

Omics Analysis of Chemoresistant Triple Negative Breast Cancer Cells Reveals Novel Metabolic Vulnerabilities

 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Generation of Paclitaxel-Resistant Cell Line
2.2. Cell Proliferation Assay
2.3. MSMO1 Knock-Down
2.4. Immunofluorescence Analysis
2.5. Quantification of Intracellular Doxorubicin by Fluorescence-Activated Cell Sorting (FACS)
2.6. RNA Extraction, cDNA Synthesis and q-RT-PCR
2.7. RNA-Sequencing (RNA-seq) and Bioinformatic Analysis
2.8. Metabolite and Lipid Extraction
2.9. 1H NMR Spectroscopy
2.10. Targeted Metabolomic Profiling and Metabolomic Data Analysis
2.11. Statistical Analysis of Lipidomic Data
3. Results
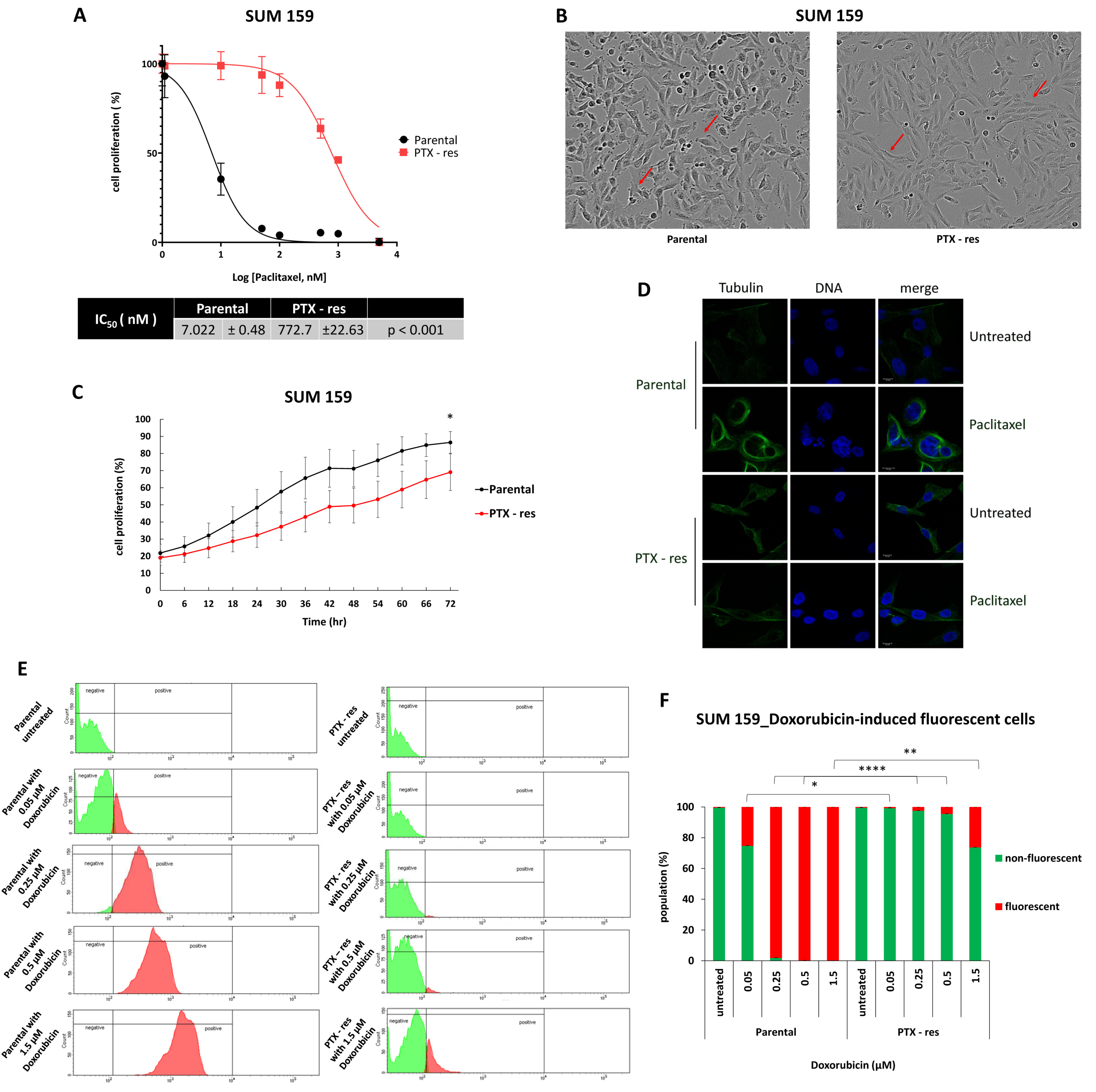
3.1. Establishment and Characterization of a Paclitaxel-Resistant TNBC Cell Line
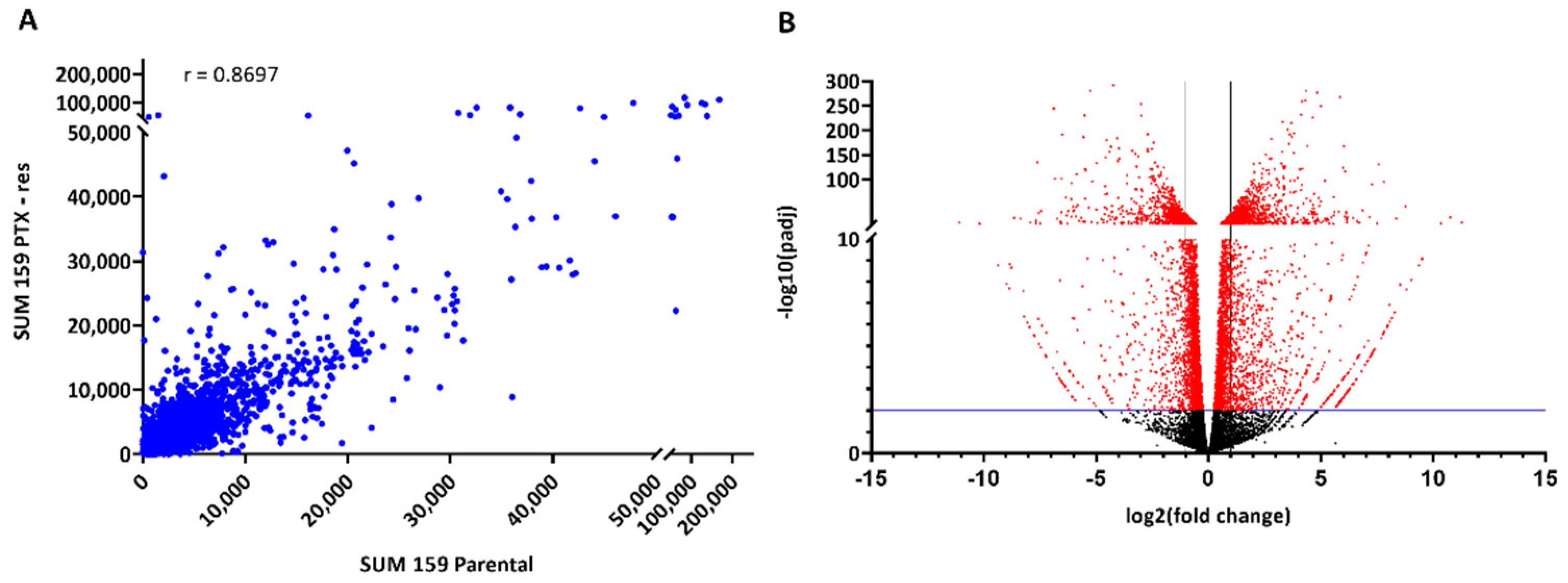
3.2. Transcriptomic Analysis of the SUM159 Parental and PTX-Res Cells
3.2.1. Identification of Differentially Expressed Genes (DEGs)
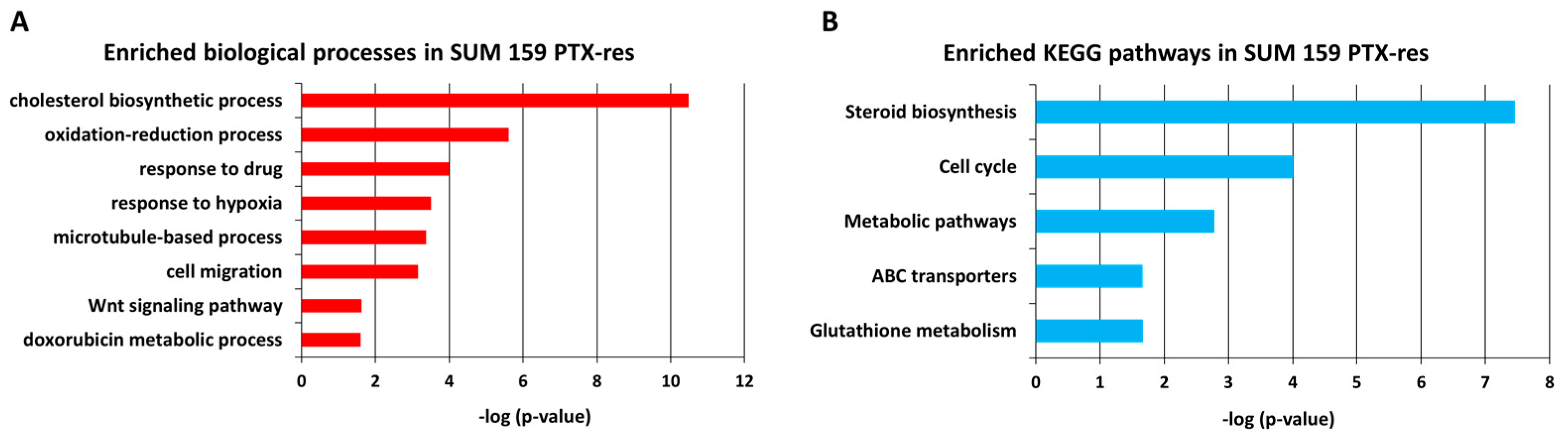
3.2.2. Gene Ontology (GO) Analysis Reveals Cholesterol Biosynthesis as an Important Process in SUM159 PTX-Res Cells
3.3. Metabolomic Analysis of the SUM159 Parental and PTX-Res Cells
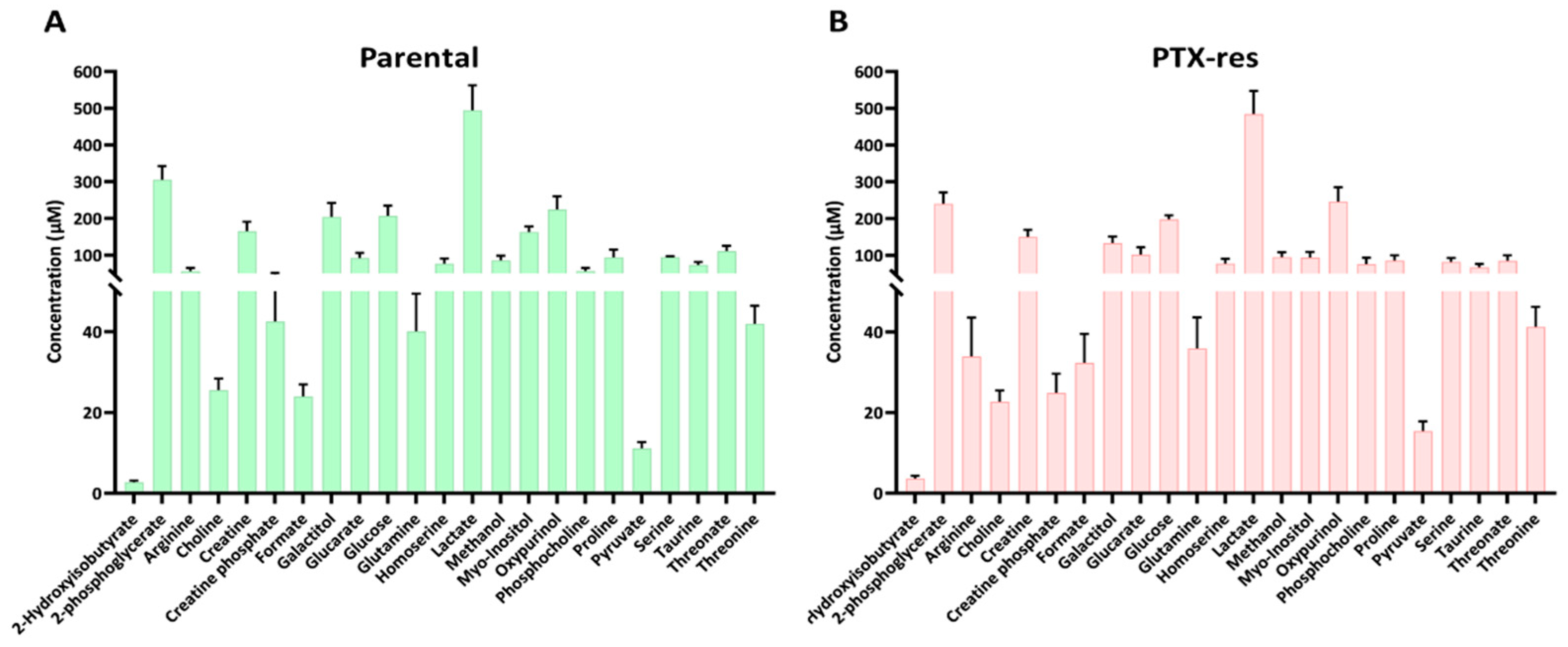
3.3.1. Metabolite Quantification
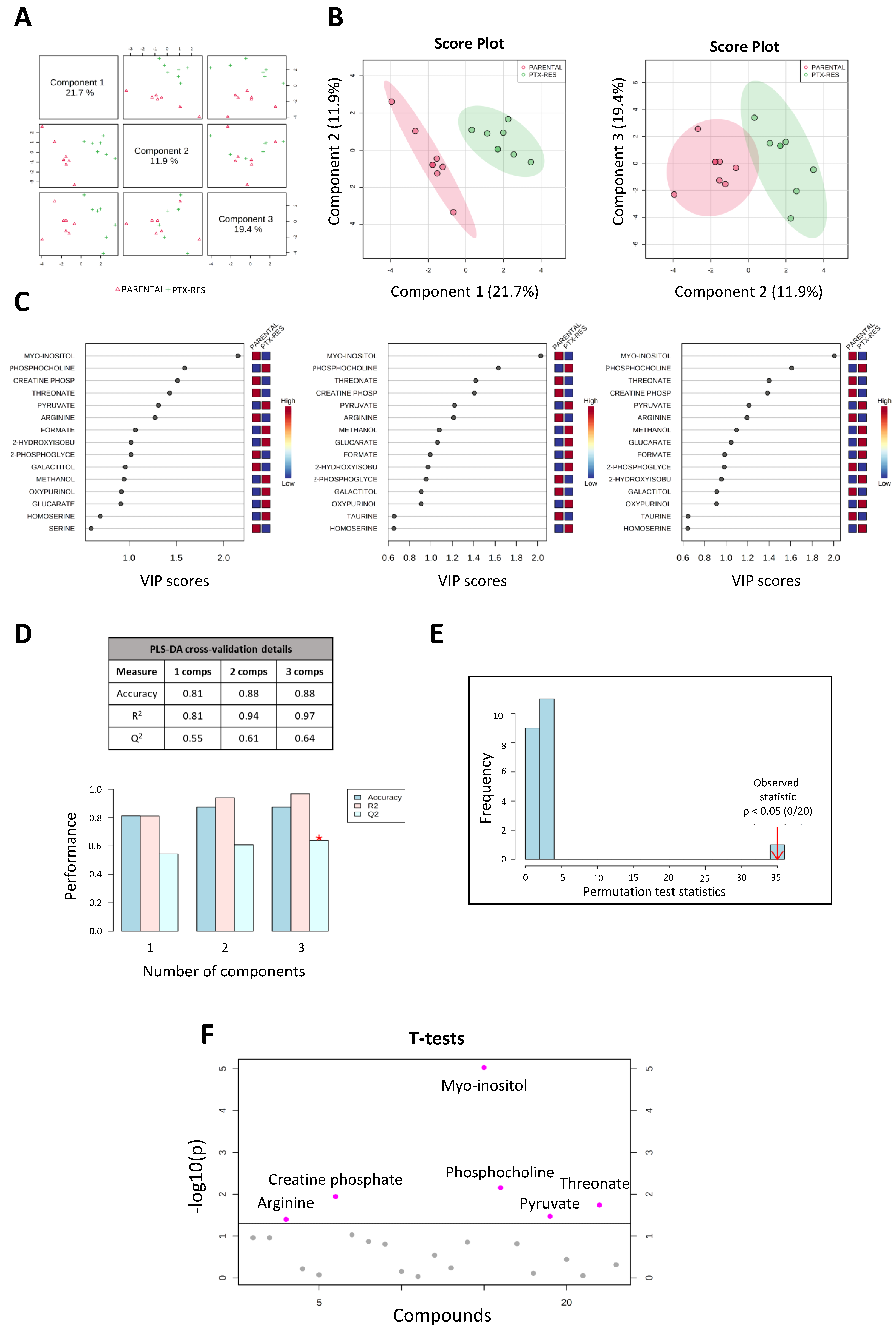
3.3.2. Metabolic Profile Analysis Reveals Significant Differences between the SUM159 Parental and PTX-Res Cells
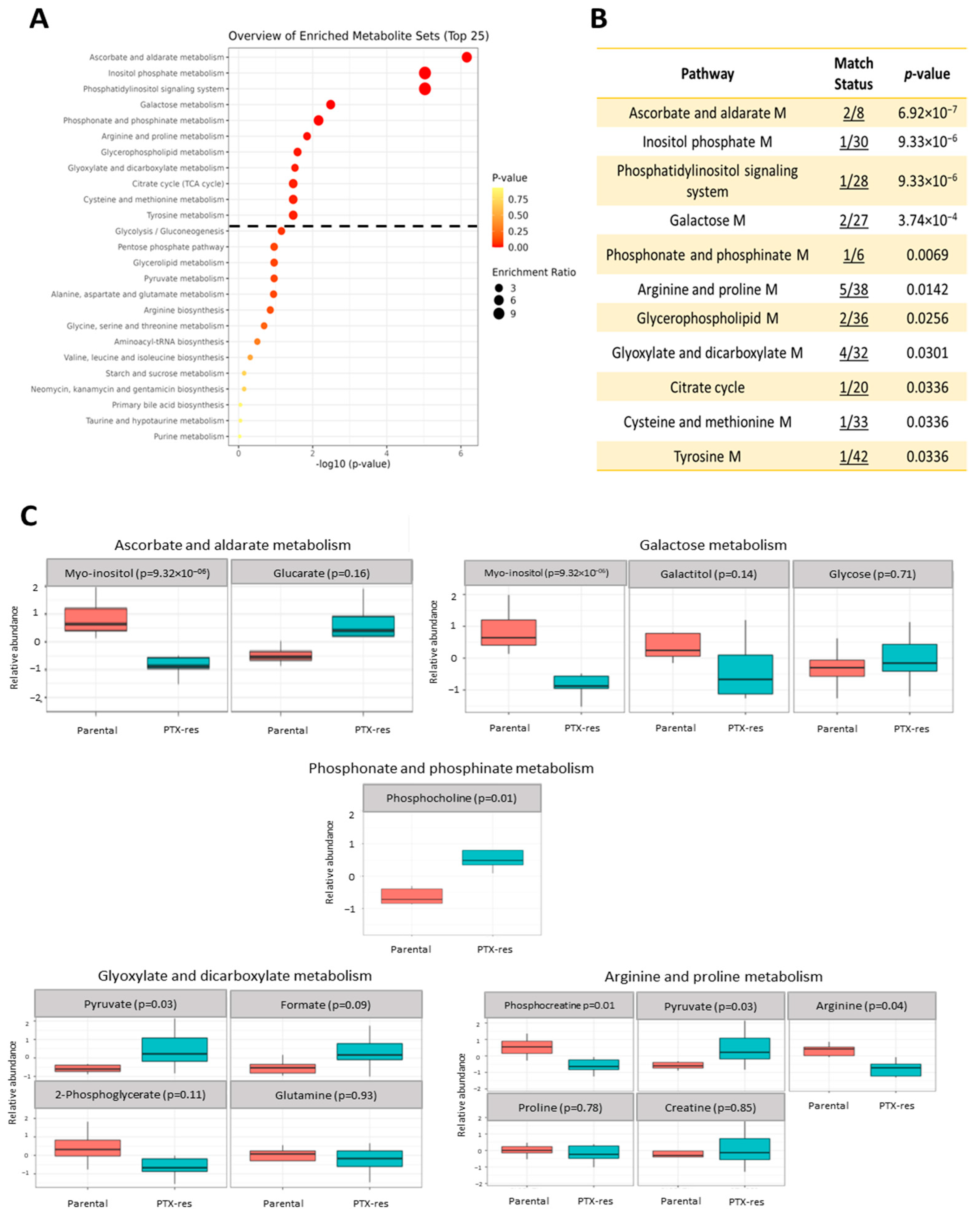
3.3.3. Metabolite Set Enrichment Analysis
3.3.4. Joint-Pathway Analysis
3.4. Lipidomic Profile Analysis
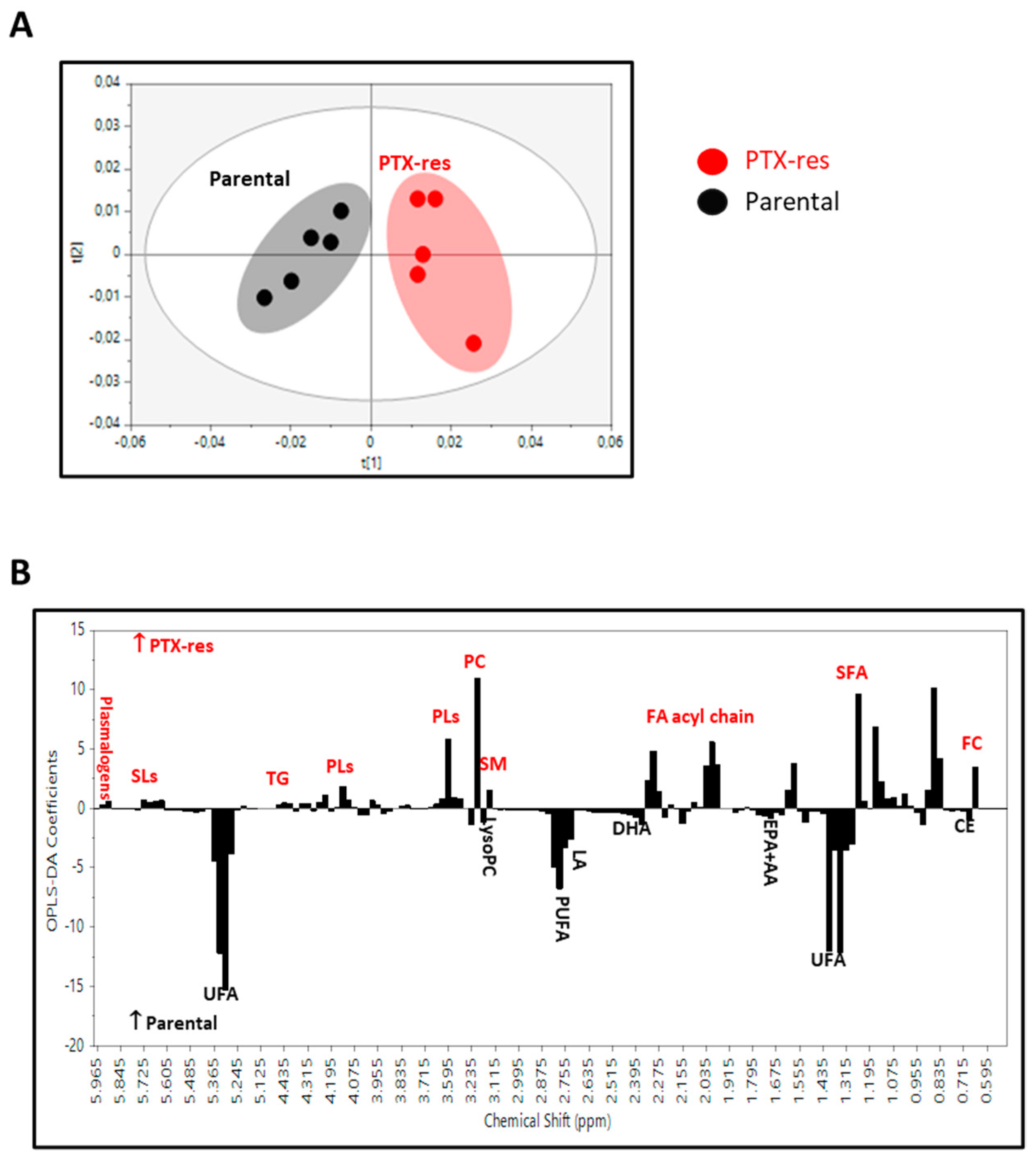
3.4.1. 1H NMR Spectra Analysis of the Lipid Extracts
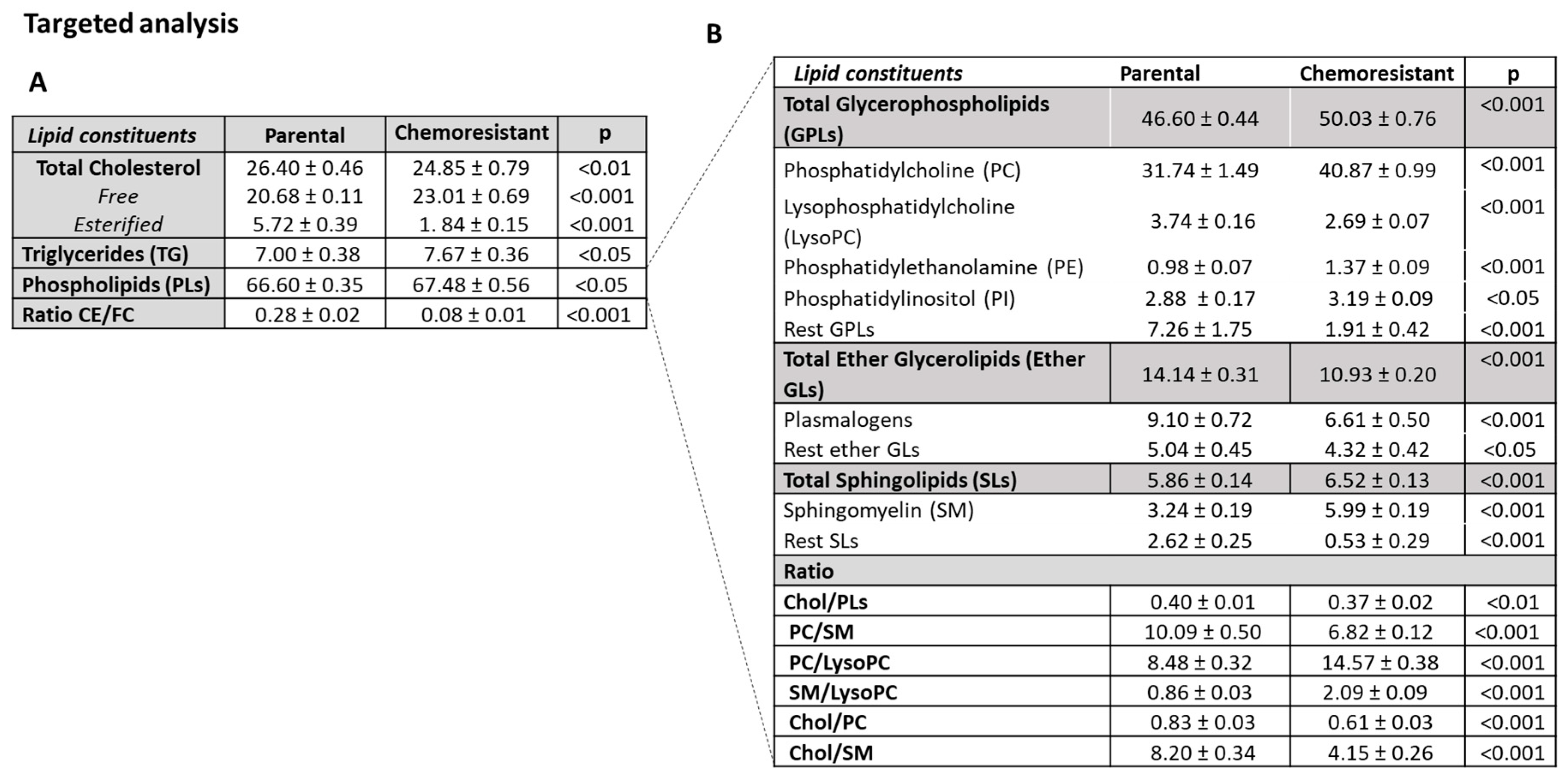
3.4.2. Lipidomic Profiles of SUM159 Parental and PTX-Res Cells
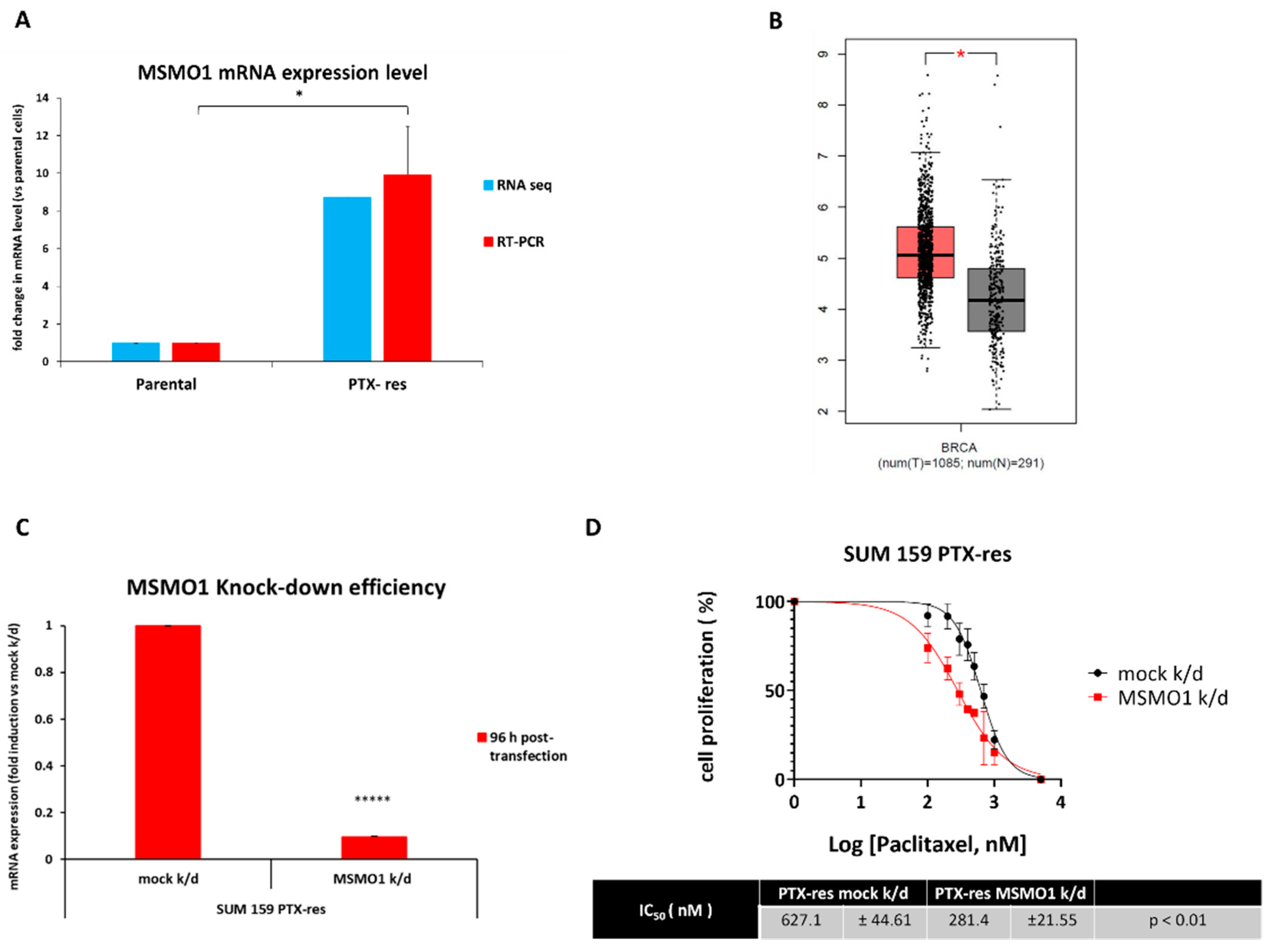
3.5. The Methylsterol Monooxygenase 1 (MSMO1) Gene Is Upregulated in the SUM159 PTX-Res Cells and Mediates Resistance to Paclitaxel
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.K.; Collier, A.L.; Lee, D.; Hoefer, R.A.; Zheleva, V.; Van Reesema, L.L.S.; Tang-Tan, A.M.; Guye, M.L.; Chang, D.Z.; Winston, J.S.; et al. Perspectives on Triple-Negative Breast Cancer: Current Treatment Strategies, Unmet Needs, and Potential Targets for Future Therapies. Cancers 2020, 12, 2392. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Thompson, C.B. Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef]
- Chen, X.; Chen, S.; Yu, D. Metabolic Reprogramming of Chemoresistant Cancer Cells and the Potential Significance of Metabolic Regulation in the Reversal of Cancer Chemoresistance. Metabolites 2020, 10, 289. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef]
- Su, Y.; Li, Y.; Guo, R.; Zhao, J.; Chi, W.; Lai, H.; Wang, J.; Wang, Z.; Li, L.; Sang, Y.; et al. Plasma extracellular vesicle long RNA profiles in the diagnosis and prediction of treatment response for breast cancer. npj Breast Cancer 2021, 7, 154. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Teng, Q.; Huang, W.; Collette, T.W.; Ekman, D.R.; Tan, C. A direct cell quenching method for cell-culture based metabolomics. Metabolomics 2009, 5, 199–208. [Google Scholar] [CrossRef]
- Rodríguez-Pérez, R.; Fernández, L.; Marco, S. Overoptimism in cross-validation when using partial least squares-discriminant analysis for omics data: A systematic study. Anal. Bioanal. Chem. 2018, 410, 5981–5992. [Google Scholar] [CrossRef]
- El Amrani, M.; Corfiotti, F.; Corvaisier, M.; Vasseur, R.; Fulbert, M.; Skrzypczyk, C.; Deshorgues, A.; Gnemmi, V.; Tulasne, D.; Lahdaoui, F.; et al. Gemcitabine-induced epithelial-mesenchymal transition-like changes sustain chemoresistance of pancreatic cancer cells of mesenchymal-like phenotype. Mol. Carcinog. 2019, 58, 1985–1997. [Google Scholar] [CrossRef] [PubMed]
- Hara, J.; Miyata, H.; Yamasaki, M.; Sugimura, K.; Takahashi, T.; Kurokawa, Y.; Nakajima, K.; Takiguchi, S.; Mori, M.; Doki, Y. Mesenchymal phenotype after chemotherapy is associated with chemoresistance and poor clinical outcome in esophageal cancer. Oncol. Rep. 2013, 31, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Dong, Y.; Kumar, R.; Jeter, C.; Tang, D.G. Slow-cycling (dormant) cancer cells in therapy resistance, cancer relapse and metastasis. Semin. Cancer Biol. 2021, 78, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Verdier-Pinard, P.; Fernandez-Fuentes, N.; Burd, B.; Angeletti, R.; Fiser, A.; Horwitz, S.B.; Orr, G.A. Insights into the mechanism of microtubule stabilization by Taxol. Proc. Natl. Acad. Sci. USA 2006, 103, 10166–10173. [Google Scholar] [CrossRef]
- Ganem, N.J.; Pellman, D. Linking abnormal mitosis to the acquisition of DNA damage. J. Cell Biol. 2012, 199, 871–881. [Google Scholar] [CrossRef]
- Giddings, E.L.; Champagne, D.P.; Wu, M.-H.; Laffin, J.M.; Thornton, T.M.; Valenca-Pereira, F.; Culp-Hill, R.; Fortner, K.A.; Romero, N.; East, J.; et al. Mitochondrial ATP fuels ABC transporter-mediated drug efflux in cancer chemoresistance. Nat. Commun. 2021, 12, 2804. [Google Scholar] [CrossRef]
- Kauffman, M.K.; Kauffman, M.E.; Zhu, H.; Jia, Z.; Li, Y.R. Fluorescence-Based Assays for Measuring Doxorubicin in Biological Systems. React. Oxyg. Species 2016, 2, 432–439. [Google Scholar] [CrossRef]
- Genovese, I.; Ilari, A.; Assaraf, Y.G.; Fazi, F.; Colotti, G. Not only P-glycoprotein: Amplification of the ABCB1- containing chromosome region 7q21 confers multidrug resistance upon cancer cells by coordinated overexpression of an assortment of resistance-related proteins. Drug Resist. Updat. 2017, 32, 23–46. [Google Scholar] [CrossRef]
- Lee, K.M.; Cao, D.; Itami, A.; Pour, P.M.; Hruban, R.H.; Maitra, A.; Ouellette, M.M. Class III ?-tubulin, a marker of resistance to paclitaxel, is overexpressed in pancreatic ductal adenocarcinoma and intraepithelial neoplasia. Histopathology 2007, 51, 539–546. [Google Scholar] [CrossRef]
- Yin, P.; Wang, W.; Gao, J.; Bai, Y.; Wang, Z.; Na, L.; Sun, Y.; Zhao, C. Fzd2 Contributes to Breast Cancer Cell Mesenchymal-Like Stemness and Drug Resistance. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2020, 28, 273–284. [Google Scholar] [CrossRef]
- Gonzalez, R.S.; Wang, J.; Kraus, T.; Sullivan, H.; Adams, A.L.; Cohen, C. GATA-3 expression in male and female breast cancers: Comparison of clinicopathologic parameters and prognostic relevance. Hum. Pathol. 2013, 44, 1065–1070. [Google Scholar] [CrossRef] [PubMed]
- Afzaljavan, F.; Sadr, A.S.; Savas, S.; Pasdar, A. GATA3 somatic mutations are associated with clinicopathological features and expression profile in TCGA breast cancer patients. Sci. Rep. 2021, 11, 1679. [Google Scholar] [CrossRef]
- Saito, M.; Goto, A.; Abe, N.; Saito, K.; Maeda, D.; Ohtake, T.; Murakami, Y.; Takenoshita, S. Decreased expression of CADM1 and CADM4 are associated with advanced stage breast cancer. Oncol. Lett. 2017, 15, 2401–2406. [Google Scholar] [CrossRef] [PubMed]
- Kanke, Y.; Saito, M.; Abe, N.; Saito, K.; Goto, A.; Ohtake, T.; Murakami, Y.; Kono, K. Expression profile of CADM1 and CADM4 in triple negative breast cancer with primary systemic therapy. Oncol. Lett. 2018, 17, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.H.; Taylor, M.G.; Robinet, P.; Smith, J.D.; Schweitzer, J.; Sehayek, E.; Falzarano, S.M.; Magi-Galluzzi, C.; Klein, E.A.; Ting, A.H. Dysregulation of Cholesterol Homeostasis in Human Prostate Cancer through Loss of ABCA1. Cancer Res. 2013, 73, 1211–1218. [Google Scholar] [CrossRef]
- Wu, Y.; Si, R.; Tang, H.; He, Z.; Zhu, H.; Wang, L.; Fan, Y.; Xia, S.; He, Z.; Wang, Q. Cholesterol reduces the sensitivity to platinum-based chemotherapy via upregulating ABCG2 in lung adenocarcinoma. Biochem. Biophys. Res. Commun. 2015, 457, 614–620. [Google Scholar] [CrossRef]
- Zhang, T.; Zhu, X.; Wu, H.; Jiang, K.; Zhao, G.; Shaukat, A.; Deng, G.; Qiu, C. Targeting the ROS/PI3K/AKT/HIF-1α/HK2 axis of breast cancer cells: Combined administration of Polydatin and 2-Deoxy-d-glucose. J. Cell. Mol. Med. 2019, 23, 3711–3723. [Google Scholar] [CrossRef]
- Oshi, M.; Gandhi, S.; Yan, L.; Tokumaru, Y.; Wu, R.; Yamada, A.; Matsuyama, R.; Endo, I.; Takabe, K. Abundance of reactive oxygen species (ROS) is associated with tumor aggressiveness, immune response, and worse survival in breast cancer. Breast Cancer Res. Treat. 2022, 194, 231–241. [Google Scholar] [CrossRef]
- Okon, I.S.; Coughlan, K.A.; Zhang, M.; Wang, Q.; Zou, M.-H. Gefitinib-mediated Reactive Oxygen Specie (ROS) Instigates Mitochondrial Dysfunction and Drug Resistance in Lung Cancer Cells. J. Biol. Chem. 2015, 290, 9101–9110. [Google Scholar] [CrossRef]
- Vaidyanathan, A.; Sawers, L.; Gannon, A.-L.; Chakravarty, P.; Scott, A.L.; E Bray, S.; Ferguson, M.J.; Smith, G. ABCB1 (MDR1) induction defines a common resistance mechanism in paclitaxel- and olaparib-resistant ovarian cancer cells. Br. J. Cancer 2016, 115, 431–441. [Google Scholar] [CrossRef] [Green Version]
- Durmus, S.; Sparidans, R.W.; van Esch, A.; Wagenaar, E.; Beijnen, J.H.; Schinkel, A.H. Breast Cancer Resistance Protein (BCRP/ABCG2) and P-glycoprotein (P-GP/ABCB1) Restrict Oral Availability and Brain Accumulation of the PARP Inhibitor Rucaparib (AG-014699). Pharm. Res. 2015, 32, 37–46. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Fortunati, E.; Liu, D.-X.; Li, Y. Pleiotropic Roles of ABC Transporters in Breast Cancer. Int. J. Mol. Sci. 2021, 22, 3199. [Google Scholar] [CrossRef] [PubMed]
- Gilkes, D.M.; Semenza, G.L. Role of hypoxia-inducible factors in breast cancer metastasis. Futur. Oncol. 2013, 9, 1623–1636. [Google Scholar] [CrossRef] [PubMed]
- Mozzetti, S.; Ferlini, C.; Concolino, P.; Filippetti, F.; Raspaglio, G.; Prislei, S.; Gallo, D.; Martinelli, E.; Ranelletti, F.O.; Ferrandina, G.; et al. Class III β-tubulin overexpression is a prominent mechanism of paclitaxel resistance in ovarian cancer patients. Clin. Cancer Res. 2005, 11, 298–305. [Google Scholar] [CrossRef]
- Zhang, H.-L.; Ruan, L.; Zheng, L.-M.; Whyte, D.; Tzeng, C.-M.; Zhou, X.-W. Association between class III β-tubulin expression and response to paclitaxel/vinorebine-based chemotherapy for non-small cell lung cancer: A meta-analysis. Lung Cancer 2012, 77, 9–15. [Google Scholar] [CrossRef]
- Lebok, P.; Ozturk, M.; Heilenkötter, U.; Jaenicke, F.; Müller, V.; Paluchowski, P.; Geist, S.; Wilke, C.; Burandt, E.; Lebeau, A.; et al. High levels of class III β-tubulin expression are associated with aggressive tumor features in breast cancer. Oncol. Lett. 2016, 11, 1987–1994. [Google Scholar] [CrossRef]
- Huang, H.-K.; Lin, Y.-H.; Chang, H.-A.; Lai, Y.-S.; Chen, Y.-C.; Huang, S.-C.; Chou, C.-Y.; Chiu, W.-T. Chemoresistant ovarian cancer enhances its migration abilities by increasing store-operated Ca2+ entry-mediated turnover of focal adhesions. J. Biomed. Sci. 2020, 27, 36:1–36:14. [Google Scholar] [CrossRef]
- Zhao, Y.; He, M.; Cui, L.; Gao, M.; Zhang, M.; Yue, F.; Shi, T.; Yang, X.; Pan, Y.; Zheng, X.; et al. Chemotherapy exacerbates ovarian cancer cell migration and cancer stem cell-like characteristics through GLI1. Br. J. Cancer 2020, 122, 1638–1648. [Google Scholar] [CrossRef]
- Sun, Z.; Zhou, D.; Yang, J.; Zhang, D. Doxorubicin promotes breast cancer cell migration and invasion via DCAF13. FEBS Open Bio 2021, 12, 221–230. [Google Scholar] [CrossRef]
- Merikhian, P.; Eisavand, M.R.; Farahmand, L. Triple-negative breast cancer: Understanding Wnt signaling in drug resistance. Cancer Cell Int. 2021, 21, 419. [Google Scholar] [CrossRef]
- Desbats, M.A.; Giacomini, I.; Prayer-Galetti, T.; Montopoli, M. Metabolic Plasticity in Chemotherapy Resistance. Front. Oncol. 2020, 10, 281. [Google Scholar] [CrossRef]
- Chhetri, D.R. Myo-Inositol and Its Derivatives: Their Emerging Role in the Treatment of Human Diseases. Front. Pharmacol. 2019, 10, 1172. [Google Scholar] [CrossRef] [PubMed]
- Kurmi, K.; Hitosugi, S.; Yu, J.; Boakye-Agyeman, F.; Wiese, E.K.; Larson, T.R.; Dai, Q.; Machida, Y.; Lou, Z.; Wang, L.; et al. Tyrosine Phosphorylation of Mitochondrial Creatine Kinase 1 Enhances a Druggable Tumor Energy Shuttle Pathway. Cell Metab. 2018, 28, 833–847.e8. [Google Scholar] [CrossRef] [PubMed]
- Smirnoff, N. Ascorbic acid metabolism and functions: A comparison of plants and mammals. Free Radic. Biol. Med. 2018, 122, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-L.; Hsu, S.-C.; Ann, D.; Yen, Y.; Kung, H.-J. Arginine Signaling and Cancer Metabolism. Cancers 2021, 13, 3541. [Google Scholar] [CrossRef] [PubMed]
- Bansal, A.; Harris, R.A.; DeGrado, T.R. Choline phosphorylation and regulation of transcription of choline kinase α in hypoxia. J. Lipid Res. 2012, 53, 149–157. [Google Scholar] [CrossRef]
- Bizzarri, M.; Dinicola, S.; Bevilacqua, A.; Cucina, A. Broad Spectrum Anticancer Activity of Myo-Inositol and Inositol Hexakisphosphate. Int. J. Endocrinol. 2016, 2016, 5616807. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M. KEGG mapping tools for uncovering hidden features in biological data. Protein Sci. 2021, 31, 47–53. [Google Scholar] [CrossRef]
- Jewison, T.; Su, Y.; Disfany, F.M.; Liang, Y.; Knox, C.; Maciejewski, A.; Poelzer, J.; Huynh, J.; Zhou, Y.; Arndt, D.; et al. SMPDB 2.0: Big Improvements to the Small Molecule Pathway Database. Nucleic Acids Res. 2013, 42, D478–D484. [Google Scholar] [CrossRef]
- Xia, J.; Wishart, D.S. Metabolomic Data Processing, Analysis, and Interpretation Using MetaboAnalyst. Curr. Protoc. Bioinform. 2011, 34, 14.10.1–14.10.48. [Google Scholar] [CrossRef]
- Ershov, P.; Kaluzhskiy, L.; Mezentsev, Y.; Yablokov, E.; Gnedenko, O.; Ivanov, A. Enzymes in the Cholesterol Synthesis Pathway: Interactomics in the Cancer Context. Biomedicines 2021, 9, 895. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.R.; Patel, R.; Kirsch, D.G.; Lewis, C.A.; Vander Heiden, M.G.; Locasale, J.W. Metabolomics in cancer research and emerging applications in clinical oncology. CA Cancer J. Clin. 2021, 71, 333–358. [Google Scholar] [CrossRef] [PubMed]
- Butler, L.M.; Perone, Y.; Dehairs, J.; Lupien, L.E.; de Laat, V.; Talebi, A.; Loda, M.; Kinlaw, W.B.; Swinnen, J.V. Lipids and cancer: Emerging roles in pathogenesis, diagnosis and therapeutic intervention. Adv. Drug Deliv. Rev. 2020, 159, 245–293. [Google Scholar] [CrossRef]
- McGuirk, S.; Audet-Delage, Y.; Annis, M.G.; Xue, Y.; Vernier, M.; Zhao, K.; St-Louis, C.; Minarrieta, L.; A Patten, D.; Morin, G.; et al. Resistance to different anthracycline chemotherapeutics elicits distinct and actionable primary metabolic dependencies in breast cancer. eLife 2021, 10, e65150. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, E.-M.; Xu, L.-Y. Guide to Metabolomics Analysis: A Bioinformatics Workflow. Metabolites 2022, 12, 357. [Google Scholar] [CrossRef]
- Schrimpe-Rutledge, A.C.; Codreanu, S.G.; Sherrod, S.D.; McLean, J.A. Untargeted Metabolomics Strategies—Challenges and Emerging Directions. J. Am. Soc. Mass Spectrom. 2016, 27, 1897–1905. [Google Scholar] [CrossRef]
- Tan, J.; Yu, C.-Y.; Wang, Z.-H.; Chen, H.-Y.; Guan, J.; Chen, Y.-X.; Fang, J.-Y. Genetic variants in the inositol phosphate metabolism pathway and risk of different types of cancer. Sci. Rep. 2015, 5, 8473. [Google Scholar] [CrossRef]
- Koguchi, T.; Tanikawa, C.; Mori, J.; Kojima, Y.; Matsuda, K. Regulation of myo-inositol biosynthesis by p53-ISYNA1 pathway. Int. J. Oncol. 2016, 48, 2415–2424. [Google Scholar] [CrossRef]
- Dinicola, S.; Fabrizi, G.; Masiello, M.G.; Proietti, S.; Palombo, A.; Minini, M.; Harrath, A.H.; Alwasel, S.H.; Ricci, G.; Catizone, A.; et al. Inositol induces mesenchymal-epithelial reversion in breast cancer cells through cytoskeleton rearrangement. Exp. Cell Res. 2016, 345, 37–50. [Google Scholar] [CrossRef]
- Bačić, I.; Družijanić, N.; Karlo, R.; Škifić, I.; Jagić, S. Efficacy of IP6 + inositol in the treatment of breast cancer patients receiving chemotherapy: Prospective, randomized, pilot clinical study. J. Exp. Clin. Cancer Res. 2010, 29, 12–15. [Google Scholar] [CrossRef] [Green Version]
- Szlasa, W.; Zendran, I.; ZalesiŃska, A.; Tarek, M.; Kulbacka, J. Lipid composition of the cancer cell membrane. J. Bioenerg. Biomembr. 2020, 52, 321–342. [Google Scholar] [CrossRef] [PubMed]
- Germain, N.; Dhayer, M.; Boileau, M.; Fovez, Q.; Kluza, J.; Marchetti, P. Lipid Metabolism and Resistance to Anticancer Treatment. Biology 2020, 9, 474. [Google Scholar] [CrossRef] [PubMed]
- Ingram, L.M.; Finnerty, M.C.; Mansoura, M.; Chou, C.-W.; Cummings, B.S. Identification of lipidomic profiles associated with drug-resistant prostate cancer cells. Lipids Health Dis. 2021, 20, 15. [Google Scholar] [CrossRef] [PubMed]
- Houthuijzen, J.M.; Daenen, L.G.M.; Roodhart, J.M.L.; Oosterom, I.; van Jaarsveld, M.T.M.; Govaert, K.M.; Smith, M.E.; Sadatmand, S.J.; Rosing, H.; Kruse, F.; et al. Lysophospholipids secreted by splenic macrophages induce chemotherapy resistance via interference with the DNA damage response. Nat. Commun. 2014, 5, 5275. [Google Scholar] [CrossRef]
- Modok, S.; Heyward, C.; Callaghan, R. P-glycoprotein retains function when reconstituted into a sphingolipid- and cholesterol-rich environment. J. Lipid Res. 2004, 45, 1910–1918. [Google Scholar] [CrossRef] [PubMed]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parental | Chemoresistant | p-value | |
|---|---|---|---|
| % Saturated | 52.95 ± 0.60 | 55.69 ± 0.60 | <0.05 |
| % Unsaturated | 47.05 ± 0.60 | 44.31 ± 2.17 | <0.05 |
| % Monounsaturated | 4.19 ± 0.38 | 4.86 ± 0.51 | NS |
| % Polyunsaturated | 42.87 ± 0.80 | 39.45 ± 2.05 | <0.05 |
| Linoleic Acid Eicosapentaenoic + Arachidonic Acid Docosahexaenoic Acid | 4.72 ± 0.11 15.58 ± 0.25 2.76 ± 0.06 | 3.93 ± 0.21 13.51 ± 1.14 2.29 ± 0.10 | <0.001 <0.05 <0.001 |
| Ratio | |||
| Saturated/Unsaturated | 1.13 ± 0.03 | 1.26 ± 0.11 | NS |
| Saturated/Polyunsaturated | 1.24 ± 0.04 | 1.42 ± 0.13 | <0.05 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kordias, D.; Kostara, C.E.; Papadaki, S.; Verigos, J.; Bairaktari, E.; Magklara, A. Omics Analysis of Chemoresistant Triple Negative Breast Cancer Cells Reveals Novel Metabolic Vulnerabilities. Cells 2022, 11, 2719. https://doi.org/10.3390/cells11172719
Kordias D, Kostara CE, Papadaki S, Verigos J, Bairaktari E, Magklara A. Omics Analysis of Chemoresistant Triple Negative Breast Cancer Cells Reveals Novel Metabolic Vulnerabilities. Cells. 2022; 11(17):2719. https://doi.org/10.3390/cells11172719
Chicago/Turabian StyleKordias, Dimitris, Christina E. Kostara, Styliani Papadaki, John Verigos, Eleni Bairaktari, and Angeliki Magklara. 2022. "Omics Analysis of Chemoresistant Triple Negative Breast Cancer Cells Reveals Novel Metabolic Vulnerabilities" Cells 11, no. 17: 2719. https://doi.org/10.3390/cells11172719
APA StyleKordias, D., Kostara, C. E., Papadaki, S., Verigos, J., Bairaktari, E., & Magklara, A. (2022). Omics Analysis of Chemoresistant Triple Negative Breast Cancer Cells Reveals Novel Metabolic Vulnerabilities. Cells, 11(17), 2719. https://doi.org/10.3390/cells11172719