
Phenotypic and Genetic Heterogeneity of Adult Patients with Hereditary Spastic Paraplegia from Serbia

 , , , , , and
, , , , , and
Abstract
1. Introduction
2. Patients and Method
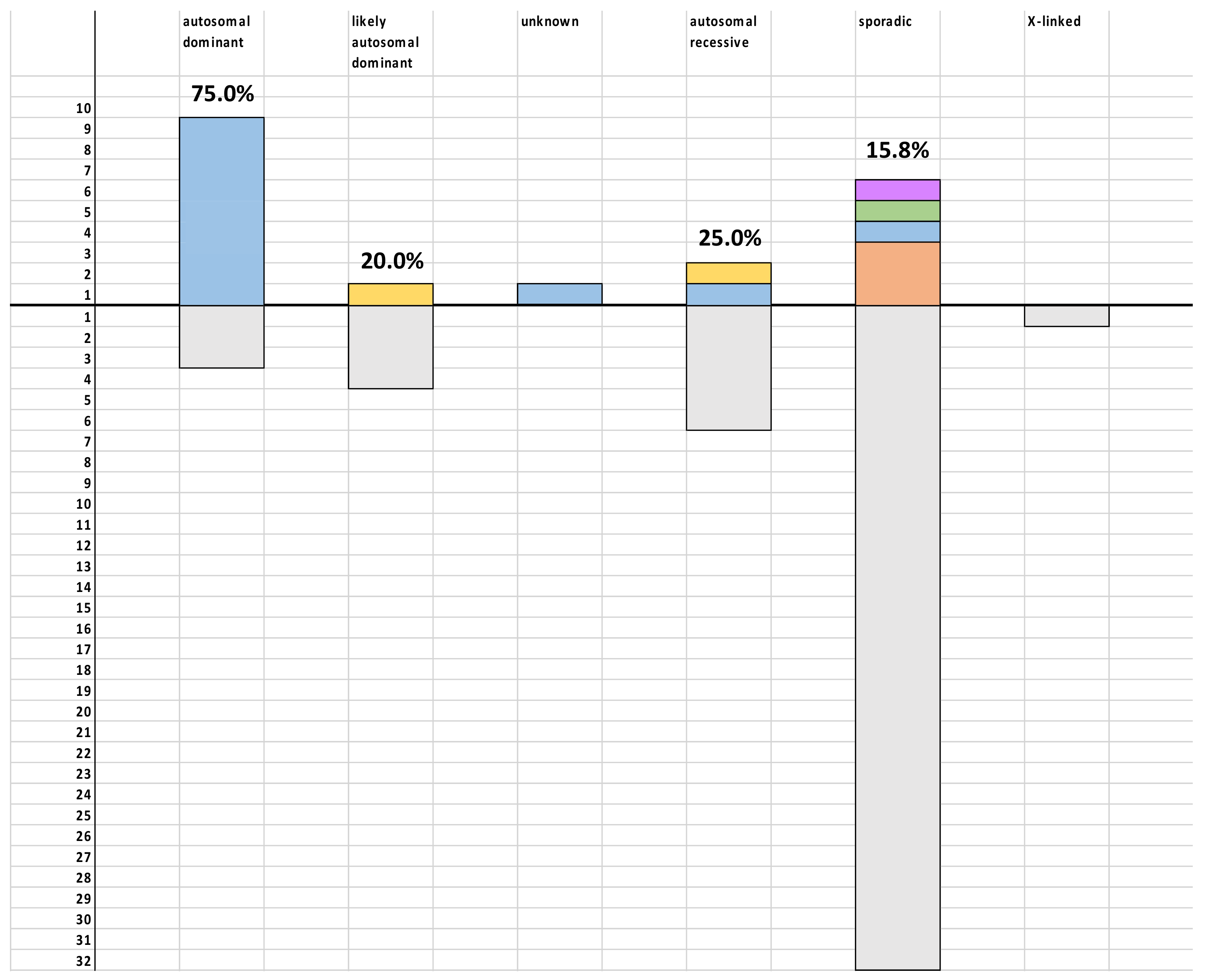
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ruano, L.; Melo, C.; Silva, M.C.; Coutinho, P. The global epidemiology of hereditary ataxia and spastic paraplegia: A systematic review of prevalence studies. Neuroepidemiology 2014, 42, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Schüle, R.; Wiethoff, S.; Martus, P.; Karle, K.N.; Otto, S.; Klebe, S.; Klimpe, S.; Gallenmüller, C.; Kurzwelly, D.; Henkel, D.; et al. Hereditary spastic paraplegia: Clinicogenetic lessons from 608 patients. Ann. Neurol. 2016, 79, 646–658. [Google Scholar] [CrossRef]
- Agosta, F.; Scarlato, M.; Spinelli, E.G.; Canu, E.; Benedetti, S.; Bassi, M.T.; Casali, C.; Sessa, M.; Copetti, M.; Pagani, E.; et al. Hereditary Spastic Paraplegia: Beyond Clinical Phenotypes toward a Unified Pattern of Central Nervous System Damage. Radiology 2015, 276, 207–218. [Google Scholar] [CrossRef]
- Meyyazhagan, A.; Kuchi Bhotla, H.; Pappuswamy, M.; Orlacchio, A. The Puzzle of Hereditary Spastic Paraplegia: From Epidemiology to Treatment. Int. J. Mol. Sci. 2022, 23, 7665. [Google Scholar] [CrossRef]
- Darios, F.; Coarelli, G.; Durr, A. Genetics in hereditary spastic paraplegias: Essential but not enough. Curr. Opin. Neurobiol. 2022, 72, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, Z.; Rydning, S.L.; Wedding, I.M.; Koht, J.; Pihlstrøm, L.; Rengmark, A.H.; Henriksen, S.P.; Tallaksen, C.M.; Toft, M. Targeted high throughput sequencing in hereditary ataxia and spastic paraplegia. PLoS ONE 2017, 12, e0174667. [Google Scholar] [CrossRef] [PubMed]
- Méreaux, J.L.; Banneau, G.; Papin, M.; Coarelli, G.; Valter, R.; Raymond, L.; Kol, B.; Ariste, O.; Parodi, L.; Tissier, L.; et al. Clinical and genetic spectra of 1550 index patients with hereditary spastic paraplegia. Brain 2022, 145, 1029–1037. [Google Scholar] [CrossRef]
- Arnoldi, A.; Tonelli, A.; Crippa, F.; Villani, G.; Pacelli, C.; Sironi, M.; Pozzoli, U.; D’Angelo, M.G.; Meola, G.; Martinuzzi, A.; et al. A clinical, genetic, and biochemical characterization of SPG7 mutations in a large cohort of patients with hereditary spastic paraplegia. Hum. Mutat. 2008, 29, 522–531. [Google Scholar] [CrossRef]
- Koh, K.; Ishiura, H.; Tsuji, S.; Takiyama, Y. JASPAC: Japan Spastic Paraplegia Research Consortium. Brain Sci. 2018, 8, 153. [Google Scholar] [CrossRef]
- Lo Giudice, T.; Lombardi, F.; Santorelli, F.M.; Kawarai, T.; Orlacchio, A. Hereditary spastic paraplegia: Clinical-genetic characteristics and evolving molecular mechanisms. Exp. Neurol. 2014, 261, 518–539. [Google Scholar] [CrossRef]
- Lan, M.Y.; Chang, Y.Y.; Yeh, T.H.; Lai, S.C.; Liou, C.W.; Kuo, H.C.; Wu, Y.R.; Lyu, R.K.; Hung, J.W.; Chang, Y.C.; et al. High frequency of SPG4 in Taiwanese families with autosomal dominant hereditary spastic paraplegia. BMC Neurol. 2014, 14, 216. [Google Scholar] [CrossRef]
- Dong, E.L.; Wang, C.; Wu, S.; Lu, Y.Q.; Lin, X.H.; Su, H.Z.; Zhao, M.; He, J.; Ma, L.X.; Wang, N.; et al. Clinical spectrum and genetic landscape for hereditary spastic paraplegias in China. Mol. Neurodegener. 2018, 13, 36. [Google Scholar] [CrossRef]
- Hazan, J.; Fonknechten, N.; Mavel, D.; Paternotte, C.; Samson, D.; Artiguenave, F.; Davoine, C.S.; Cruaud, C.; Dürr, A.; Wincker, P.; et al. Spastin, a new AAA protein, is altered in the most frequent form of autosomal dominant spastic paraplegia. Nat. Genet. 1999, 23, 296–303. [Google Scholar] [CrossRef] [PubMed]
- McDermott, C.J.; Burness, C.E.; Kirby, J.; Cox, L.E.; Rao, D.G.; Hewamadduma, C.; Sharrack, B.; Hadjivassiliou, M.; Chinnery, P.F.; Dalton, A. UK and Irish HSP Consortium: Clinical features of hereditary spastic paraplegia due to spastin mutation. Neurology 2006, 67, 45–51. [Google Scholar] [CrossRef]
- Orlacchio, A.; Patrono, C.; Gaudiello, F.; Rocchi, C.; Moschella, V.; Floris, R.; Bernardi, G.; Kawarai, T. Silver syndrome variant of hereditary spastic paraplegia: A locus to 4p and allelism with SPG4. Neurology 2008, 70, 1959–1966. [Google Scholar] [CrossRef] [PubMed]
- De Souza, P.V.; Bortholin, T.; Naylor, F.G.; de Rezende Pinto, W.B.; Oliveira, A.S. Infantile-onset ascending spastic paraplegia phenotype associated with SPAST mutation. J. Neurol. Sci. 2016, 371, 34–35. [Google Scholar] [CrossRef]
- Klebe, S.; Stevanin, G.; Depienne, C. Clinical and genetic heterogeneity in hereditary spastic paraplegias: From SPG1 to SPG72 and still counting. Rev. Neurol. 2015, 171, 505–530. [Google Scholar] [CrossRef]
- Nielsen, J.E.; Johnsen, B.; Koefoed, P.; Scheuer, K.H.; Grønbech-Jensen, M.; Law, I.; Krabbe, K.; Nørremølle, A.; Eiberg, H.; Søndergård, H.; et al. Hereditary spastic paraplegia with cerebellar ataxia: A complex phenotype associated with a new SPG4 gene mutation. Eur. J. Neurol. 2004, 11, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Orlacchio, A.; Kawarai, T.; Totaro, A.; Errico, A.; St George-Hyslop, P.H.; Rugarli, E.I.; Bernardi, G. Hereditary spastic paraplegia: Clinical genetic study of 15 families. Arch. Neurol. 2004, 61, 849–855. [Google Scholar] [CrossRef]
- Ribaï, P.; Depienne, C.; Fedirko, E.; Jothy, A.C.; Viveweger, C.; Hahn-Barma, V.; Brice, A.; Durr, A. Mental deficiency in three families with SPG4 spastic paraplegia. Eur. J. Hum. Genet. 2008, 16, 97–104. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Parodi, L.; Fenu, S.; Stevanin, G.; Durr, A. Hereditary spastic paraplegia: More than an upper motor neuron disease. Rev. Neurol. 2017, 173, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Lindig, T.; Bender, B.; Hauser, T.K.; Mang, S.; Schweikardt, D.; Klose, U.; Karle, K.N.; Schüle, R.; Schöls, L.; Rattay, T.W. Gray and white matter alterations in hereditary spastic paraplegia type SPG4 and clinical correlations. J. Neurol. 2015, 262, 1961–1971. [Google Scholar] [CrossRef] [PubMed]
- Kara, E.; Tucci, A.; Manzoni, C.; Lynch, D.S.; Elpidorou, M.; Bettencourt, C.; Chelban, V.; Manole, A.; Hamed, S.A.; Haridy, N.A.; et al. Genetic and phenotypic characterization of complex hereditary spastic paraplegia. Brain 2016, 139 Pt 7, 1904–1918. [Google Scholar] [CrossRef]
- Rubegni, A.; Storti, E.; Tessa, A.; Federico, A.; Santorelli, F.M. Hereditary spastic paraplegia type 11 with a very late onset. J. Neurol. 2015, 262, 1987–1989. [Google Scholar] [CrossRef]
- Kawarai, T.; Miyamoto, R.; Mori, A.; Oki, R.; Tsukamoto-Miyashiro, A.; Matsui, N.; Miyazaki, Y.; Orlacchio, A.; Izumi, Y.; Nishida, Y.; et al. Late-onset spastic paraplegia: Aberrant SPG11 transcripts generated by a novel splice site donor mutation. J. Neurol. Sci. 2015, 359, 250–255. [Google Scholar] [CrossRef]
- Doleckova, K.; Roth, J.; Stellmachova, J.; Gescheidt, T.; Sigut, V.; Houska, P.; Jech, R.; Zech, M.; Vyhnalek, M.; Vyhnalkova, E.; et al. SPG11: Clinical and genetic features of seven Czech patients and literature review. Neurol. Res. 2022, 44, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Del Bo, R.; Di Fonzo, A.; Ghezzi, S.; Locatelli, F.; Stevanin, G.; Costa, A.; Corti, S.; Bresolin, N.; Comi, G.P. SPG11: A consistent clinical phenotype in a family with homozygous spatacsin truncating mutation. Neurogenetics 2007, 8, 301–305. [Google Scholar] [CrossRef]
- Pippucci, T.; Panza, E.; Pompilii, E.; Donadio, V.; Borreca, A.; Babalini, C.; Patrono, C.; Zuntini, R.; Kawarai, T.; Bernardi, G.; et al. Autosomal recessive hereditary spastic paraplegia with thin corpus callosum: A novel mutation in the SPG11 gene and further evidence for genetic heterogeneity. Eur. J. Neurol. 2009, 16, 121–126. [Google Scholar] [CrossRef]
- Chrestian, N.; Dupré, N.; Gan-Or, Z.; Szuto, A.; Chen, S.; Venkitachalam, A.; Brisson, J.D.; Warman-Chardon, J.; Ahmed, S.; Ashtiani, S.; et al. Clinical and genetic study of hereditary spastic paraplegia in Canada. Neurol. Genet. 2016, 3, e122. [Google Scholar] [CrossRef]
- Wagner, F.; Titelbaum, D.S.; Engisch, R.; Coskun, E.K.; Waugh, J.L. Subtle Imaging Findings Aid the Diagnosis of Adolescent Hereditary Spastic Paraplegia and Ataxia. Clin. Neuroradiol. 2019, 29, 215–221. [Google Scholar] [CrossRef]
- Stevanin, G.; Azzedine, H.; Denora, P.; Boukhris, A.; Tazir, M.; Lossos, A.; Rosa, A.L.; Lerer, I.; Hamri, A.; Alegria, P.; et al. SPATAX consortium. Mutations in SPG11 are frequent in autosomal recessive spastic paraplegia with thin corpus callosum, cognitive decline and lower motor neuron degeneration. Brain 2008, 131 Pt 3, 772–784. [Google Scholar] [CrossRef] [PubMed]
- Klebe, S.; Depienne, C.; Gerber, S.; Challe, G.; Anheim, M.; Charles, P.; Fedirko, E.; Lejeune, E.; Cottineau, J.; Brusco, A.; et al. Spastic paraplegia gene 7 in patients with spasticity and/or optic neuropathy. Brain 2012, 135 Pt 10, 2980–2993. [Google Scholar] [CrossRef]
- Yahikozawa, H.; Yoshida, K.; Sato, S.; Hanyu, N.; Doi, H.; Miyatake, S.; Matsumoto, N. Predominant cerebellar phenotype in spastic paraplegia 7 (SPG7). Hum. Genome Var. 2015, 2, 15012. [Google Scholar] [CrossRef] [PubMed]
- Van Gassen, K.L.; van der Heijden, C.D.; de Bot, S.T.; den Dunnen, W.F.; van den Berg, L.H.; Verschuuren-Bemelmans, C.C.; Kremer, H.P.; Veldink, J.H.; Kamsteeg, E.J.; Scheffer, H.; et al. Genotype-phenotype correlations in spastic paraplegia type 7: A study in a large Dutch cohort. Brain 2012, 135 Pt 10, 2994–3004. [Google Scholar] [CrossRef]
- Sánchez-Ferrero, E.; Coto, E.; Beetz, C.; Gámez, J.; Corao, A.I.; Díaz, M.; Esteban, J.; del Castillo, E.; Moris, G.; Infante, J.; et al. SPG7 mutational screening in spastic paraplegia patients supports a dominant effect for some mutations and a pathogenic role for p.A510V. Clin. Genet. 2013, 83, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Orsucci, D.; Petrucci, L.; Ienco, E.C.; Chico, L.; Simi, P.; Fogli, A.; Baldinotti, F.; Simoncini, C.; LoGerfo, A.; Carlesi, C.; et al. Hereditary spastic paraparesis in adults. A clinical and genetic perspective from Tuscany. Clin. Neurol. Neurosurg. 2014, 120, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, G.; Gorman, G.S.; Griffin, H.; Kurzawa-Akanbi, M.; Blakely, E.L.; Wilson, I.; Sitarz, K.; Moore, D.; Murphy, J.L.; Alston, C.L.; et al. Mutations in the SPG7 gene cause chronic progressive external ophthalmoplegia through disordered mitochondrial DNA maintenance. Brain 2014, 137 Pt 5, 1323–1336. [Google Scholar] [CrossRef]
- Goizet, C.; Boukhris, A.; Mundwiller, E.; Tallaksen, C.; Forlani, S.; Toutain, A.; Carriere, N.; Paquis, V.; Depienne, C.; Durr, A.; et al. Complicated forms of autosomal dominant hereditary spastic paraplegia are frequent in SPG10. Hum. Mutat. 2009, 30, E376–E385. [Google Scholar] [CrossRef]
- Crimella, C.; Baschirotto, C.; Arnoldi, A.; Tonelli, A.; Tenderini, E.; Airoldi, G.; Martinuzzi, A.; Trabacca, A.; Losito, L.; Scarlato, M.; et al. Mutations in the motor and stalk domains of KIF5A in spastic paraplegia type 10 and in axonal Charcot-Marie-Tooth type 2. Clin. Genet. 2012, 82, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Hedera, P. Hereditary Spastic Paraplegia Overview. 2000 August 15 [Updated 2021 Feb 11]. In GeneReviews® [Internet]; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallance, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1509/ (accessed on 6 September 2022).


{kind=link}
| Family ID | Patient ID | Presumed Inheritance | Disease Subtype | Gene Name | Transcript | Variant | Zygosity | HGMD Identifier # | gnomAD Frequency | ACMG Pathogenicity Class | ACMG Pathogenicity Evidence |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | HSP24 | AD | SPG4 | SPAST | NM_014946.3 | c.1245+1delG | het | - | 0 | Likely pathogenic (novel) | PM2, PM5, PP1, PP3, PP4 |
| HSP94 | AD | SPG4 | SPAST | NM_014946.3 | c.1245+1delG | het | |||||
| 2 | HSP32 | AD | SPG4 | SPAST | NM_014946.3 | deletion of exon 15 and 16 | het | CG1415382 | - | Pathogenic (recurrent) | |
| 3 | HSP33 | AD | SPG4 | SPAST | NM_014946.3 | c.425delA (p.Lys142Argfs*19) | het | - | 0 | Pathogenic (novel) | PVS1, PM2, PM4, PP1 |
| 4 | HSP42 | AD | SPG4 | SPAST | NM_014946.3 | c.1069_1070insA (p.I357Nfs*10) | het | - | 0 | Pathogenic (novel) | PVS1, PM2, PM4, PP1 |
| 5 | HSP4 | AR | SPG4 | SPAST | NM_014946.3 | c.1351A>G (p.R451G) | het | - | 0 | Likely pathogenic (novel) | PVS1, PM2, PM4, PP1 |
| 6 | HSP68 | sporadic | SPG4 | SPAST | NM_014946.3 | deletion exons 8–17 and further downstream | het | CG1415377 | - | Pathogenic (recurrent) | |
| 7 | HSP52 | AD | SPG4 | SPAST | NM_014946.3 | c.820_827del (p.M274Wfs*14) | het | - | 0 | Pathogenic (novel) | PVS1, PM2, PM4, PP1 |
| 8 | HSP80 | AD | SPG4 | SPAST | NM_014946.3 | c.1494G>T (p.R498S) | het | CM1512265 | 0 | Pathogenic (recurrent) | |
| HSP81 | AD | SPG4 | SPAST | NM_014946.3 | c.1494G>T (p.R498S) | het | |||||
| 9 | HSP114 | unknown (adopted) | SPG4 | SPAST | NM_014946.3 | c.308C>A (p.S103*) | het | - | 0 | Pathogenic (novel) | PVS1, PM2, PM4, PP1 |
| 10 | HSP9 | AD | SPG4 | SPAST | NM_014946.3 | c.1495C>T (p.R499C) | het | CM992676 | 0 | Pathogenic (recurrent) | |
| HSP10 | AD | SPG4 | SPAST | NM_014946.3 | c.1495C>T (p.R499C) | het | |||||
| 11 | HSP11 | AD | SPG4 | SPAST | NM_014946.3 | c.1672_1673del (p.L558Gfs*18) | het | CD021858 | 0 | Pathogenic (recurrent) | |
| HSP12 | AD | SPG4 | SPAST | NM_014946.3 | c.1672_1673del (p.L558Gfs*18) | het | |||||
| 12 | HSP112 | AD | SPG4 | SPAST | NM_014946.3 | deletion of 5’UTR-ex1 | het | CG052756 | - | Pathogenic (recurrent) | |
| 13 | HSP118 | AR | SPG7 | SPG7 | NM_003119.3 | c.233T>A (p.L78*) | hom | CM081826 | 0 | Pathogenic (recurrent) | |
| 14 | HSP119 | likely AD | SPG7 | SPG7 | NM_003119.3 | c.233T>A (p.L78*) | hom | CM081826 | 0 | Pathogenic (recurrent) | |
| 15 | HSP113 | sporadic | SPG11 | SPG11 | NM_025137.4 | c.5381T>C (p.L1794P) | hom | CM166061 | 0 | Pathogenic (recurrent) | |
| 16 | HSP128 | sporadic | SPG11 | SPG11 | NM_025137.4 | c.5381T>C (p.L1794P) | comp het | CM166061 | 0 | Pathogenic (recurrent) | |
| duplication spanning intron27-ex29 | CN166911 | 0 | Pathogenic (recurrent) | ||||||||
| 17 | HSP35 | sporadic | SPG11 | SPG11 | NM_025137.4 | duplication spanning intron27-ex29 | hom | CN166911 | - | Pathogenic (recurrent) | |
| 18 | HSP34_HSP69 | sporadic | SPG10 | KIF5A | NM_004984.4 | c.746T>C (p.Leu249Pro) | het | - | 0 | Likely pathogenic (novel) | PM2, PM5, PP1, PP3, PP4 |
| 19 | DIST690 | sporadic | SPG15 | ZFYVE26 | NM_015346.4 | c.2114dupC (p.E706*) | comp het | - | 0 | Pathogenic (novel) | PVS1, PM2, PM4, PP1, PP3, PP4, PP5 |
| c.2357delC (p.P786Hfs*10) | - | 0 | Pathogenic (novel) | PVS1, PM2, PM4, PP1, PP3, PP4 |
| Features | Number of Patients |
|---|---|
| % females | 50% |
| Age at onset | <1–6.2% 6–10–18.8% 11–20–6.2% 21–40–31.2% 41–60–31.2% >60–6.2% |
| Age | 42.9 ± 11. |
| Mobility | normal—6.2% abnormal, but no aids—93.8% |
| Lower limbs | hyperreflexia—31.2% hyperreflexia, weakness—56.2% hyperreflexia, weakness, distal muscle atrophy—12.5% |
| Upper limbs | normal—18.8% hyperreflexia—75.0% hyperreflexia, distal muscle weakness—6.2% |
| Sphincters | normal—50.0% bladder impairment—12.5% bladder and bowel impairment—25.0% |
| Sensibility | normal—56.2% vibration impaired in lower limbs—37.5% vibration and touch impaired in lower limbs—6.2% |
| Additional features | 56.2% |
| Patient ID | HSP113 | HSP128 | HSP35 |
|---|---|---|---|
| Gender | female | male | male |
| Age at onset | 41–60 | 41–60 | 21–40 |
| Age | 52 | 53 | 25 |
| Mobility | abnormal, but no aids | abnormal, but no aids | abnormal, but no aids |
| LL | hyperreflexia, weakness | asymmetric hyperreflexia | hyperreflexia |
| UL | normal | normal | hyperreflexia |
| Sphincter dysfunction | no | no | no |
| Sensibility impairment | normal | normal | normal |
| Additional features | foot dystonia | no | mild mental retardation (IQ 78), mild dysarthria, mild postural tremor |
| NCS | normal | bilateral carpal tunnel syndrome | normal |
| SSEP | not done | not done | abnormal |
| Brain MRI | normal | temporal arachnoid cyst | thin corpus callosum, mild periventricular WMHLs |
| Spine MRI | normal | normal | normal |
| Serbian p.L78* Patients | Italian p.L78* Patients | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| HSP118 | HSP119 | P1-EM9-06 | P2-EM18-08 | ||||||
| Marker | Position (Mb) | ||||||||
| D16S3123 | 87.6500 | 99 | 101 | 101 | 109 | 101 | 109 | 101 | 109 |
| rs8191483 | 88.8770 | C | C | C | C | C | C | C | C |
| D16S3026 | 89.4930 | 202 | 202 | 202 | 202 | 202 | 202 | 202 | 202 |
| D16S3121 | 89.4985 | 73 | 73 | 73 | 73 | 73 | 73 | 73 | 73 |
| p.L78* | 89.5769 | A | A | A | A | A | A | A | A |
| rs12960 | 89.6203 | G | G | G | G | G | G | G | G |
| rs174035 | 89.6277 | C | C | C | C | C | C | C | C |
| rs455527 | 89.6440 | T | T | T | T | T | T | T | T |
| rs352935 | 89.6486 | C | C | C | C | C | C | C | C |
| rs2162943 | 89.7607 | C | C | C | C | C | C | C | C |
| Patient ID | HSP118 | HSP119 |
|---|---|---|
| Gender | male | male |
| Age at onset | 41–60 | 41–60 |
| Age | 54 | 57 |
| Mobility | unilateral support | abnormal, but no aids |
| LL | hyperreflexia, weakness | hyperreflexia, weakness |
| UL | hyperreflexia | hyperreflexia |
| Sphincter dysfunction | no | bladder |
| Sensibility impairment | no | vibration in LL |
| Additional features | postural tremor | torticollis |
| NCS | not done | normal |
| SSEP | not done | not done |
| Brain MRI | one small supratentorial WMHLs | severe cortical and cerebellar atrophy |
| Spine MRI | normal | normal |
| Patient ID | HSP34 | DIST690 |
|---|---|---|
| HSP type | SPG10 | SPG15 |
| Gender | female | female |
| Age at onset | 6–10 | 11–20 |
| Age | 20 | 23 |
| Mobility | abnormal, but no aids | abnormal, but no aids |
| LL | hyperreflexia, weakness | hyperreflexia, weakness |
| UL | hyperreflexia | normal |
| Sphincter dysfunction | bladder | no |
| Sensibility impairment | vibration and touch in LL | normal |
| Additional features | foot deformities, mild static hand tremor | foot dystonia, mild dysarthria, mild limb ataxia and tremor |
| NCS | predominantly motor, axonal polyneuropathy | normal |
| SSEP | abnormal | normal |
| Brain MRI | normal | normal |
| Spine MRI | normal | normal |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perić, S.; Marković, V.; Candayan, A.; De Vriendt, E.; Momčilović, N.; Savić, A.; Dragašević-Mišković, N.; Svetel, M.; Stević, Z.; Božović, I.; et al. Phenotypic and Genetic Heterogeneity of Adult Patients with Hereditary Spastic Paraplegia from Serbia. Cells 2022, 11, 2804. https://doi.org/10.3390/cells11182804
Perić S, Marković V, Candayan A, De Vriendt E, Momčilović N, Savić A, Dragašević-Mišković N, Svetel M, Stević Z, Božović I, et al. Phenotypic and Genetic Heterogeneity of Adult Patients with Hereditary Spastic Paraplegia from Serbia. Cells. 2022; 11(18):2804. https://doi.org/10.3390/cells11182804
Chicago/Turabian StylePerić, Stojan, Vladana Marković, Ayşe Candayan, Els De Vriendt, Nikola Momčilović, Andrija Savić, Nataša Dragašević-Mišković, Marina Svetel, Zorica Stević, Ivo Božović, and et al. 2022. "Phenotypic and Genetic Heterogeneity of Adult Patients with Hereditary Spastic Paraplegia from Serbia" Cells 11, no. 18: 2804. https://doi.org/10.3390/cells11182804
APA StylePerić, S., Marković, V., Candayan, A., De Vriendt, E., Momčilović, N., Savić, A., Dragašević-Mišković, N., Svetel, M., Stević, Z., Božović, I., Mesaroš, Š., Drulović, J., Basta, I., Petrović, I., Tamaš, O., Mijajlović, M., Novaković, I., Sokić, D., & Jordanova, A. (2022). Phenotypic and Genetic Heterogeneity of Adult Patients with Hereditary Spastic Paraplegia from Serbia. Cells, 11(18), 2804. https://doi.org/10.3390/cells11182804