib: Effects on Immune Function and in Combination with Immunomodulatory Antibodies Targeting PD-1, PD-L1, and CTLA-4. Clin. Cancer Res. 2015, 21, 1639–1651. [Google Scholar] [CrossRef]
- Antoniotti, C.; Rossini, D.; Pietrantonio, F.; Catteau, A.; Salvatore, L.; Lonardi, S.; Boquet, I.; Tamberi, S.; Marmorino, F.; Moretto, R.; et al. Upfront FOLFOXIRI plus bevacizumab with or without atezolizumab in the treatment of patients with metastatic colorectal cancer (AtezoTRIBE): A multicentre, open-label, randomised, controlled, phase 2 trial. Lancet Oncol. 2022, 23, 876–887. [Google Scholar] [CrossRef]
- Guan, Y.; Kraus, S.G.; Quaney, M.J.; Daniels, M.A.; Mitchem, J.B.; Teixeiro, E. FOLFOX Chemotherapy Ameliorates CD8 T Lymphocyte Exhaustion and Enhances Checkpoint Blockade Efficacy in Colorectal Cancer. Front. Oncol. 2020, 10, 586. [Google Scholar] [CrossRef]
- Grapin, M.; Richard, C.; Limagne, E.; Boidot, R.; Morgand, V.; Bertaut, A.; Derangere, V.; Laurent, P.-A.; Thibaudin, M.; Fumet, J.D.; et al. Optimized fractionated radiotherapy with anti-PD-L1 and anti-TIGIT: A promising new combination. J. Immunother. Cancer 2019, 7, 160. [Google Scholar] [CrossRef]
- Yuan, Z.; Fan, G.; Wu, H.; Liu, C.; Zhan, Y.; Qiu, Y.; Shou, C.; Gao, F.; Zhang, J.; Yin, P.; et al. Photodynamic therapy synergizes with PD-L1 checkpoint blockade for immunotherapy of CRC by multifunctional nanoparticles. Mol. Ther. 2021, 29, 2931–2948. [Google Scholar] [CrossRef]
- Ganesh, K.; Stadler, Z.K.; Cercek, A.; Mendelsohn, R.B.; Shia, J.; Segal, N.H.; Diaz, L.A., Jr. Immunotherapy in colorectal cancer: Rationale, challenges and potential. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 361–375. [Google Scholar] [CrossRef]
- Golchin, S.; Alimohammadi, R.; Nejad, M.R.; Jalali, S.A. Synergistic antitumor effect of anti-PD-L1 combined with oxaliplatin on a mouse tumor model. J. Cell. Physiol. 2019, 234, 19866–19874. [Google Scholar] [CrossRef]
- Cubas, R.; Moskalenko, M.; Cheung, J.; Yang, M.; McNamara, E.; Xiong, H.; Hoves, S.; Ries, C.H.; Kim, J.; Gould, S. Chemotherapy Combines Effectively with Anti–PD-L1 Treatment and Can Augment Antitumor Responses. J. Immunol. 2018, 201, 2273–2286. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell 2015, 28, 690–714. [Google Scholar] [CrossRef]
- Ogu, C.C.; Maxa, J.L. Drug Interactions Due to Cytochrome P450. Bayl. Univ. Med Cent. Proc. 2000, 13, 421–423. [Google Scholar] [CrossRef]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, E.-J.; Yang, J.-H.; Choi, J.-G.; Chung, H.-S. Augmented Antitumor Effect of Unripe Rubus coreanus Miquel Combined with Oxaliplatin in a Humanized PD-1/PD-L1 Knock-In Colorectal Cancer Mouse Model. Cells 2022, 11, 2876. https://doi.org/10.3390/cells11182876
Lee E-J, Yang J-H, Choi J-G, Chung H-S. Augmented Antitumor Effect of Unripe Rubus coreanus Miquel Combined with Oxaliplatin in a Humanized PD-1/PD-L1 Knock-In Colorectal Cancer Mouse Model. Cells. 2022; 11(18):2876. https://doi.org/10.3390/cells11182876
Chicago/Turabian StyleLee, Eun-Ji, Ju-Hye Yang, Jang-Gi Choi, and Hwan-Suck Chung. 2022. "Augmented Antitumor Effect of Unripe Rubus coreanus Miquel Combined with Oxaliplatin in a Humanized PD-1/PD-L1 Knock-In Colorectal Cancer Mouse Model" Cells 11, no. 18: 2876. https://doi.org/10.3390/cells11182876
APA StyleLee, E.-J., Yang, J.-H., Choi, J.-G., & Chung, H.-S. (2022). Augmented Antitumor Effect of Unripe Rubus coreanus Miquel Combined with Oxaliplatin in a Humanized PD-1/PD-L1 Knock-In Colorectal Cancer Mouse Model. Cells, 11(18), 2876. https://doi.org/10.3390/cells11182876