
The Prostaglandin E2 Receptor EP4 Promotes Vascular Neointimal Hyperplasia through Translational Control of Tenascin C via the cAMP/PKA/mTORC1/rpS6 Pathway
 ,
,  , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Femoral Artery Wire Injury and Adenovirus-Mediated Gene Delivery
2.3. Carotid Artery Ligation
2.4. Morphometric and Immunohistological Analysis
2.5. RNA Extraction and Real-Time PCR
2.6. Western Blot Assay
2.7. Cell Culture
2.8. Scratch-Induced Wound Healing Assay
2.9. Transwell Migration Assay
2.10. Cell Proliferation Analysis
2.11. Immunocytochemistry
2.12. MTS Assay
2.13. Gene Silencing by siRNA
2.14. Statistics
3. Results
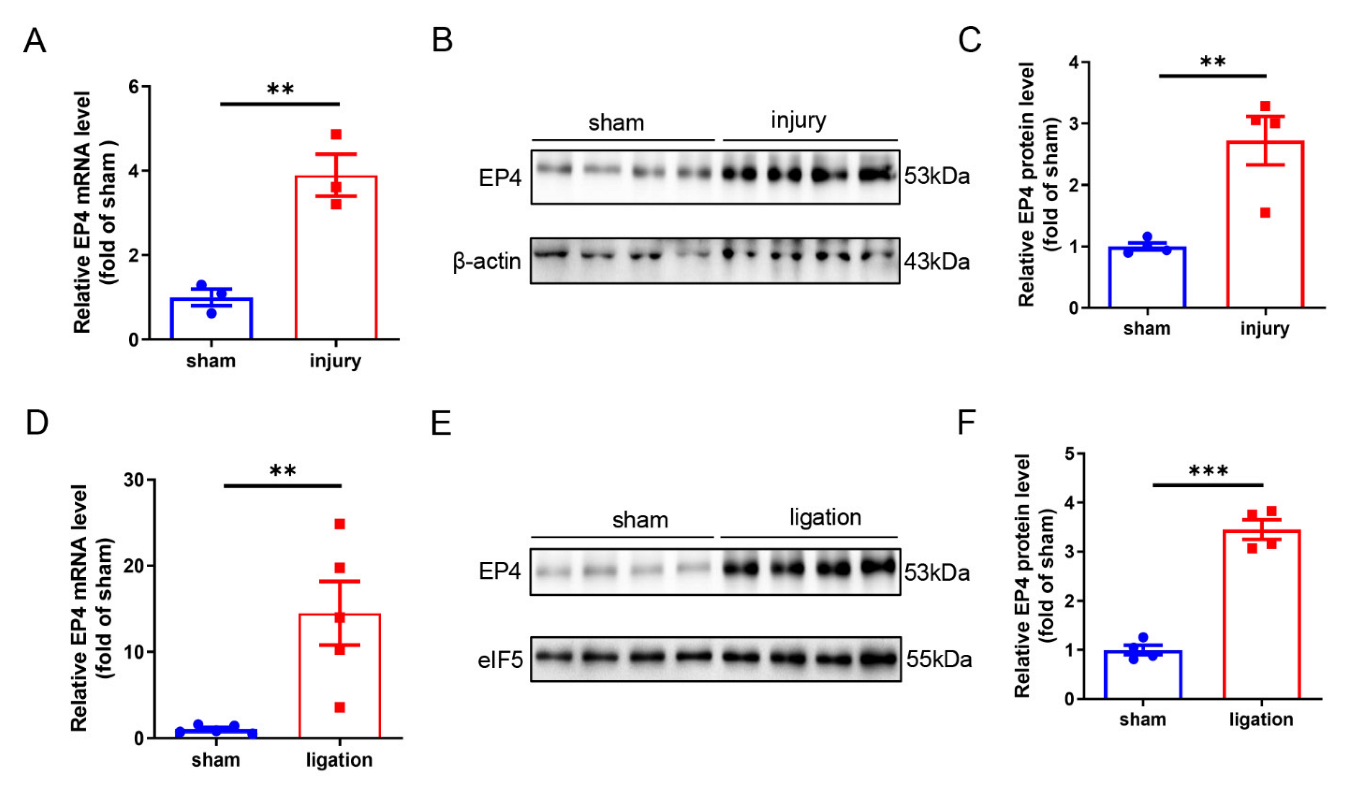
3.1. Vascular EP4 Expression Is Increased in Injured Arteries
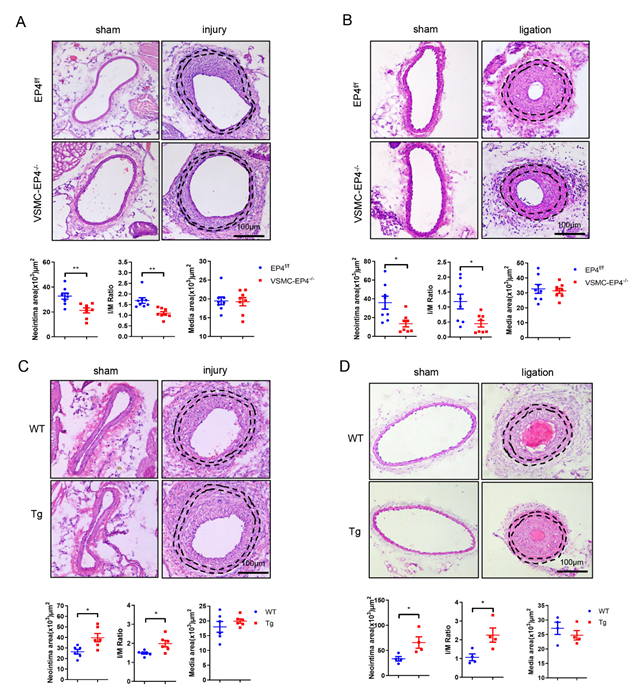
3.2. Specific Deletion of EP4 in VSMCs Ameliorates Neointimal Hyperplasia
3.3. Overexpression of Human EP4 in VSMCs Exacerbates Neointimal Formation in Mice
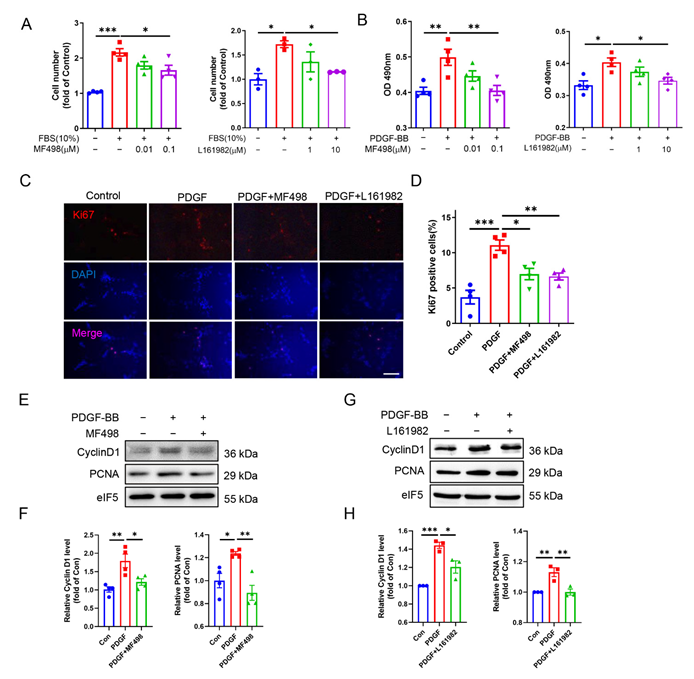
3.4. EP4 Facilitates VSMC Proliferation In Vivo and In Vitro
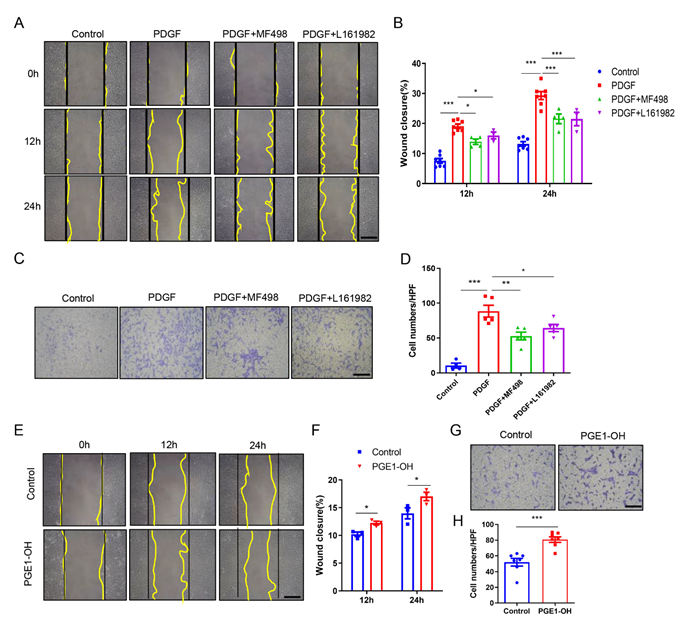
3.5. Pharmacological Activation of EP4 Promotes VSMCs Migration In Vitro
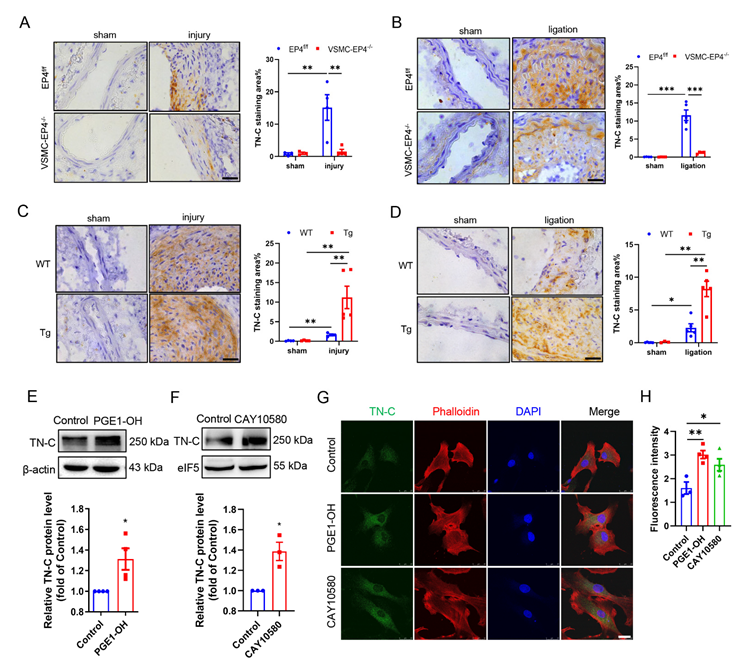
3.6. EP4 Induces Proteoglycan Tenascin Expression in Injured Arteries and in Cultured VSMCs
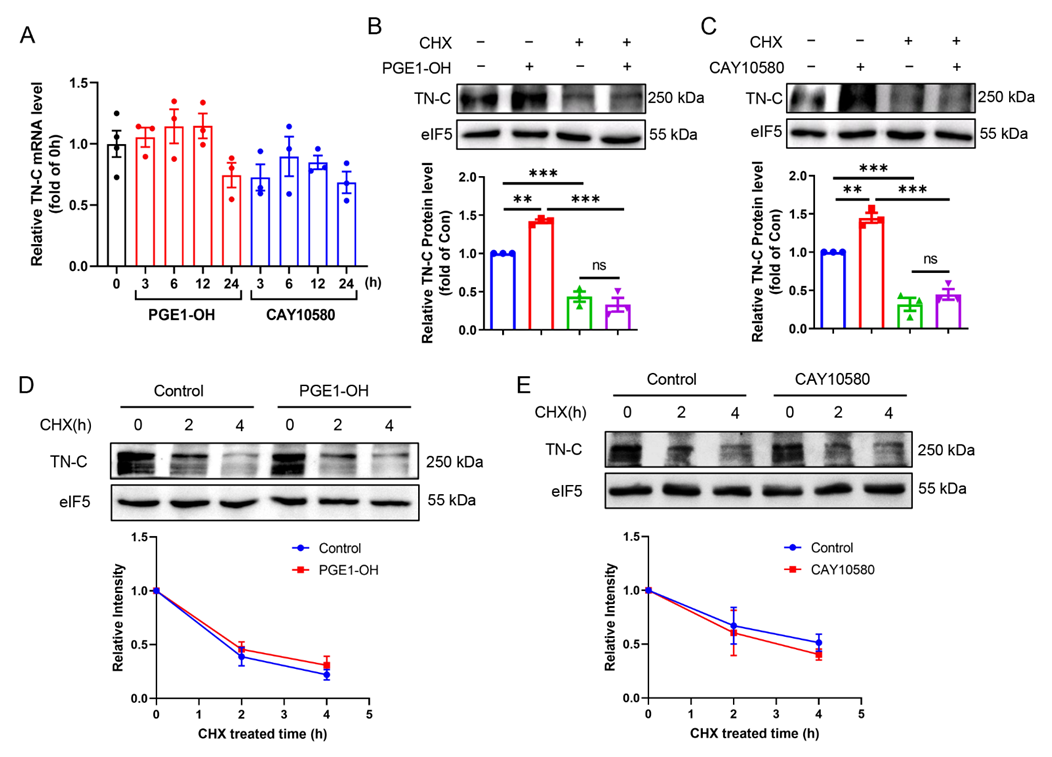
3.7. EP4 Upregulates TN-C Protein Expression at Translational Level
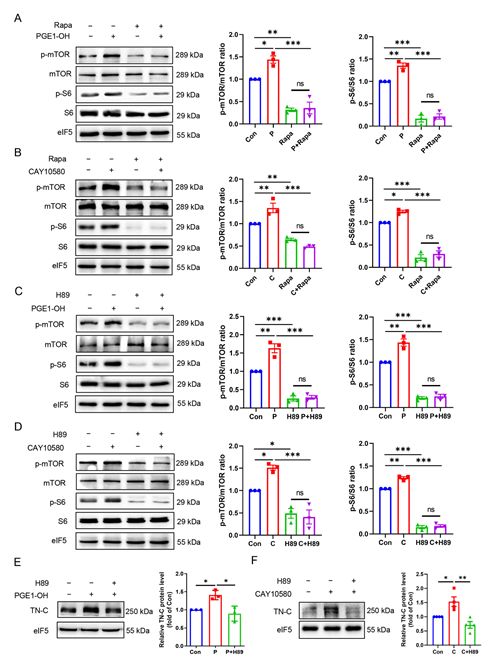
3.8. EP4 Upregulates TN-C Protein Expression via the PKA/mTOR/rpS6 Pathway
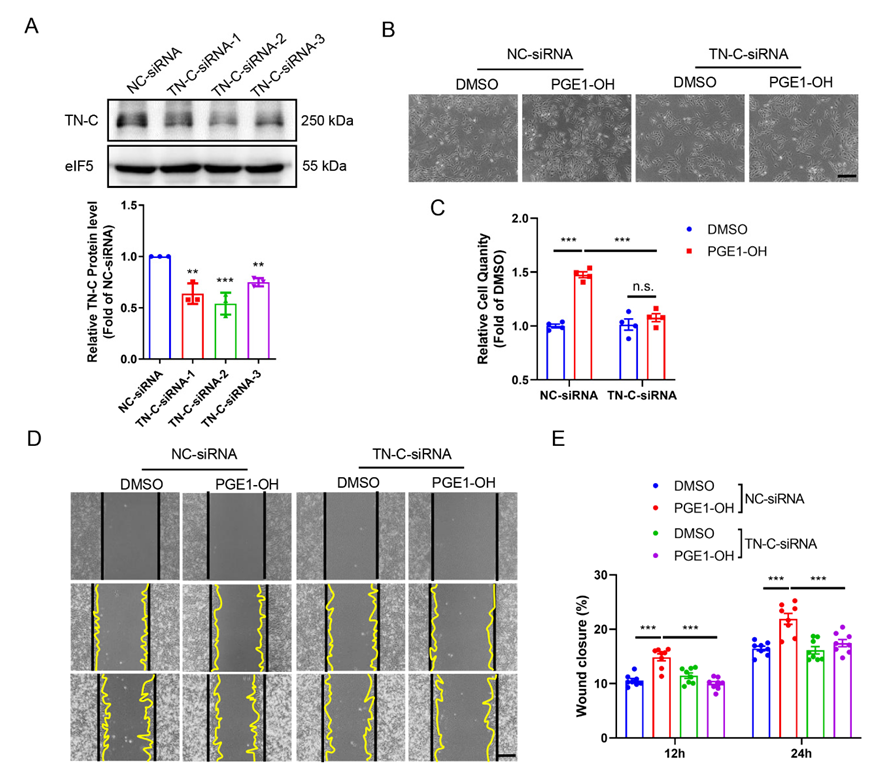
3.9. TN-C Mediates EP4-Elicited VSMC Proliferation and Migration
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Timmis, A.; Townsend, N.; Gale, C.P.; Torbica, A.; Lettino, M.; Petersen, S.E.; Mossialos, E.A.; Maggioni, A.P.; Kazakiewicz, D.; May, H.T.; et al. European Society of Cardiology: Cardiovascular Disease Statistics 2019. Eur. Heart J. 2020, 41, 12–85. [Google Scholar] [CrossRef] [PubMed]
- Wierer, M.; Werner, J.; Wobst, J.; Kastrati, A.; Cepele, G.; Aherrahrou, R.; Sager, H.B.; Erdmann, J.; Dichgans, M.; Flockerzi, V.; et al. A proteomic atlas of the neointima identifies novel druggable targets for preventive therapy. Eur. Heart J. 2021, 42, 1773–1785. [Google Scholar] [CrossRef] [PubMed]
- Canfield, J.; Totary-Jain, H. 40 Years of Percutaneous Coronary Intervention: History and Future Directions. J. Pers. Med. 2018, 8, 33. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular Smooth Muscle Cells in Atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Zhang, L.; Peppel, K.; Wu, J.H.; Zidar, D.A.; Brian, L.; DeWire, S.M.; Exum, S.T.; Lefkowitz, R.J.; Freedman, N.J. Beta-arrestins regulate atherosclerosis and neointimal hyperplasia by controlling smooth muscle cell proliferation and migration. Circ. Res. 2008, 103, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Rzucidlo, E.M.; Martin, K.A.; Powell, R.J. Regulation of vascular smooth muscle cell differentiation. J. Vasc. Surg. 2007, 45, A25–A32. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Vascular smooth muscle cell in atherosclerosis. Acta Physiol. 2015, 214, 33–50. [Google Scholar] [CrossRef]
- Costa, M.A.; Simon, D.I. Molecular basis of restenosis and drug-eluting stents. Circulation 2005, 111, 2257–2273. [Google Scholar] [CrossRef]
- Wang, M.; Ihida-Stansbury, K.; Kothapalli, D.; Tamby, M.C.; Yu, Z.; Chen, L.; Grant, G.; Cheng, Y.; Lawson, J.A.; Assoian, R.K.; et al. Microsomal prostaglandin e2 synthase-1 modulates the response to vascular injury. Circulation 2011, 123, 631–639. [Google Scholar] [CrossRef]
- Chen, L.; Yang, G.; Xu, X.; Grant, G.; Lawson, J.A.; Bohlooly, Y.M.; FitzGerald, G.A. Cell selective cardiovascular biology of microsomal prostaglandin E synthase-1. Circulation 2013, 127, 233–243. [Google Scholar] [CrossRef]
- Zhang, J.; Zou, F.; Tang, J.; Zhang, Q.; Gong, Y.; Wang, Q.; Shen, Y.; Xiong, L.; Breyer, R.M.; Lazarus, M.; et al. Cyclooxygenase-2-derived prostaglandin E(2) promotes injury-induced vascular neointimal hyperplasia through the E-prostanoid 3 receptor. Circ. Res. 2013, 113, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.M.; Kim, H.S.; Park, K.W.; You, H.J.; Jeon, S.I.; Youn, S.W.; Kim, S.H.; Oh, B.H.; Lee, M.M.; Park, Y.B.; et al. Celecoxib, a cyclooxygenase-2 inhibitor, reduces neointimal hyperplasia through inhibition of Akt signaling. Circulation 2004, 110, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Koo, B.K.; Kim, Y.S.; Park, K.W.; Yang, H.M.; Kwon, D.A.; Chung, J.W.; Hahn, J.Y.; Lee, H.Y.; Park, J.S.; Kang, H.J.; et al. Effect of celecoxib on restenosis after coronary angioplasty with a Taxus stent (COREA-TAXUS trial): An open-label randomised controlled study. Lancet 2007, 370, 567–574. [Google Scholar] [CrossRef]
- Yu, Y.; Ricciotti, E.; Scalia, R.; Tang, S.Y.; Grant, G.; Yu, Z.; Landesberg, G.; Crichton, I.; Wu, W.; Puré, E.; et al. Vascular COX-2 modulates blood pressure and thrombosis in mice. Sci. Transl. Med. 2012, 4, 132ra154. [Google Scholar] [CrossRef]
- Kang, H.J.; Oh, I.Y.; Chung, J.W.; Yang, H.M.; Suh, J.W.; Park, K.W.; Kwon, T.K.; Lee, H.Y.; Cho, Y.S.; Youn, T.J.; et al. Effects of celecoxib on restenosis after coronary intervention and evolution of atherosclerosis (Mini-COREA) trial: Celecoxib, a double-edged sword for patients with angina. Eur. Heart J. 2012, 33, 2653–2661. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, D.J.; Chen, L.H.; Zhang, Y.H.; Yang, G.R.; Dou, D.; Gao, Y.S.; Zhang, X.Y.; Kong, X.M.; Zhao, P.; Pu, D.; et al. Enhanced pressor response to acute Ang II infusion in mice lacking membrane-associated prostaglandin E2 synthase-1. Acta Pharmacol. Sin. 2010, 31, 1284–1292. [Google Scholar] [CrossRef]
- Tsuboi, K.; Sugimoto, Y.; Ichikawa, A. Prostanoid receptor subtypes. Prostaglandins Other Lipid Mediat. 2002, 68-69, 535–556. [Google Scholar] [CrossRef]
- Zhu, S.; Xue, R.; Zhao, P.; Fan, F.L.; Kong, X.; Zheng, S.; Han, Q.; Zhu, Y.; Wang, N.; Yang, J.; et al. Targeted disruption of the prostaglandin E2 E-prostanoid 2 receptor exacerbates vascular neointimal formation in mice. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1739–1747. [Google Scholar] [CrossRef]
- Xu, H.; Du, S.; Fang, B.; Li, C.; Jia, X.; Zheng, S.; Wang, S.; Li, Q.; Su, W.; Wang, N.; et al. VSMC-specific EP4 deletion exacerbates angiotensin II-induced aortic dissection by increasing vascular inflammation and blood pressure. Proc. Natl. Acad. Sci. USA 2019, 116, 8457–8462. [Google Scholar] [CrossRef]
- Hao, H.; Hu, S.; Wan, Q.; Xu, C.; Chen, H.; Zhu, L.; Xu, Z.; Meng, J.; Breyer, R.M.; Li, N.; et al. Protective Role of mPGES-1 (Microsomal Prostaglandin E Synthase-1)-Derived PGE2 (Prostaglandin E2) and the Endothelial EP4 (Prostaglandin E Receptor) in Vascular Responses to Injury. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1115–1124. [Google Scholar] [CrossRef]
- Xu, H.; Fang, B.; Du, S.; Wang, S.; Li, Q.; Jia, X.; Bao, C.; Ye, L.; Sui, X.; Qian, L.; et al. Endothelial cell prostaglandin E2 receptor EP4 is essential for blood pressure homeostasis. JCI Insight 2020, 5, e138505. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.; Camenisch, T.; Snouwaert, J.N.; Hicks, E.; Coffman, T.M.; Anderson, P.A.; Malouf, N.N.; Koller, B.H. The prostaglandin receptor EP4 triggers remodelling of the cardiovascular system at birth. Nature 1997, 390, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, U.; Ishiwata, R.; Jin, M.H.; Kato, Y.; Suzuki, O.; Jin, H.; Ichikawa, Y.; Kumagaya, S.; Katayama, Y.; Fujita, T.; et al. Inhibition of EP4 signaling attenuates aortic aneurysm formation. PLoS ONE 2012, 7, e36724. [Google Scholar] [CrossRef] [PubMed]
- Mamun, A.; Yokoyama, U. A selective antagonist of prostaglandin E receptor subtype 4 attenuates abdominal aortic aneurysm. Physiol. Rep. 2018, 6, e13878. [Google Scholar] [CrossRef]
- Hedin, U.; Holm, J.; Hansson, G.K. Induction of tenascin in rat arterial injury. Relationship to altered smooth muscle cell phenotype. Am. J. Pathol. 1991, 139, 649–656. [Google Scholar]
- Davies, M.G.; Hagen, P.O. Pathobiology of intimal hyperplasia. Br. J. Surg 1994, 81, 1254–1269. [Google Scholar] [CrossRef]
- Yamamoto, K.; Onoda, K.; Sawada, Y.; Fujinaga, K.; Imanaka-Yoshida, K.; Shimpo, H.; Yoshida, T.; Yada, I. Tenascin-C is an essential factor for neointimal hyperplasia after aortotomy in mice. Cardiovasc. Res. 2005, 65, 737–742. [Google Scholar] [CrossRef]
- Xu, H.; Wang, S.L.; Bao, C.Z.; Ye, L.; Guan, Y.F.; Zhang, X.Y. Overexpression of human EP4 receptor in vascular smooth muscle cells attenuates angiotensin II-induced hypertension in mice. Sheng Li Xue Bao Acta Physiol. Sin. 2021, 73, 597–605. [Google Scholar]
- Sata, M.; Maejima, Y.; Adachi, F.; Fukino, K.; Saiura, A.; Sugiura, S.; Aoyagi, T.; Imai, Y.; Kurihara, H.; Kimura, K.; et al. A mouse model of vascular injury that induces rapid onset of medial cell apoptosis followed by reproducible neointimal hyperplasia. J. Mol. Cell. Cardiol. 2000, 32, 2097–2104. [Google Scholar] [CrossRef]
- Jovinge, S.; Hultgardh-Nilsson, A.; Regnstrom, J.; Nilsson, J. Tumor necrosis factor-alpha activates smooth muscle cell migration in culture and is expressed in the balloon-injured rat aorta. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 490–497. [Google Scholar] [CrossRef]
- Liang, C.C.; Park, A.Y.; Guan, J.L. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007, 2, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Newby, A.C.; Zaltsman, A.B. Molecular mechanisms in intimal hyperplasia. J. Pathol. 2000, 190, 300–309. [Google Scholar] [CrossRef]
- Meyuhas, O. Ribosomal Protein S6 Phosphorylation: Four Decades of Research. Int. Rev. Cell Mol. Biol. 2015, 320, 41–73. [Google Scholar] [CrossRef] [PubMed]
- Regan, J.W. EP2 and EP4 prostanoid receptor signaling. Life Sci. 2003, 74, 143–153. [Google Scholar] [CrossRef]
- Melnik, T.; Jordan, O.; Corpataux, J.M.; Delie, F.; Saucy, F. Pharmacological prevention of intimal hyperplasia: A state-of-the-art review. Pharmacol. Ther. 2022, 235, 108157. [Google Scholar] [CrossRef] [PubMed]
- Fujinaga, K.; Onoda, K.; Yamamoto, K.; Imanaka-Yoshida, K.; Takao, M.; Shimono, T.; Shimpo, H.; Yoshida, T.; Yada, I. Locally applied cilostazol suppresses neointimal hyperplasia by inhibiting tenascin-C synthesis and smooth muscle cell proliferation in free artery grafts. J. Thorac. Cardiovasc. Surg. 2004, 128, 357–363. [Google Scholar] [CrossRef]
- Jones, P.L.; Rabinovitch, M. Tenascin-C is induced with progressive pulmonary vascular disease in rats and is functionally related to increased smooth muscle cell proliferation. Circ. Res. 1996, 79, 1131–1142. [Google Scholar] [CrossRef]
- Yi, Y.W.; You, K.S.; Park, J.S.; Lee, S.G.; Seong, Y.S. Ribosomal Protein S6: A Potential Therapeutic Target against Cancer? Int. J. Mol. Sci. 2021, 23, 48. [Google Scholar] [CrossRef]
- Yokoyama, U.; Iwatsubo, K.; Umemura, M.; Fujita, T.; Ishikawa, Y. The prostanoid EP4 receptor and its signaling pathway. Pharmacol. Rev. 2013, 65, 1010–1052. [Google Scholar] [CrossRef]
- Moore, C.E.; Xie, J.; Gomez, E.; Herbert, T.P. Identification of cAMP-dependent kinase as a third in vivo ribosomal protein S6 kinase in pancreatic beta-cells. J. Mol. Biol. 2009, 389, 480–494. [Google Scholar] [CrossRef]
- Chowdhury, T.; Köhler, J.R. Ribosomal protein S6 phosphorylation is controlled by TOR and modulated by PKA in Candida albicans. Mol. Microbiol. 2015, 98, 384–402. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.Q.; Zou, Y.; Tian, Y.; Ma, X.F.; Zhou, Q.Y.; Li, Z.Y.; Gong, S.X.; Wang, A.P. mTOR as the therapeutic target of vascular proliferative diseases: Past, present, and future. J. Cardiovasc. Pharmacol. 2022, 79, 444–455. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Parent, C.A. Review series: TOR kinase complexes and cell migration. J. Cell Biol. 2011, 194, 815–824. [Google Scholar] [CrossRef]
- Giordano, A.; Romano, A. Inhibition of human in-stent restenosis: A molecular view. Curr. Opin. Pharmacol. 2011, 11, 372–377. [Google Scholar] [CrossRef]
- Valdes, P.J.; Akbar, H.; Kahloon, R.A.; Diaz, M.A. Intracoronary Stents. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2022. [Google Scholar]
- Santulli, G.; Totary-Jain, H. Tailoring mTOR-based therapy: Molecular evidence and clinical challenges. Pharmacogenomics 2013, 14, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Habib, A.; Finn, A.V. Antiproliferative Drugs for Restenosis Prevention. Interv. Cardiol. Clin. 2016, 5, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Imanaka-Yoshida, K.; Tawara, I.; Yoshida, T. Tenascin-C in cardiac disease: A sophisticated controller of inflammation, repair, and fibrosis. Am. J. Physiol. Cell Physiol. 2020, 319, C781–C796. [Google Scholar] [CrossRef]
- McKean, J.S.; Murray, F.; Gibson, G.; Shewan, D.A.; Tucker, S.J.; Nixon, G.F. The cAMP-producing agonist beraprost inhibits human vascular smooth muscle cell migration via exchange protein directly activated by cAMP. Cardiovasc. Res. 2015, 107, 546–555. [Google Scholar] [CrossRef]
- Smith, S.A.; Newby, A.C.; Bond, M. Ending Restenosis: Inhibition of Vascular Smooth Muscle Cell Proliferation by cAMP. Cells 2019, 8, 1447. [Google Scholar] [CrossRef]
- Rao, R.; Redha, R.; Macias-Perez, I.; Su, Y.; Hao, C.; Zent, R.; Breyer, M.D.; Pozzi, A. Prostaglandin E2-EP4 receptor promotes endothelial cell migration via ERK activation and angiogenesis in vivo. J. Biol. Chem. 2007, 282, 16959–16968. [Google Scholar] [CrossRef]
- Zhang, Y.; Daaka, Y. PGE2 promotes angiogenesis through EP4 and PKA Cgamma pathway. Blood 2011, 118, 5355–5364. [Google Scholar] [CrossRef] [PubMed]
- Maguire, E.M.; Xiao, Q. Noncoding RNAs in vascular smooth muscle cell function and neointimal hyperplasia. FEBS J. 2020, 287, 5260–5283. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, H.; Fang, B.; Bao, C.; Mao, X.; Zhu, C.; Ye, L.; Liu, Q.; Li, Y.; Du, C.; Qi, H.; et al. The Prostaglandin E2 Receptor EP4 Promotes Vascular Neointimal Hyperplasia through Translational Control of Tenascin C via the cAMP/PKA/mTORC1/rpS6 Pathway. Cells 2022, 11, 2720. https://doi.org/10.3390/cells11172720
Xu H, Fang B, Bao C, Mao X, Zhu C, Ye L, Liu Q, Li Y, Du C, Qi H, et al. The Prostaglandin E2 Receptor EP4 Promotes Vascular Neointimal Hyperplasia through Translational Control of Tenascin C via the cAMP/PKA/mTORC1/rpS6 Pathway. Cells. 2022; 11(17):2720. https://doi.org/10.3390/cells11172720
Chicago/Turabian StyleXu, Hu, Bingying Fang, Chengzhen Bao, Xiuhui Mao, Chunhua Zhu, Lan Ye, Qian Liu, Yaqing Li, Chunxiu Du, Hang Qi, and et al. 2022. "The Prostaglandin E2 Receptor EP4 Promotes Vascular Neointimal Hyperplasia through Translational Control of Tenascin C via the cAMP/PKA/mTORC1/rpS6 Pathway" Cells 11, no. 17: 2720. https://doi.org/10.3390/cells11172720
APA StyleXu, H., Fang, B., Bao, C., Mao, X., Zhu, C., Ye, L., Liu, Q., Li, Y., Du, C., Qi, H., Zhang, X., & Guan, Y. (2022). The Prostaglandin E2 Receptor EP4 Promotes Vascular Neointimal Hyperplasia through Translational Control of Tenascin C via the cAMP/PKA/mTORC1/rpS6 Pathway. Cells, 11(17), 2720. https://doi.org/10.3390/cells11172720