
Nicotinamide Adenine Dinucleotide (NAD) Metabolism as a Relevant Target in Cancer



Abstract
:1. Nicotinamide Adenine Dinucleotide (NAD+)
2. Redox Functions of NAD+
3. NAD+ as a Substrate in Nonredox Reactions
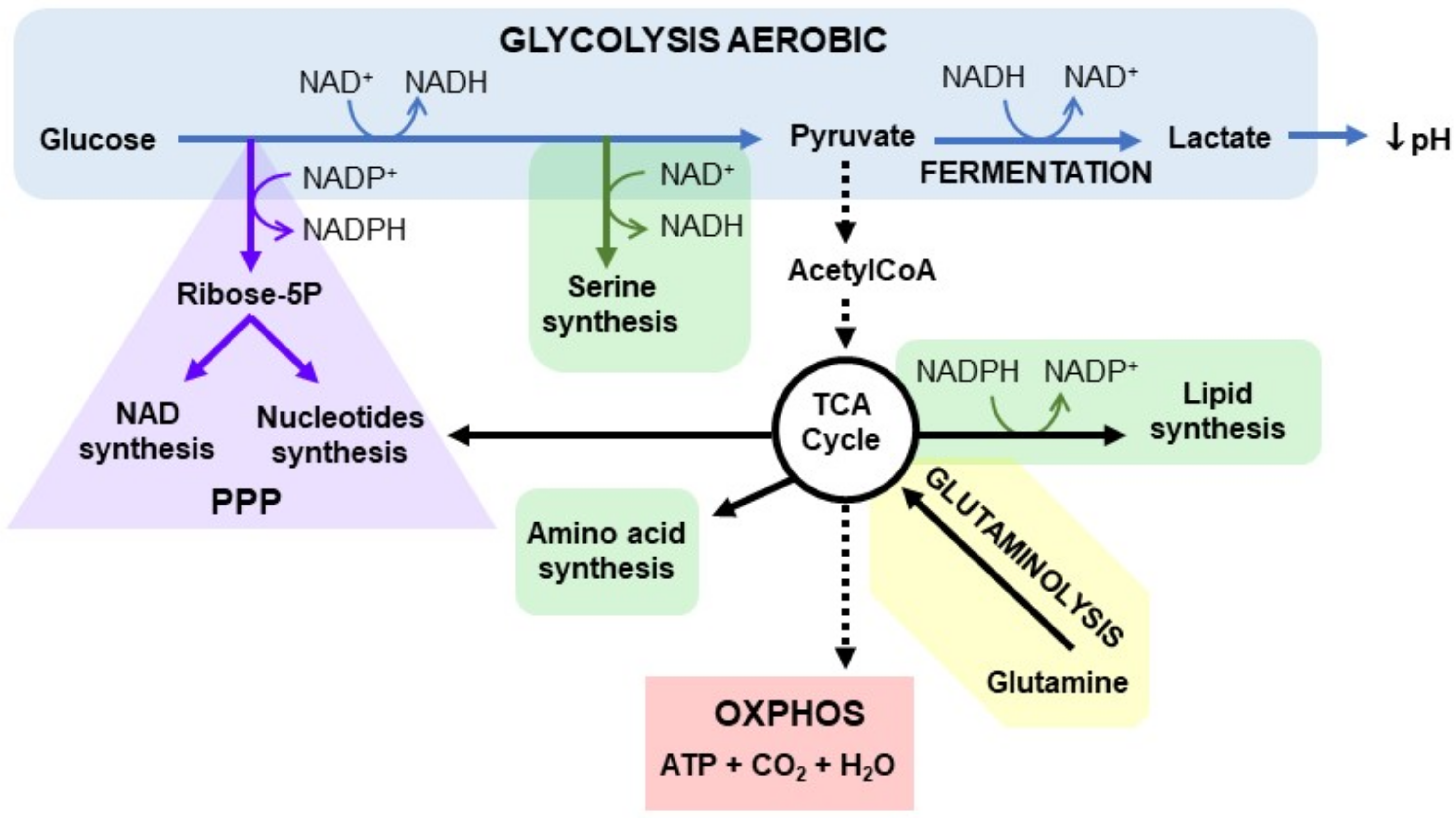
4. Cancer and NAD+ Metabolism
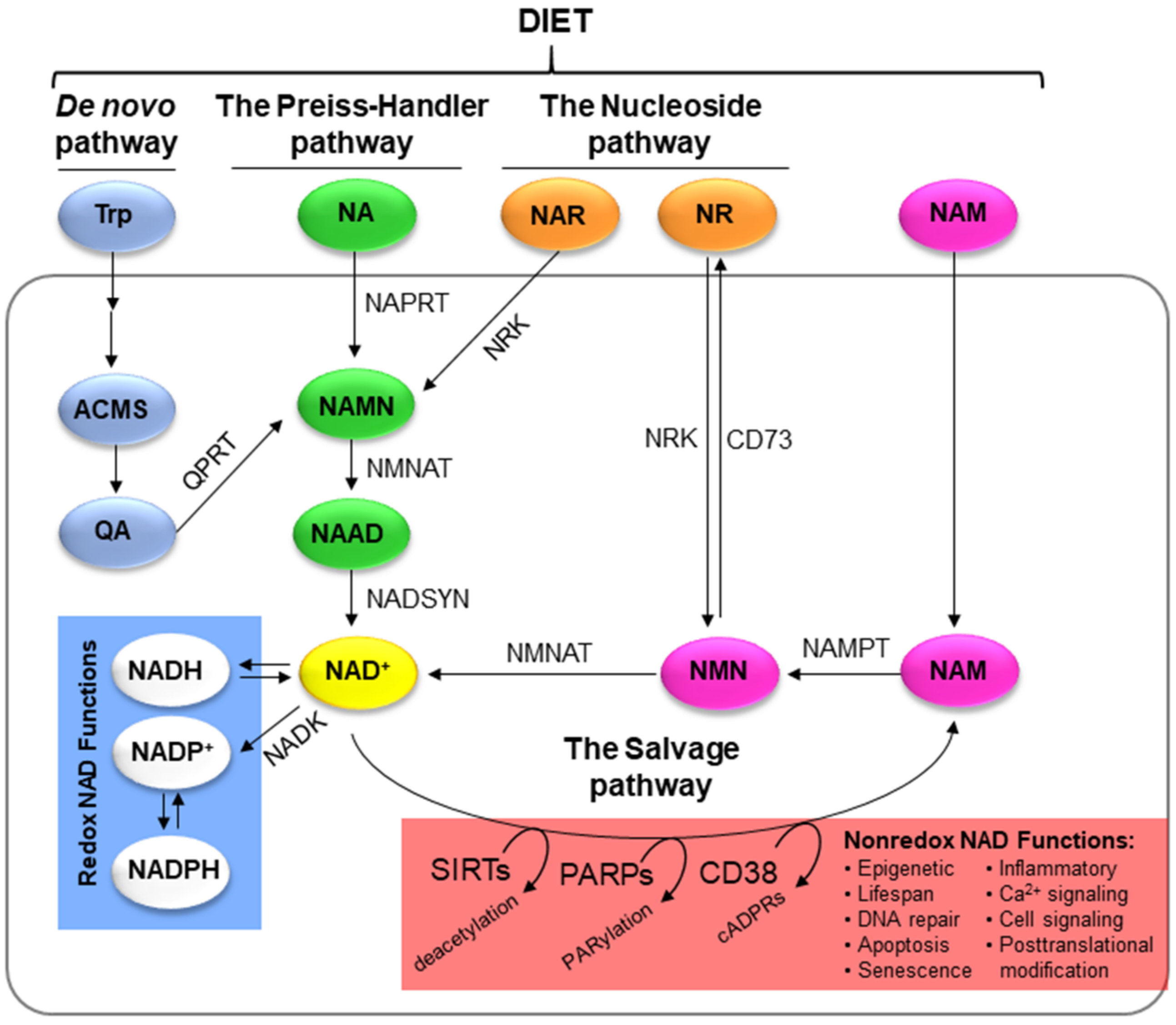
5. NAD+ Biosynthesis
5.1. De Novo Pathway
5.2. The Preiss–Handler Pathway
5.3. The Salvage Pathway
5.4. The Nucleoside Pathway
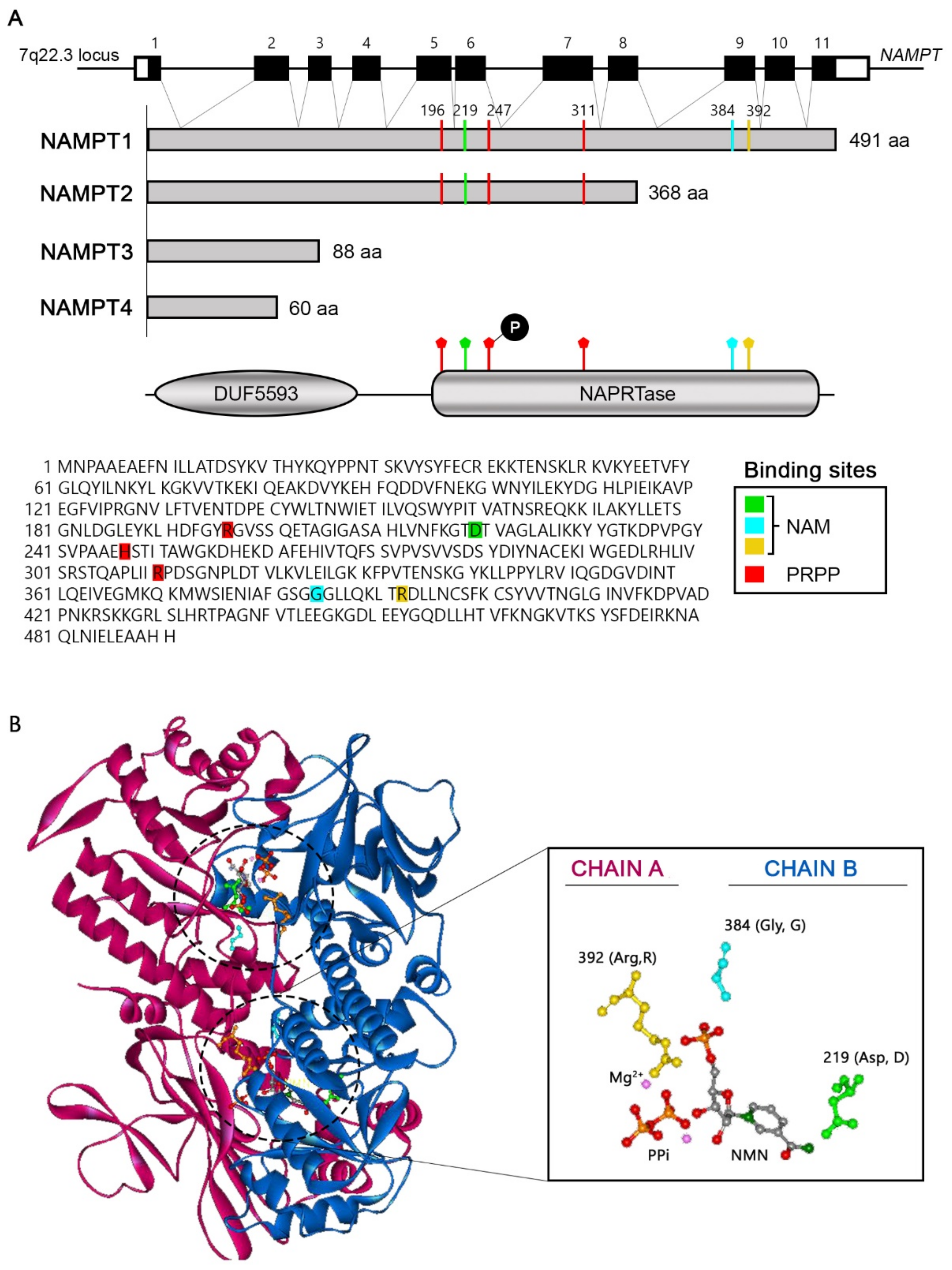
6. Nicotinamide Phosphoribosyltransferase (NAMPT)
7. NAMPT in Cancer
8. Other NAD+-Dependent Enzymes in Cancer
8.1. SIRTs in Cancer
8.2. PARPs in Cancer
8.3. cADPRSs in Cancer
8.4. NAPRT in Cancer
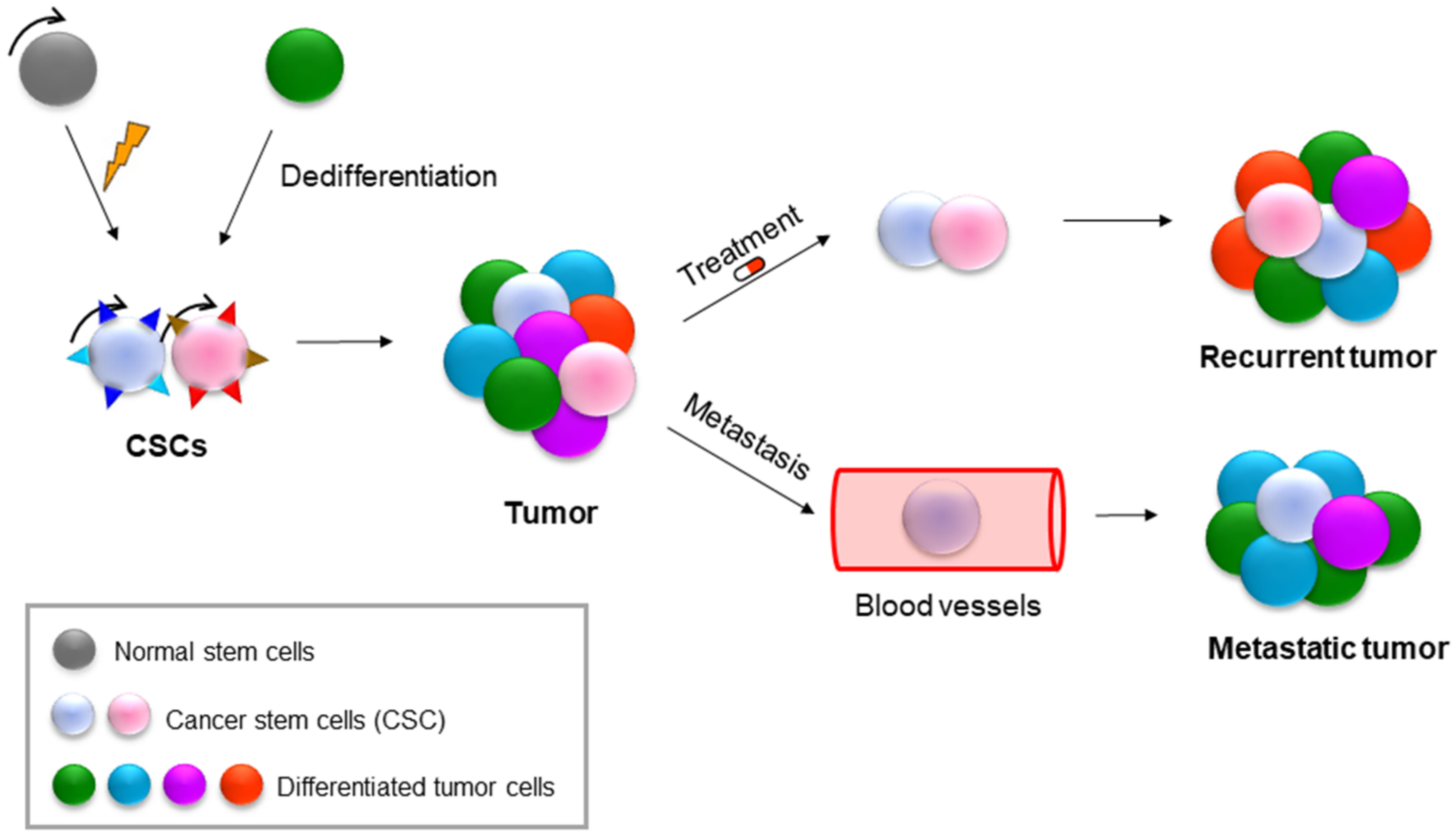
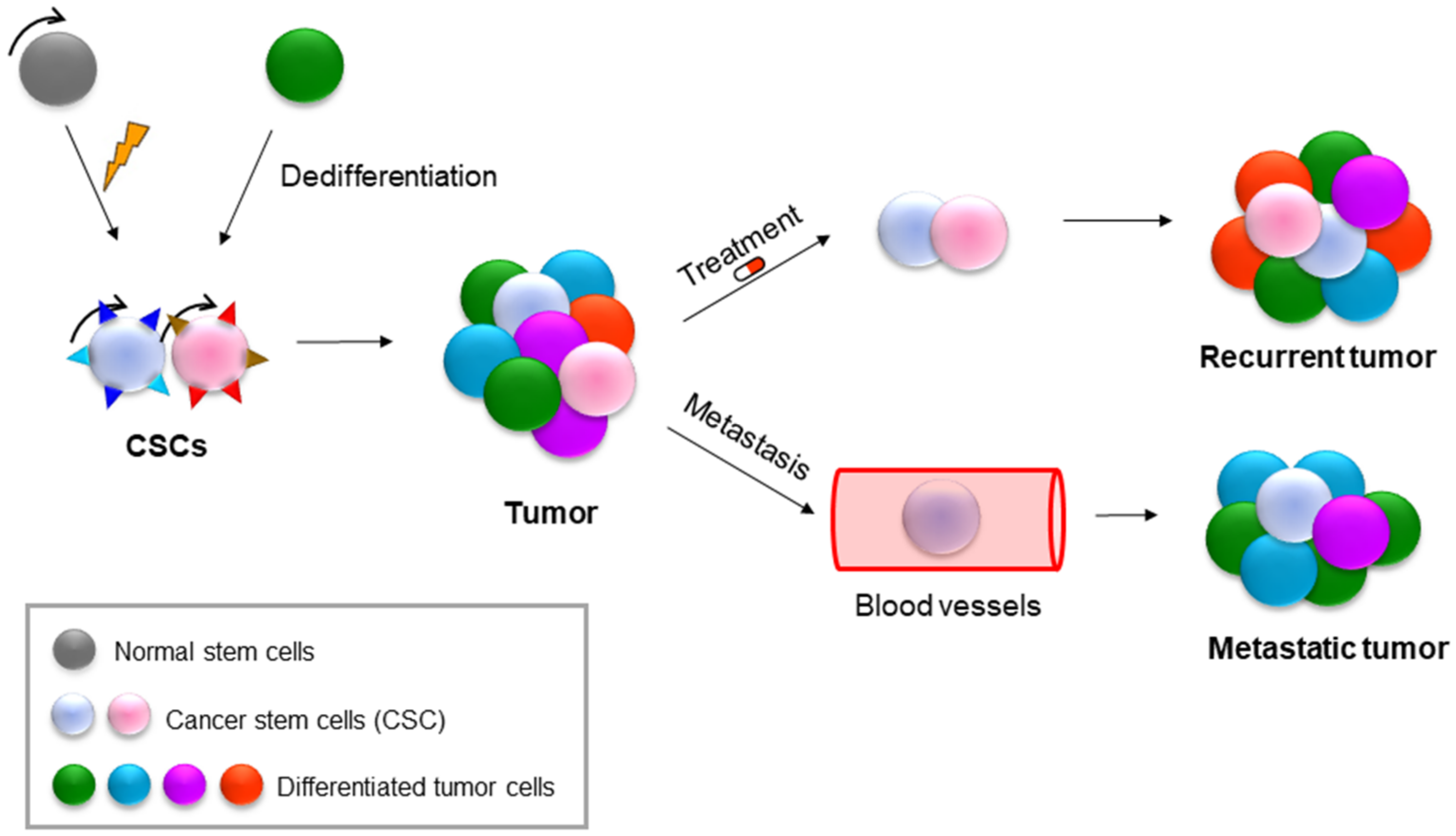
9. NAMPT in Cancer Stem Cells (CSCs)
10. NAMPT as a Therapeutic Strategy
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Navas, L.E.; Carnero, A. NAD(+) metabolism, stemness, the immune response, and cancer. Signal Transduct. Target Ther. 2021, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Poljsak, B. NAD+ in Cancer Prevention and Treatment: Pros and Cons. J. Clin. Exp. Oncol. 2016, 4, 2. [Google Scholar] [CrossRef]
- Yang, Y.; Sauve, A.A. NAD(+) metabolism: Bioenergetics, signaling and manipulation for therapy. Biochim. Biophys. Acta 2016, 1864, 1787–1800. [Google Scholar] [CrossRef]
- Nakamura, M.; Bhatnagar, A.; Sadoshima, J. Overview of pyridine nucleotides review series. Circ. Res. 2012, 111, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Guillot, B.; Lecomte, C.; Cousson, A.; Scherf, C.; Jelsch, C. High-resolution neutron structure of nicotinamide adenine dinucleotide. Acta Crystallogr. D. Biol. Crystallogr. 2001, 57, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Htet, Y.; Tennyson, A.G. NAD(+) as a Hydride Donor and Reductant. J. Am. Chem. Soc. 2016, 138, 15833–15836. [Google Scholar] [CrossRef]
- Fletcher, R.S.; Lavery, G.G. The emergence of the nicotinamide riboside kinases in the regulation of NAD+ metabolism. J. Mol. Endocrinol. 2018, 61, R107–R121. [Google Scholar] [CrossRef]
- Kulkarni, C.A.; Brookes, P.S. Cellular Compartmentation and the Redox/Nonredox Functions of NAD(+). Antioxid. Redox Signal 2019, 31, 623–642. [Google Scholar] [CrossRef]
- Opitz, C.A.; Heiland, I. Dynamics of NAD-metabolism: Everything but constant. Biochem. Soc. Trans. 2015, 43, 1127–1132. [Google Scholar] [CrossRef]
- Nakahata, Y.; Sahar, S.; Astarita, G.; Kaluzova, M.; Sassone-Corsi, P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science 2009, 324, 654–657. [Google Scholar] [CrossRef]
- Masri, S. Sirtuin-dependent clock control: New advances in metabolism, aging and cancer. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, K.M.; Yoshino, J.; Brace, C.S.; Abrassart, D.; Kobayashi, Y.; Marcheva, B.; Hong, H.K.; Chong, J.L.; Buhr, E.D.; Lee, C.; et al. Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science 2009, 324, 651–654. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Dan Dunn, J.; Alvarez, L.A.; Zhang, X.; Soldati, T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. 2015, 6, 472–485. [Google Scholar] [CrossRef]
- Lee, B.W.L.; Ghode, P.; Ong, D.S.T. Redox regulation of cell state and fate. Redox Biol. 2019, 25, 101056. [Google Scholar] [CrossRef]
- Chiarugi, A.; Dolle, C.; Felici, R.; Ziegler, M. The NAD metabolome—A key determinant of cancer cell biology. Nat. Rev. Cancer 2012, 12, 741–752. [Google Scholar] [CrossRef]
- Braidy, N.; Berg, J.; Clement, J.; Khorshidi, F.; Poljak, A.; Jayasena, T.; Grant, R.; Sachdev, P. Role of Nicotinamide Adenine Dinucleotide and Related Precursors as Therapeutic Targets for Age-Related Degenerative Diseases: Rationale, Biochemistry, Pharmacokinetics, and Outcomes. Antioxid. Redox Signal 2019, 30, 251–294. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Rajman, L.; Chwalek, K.; Sinclair, D.A. Therapeutic Potential of NAD-Boosting Molecules: The In Vivo Evidence. Cell Metab. 2018, 27, 529–547. [Google Scholar] [CrossRef]
- Kim, M.Y.; Zhang, T.; Kraus, W.L. Poly(ADP-ribosyl)ation by PARP-1: ‘PAR-laying’ NAD+ into a nuclear signal. Genes Dev. 2005, 19, 1951–1967. [Google Scholar] [CrossRef] [Green Version]
- Langelier, M.F.; Zandarashvili, L.; Aguiar, P.M.; Black, B.E.; Pascal, J.M. NAD(+) analog reveals PARP-1 substrate-blocking mechanism and allosteric communication from catalytic center to DNA-binding domains. Nat. Commun. 2018, 9, 844. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Canto, C.; Wanders, R.J.; Auwerx, J. The secret life of NAD+: An old metabolite controlling new metabolic signaling pathways. Endocr. Rev. 2010, 31, 194–223. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Menzies, K.J.; Auwerx, J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.P.; Price, N.L.; Ling, A.J.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 2013, 155, 1624–1638. [Google Scholar] [CrossRef]
- Moreschi, I.; Bruzzone, S.; Melone, L.; De Flora, A.; Zocchi, E. NAADP+ synthesis from cADPRP and nicotinic acid by ADP-ribosyl cyclases. Biochem. Biophys. Res. Commun. 2006, 345, 573–580. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Moreira, J.D.; Hamraz, M.; Abolhassani, M.; Bigan, E.; Peres, S.; Pauleve, L.; Nogueira, M.L.; Steyaert, J.M.; Schwartz, L. The Redox Status of Cancer Cells Supports Mechanisms behind the Warburg Effect. Metabolites 2016, 6, 33. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Cantor, J.R.; Sabatini, D.M. Cancer cell metabolism: One hallmark, many faces. Cancer Discov. 2012, 2, 881–898. [Google Scholar] [CrossRef]
- Yaku, K.; Okabe, K.; Hikosaka, K.; Nakagawa, T. NAD Metabolism in Cancer Therapeutics. Front. Oncol. 2018, 8, 622. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Mates, J.M.; Di Paola, F.J.; Campos-Sandoval, J.A.; Mazurek, S.; Marquez, J. Therapeutic targeting of glutaminolysis as an essential strategy to combat cancer. Semin. Cell Dev. Biol. 2020, 98, 34–43. [Google Scholar] [CrossRef]
- Bott, A.J.; Maimouni, S.; Zong, W.X. The Pleiotropic Effects of Glutamine Metabolism in Cancer. Cancers 2019, 11, 770. [Google Scholar] [CrossRef] [PubMed]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Amelio, I.; Cutruzzola, F.; Antonov, A.; Agostini, M.; Melino, G. Serine and glycine metabolism in cancer. Trends Biochem. Sci. 2014, 39, 191–198. [Google Scholar] [CrossRef]
- Mattaini, K.R.; Sullivan, M.R.; Vander Heiden, M.G. The importance of serine metabolism in cancer. J. Cell Biol. 2016, 214, 249–257. [Google Scholar] [CrossRef]
- Cambronne, X.A.; Stewart, M.L.; Kim, D.; Jones-Brunette, A.M.; Morgan, R.K.; Farrens, D.L.; Cohen, M.S.; Goodman, R.H. Biosensor reveals multiple sources for mitochondrial NAD(+). Science 2016, 352, 1474–1477. [Google Scholar] [CrossRef]
- Hara, N.; Yamada, K.; Terashima, M.; Osago, H.; Shimoyama, M.; Tsuchiya, M. Molecular identification of human glutamine- and ammonia-dependent NAD synthetases. Carbon-nitrogen hydrolase domain confers glutamine dependency. J. Biol. Chem. 2003, 278, 10914–10921. [Google Scholar] [CrossRef]
- Lucena-Cacace, A.; Carnero, A. Nicotinamide phosphoribosyltransferase: Biology, role in cancer, and novel drug target. Cancer Transl. Med. 2018, 4, 109–116. [Google Scholar] [CrossRef]
- Lucena-Cacace, A.; Otero-Albiol, D.; Jimenez-Garcia, M.P.; Munoz-Galvan, S.; Carnero, A. NAMPT Is a Potent Oncogene in Colon Cancer Progression that Modulates Cancer Stem Cell Properties and Resistance to Therapy through Sirt1 and PARP. Clin. Cancer Res. 2018, 24, 1202–1215. [Google Scholar] [CrossRef] [Green Version]
- Lucena-Cacace, A.; Otero-Albiol, D.; Jimenez-Garcia, M.P.; Peinado-Serrano, J.; Carnero, A. NAMPT overexpression induces cancer stemness and defines a novel tumor signature for glioma prognosis. Oncotarget 2017, 8, 99514–99530. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-García, M.P.; Verdugo-Sivianes, E.M.; Lucena-Cacace, A.J.C.T.M. Nicotinamide adenine dinucleotide+ metabolism biomarkers in malignant gliomas. Cancers 2016, 2, 189–196. [Google Scholar]
- Lucena-Cacace, A.; Umeda, M.; Navas, L.E.; Carnero, A. NAMPT as a Dedifferentiation-Inducer Gene: NAD(+) as Core Axis for Glioma Cancer Stem-Like Cells Maintenance. Front. Oncol. 2019, 9, 292. [Google Scholar] [CrossRef] [PubMed]
- Grozio, A.; Mills, K.F.; Yoshino, J.; Bruzzone, S.; Sociali, G.; Tokizane, K.; Lei, H.C.; Cunningham, R.; Sasaki, Y.; Migaud, M.E.; et al. Slc12a8 is a nicotinamide mononucleotide transporter. Nat. Metab. 2019, 1, 47–57. [Google Scholar] [CrossRef]
- Wu, L.E.; Sinclair, D.A. The elusive NMN transporter is found. Nat. Metab. 2019, 1, 8–9. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Hung, A.C.; Lo, S.; Yuan, S.F. Adipocytokines visfatin and resistin in breast cancer: Clinical relevance, biological mechanisms, and therapeutic potential. Cancer Lett. 2021, 498, 229–239. [Google Scholar] [CrossRef]
- Grolla, A.A.; Travelli, C.; Genazzani, A.A.; Sethi, J.K. Extracellular nicotinamide phosphoribosyltransferase, a new cancer metabokine. Br. J. Pharmacol. 2016, 173, 2182–2194. [Google Scholar] [CrossRef]
- Sun, Z.; Lei, H.; Zhang, Z. Pre-B cell colony enhancing factor (PBEF), a cytokine with multiple physiological functions. Cytokine Growth Factor Rev. 2013, 24, 433–442. [Google Scholar] [CrossRef]
- Ognjanovic, S.; Bao, S.; Yamamoto, S.Y.; Garibay-Tupas, J.; Samal, B.; Bryant-Greenwood, G.D. Genomic organization of the gene coding for human pre-B-cell colony enhancing factor and expression in human fetal membranes. J. Mol. Endocrinol. 2001, 26, 107–117. [Google Scholar] [CrossRef]
- Samal, B.; Sun, Y.; Stearns, G.; Xie, C.; Suggs, S.; McNiece, I. Cloning and characterization of the cDNA encoding a novel human pre-B-cell colony-enhancing factor. Mol. Cell Biol. 1994, 14, 1431–1437. [Google Scholar] [CrossRef]
- Fukuhara, A.; Matsuda, M.; Nishizawa, M.; Segawa, K.; Tanaka, M.; Kishimoto, K.; Matsuki, Y.; Murakami, M.; Ichisaka, T.; Murakami, H.; et al. Visfatin: A protein secreted by visceral fat that mimics the effects of insulin. Science 2005, 307, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Dakroub, A.; Nasser, S.A.; Younis, N.; Bhagani, H.; Al-Dhaheri, Y.; Pintus, G.; Eid, A.A.; El-Yazbi, A.F.; Eid, A.H. Visfatin: A Possible Role in Cardiovasculo-Metabolic Disorders. Cells 2020, 9, 2444. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.Q.; Heruth, D.P.; Ye, S.Q. Nicotinamide Phosphoribosyltransferase in Human Diseases. J. Bioanal Biomed. 2011, 3, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Xia, T.; Zhou, L.; Chen, X.; Gan, L.; Yao, W.; Peng, Y.; Yang, Z. Gene organization, alternate splicing and expression pattern of porcine visfatin gene. Domest. Anim. Endocrinol. 2007, 32, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.; Nakamura, S.; Nakazawa, T.; Minoura, K.; Yoshida, T.; Nishi, Y.; Kobayashi, Y.; Ohkubo, T. Structure and reaction mechanism of human nicotinamide phosphoribosyltransferase. J. Biochem. 2010, 147, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Dulyaninova, N.G.; Podlepa, E.M.; Toulokhonova, L.V.; Bykhovsky, V.Y. Salvage pathway for NAD biosynthesis in Brevibacterium ammoniagenes: Regulatory properties of triphosphate-dependent nicotinate phosphoribosyltransferase. Biochim. Biophys. Acta 2000, 1478, 211–220. [Google Scholar] [CrossRef]
- Kim, M.K.; Lee, J.H.; Kim, H.; Park, S.J.; Kim, S.H.; Kang, G.B.; Lee, Y.S.; Kim, J.B.; Kim, K.K.; Suh, S.W.; et al. Crystal structure of visfatin/pre-B cell colony-enhancing factor 1/nicotinamide phosphoribosyltransferase, free and in complex with the anti-cancer agent FK-866. J. Mol. Biol 2006, 362, 66–77. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, X.; Bheda, P.; Revollo, J.R.; Imai, S.; Wolberger, C. Structure of Nampt/PBEF/visfatin, a mammalian NAD+ biosynthetic enzyme. Nat. Struct. Mol. Biol. 2006, 13, 661–662. [Google Scholar] [CrossRef]
- Khan, J.A.; Tao, X.; Tong, L. Molecular basis for the inhibition of human NMPRTase, a novel target for anticancer agents. Nat. Struct. Mol. Biol. 2006, 13, 582–588. [Google Scholar] [CrossRef]
- Burgos, E.S.; Ho, M.C.; Almo, S.C.; Schramm, V.L. A phosphoenzyme mimic, overlapping catalytic sites and reaction coordinate motion for human NAMPT. Proc. Natl. Acad. Sci. USA 2009, 106, 13748–13753. [Google Scholar] [CrossRef]
- Burgos, E.S.; Schramm, V.L. Weak coupling of ATP hydrolysis to the chemical equilibrium of human nicotinamide phosphoribosyltransferase. Biochemistry 2008, 47, 11086–11096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, T.Q.; Che, X.M. Nampt/PBEF/visfatin and cancer. Cancer Biol. Ther. 2010, 10, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Olesen, U.H.; Hastrup, N.; Sehested, M. Expression patterns of nicotinamide phosphoribosyltransferase and nicotinic acid phosphoribosyltransferase in human malignant lymphomas. APMIS 2011, 119, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Hasan, M.K.; Alvarado, E.; Yuan, H.; Wu, H.; Chen, W.Y. NAMPT overexpression in prostate cancer and its contribution to tumor cell survival and stress response. Oncogene 2011, 30, 907–921. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, N.; Liu, Y.; Su, P.; Liang, Y.; Li, Y.; Wang, X.; Chen, T.; Song, X.; Sang, Y.; et al. Epigenetic Regulation of NAMPT by NAMPT-AS Drives Metastatic Progression in Triple-Negative Breast Cancer. Cancer Res. 2019, 79, 3347–3359. [Google Scholar] [CrossRef] [PubMed]
- Sawicka-Gutaj, N.; Waligorska-Stachura, J.; Andrusiewicz, M.; Biczysko, M.; Sowinski, J.; Skrobisz, J.; Ruchala, M. Nicotinamide phosphorybosiltransferase overexpression in thyroid malignancies and its correlation with tumor stage and with survivin/survivin DEx3 expression. Tumour Biol. 2015, 36, 7859–7863. [Google Scholar] [CrossRef]
- Bi, T.Q.; Che, X.M.; Liao, X.H.; Zhang, D.J.; Long, H.L.; Li, H.J.; Zhao, W. Overexpression of Nampt in gastric cancer and chemopotentiating effects of the Nampt inhibitor FK866 in combination with fluorouracil. Oncol. Rep. 2011, 26, 1251–1257. [Google Scholar] [CrossRef]
- Kim, J.Y.; Bae, Y.H.; Bae, M.K.; Kim, S.R.; Park, H.J.; Wee, H.J.; Bae, S.K. Visfatin through STAT3 activation enhances IL-6 expression that promotes endothelial angiogenesis. Biochim. Biophys. Acta 2009, 1793, 1759–1767. [Google Scholar] [CrossRef]
- Kim, S.R.; Bae, Y.H.; Bae, S.K.; Choi, K.S.; Yoon, K.H.; Koo, T.H.; Jang, H.O.; Yun, I.; Kim, K.W.; Kwon, Y.G.; et al. Visfatin enhances ICAM-1 and VCAM-1 expression through ROS-dependent NF-kappaB activation in endothelial cells. Biochim. Biophys. Acta 2008, 1783, 886–895. [Google Scholar] [CrossRef]
- Cymbaluk-Ploska, A.; Chudecka-Glaz, A.; Pius-Sadowska, E.; Sompolska-Rzechula, A.; Machalinski, B.; Menkiszak, J. Circulating Serum Level of Visfatin in Patients with Endometrial Cancer. Biomed. Res. Int. 2018, 2018, 8576179. [Google Scholar] [CrossRef]
- Pazgan-Simon, M.; Kukla, M.; Zuwala-Jagiello, J.; Derra, A.; Bator, M.; Menzyk, T.; Lekstan, A.; Grzebyk, E.; Simon, K. Serum visfatin and vaspin levels in hepatocellular carcinoma (HCC). PLoS ONE 2020, 15, e0227459. [Google Scholar] [CrossRef] [Green Version]
- Lu, G.W.; Wang, Q.J.; Xia, M.M.; Qian, J. Elevated plasma visfatin levels correlate with poor prognosis of gastric cancer patients. Peptides 2014, 58, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.C. The role of visfatin in cancer proliferation, angiogenesis, metastasis, drug resistance and clinical prognosis. Cancer Manag. Res. 2019, 11, 3481–3491. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.B.; Chen, C.X.; Huang, J.; Tian, Y.X.; Xie, X.; Yang, P.; Wu, M.; Tang, C.; Zhang, W.P. Nicotinamide phosphoribosyltransferase secreted from microglia via exosome during ischemic injury. J. Neurochem. 2019, 150, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Audrito, V.; Messana, V.G.; Brandimarte, L.; Deaglio, S. The Extracellular NADome Modulates Immune Responses. Front. Immunol. 2021, 12, 704779. [Google Scholar] [CrossRef]
- Vona-Davis, L.; Rose, D.P. Adipokines as endocrine, paracrine, and autocrine factors in breast cancer risk and progression. Endocr. Relat. Cancer 2007, 14, 189–206. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Chen, H.D.; Lo, S.; Chen, Y.K.; Huang, Y.C.; Hu, S.C.; Hsieh, Y.C.; Hung, A.C.; Hou, M.F.; Yuan, S.F. Visfatin Enhances Breast Cancer Progression through CXCL1 Induction in Tumor-Associated Macrophages. Cancers 2020, 12, 3526. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Y.; Dorweiler, B.; Cui, D.; Wang, T.; Woo, C.W.; Brunkan, C.S.; Wolberger, C.; Imai, S.; Tabas, I. Extracellular Nampt promotes macrophage survival via a nonenzymatic interleukin-6/STAT3 signaling mechanism. J. Biol. Chem. 2008, 283, 34833–34843. [Google Scholar] [CrossRef]
- Aman, Y.; Qiu, Y.; Tao, J.; Fang, E.F. Therapeutic potential of boosting NAD+ in aging and age-related diseases. Transl. Med. Aging 2018, 2, 30–37. [Google Scholar] [CrossRef]
- Hwang, E.S.; Song, S.B. Nicotinamide is an inhibitor of SIRT1 in vitro, but can be a stimulator in cells. Cell Mol. Life Sci. 2017, 74, 3347–3362. [Google Scholar] [CrossRef]
- Menssen, A.; Hydbring, P.; Kapelle, K.; Vervoorts, J.; Diebold, J.; Luscher, B.; Larsson, L.G.; Hermeking, H. The c-MYC oncoprotein, the NAMPT enzyme, the SIRT1-inhibitor DBC1, and the SIRT1 deacetylase form a positive feedback loop. Proc. Natl. Acad. Sci. USA 2012, 109, E187–E196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behrouzfar, K.; Alaee, M.; Nourbakhsh, M.; Gholinejad, Z.; Golestani, A. Extracellular NAMPT/visfatin causes p53 deacetylation via NAD production and SIRT1 activation in breast cancer cells. Cell Biochem. Funct. 2017, 35, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, B.E.; Sharif, T.; Martell, E.; Dai, C.; Kim, Y.; Lee, P.W.; Gujar, S.A. NAD(+) salvage pathway in cancer metabolism and therapy. Pharmacol. Res. 2016, 114, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liang, C.; Li, F.; Guan, D.; Wu, X.; Fu, X.; Lu, A.; Zhang, G. PARP1 in Carcinomas and PARP1 Inhibitors as Antineoplastic Drugs. Int. J. Mol. Sci. 2017, 18, 2111. [Google Scholar] [CrossRef]
- Dorsam, B.; Seiwert, N.; Foersch, S.; Stroh, S.; Nagel, G.; Begaliew, D.; Diehl, E.; Kraus, A.; McKeague, M.; Minneker, V.; et al. PARP-1 protects against colorectal tumor induction, but promotes inflammation-driven colorectal tumor progression. Proc. Natl. Acad. Sci. USA 2018, 115, E4061–E4070. [Google Scholar] [CrossRef]
- Schiewer, M.J.; Goodwin, J.F.; Han, S.; Brenner, J.C.; Augello, M.A.; Dean, J.L.; Liu, F.; Planck, J.L.; Ravindranathan, P.; Chinnaiyan, A.M.; et al. Dual roles of PARP-1 promote cancer growth and progression. Cancer Discov. 2012, 2, 1134–1149. [Google Scholar] [CrossRef]
- Schiewer, M.J.; Mandigo, A.C.; Gordon, N.; Huang, F.; Gaur, S.; de Leeuw, R.; Zhao, S.G.; Evans, J.; Han, S.; Parsons, T.; et al. PARP-1 regulates DNA repair factor availability. EMBO Mol. Med. 2018, 10, e8816. [Google Scholar] [CrossRef]
- Chini, E.N. CD38 as a regulator of cellular NAD: A novel potential pharmacological target for metabolic conditions. Curr. Pharm. Des. 2009, 15, 57–63. [Google Scholar] [CrossRef]
- Camacho-Pereira, J.; Tarrago, M.G.; Chini, C.C.S.; Nin, V.; Escande, C.; Warner, G.M.; Puranik, A.S.; Schoon, R.A.; Reid, J.M.; Galina, A.; et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016, 23, 1127–1139. [Google Scholar] [CrossRef]
- Zhao, Y.J.; Lam, C.M.; Lee, H.C. The membrane-bound enzyme CD38 exists in two opposing orientations. Sci. Signal. 2012, 5, ra67. [Google Scholar] [CrossRef]
- Malavasi, F.; Deaglio, S.; Damle, R.; Cutrona, G.; Ferrarini, M.; Chiorazzi, N. CD38 and chronic lymphocytic leukemia: A decade later. Blood 2011, 118, 3470–3478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chini, C.C.; Guerrico, A.M.; Nin, V.; Camacho-Pereira, J.; Escande, C.; Barbosa, M.T.; Chini, E.N. Targeting of NAD metabolism in pancreatic cancer cells: Potential novel therapy for pancreatic tumors. Clin. Cancer Res. 2014, 20, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Grozio, A.; Sociali, G.; Sturla, L.; Caffa, I.; Soncini, D.; Salis, A.; Raffaelli, N.; De Flora, A.; Nencioni, A.; Bruzzone, S. CD73 protein as a source of extracellular precursors for sustained NAD+ biosynthesis in FK866-treated tumor cells. J. Biol. Chem. 2013, 288, 25938–25949. [Google Scholar] [CrossRef] [PubMed]
- Sadej, R.; Skladanowski, A.C. Dual, enzymatic and non-enzymatic, function of ecto-5′-nucleotidase (eN, CD73) in migration and invasion of A375 melanoma cells. Acta Biochim. Pol. 2012, 59, 647–652. [Google Scholar]
- Cerna, D.; Li, H.; Flaherty, S.; Takebe, N.; Coleman, C.N.; Yoo, S.S. Inhibition of nicotinamide phosphoribosyltransferase (NAMPT) activity by small molecule GMX1778 regulates reactive oxygen species (ROS)-mediated cytotoxicity in a p53- and nicotinic acid phosphoribosyltransferase1 (NAPRT1)-dependent manner. J. Biol. Chem. 2012, 287, 22408–22417. [Google Scholar] [CrossRef]
- Audrito, V.; Messana, V.G.; Deaglio, S. NAMPT and NAPRT: Two Metabolic Enzymes with Key Roles in Inflammation. Front. Oncol. 2020, 10, 358. [Google Scholar] [CrossRef]
- Watson, M.; Roulston, A.; Belec, L.; Billot, X.; Marcellus, R.; Bedard, D.; Bernier, C.; Branchaud, S.; Chan, H.; Dairi, K.; et al. The small molecule GMX1778 is a potent inhibitor of NAD+ biosynthesis: Strategy for enhanced therapy in nicotinic acid phosphoribosyltransferase 1-deficient tumors. Mol. Cell Biol. 2009, 29, 5872–5888. [Google Scholar] [CrossRef]
- Peterse, E.F.P.; van den Akker, B.; Niessen, B.; Oosting, J.; Suijker, J.; de Jong, Y.; Danen, E.H.J.; Cleton-Jansen, A.M.; Bovee, J. NAD Synthesis Pathway Interference Is a Viable Therapeutic Strategy for Chondrosarcoma. Mol. Cancer Res. 2017, 15, 1714–1721. [Google Scholar] [CrossRef]
- Lee, J.; Kim, H.; Lee, J.E.; Shin, S.J.; Oh, S.; Kwon, G.; Kim, H.; Choi, Y.Y.; White, M.A.; Paik, S.; et al. Selective Cytotoxicity of the NAMPT Inhibitor FK866 toward Gastric Cancer Cells with Markers of the Epithelial-Mesenchymal Transition, Due to Loss of NAPRT. Gastroenterology 2018, 155, 799–814.e13. [Google Scholar] [CrossRef]
- Li, X.Q.; Lei, J.; Mao, L.H.; Wang, Q.L.; Xu, F.; Ran, T.; Zhou, Z.H.; He, S. NAMPT and NAPRT, Key Enzymes in NAD Salvage Synthesis Pathway, Are of Negative Prognostic Value in Colorectal Cancer. Front. Oncol. 2019, 9, 736. [Google Scholar] [CrossRef]
- Duarte-Pereira, S.; Pereira-Castro, I.; Silva, S.S.; Correia, M.G.; Neto, C.; da Costa, L.T.; Amorim, A.; Silva, R.M. Extensive regulation of nicotinate phosphoribosyltransferase (NAPRT) expression in human tissues and tumors. Oncotarget 2016, 7, 1973–1983. [Google Scholar] [CrossRef] [PubMed]
- Chowdhry, S.; Zanca, C.; Rajkumar, U.; Koga, T.; Diao, Y.; Raviram, R.; Liu, F.; Turner, K.; Yang, H.; Brunk, E.; et al. NAD metabolic dependency in cancer is shaped by gene amplification and enhancer remodelling. Nature 2019, 569, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Tateishi, K.; Wakimoto, H.; Iafrate, A.J.; Tanaka, S.; Loebel, F.; Lelic, N.; Wiederschain, D.; Bedel, O.; Deng, G.; Zhang, B.; et al. Extreme Vulnerability of IDH1 Mutant Cancers to NAD+ Depletion. Cancer Cell 2015, 28, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Pramono, A.A.; Rather, G.M.; Herman, H.; Lestari, K.; Bertino, J.R. NAD- and NADPH-Contributing Enzymes as Therapeutic Targets in Cancer: An Overview. Biomolecules 2020, 10, 358. [Google Scholar] [CrossRef]
- Fons, N.R.; Sundaram, R.K.; Breuer, G.A.; Peng, S.; McLean, R.L.; Kalathil, A.N.; Schmidt, M.S.; Carvalho, D.M.; Mackay, A.; Jones, C.; et al. PPM1D mutations silence NAPRT gene expression and confer NAMPT inhibitor sensitivity in glioma. Nat. Commun. 2019, 10, 3790. [Google Scholar] [CrossRef]
- Piacente, F.; Caffa, I.; Ravera, S.; Sociali, G.; Passalacqua, M.; Vellone, V.G.; Becherini, P.; Reverberi, D.; Monacelli, F.; Ballestrero, A.; et al. Nicotinic Acid Phosphoribosyltransferase Regulates Cancer Cell Metabolism, Susceptibility to NAMPT Inhibitors, and DNA Repair. Cancer Res. 2017, 77, 3857–3869. [Google Scholar] [CrossRef]
- Hara, N.; Yamada, K.; Shibata, T.; Osago, H.; Hashimoto, T.; Tsuchiya, M. Elevation of cellular NAD levels by nicotinic acid and involvement of nicotinic acid phosphoribosyltransferase in human cells. J. Biol. Chem. 2007, 282, 24574–24582. [Google Scholar] [CrossRef]
- Ghanem, M.S.; Monacelli, F.; Nencioni, A. Advances in NAD-Lowering Agents for Cancer Treatment. Nutrients 2021, 13, 1665. [Google Scholar] [CrossRef]
- Gaut, Z.N.; Solomon, H.M. Uptake and metabolism of nicotinic acid by human blood platelets. Effects of structure analogs and metabolic inhibitors. Biochim. Biophys. Acta 1970, 201, 316–322. [Google Scholar] [CrossRef]
- Carnero, A.; Lleonart, M. The hypoxic microenvironment: A determinant of cancer stem cell evolution. Bioessays 2016, 38 (Suppl. S1), S65–S74. [Google Scholar] [CrossRef]
- Peiris-Pages, M.; Martinez-Outschoorn, U.E.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer stem cell metabolism. Breast Cancer Res. 2016, 18, 55. [Google Scholar] [CrossRef] [PubMed]
- Bao, B.; Ahmad, A.; Azmi, A.S.; Ali, S.; Sarkar, F.H. Overview of cancer stem cells (CSCs) and mechanisms of their regulation: Implications for cancer therapy. Curr. Protoc. Pharmacol. 2013, 14, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espinosa-Sanchez, A.; Suarez-Martinez, E.; Sanchez-Diaz, L.; Carnero, A. Therapeutic Targeting of Signaling Pathways Related to Cancer Stemness. Front. Oncol. 2020, 10, 1533. [Google Scholar] [CrossRef]
- Korotchkina, L.; Kazyulkin, D.; Komarov, P.G.; Polinsky, A.; Andrianova, E.L.; Joshi, S.; Gupta, M.; Vujcic, S.; Kononov, E.; Toshkov, I.; et al. OT-82, a novel anticancer drug candidate that targets the strong dependence of hematological malignancies on NAD biosynthesis. Leukemia 2020, 34, 1828–1839. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Fessler, E.; Dijkgraaf, F.E.; De Sousa, E.M.F.; Medema, J.P. Cancer stem cell dynamics in tumor progression and metastasis: Is the microenvironment to blame? Cancer Lett. 2013, 341, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Snyder, V.; Reed-Newman, T.C.; Arnold, L.; Thomas, S.M.; Anant, S. Cancer Stem Cell Metabolism and Potential Therapeutic Targets. Front. Oncol. 2018, 8, 203. [Google Scholar] [CrossRef]
- Teslaa, T.; Teitell, M.A. Pluripotent stem cell energy metabolism: An update. EMBO J. 2015, 34, 138–153. [Google Scholar] [CrossRef]
- Sancho, P.; Barneda, D.; Heeschen, C. Hallmarks of cancer stem cell metabolism. Br. J. Cancer 2016, 114, 1305–1312. [Google Scholar] [CrossRef]
- Deshmukh, A.; Deshpande, K.; Arfuso, F.; Newsholme, P.; Dharmarajan, A. Cancer stem cell metabolism: A potential target for cancer therapy. Mol. Cancer 2016, 15, 69. [Google Scholar] [CrossRef]
- Karsten, U.; Goletz, S. What makes cancer stem cell markers different? Springerplus 2013, 2, 301. [Google Scholar] [CrossRef] [PubMed]
- Son, M.J.; Son, M.Y.; Seol, B.; Kim, M.J.; Yoo, C.H.; Han, M.K.; Cho, Y.S. Nicotinamide overcomes pluripotency deficits and reprogramming barriers. Stem Cells 2013, 31, 1121–1135. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Yan, Z.; Miao, J.; Cai, R.; Zhang, M.; Wang, Y.; Wang, L.; Dang, W.; Wang, D.; Xiang, D.; et al. Autofluorescence of NADH is a new biomarker for sorting and characterizing cancer stem cells in human glioma. Stem Cell Res. Ther. 2019, 10, 330. [Google Scholar] [CrossRef] [PubMed]
- Bonuccelli, G.; De Francesco, E.M.; de Boer, R.; Tanowitz, H.B.; Lisanti, M.P. NADH autofluorescence, a new metabolic biomarker for cancer stem cells: Identification of Vitamin C and CAPE as natural products targeting “stemness”. Oncotarget 2017, 8, 20667–20678. [Google Scholar] [CrossRef]
- Cui, Q.; Wang, J.Q.; Assaraf, Y.G.; Ren, L.; Gupta, P.; Wei, L.; Ashby, C.R., Jr.; Yang, D.H.; Chen, Z.S. Modulating ROS to overcome multidrug resistance in cancer. Drug Resist. Updat. 2018, 41, 1–25. [Google Scholar] [CrossRef]
- Panieri, E.; Santoro, M.M. ROS homeostasis and metabolism: A dangerous liason in cancer cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef]
- Von Heideman, A.; Berglund, A.; Larsson, R.; Nygren, P. Safety and efficacy of NAD depleting cancer drugs: Results of a phase I clinical trial of CHS 828 and overview of published data. Cancer Chemother. Pharmacol. 2010, 65, 1165–1172. [Google Scholar] [CrossRef]
- Skelton, L.A.; Ormerod, M.G.; Titley, J.C.; Jackman, A.L. Cell cycle effects of CB30865, a lipophilic quinazoline-based analogue of the antifolate thymidylate synthase inhibitor ICI 198583 with an undefined mechanism of action. Cytometry 1998, 33, 56–66. [Google Scholar] [CrossRef]
- Fleischer, T.C.; Murphy, B.R.; Flick, J.S.; Terry-Lorenzo, R.T.; Gao, Z.H.; Davis, T.; McKinnon, R.; Ostanin, K.; Willardsen, J.A.; Boniface, J.J. Chemical proteomics identifies Nampt as the target of CB30865, an orphan cytotoxic compound. Chem. Biol. 2010, 17, 659–664. [Google Scholar] [CrossRef]
- Bavetsias, V.; Skelton, L.A.; Yafai, F.; Mitchell, F.; Wilson, S.C.; Allan, B.; Jackman, A.L. The design and synthesis of water-soluble analogues of CB30865, a quinazolin-4-one-based antitumor agent. J. Med. Chem. 2002, 45, 3692–3702. [Google Scholar] [CrossRef]
- O’Brien, T.; Oeh, J.; Xiao, Y.; Liang, X.; Vanderbilt, A.; Qin, A.; Yang, L.; Lee, L.B.; Ly, J.; Cosino, E.; et al. Supplementation of nicotinic acid with NAMPT inhibitors results in loss of in vivo efficacy in NAPRT1-deficient tumor models. Neoplasia 2013, 15, 1314–1329. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Elkins, K.; Durieux, J.K.; Lee, L.; Oeh, J.; Yang, L.X.; Liang, X.; DelNagro, C.; Tremayne, J.; Kwong, M.; et al. Dependence of tumor cell lines and patient-derived tumors on the NAD salvage pathway renders them sensitive to NAMPT inhibition with GNE-618. Neoplasia 2013, 15, 1151–1160. [Google Scholar] [CrossRef] [PubMed]
- Zabka, T.S.; Singh, J.; Dhawan, P.; Liederer, B.M.; Oeh, J.; Kauss, M.A.; Xiao, Y.; Zak, M.; Lin, T.; McCray, B.; et al. Retinal toxicity, in vivo and in vitro, associated with inhibition of nicotinamide phosphoribosyltransferase. Toxicol. Sci. 2015, 144, 163–172. [Google Scholar] [CrossRef]
- Baichwal, V.R.; Willardsen, J.A.; Lockman, J.W.; Murphy, B.R.; Gordillo, R.; Fleischer, T.C.; Bradford, C.; Papac, D.I.; Mather, G.G.; Carlson, R.O. Activity of the cancer metabolism inhibitor MPC-9528 in xenograft models: Comparison of different dosing schedules. J. Clin. Oncol. 2011, 29, 10529. [Google Scholar] [CrossRef]
- Matheny, C.J.; Wei, M.C.; Bassik, M.C.; Donnelly, A.J.; Kampmann, M.; Iwasaki, M.; Piloto, O.; Solow-Cordero, D.E.; Bouley, D.M.; Rau, R.; et al. Next-generation NAMPT inhibitors identified by sequential high-throughput phenotypic chemical and functional genomic screens. Chem. Biol. 2013, 20, 1352–1363. [Google Scholar] [CrossRef]
- Olesen, U.H.; Petersen, J.G.; Garten, A.; Kiess, W.; Yoshino, J.; Imai, S.; Christensen, M.K.; Fristrup, P.; Thougaard, A.V.; Bjorkling, F.; et al. Target enzyme mutations are the molecular basis for resistance towards pharmacological inhibition of nicotinamide phosphoribosyltransferase. BMC Cancer 2010, 10, 677. [Google Scholar] [CrossRef]
- Zhao, G.; Green, C.F.; Hui, Y.H.; Prieto, L.; Shepard, R.; Dong, S.; Wang, T.; Tan, B.; Gong, X.; Kays, L.; et al. Discovery of a Highly Selective NAMPT Inhibitor That Demonstrates Robust Efficacy and Improved Retinal Toxicity with Nicotinic Acid Coadministration. Mol. Cancer Ther. 2017, 16, 2677–2688. [Google Scholar] [CrossRef]
- Estoppey, D.; Hewett, J.W.; Guy, C.T.; Harrington, E.; Thomas, J.R.; Schirle, M.; Cuttat, R.; Waldt, A.; Gerrits, B.; Yang, Z.; et al. Identification of a novel NAMPT inhibitor by CRISPR/Cas9 chemogenomic profiling in mammalian cells. Sci. Rep. 2017, 7, 42728. [Google Scholar] [CrossRef]
- Esposito, E.; Impellizzeri, D.; Mazzon, E.; Fakhfouri, G.; Rahimian, R.; Travelli, C.; Tron, G.C.; Genazzani, A.A.; Cuzzocrea, S. The NAMPT inhibitor FK866 reverts the damage in spinal cord injury. J. Neuroinflammation 2012, 9, 66. [Google Scholar] [CrossRef]
- Bai, J.; Liao, C.; Liu, Y.; Qin, X.; Chen, J.; Qiu, Y.; Qin, D.; Li, Z.; Tu, Z.C.; Jiang, S. Structure-Based Design of Potent Nicotinamide Phosphoribosyltransferase Inhibitors with Promising In Vitro and In Vivo Antitumor Activities. J. Med. Chem. 2016, 59, 5766–5779. [Google Scholar] [CrossRef]
- Travelli, C.; Aprile, S.; Mattoteia, D.; Colombo, G.; Clemente, N.; Scanziani, E.; Terrazzino, S.; Alisi, M.A.; Polenzani, L.; Grosa, G.; et al. Identification of potent triazolylpyridine nicotinamide phosphoribosyltransferase (NAMPT) inhibitors bearing a 1,2,3-triazole tail group. Eur. J. Med. Chem. 2019, 181, 111576. [Google Scholar] [CrossRef]
- Zhang, K.; Ni, Y.; Chen, J.; Tu, Z.; Wu, X.; Chen, D.; Yao, H.; Jiang, S. Discovery of trans-3-(pyridin-3-yl)acrylamide-derived sulfamides as potent nicotinamide phosphoribosyltransferase (NAMPT) inhibitors for the potential treatment of cancer. Bioorg. Med. Chem. Lett. 2019, 29, 1502–1506. [Google Scholar] [CrossRef] [PubMed]
- Wilsbacher, J.L.; Cheng, M.; Cheng, D.; Trammell, S.A.J.; Shi, Y.; Guo, J.; Koeniger, S.L.; Kovar, P.J.; He, Y.; Selvaraju, S.; et al. Discovery and Characterization of Novel Nonsubstrate and Substrate NAMPT Inhibitors. Mol. Cancer Ther. 2017, 16, 1236–1245. [Google Scholar] [CrossRef] [PubMed]
- Mudd, S.R.; Voorbach, M.J.; Cheng, D.; Cheng, M.; Guo, J.; Gao, W.; Buchanan, F.G.; Tse, C.; Wilsbacher, J. Utilization of (18)F-Fluorodeoxyglucose-Positron Emission Tomography to Understand the Mechanism of Nicotinamide Phosphoribosyltransferase Inhibitors In Vivo. J. Pharmacol. Exp. Ther. 2019, 371, 583–589. [Google Scholar] [CrossRef]
- Zhang, S.L.; Xu, T.Y.; Yang, Z.L.; Han, S.; Zhao, Q.; Miao, C.Y. Crystal structure-based comparison of two NAMPT inhibitors. Acta Pharmacol. Sin. 2018, 39, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.Y.; Zhang, S.L.; Dong, G.Q.; Liu, X.Z.; Wang, X.; Lv, X.Q.; Qian, Q.J.; Zhang, R.Y.; Sheng, C.Q.; Miao, C.Y. Discovery and characterization of novel small-molecule inhibitors targeting nicotinamide phosphoribosyltransferase. Sci. Rep. 2015, 5, 10043. [Google Scholar] [CrossRef] [PubMed]
- Kraus, D.; Reckenbeil, J.; Veit, N.; Kuerpig, S.; Meisenheimer, M.; Beier, I.; Stark, H.; Winter, J.; Probstmeier, R. Targeting glucose transport and the NAD pathway in tumor cells with STF-31: A re-evaluation. Cell Oncol. 2018, 41, 485–494. [Google Scholar] [CrossRef]
- Dong, G.; Chen, W.; Wang, X.; Yang, X.; Xu, T.; Wang, P.; Zhang, W.; Rao, Y.; Miao, C.; Sheng, C. Small Molecule Inhibitors Simultaneously Targeting Cancer Metabolism and Epigenetics: Discovery of Novel Nicotinamide Phosphoribosyltransferase (NAMPT) and Histone Deacetylase (HDAC) Dual Inhibitors. J. Med. Chem. 2017, 60, 7965–7983. [Google Scholar] [CrossRef]
- Palacios, D.S.; Meredith, E.L.; Kawanami, T.; Adams, C.M.; Chen, X.; Darsigny, V.; Palermo, M.; Baird, D.; George, E.L.; Guy, C.; et al. Scaffold Morphing Identifies 3-Pyridyl Azetidine Ureas as Inhibitors of Nicotinamide Phosphoribosyltransferase (NAMPT). ACS Med. Chem. Lett. 2019, 10, 1524–1529. [Google Scholar] [CrossRef]
- Karpov, A.S.; Abrams, T.; Clark, S.; Raikar, A.; D’Alessio, J.A.; Dillon, M.P.; Gesner, T.G.; Jones, D.; Lacaud, M.; Mallet, W.; et al. Nicotinamide Phosphoribosyltransferase Inhibitor as a Novel Payload for Antibody-Drug Conjugates. ACS Med. Chem. Lett. 2018, 9, 838–842. [Google Scholar] [CrossRef]
- Neumann, C.S.; Olivas, K.C.; Anderson, M.E.; Cochran, J.H.; Jin, S.; Li, F.; Loftus, L.V.; Meyer, D.W.; Neale, J.; Nix, J.C.; et al. Targeted Delivery of Cytotoxic NAMPT Inhibitors Using Antibody-Drug Conjugates. Mol. Cancer Ther. 2018, 17, 2633–2642. [Google Scholar] [CrossRef] [PubMed]
- Lameijer, L.N.; Ernst, D.; Hopkins, S.L.; Meijer, M.S.; Askes, S.H.C.; Le Dévédec, S.E.; Bonnet, S. A Red-Light-Activated Ruthenium-Caged NAMPT Inhibitor Remains Phototoxic in Hypoxic Cancer Cells. Angew. Chem. 2017, 56, 11549–11553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heske, C.M.; Davis, M.I.; Baumgart, J.T.; Wilson, K.; Gormally, M.V.; Chen, L.; Zhang, X.; Ceribelli, M.; Duveau, D.Y.; Guha, R.; et al. Matrix Screen Identifies Synergistic Combination of PARP Inhibitors and Nicotinamide Phosphoribosyltransferase (NAMPT) Inhibitors in Ewing Sarcoma. Clin. Cancer Res. 2017, 23, 7301–7311. [Google Scholar] [CrossRef]
- Galli, U.; Colombo, G.; Travelli, C.; Tron, G.C.; Genazzani, A.A.; Grolla, A.A. Recent Advances in NAMPT Inhibitors: A Novel Immunotherapic Strategy. Front. Pharmacol. 2020, 11, 656. [Google Scholar] [CrossRef] [PubMed]
- Aprile, S.; Galli, U.; Tron, G.C.; Del Grosso, E.; Travelli, C.; Grosa, G. Data on metabolic stability, aqueous solubility and CYP inhibition of novel triazole-based nicotinamide phosphoribosyltransferase (NAMPT) inhibitors. Data Brief. 2020, 28, 105034. [Google Scholar] [CrossRef]
- Adams, D.J.; Ito, D.; Rees, M.G.; Seashore-Ludlow, B.; Puyang, X.; Ramos, A.H.; Cheah, J.H.; Clemons, P.A.; Warmuth, M.; Zhu, P.; et al. NAMPT is the cellular target of STF-31-like small-molecule probes. ACS Chem. Biol. 2014, 9, 2247–2254. [Google Scholar] [CrossRef]
- Abu Aboud, O.; Chen, C.H.; Senapedis, W.; Baloglu, E.; Argueta, C.; Weiss, R.H. Dual and Specific Inhibition of NAMPT and PAK4 By KPT-9274 Decreases Kidney Cancer Growth. Mol. Cancer Ther. 2016, 15, 2119–2129. [Google Scholar] [CrossRef]
- Aboukameel, A.; Muqbil, I.; Senapedis, W.; Baloglu, E.; Landesman, Y.; Shacham, S.; Kauffman, M.; Philip, P.A.; Mohammad, R.M.; Azmi, A.S. Novel p21-Activated Kinase 4 (PAK4) Allosteric Modulators Overcome Drug Resistance and Stemness in Pancreatic Ductal Adenocarcinoma. Mol. Cancer Ther. 2017, 16, 76–87. [Google Scholar] [CrossRef]
- Rane, C.; Senapedis, W.; Baloglu, E.; Landesman, Y.; Crochiere, M.; Das-Gupta, S.; Minden, A. A novel orally bioavailable compound KPT-9274 inhibits PAK4, and blocks triple negative breast cancer tumor growth. Sci. Rep. 2017, 7, 42555. [Google Scholar] [CrossRef]
- Radu, M.; Semenova, G.; Kosoff, R.; Chernoff, J. PAK signalling during the development and progression of cancer. Nat. Rev. Cancer 2014, 14, 13–25. [Google Scholar] [CrossRef]
- Somers, K.; Evans, K.; Cheung, L.; Karsa, M.; Pritchard, T.; Kosciolek, A.; Bongers, A.; El-Ayoubi, A.; Forgham, H.; Middlemiss, S.; et al. Effective targeting of NAMPT in patient-derived xenograft models of high-risk pediatric acute lymphoblastic leukemia. Leukemia 2020, 34, 1524–1539. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
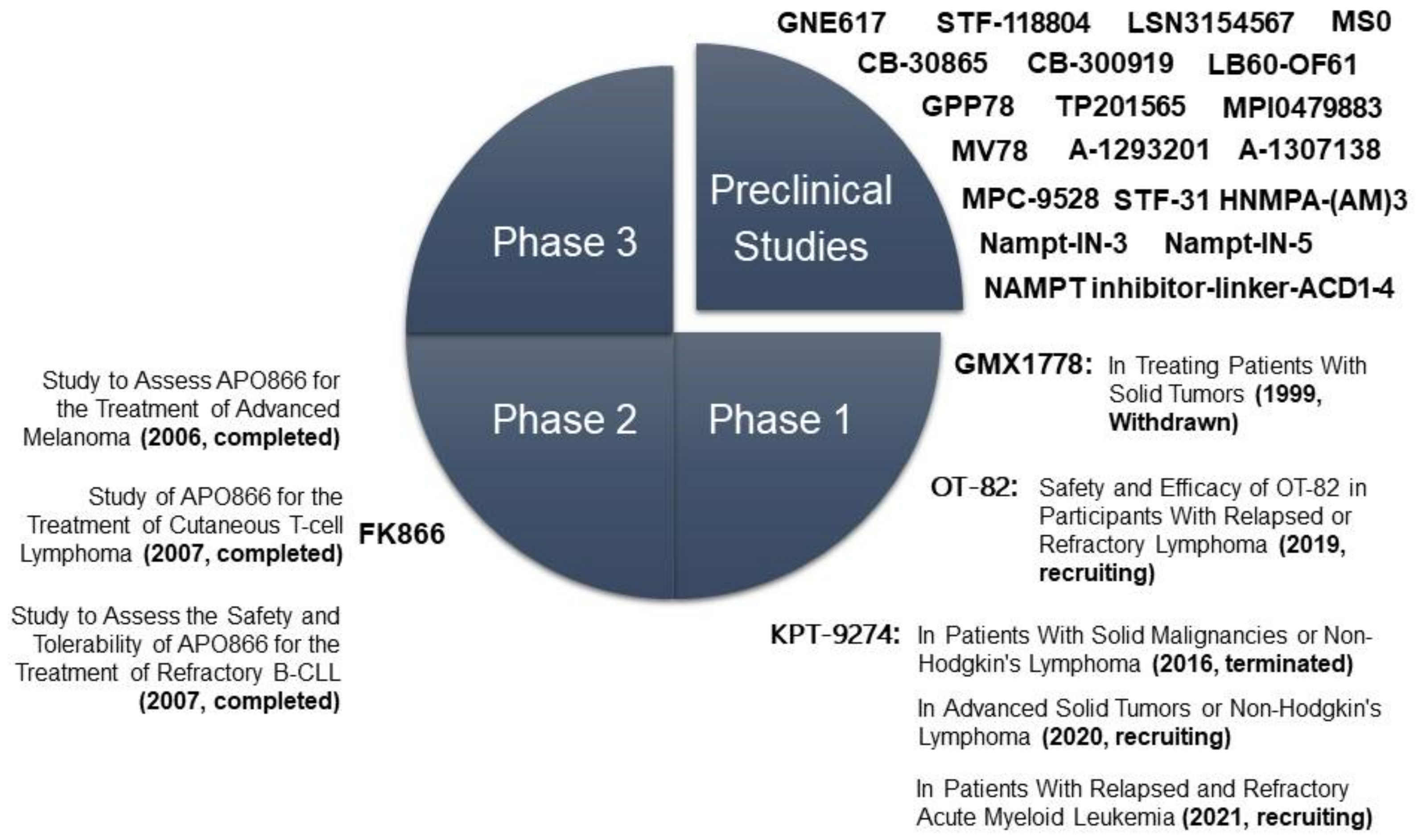
| Ph | Drug | Type | Condition | ClinicalTrials.gov Identifier | Treatment | Name of the Study |
|---|---|---|---|---|---|---|
| II | FK866 | Non- competitive | Melanoma | NCT00432107 | Alone | Study to Assess APO866 for the Treatment of Advanced Melanoma (2006, completed) |
| Cutaneous T-cell Lymphoma | NCT00431912 | Alone | Study of APO866 for the Treatment of Cutaneous T-cell Lymphoma (2007, completed) | |||
| B-cell Chronic Lymphocytic Leukemia | NCT00435084 | Alone | Study to Assess the Safety and Tolerability of APO866 for the Treatment of Refractory B-CLL (2007, completed) | |||
| I | GMX1778/CHS-828 | Oral competitive | Solid tumors | NCT00003979 | Alone | CHS 828 in Treating Patients with Solid Tumors (1999, Withdrawn) |
| I | OT-82 | Oral | Relapsed or refractory lymphoma | NCT03921879 | Dose escalation and expansion | Safety and Efficacy of OT-82 in Participants with Relapsed or Refractory Lymphoma (2019, recruiting) |
| I | KPT-9274/ATG-019 | Non- competitive oral dual inhibitor of PAK4 and NAMPT | Solid tumors, non-Hodgkin’s lymphoma | NCT02702492 | Alone or co-administered with Niacin or Nivolumab | PAK4 and NAMPT in Patients with Solid Malignancies or Non-Hodgkin’s Lymphoma (2016, terminated) |
| Solid tumors, non-Hodgkin’s lymphoma | NCT04281420 | Alone or co-administered with Niacin | Study of Evaluating Dual Inhibitor of PAK4 and NAMPT ATG-019 in Advanced Solid Tumors or Non-Hodgkin’s Lymphoma (2020, recruiting) | |||
| Acute Myeloid Leukemia | NCT04914845 | Alone | KPT-9274 in Patients with Relapsed and Refractory Acute Myeloid Leukemia (2021, recruiting) |
| Preclinical Drugs | Type | IC50 | In Vivo Treatments |
|---|---|---|---|
| GNE617 GNE618 GNE643 GNE875 | Oral competitive | 5 nM | 20–30 mg/kg orally in mice [131,133] |
| STF-118804 | Competitive | <10 nM | 50 mg/kg by subcutaneous injections in mice [135] |
| Nampt-IN-1/LSN3154567 | Competitive | 3.1 nM | 2 mg/kg in mice, 1–2.5 mg/kg or 5 mg/kg (with NA) in dogs [137] |
| CB30865 CB300919 | Competitive | 1–10 nM | 0.25 mg/kg by intraperitoneal injection in mice [130] |
| LB-60-OF61 | Competitive | 30 nM | In MYC-overexpressing cell lines [138] |
| GPP78/CAY10618 | Competitive | 3 nM | 10 mg/kg by intraperitoneal injection in mice [139] |
| Compound 30 | Competitive | 0.13–25.3 nM | 15 mg/kg by intravenous injection [140] |
| TP201565 | Competitive | In several human cell lines [136] | |
| MV78 | Competitive | 3.1 nM | [141] |
| trans-3-(pyridin-3-yl) acrylamide- sulfamides | Competitive | 0.2–5 nM | In several human cell lines [142] |
| MPC-9528 MPI0479883 | Competitive | 0.06 nM | 75 mg/kg in mice [134] |
| A-1293201 A-1307138 | Oral Competitive | 11–900 nM | 7.5, 15 or 30 mg/kg orally in mice [143,144] |
| MS0-MS31 | Competitive | 0.9–96 nM | In some human cell lines [145,146] |
| STF-31 | Dual inhibitor of GLUT1 and NAMPT | 1 µM | In several human cell lines [147] |
| Nampt-IN-3 | Dual inhibitor of NAMPT and HDAC | 31–55 nM | 25 mg/kg by intraperitoneal injection in mice [148] |
| Nampt-IN-5 | Dual inhibitor of NAMPT and CYP3A4 | 0.7–3.9 nM | 5–30 mg/kg oral gavage in mice [149] |
| NAMPT inhibitor-ADC1–4 | Drug-linker conjugates for ADC (anti-c-Kit) | 0.1 pM– 10 nM | 3–10 mg/kg by intraperitoneal injection in mice [150,151] |
| Water-soluble ruthenium complexes | Pro-drug photoactivated chemotherapy (PACT) | In skin and lung human tumour cell lines [152] | |
| Niraparib/Olaparib +NAMPT inhibitors | Combination with synergistic effect | 50 mg/kg orally in mice (PARPi) [153] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Navas, L.E.; Carnero, A. Nicotinamide Adenine Dinucleotide (NAD) Metabolism as a Relevant Target in Cancer. Cells 2022, 11, 2627. https://doi.org/10.3390/cells11172627
Navas LE, Carnero A. Nicotinamide Adenine Dinucleotide (NAD) Metabolism as a Relevant Target in Cancer. Cells. 2022; 11(17):2627. https://doi.org/10.3390/cells11172627
Chicago/Turabian StyleNavas, Lola E., and Amancio Carnero. 2022. "Nicotinamide Adenine Dinucleotide (NAD) Metabolism as a Relevant Target in Cancer" Cells 11, no. 17: 2627. https://doi.org/10.3390/cells11172627
APA StyleNavas, L. E., & Carnero, A. (2022). Nicotinamide Adenine Dinucleotide (NAD) Metabolism as a Relevant Target in Cancer. Cells, 11(17), 2627. https://doi.org/10.3390/cells11172627