
CAR-T-Cell Therapy for Solid Tumors Positive for Fibronectin Extra Domain B

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture
2.2. Lentivirus Production
2.3. Transduction and Culture of Primary T-Cells
2.4. Flow Cytometric Analysis
2.5. CD107a Degranulation Assay
2.6. Quantitative Real-Time PCR
2.7. Cytotoxicity and Cytokine Release Assays In Vitro
2.8. Animal Models
2.9. Immunohistochemistry (IHC) Analysis
2.10. Statistical Analysis
3. Results
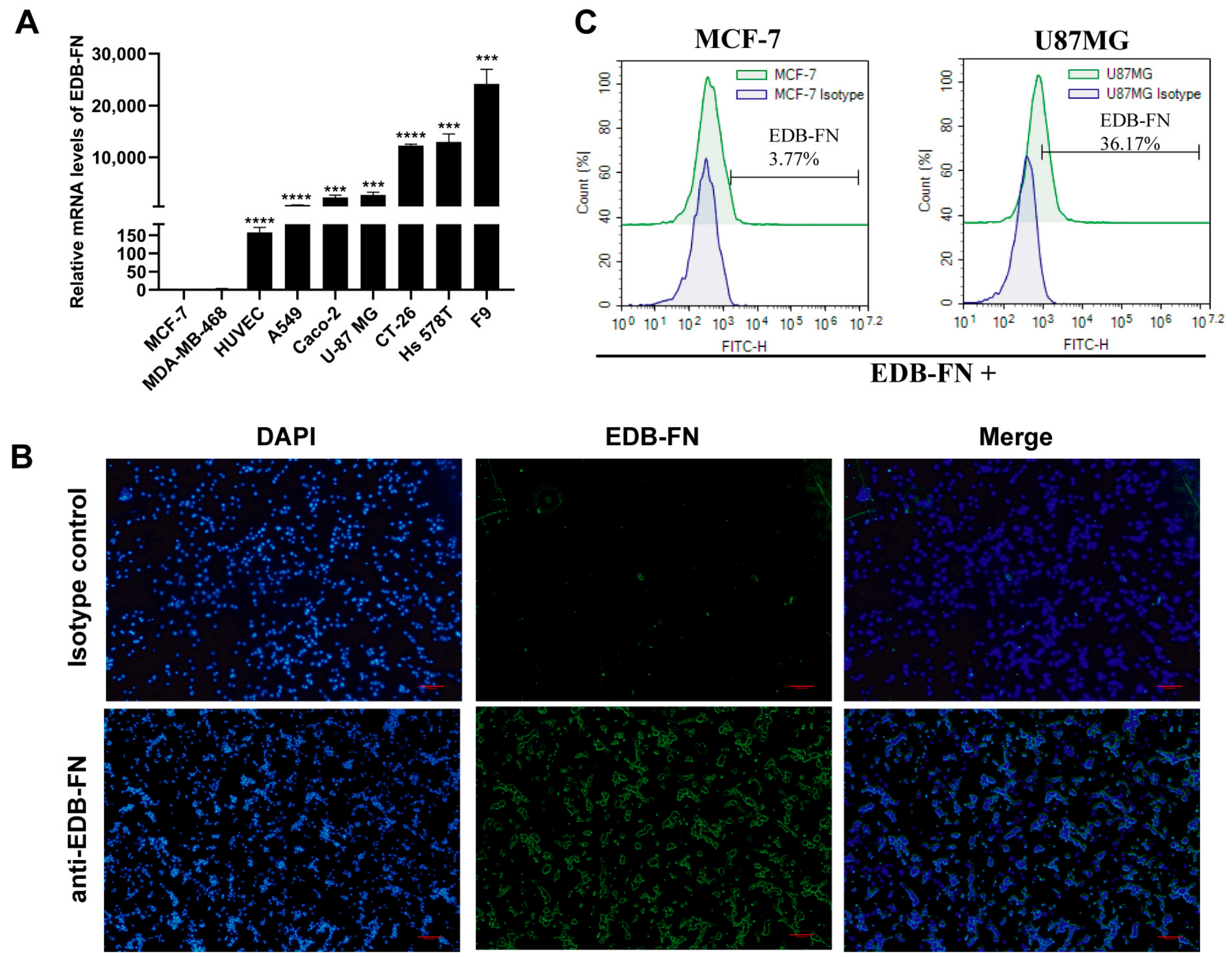
3.1. EDB-FN Antigen Is Expressed in Various Tumors and Related Cells
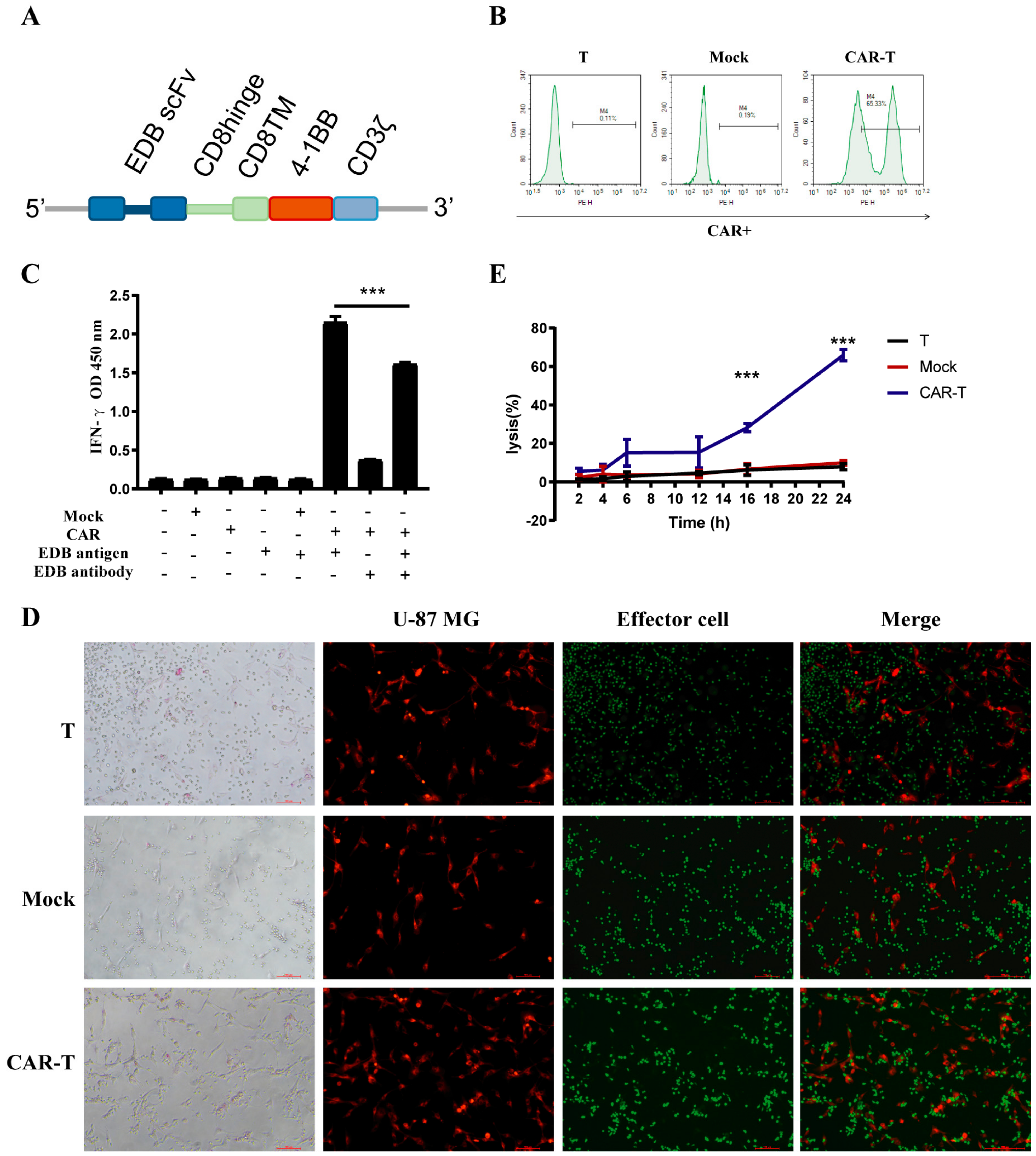
3.2. CAR Construction and Characteristics
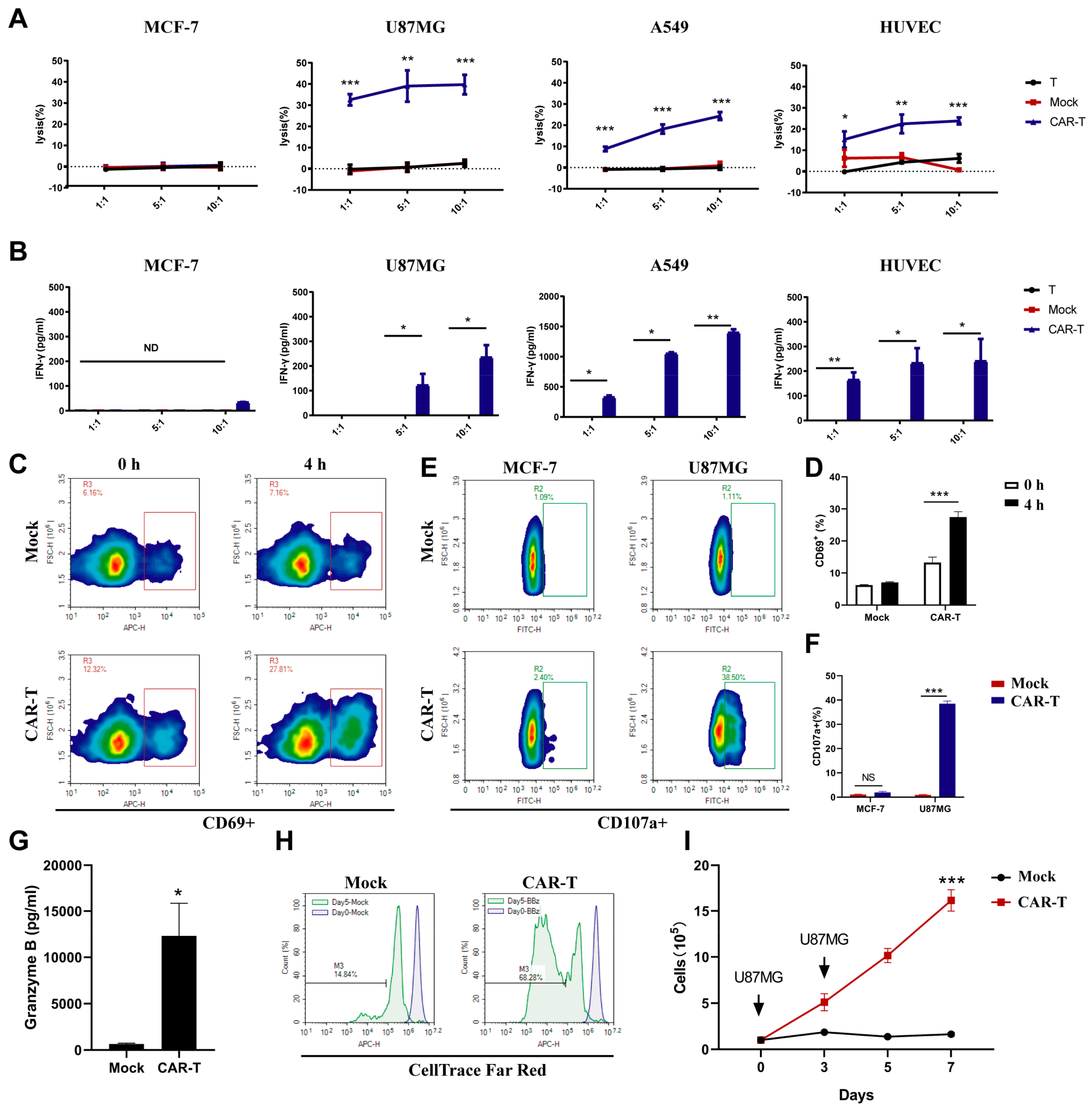
3.3. CAR-T-Cells Exert Antigen-Specific Cytotoxic Potency and Cytokine Secretion In Vitro
3.4. CAR-T-Cells Inhibit U-87 MG Tumor Progression In Vivo
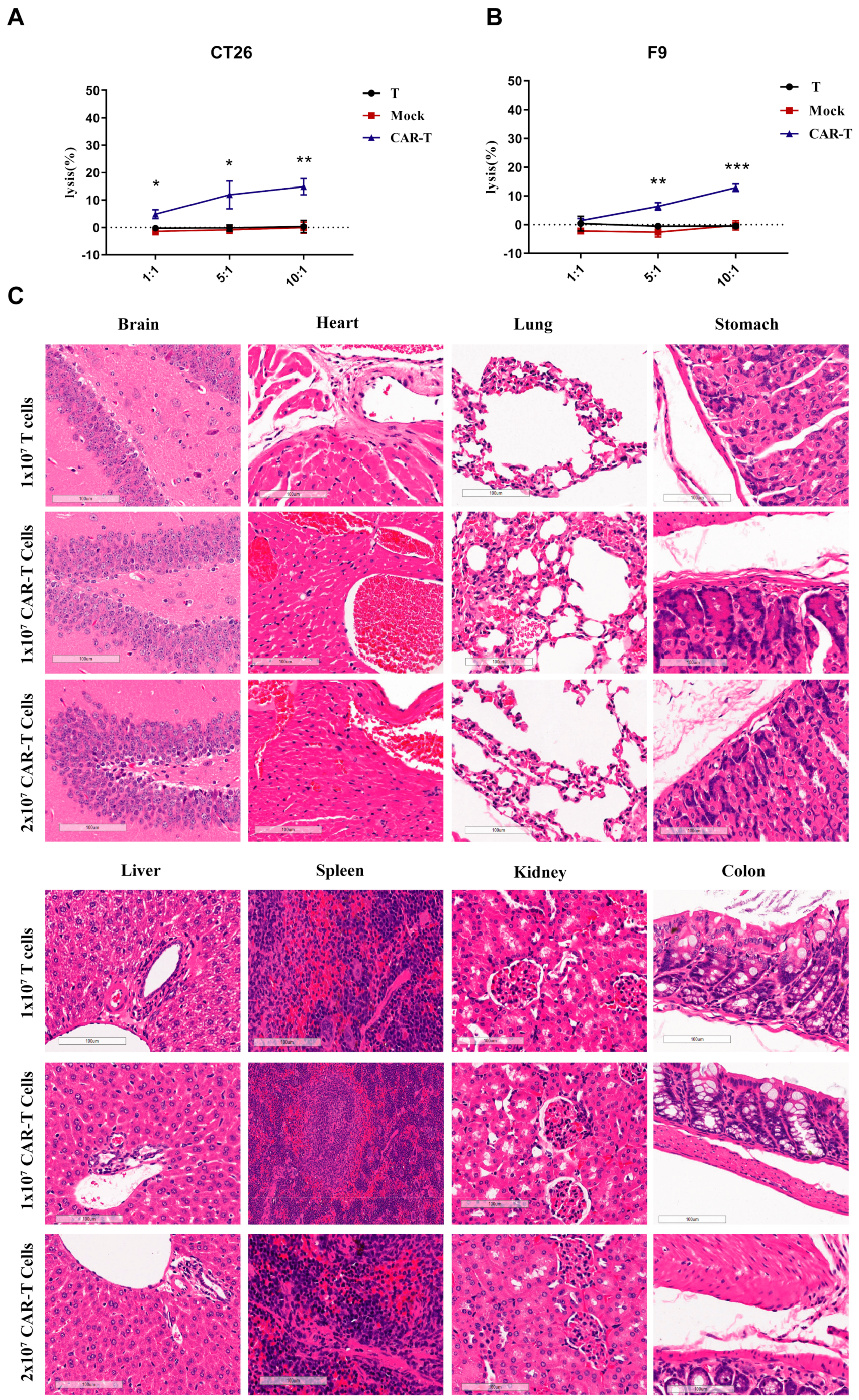
3.5. On-Target Off-Tumor Toxicities in Mouse Models Treated with CAR-T-Cells
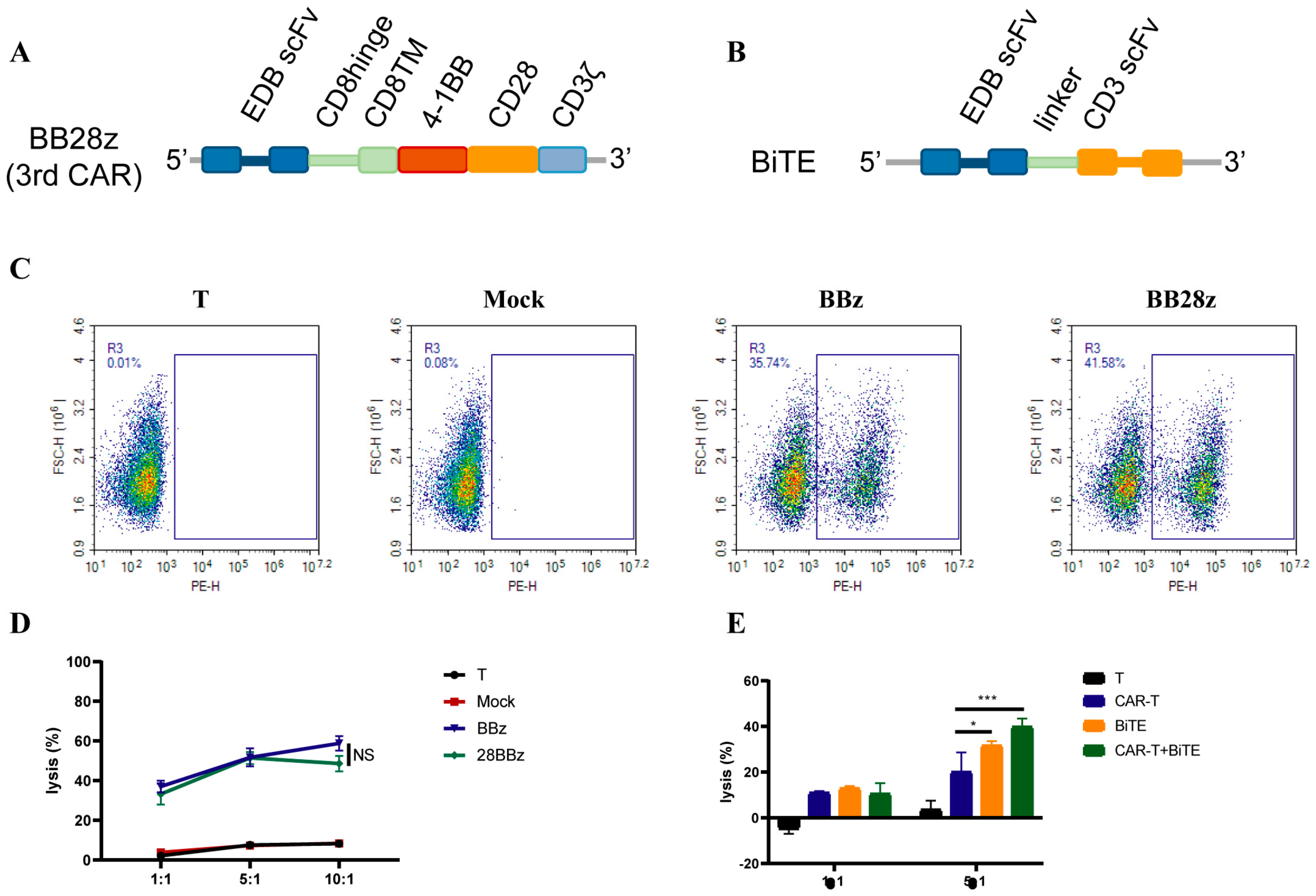
3.6. Enhanced Signaling Improves CAR-T-Cell Cytotoxicity against U87MG Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hong, M.; Clubb, J.D.; Chen, Y.Y. Engineering CAR-T cells for next-generation cancer therapy. Cancer Cell 2020, 38, 473–488. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- D’Aloia, M.M.; Zizzari, I.G.; Sacchetti, B.; Pierelli, L.; Alimandi, M. CAR-T cells: The long and winding road to solid tumors. Cell Death Dis. 2018, 9, 282. [Google Scholar] [CrossRef] [PubMed]
- Straathof, K.; Flutter, B.; Wallace, R.; Jain, N.; Loka, T.; Depani, S.; Wright, G.; Thomas, S.; Cheung, G.W.-K.; Gileadi, T. Antitumor activity without on-target off-tumor toxicity of GD2–chimeric antigen receptor T cells in patients with neuroblastoma. Sci. Transl. Med. 2020, 12, eabd6169. [Google Scholar] [CrossRef] [PubMed]
- Marofi, F.; Motavalli, R.; Safonov, V.A.; Thangavelu, L.; Yumashev, A.V.; Alexander, M.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; Jarahian, M. CAR T cells in solid tumors: Challenges and opportunities. Stem. Cell Res. Ther. 2021, 12, 81. [Google Scholar] [CrossRef] [PubMed]
- Klampatsa, A.; Dimou, V.; Albelda, S.M. Mesothelin-targeted CAR-T cell therapy for solid tumors. Expert Opin. Biol. Ther. 2021, 21, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.; Mukherjee, M.; Grada, Z.; Pignata, A.; Landi, D.; Navai, S.A.; Wakefield, A.; Fousek, K.; Bielamowicz, K.; Chow, K.K. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J. Clin. Investig. 2016, 126, 3036–3052. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, Z.; Yang, Z.; Wang, M.; Li, S.; Li, Y.; Zhang, R.; Xiong, Z.; Wei, Z.; Shen, J. Phase I escalating-dose trial of CAR-T therapy targeting CEA+ metastatic colorectal cancers. Mol. Ther. 2017, 25, 1248–1258. [Google Scholar] [CrossRef]
- White, E.S.; Muro, A.F. Fibronectin splice variants: Understanding their multiple roles in health and disease using engineered mouse models. IUBMB Life 2011, 63, 538–546. [Google Scholar] [CrossRef]
- Ebbinghaus, C.; Ronca, R.; Kaspar, M.; Grabulovski, D.; Berndt, A.; Kosmehl, H.; Zardi, L.; Neri, D. Engineered vascular-targeting antibody-interferon-γ fusion protein for cancer therapy. Int. J. Cancer 2005, 116, 304–313. [Google Scholar] [CrossRef]
- Schiefner, A.; Gebauer, M.; Skerra, A. Extra-domain B in oncofetal fibronectin structurally promotes fibrillar head-to-tail dimerization of extracellular matrix protein. J. Biol. Chem. 2012, 287, 17578–17588. [Google Scholar] [CrossRef]
- Zardi, L.; Carnemolla, B.; Siri, A.; Petersen, T.; Paolella, G.; Sebastio, G.; Baralle, F. Transformed human cells produce a new fibronectin isoform by preferential alternative splicing of a previously unobserved exon. EMBO J. 1987, 6, 2337–2342. [Google Scholar] [CrossRef]
- Kumra, H.; Reinhardt, D.P. Fibronectin-targeted drug delivery in cancer. Adv. Drug Deliv. Rev. 2016, 97, 101–110. [Google Scholar] [CrossRef]
- Borsi, L.; Allemanni, G.; Castellani, P.; Rosellini, C.; Di Vinci, A.; Zardi, L. Structural differences in the cell binding region of human fibronectin molecules isolated from cultured normal and tumor-derived human cells. FEBS Lett. 1985, 192, 71–74. [Google Scholar] [CrossRef]
- Saw, P.E.; Xu, X.; Kang, B.R.; Lee, J.; Lee, Y.S.; Kim, C.; Kim, H.; Kang, S.-H.; Na, Y.J.; Moon, H.J. Extra-domain B of fibronectin as an alternative target for drug delivery and a cancer diagnostic and prognostic biomarker for malignant glioma. Theranostics 2021, 11, 941. [Google Scholar] [CrossRef]
- Birchler, M.T.; Milisavlijevic, D.; Pfaltz, M.; Neri, D.; Odermatt, B.; Schmid, S.; Stoeckli, S.J. Expression of the extra domain B of fibronectin, a marker of angiogenesis, in head and neck tumors. Laryngoscope 2003, 113, 1231–1237. [Google Scholar] [CrossRef]
- Castellani, P.; Borsi, L.; Carnemolla, B.; Biro, A.; Dorcaratto, A.; Viale, G.L.; Neri, D.; Zardi, L. Differentiation between high-and low-grade astrocytoma using a human recombinant antibody to the extra domain-B of fibronectin. Am. J. Pathol. 2002, 161, 1695–1700. [Google Scholar] [CrossRef]
- Pini, A.; Viti, F.; Santucci, A.; Carnemolla, B.; Zardi, L.; Neri, P.; Neri, D. Design and use of a phage display library: Human antibodies with subnanomolar affinity against a marker of angiogenesis eluted from a two-dimensional gel. J. Biol. Chem. 1998, 273, 21769–21776. [Google Scholar] [CrossRef]
- Lieverse, R.I.; Marcus, D.; van der Wiel, A.M.; Van Limbergen, E.J.; Theys, J.; Yaromina, A.; Lambin, P.; Dubois, L.J. Human fibronectin extra domain B as a biomarker for targeted therapy in cancer. Mol. Oncol. 2020, 14, 1555–1568. [Google Scholar] [CrossRef]
- Wagner, J.; Wickman, E.; Shaw, T.I.; Anido, A.A.; Langfitt, D.; Zhang, J.; Porter, S.N.; Pruett-Miller, S.M.; Tillman, H.; Krenciute, G. Antitumor effects of CAR T cells redirected to the EDB splice variant of fibronectin. Cancer Immunol. Res. 2021, 9, 279–290. [Google Scholar] [CrossRef]
- Früh, S.M.; Schoen, I.; Ries, J.; Vogel, V. Molecular architecture of native fibronectin fibrils. Nat. Commun. 2015, 6, 1–10. [Google Scholar] [CrossRef]
- Qi, C.; Gong, J.; Li, J.; Liu, D.; Qin, Y.; Ge, S.; Zhang, M.; Peng, Z.; Zhou, J.; Cao, Y. Claudin18. 2-specific CAR T cells in gastrointestinal cancers: Phase 1 trial interim results. Nat. Med. 2022, 28, 1–10. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Liu, C.; Yang, Z.; Yin, H. CAR-T-Cell Therapy for Solid Tumors Positive for Fibronectin Extra Domain B. Cells 2022, 11, 2863. https://doi.org/10.3390/cells11182863
Zhang Z, Liu C, Yang Z, Yin H. CAR-T-Cell Therapy for Solid Tumors Positive for Fibronectin Extra Domain B. Cells. 2022; 11(18):2863. https://doi.org/10.3390/cells11182863
Chicago/Turabian StyleZhang, Zhijie, Chang Liu, Zhe Yang, and Hongping Yin. 2022. "CAR-T-Cell Therapy for Solid Tumors Positive for Fibronectin Extra Domain B" Cells 11, no. 18: 2863. https://doi.org/10.3390/cells11182863
APA StyleZhang, Z., Liu, C., Yang, Z., & Yin, H. (2022). CAR-T-Cell Therapy for Solid Tumors Positive for Fibronectin Extra Domain B. Cells, 11(18), 2863. https://doi.org/10.3390/cells11182863