
A Comprehensive Transcriptomic Analysis of Arsenic-Induced Bladder Carcinogenesis


 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Reagents
2.2. Cell Proliferation in Monolayer Culture
2.3. Anchorage-Independent (Colony Formation) Growth Assay
2.4. Spheroid Formation
2.5. Library Construction and RNA Sequencing
2.6. Computational Data Analysis
2.7. Reverse Transcription and Real-Time PCR Quantitation
2.8. Functional Enrichment Analysis
2.9. Statistical Analysis
3. Results
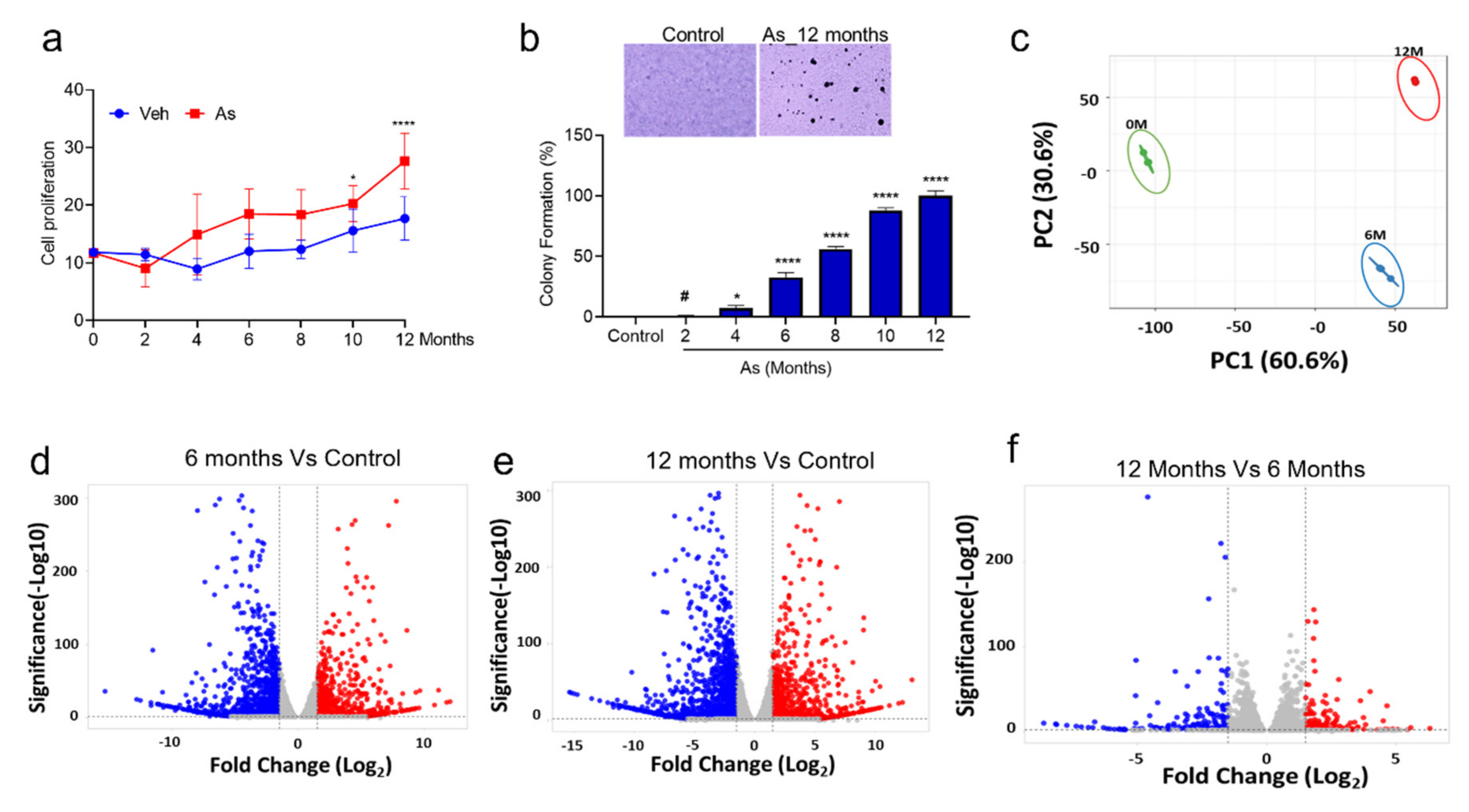
3.1. Chronic Exposure to As Induces Malignant Transformation of TRT-HU1 Cells
3.2. Identification of Differentially Expressed Genes (DEGs) in As-Induced Transformation
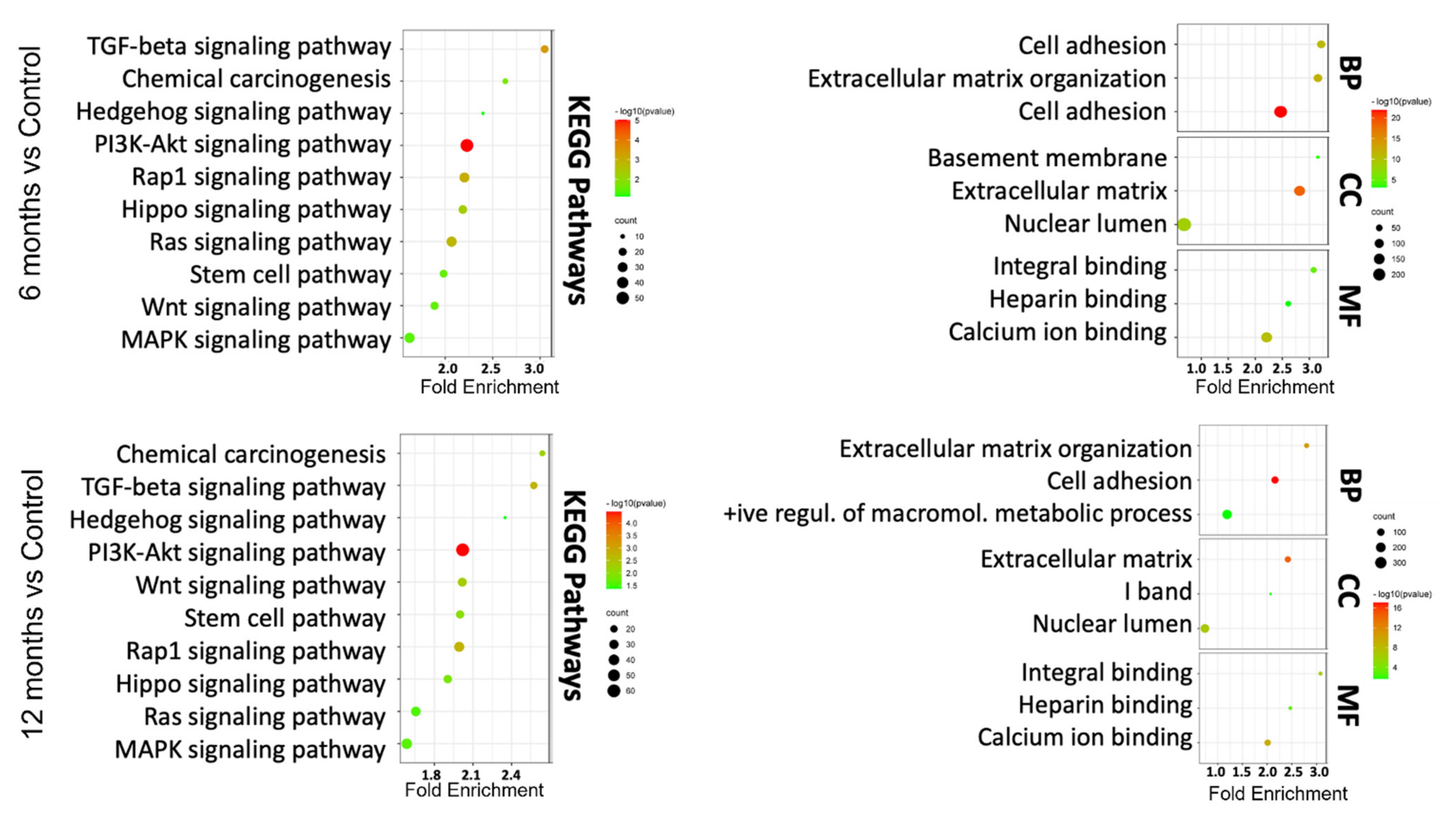
3.3. In Silico Functional Analysis of DEGs
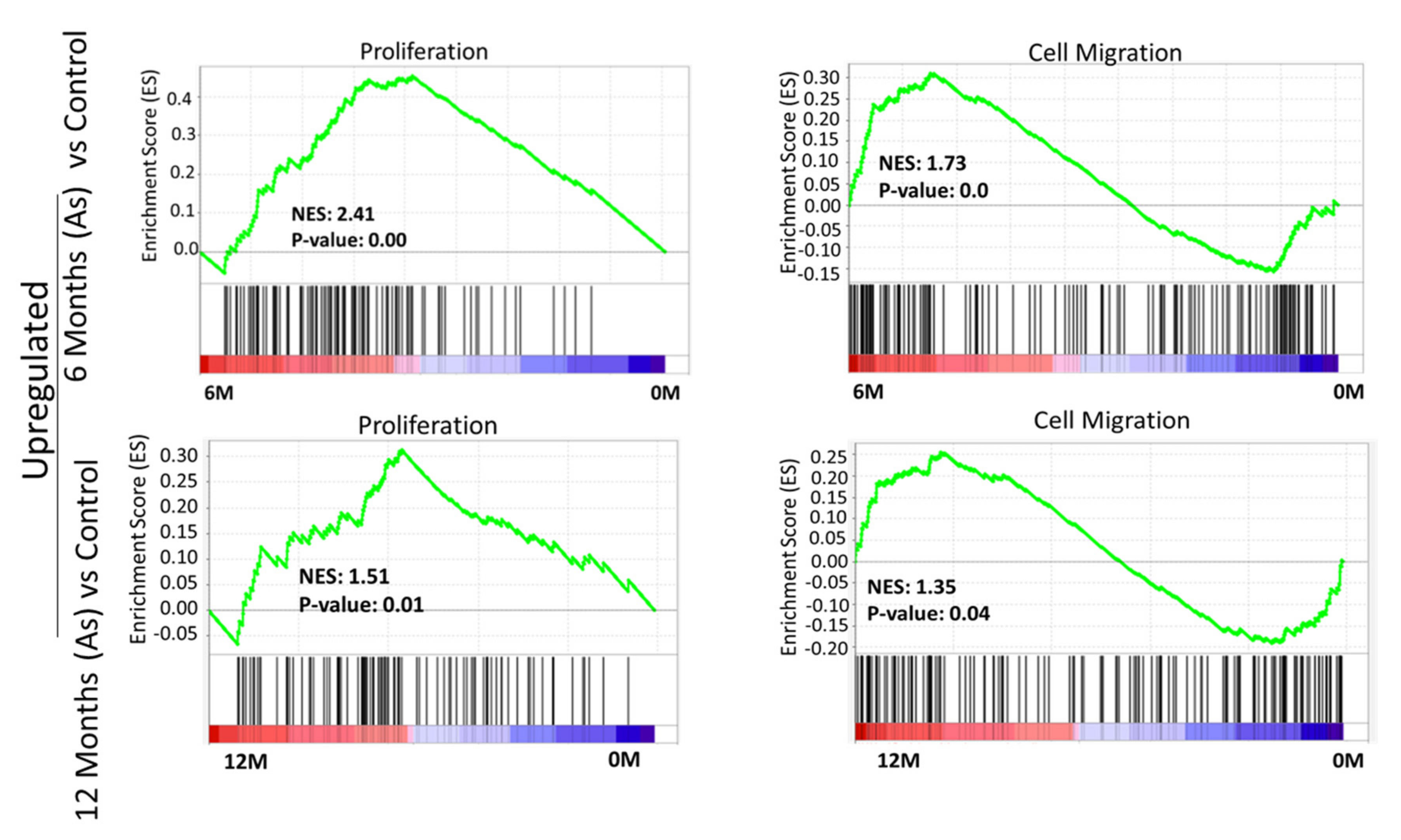
3.4. GSEA Confirmed Functional Changes at the Transcripts Level during Transformation in the As Exposed Cells
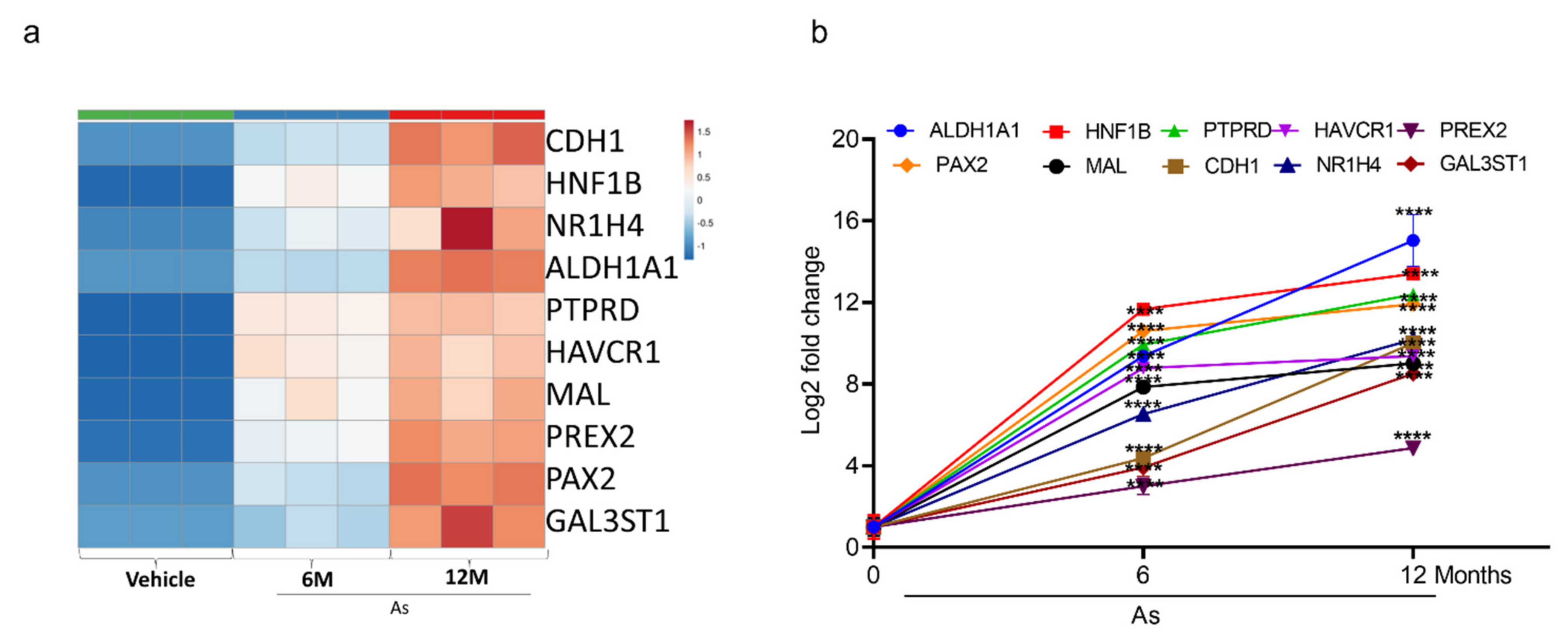
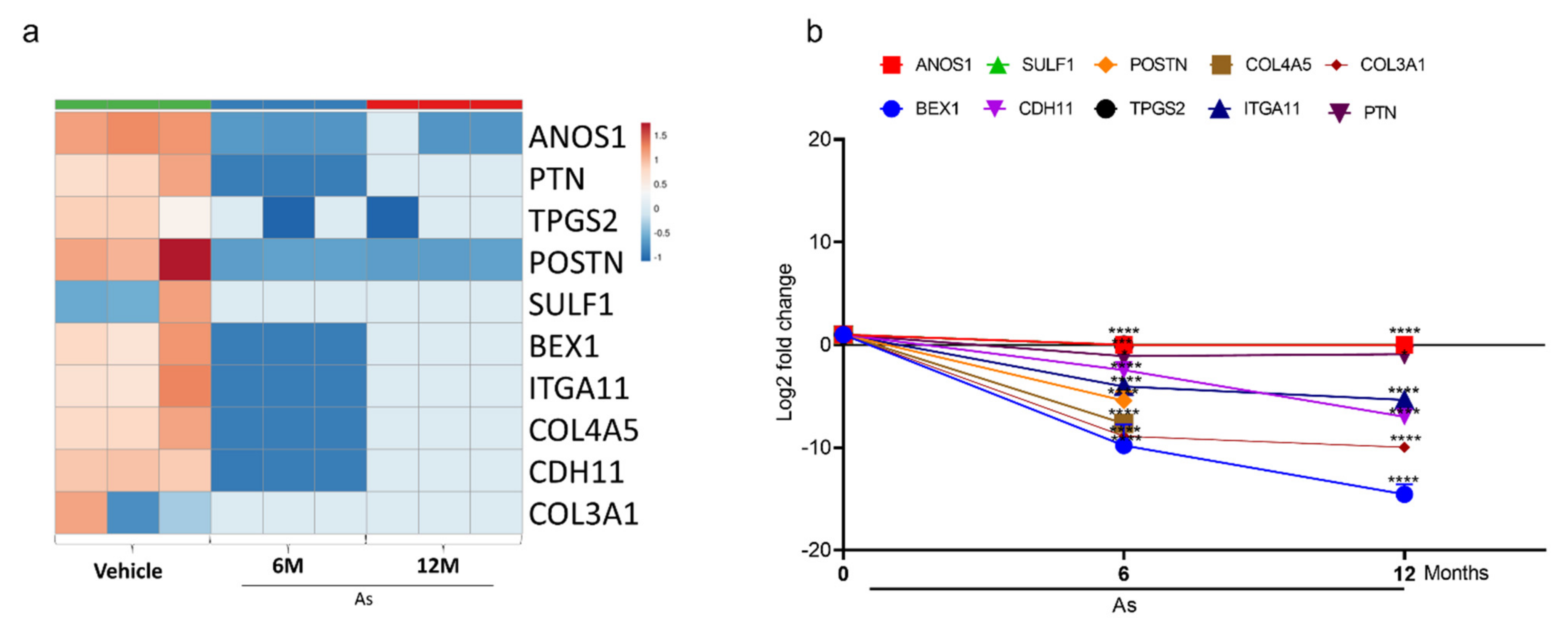
3.5. Validation of Transcriptome Results by qRT-PCR
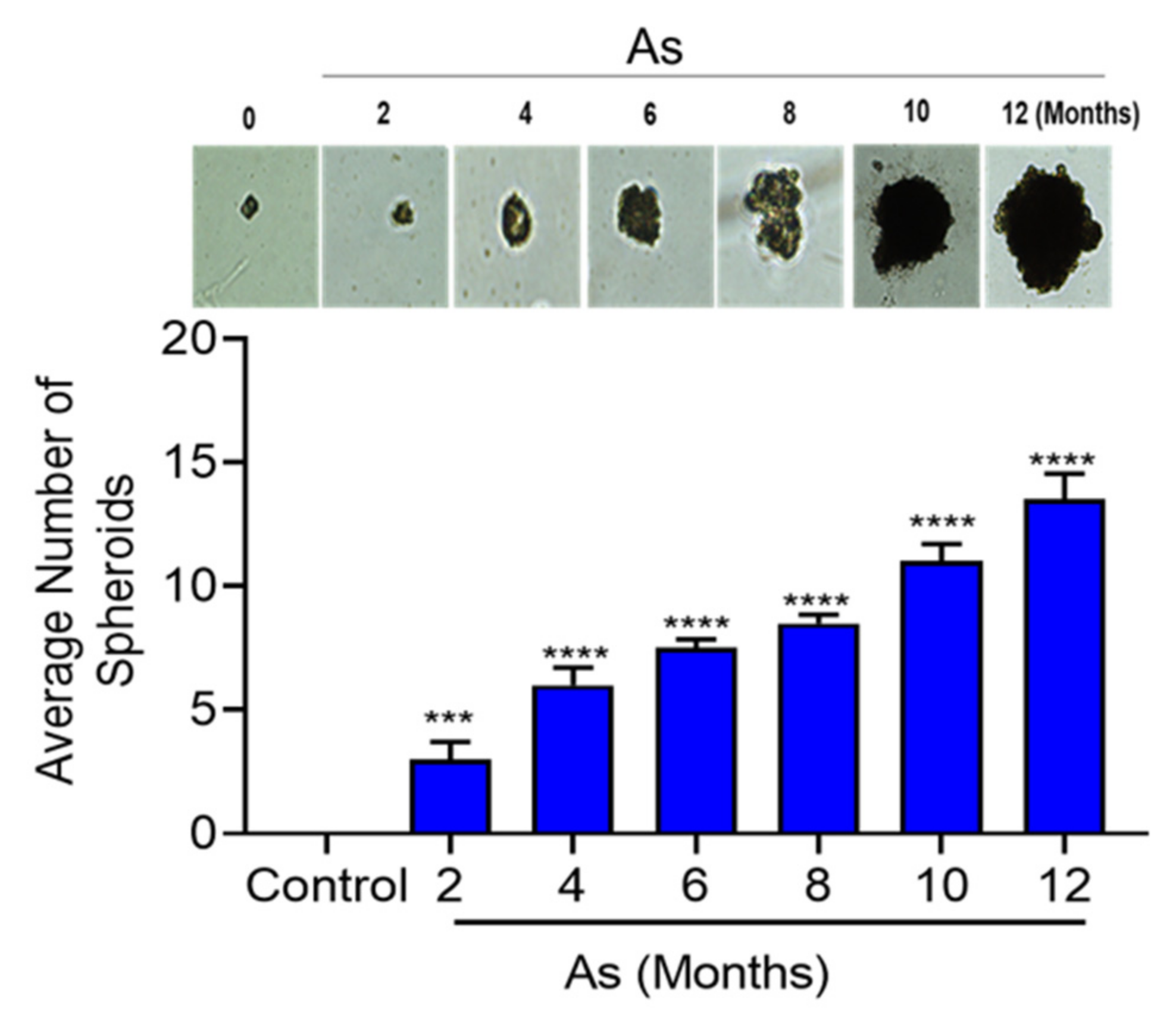
3.6. Characterization of Stem Cells Properties
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Choudhury, M.I.M.; Shabnam, N.; Ahsan, T.; Ahsan, S.M.A.; Kabir, M.S.; Khan, R.M.; Miah, M.A.; Uddin, M.K.; Liton, M.A.R.; Chakraborty, S. Cutaneous Malignancy due to Arsenicosis in Bangladesh: 12-Year Study in Tertiary Level Hospital. Biomed Res. Int. 2018, 2018, 4678362. [Google Scholar] [CrossRef] [PubMed]
- Cutaneous manifestations of chronic arsenicism: Review of seventeen cases. J. Am. Acad. Dermatol. 1998, 38, 179–185. [CrossRef]
- Tsai, S.M.; Wang, T.N.; Ko, Y.C. Mortality for certain diseases in areas with high levels of arsenic in drinking water. Arch. Environ. Health 1999, 54, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Ferreccio, C.; González, C.; Milosavjlevic, V.; Marshall, G.; Sancha, A.M.; Smith, A.H. Lung cancer and arsenic concentrations in drinking water in Chile. Epidemiology 2000, 11, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Mendez, W.M.; Eftim, S.; Cohen, J.; Warren, I.; Cowden, J.; Lee, J.S.; Sams, R. Relationships between arsenic concentrations in drinking water and lung and bladder cancer incidence in U.S. counties. J. Expo. Sci. Environ. Epidemiol. 2017, 27, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, J.S.; Alexander, D.D.; Perez, V.; Mink, P.J. Arsenic exposure and bladder cancer: Quantitative assessment of studies in human populations to detect risks at low doses. Toxicology 2014, 317, 17–30. [Google Scholar] [CrossRef]
- Khairul, I.; Wang, Q.Q.; Jiang, Y.H.; Wang, C.; Naranmandura, H. Metabolism, toxicity and anticancer activities of arsenic compounds. Oncotarget 2017, 8, 23905–23926. [Google Scholar] [CrossRef]
- Clapp, R.W.; Jacobs, M.M.; Loechler, E.L. Environmental and occupational causes of cancer: New evidence 2005–2007. Rev. Environ. Health 2008, 23, 1–37. [Google Scholar] [CrossRef]
- Cohen, S.M.; Arnold, L.L.; Beck, B.D.; Lewis, A.S.; Eldan, M. Evaluation of the carcinogenicity of inorganic arsenic. Crit. Rev. Toxicol. 2013, 43, 711–752. [Google Scholar] [CrossRef]
- Mass, M.J.; Wang, L. Arsenic alters cytosine methylation patterns of the promoter of the tumor suppressor gene p53 in human lung cells: A model for a mechanism of carcinogenesis. Mutat. Res. 1997, 386, 263–277. [Google Scholar] [CrossRef]
- Marsit, C.J.; Karagas, M.R.; Danaee, H.; Liu, M.; Andrew, A.; Schned, A.; Nelson, H.H.; Kelsey, K.T. Carcinogen exposure and gene promoter hypermethylation in bladder cancer. Carcinogenesis 2006, 27, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Pierce, B.L.; Kibriya, M.G.; Tong, L.; Jasmine, F.; Argos, M.; Roy, S.; Paul-Brutus, R.; Rahaman, R.; Rakibuz-Zaman, M.; Parvez, F.; et al. Genome-wide association study identifies chromosome 10q24.32 variants associated with arsenic metabolism and toxicity phenotypes in Bangladesh. PLoS Genet. 2012, 8, e1002522. [Google Scholar] [CrossRef]
- Martinez, V.D.; Vucic, E.A.; Adonis, M.; Gil, L.; Lam, W.L. Arsenic biotransformation as a cancer promoting factor by inducing DNA damage and disruption of repair mechanisms. Mol. Biol. Int. 2011, 2011, 718974. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.P.; Kumari, R.; Treas, J.; Dumond, J.W. Chronic exposure to arsenic causes increased cell survival, DNA damage, and increased expression of mitochondrial transcription factor A (mtTFA) in human prostate epithelial cells. Chem. Res. Toxicol. 2011, 24, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Tokar, E.J.; Diwan, B.A.; Waalkes, M.P. Arsenic exposure transforms human epithelial stem/progenitor cells into a cancer stem-like phenotype. Environ. Health Perspect. 2010, 118, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Tokar, E.J.; Qu, W.; Liu, J.; Liu, W.; Webber, M.M.; Phang, J.M.; Waalkes, M.P. Arsenic-specific stem cell selection during malignant transformation. J. Natl. Cancer Inst. 2010, 102, 638–649. [Google Scholar] [CrossRef]
- Chang, Q.; Bi, Z.; Fu, Y.; Rice, M.K.A.; Zhang, Q.; Wadgaonkar, P.; Almutairy, B.; Zhang, W.; Lu, Y.; Xu, L.; et al. Characterization of Arsenic-Induced Cancer Stem-Like Cells. Methods Mol. Biol. 2020, 2117, 293–303. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef] [PubMed]
- Goedhart, J.; Luijsterburg, M.S. VolcaNoseR is a web app for creating, exploring, labeling and sharing volcano plots. Sci. Rep. 2020, 10, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Ge, S.X.; Jung, D.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P.D. PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019, 47, D419–D426. [Google Scholar] [CrossRef]
- Nie, J.; Gong, L.; Li, Z.; Ou, D.; Zhang, L.; Liu, Y.; Zhang, J.; Liu, D. Bioinformatics Analysis of mRNAs and miRNAs for Identifying Potential Biomarkers in Lung Adenosquamous Carcinoma. Comput. Math. Methods Med. 2022, 2022, 5851269. [Google Scholar] [CrossRef]
- Liu, C.J.; Hu, F.F.; Xia, M.X.; Han, L.; Zhang, Q.; Guo, A.Y. GSCALite: A web server for gene set cancer analysis. Bioinformatics 2018, 34, 3771–3772. [Google Scholar] [CrossRef]
- Xiao, J.; Qiu, Y. Bioinformatics analysis of the gene expression profile in Bladder carcinoma. Genet. Mol. Biol. 2013, 36, 287–291. [Google Scholar] [CrossRef]
- Gao, X.; Chen, Y.; Chen, M.; Wang, S.; Wen, X.; Zhang, S. Identification of key candidate genes and biological pathways in bladder cancer. PeerJ 2018, 6, e6036. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Qiu, Q.; Zhang, X.; Jiang, Z.; Leng, Q.; Liu, Z.; Stass, S.A.; Jiang, F. Aldehyde dehydrogenase 1 A1-positive cell population is enriched in tumor-initiating cells and associated with progression of bladder cancer. Cancer Epidemiol. Biomark. Prev. 2010, 19, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Namekawa, T.; Ikeda, K.; Horie-Inoue, K.; Suzuki, T.; Okamoto, K.; Ichikawa, T.; Yano, A.; Kawakami, S.; Inoue, S. ALDH1A1 in patient-derived bladder cancer spheroids activates retinoic acid signaling leading to TUBB3 overexpression and tumor progression. Int. J. Cancer 2020, 146, 1099–1113. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Li, Y.; Pang, Y.; Ling, M.; Shen, L.; Yang, X.; Zhang, J.; Zhou, J.; Wang, X.; Liu, Q. EMT and stem cell-like properties associated with HIF-2α are involved in arsenite-induced transformation of human bronchial epithelial cells. PLoS ONE 2012, 7, e37765. [Google Scholar] [CrossRef] [PubMed]
- Rodova, M.; Fu, J.; Watkins, D.N.; Srivastava, R.K.; Shankar, S. Sonic hedgehog signaling inhibition provides opportunities for targeted therapy by sulforaphane in regulating pancreatic cancer stem cell self-renewal. PLoS ONE 2012, 7, e46083. [Google Scholar] [CrossRef]
- Zhu, J.N.; Jiang, L.; Jiang, J.H.; Yang, X.; Li, X.Y.; Zeng, J.X.; Shi, R.Y.; Shi, Y.; Pan, X.R.; Han, Z.P.; et al. Hepatocyte nuclear factor-1beta enhances the stemness of hepatocellular carcinoma cells through activation of the Notch pathway. Sci. Rep. 2017, 7, 4793. [Google Scholar] [CrossRef]
- Zanotti, L.; Romani, C.; Tassone, L.; Todeschini, P.; Tassi, R.A.; Bandiera, E.; Damia, G.; Ricci, F.; Ardighieri, L.; Calza, S.; et al. MAL gene overexpression as a marker of high-grade serous ovarian carcinoma stem-like cells that predicts chemoresistance and poor prognosis. BMC Cancer 2017, 17, 366. [Google Scholar] [CrossRef]
- Fu, T.; Coulter, S.; Yoshihara, E.; Oh, T.G.; Fang, S.; Cayabyab, F.; Zhu, Q.; Zhang, T.; Leblanc, M.; Liu, S.; et al. FXR Regulates Intestinal Cancer Stem Cell Proliferation. Cell 2019, 176, 1098–1112.e18. [Google Scholar] [CrossRef]
- Ye, T.; Li, J.; Sun, Z.; Liu, D.; Zeng, B.; Zhao, Q.; Wang, J.; Rosie Xing, H. Cdh1 functions as an oncogene by inducing self-renewal of lung cancer stem-like cells via oncogenic pathways. Int. J. Biol. Sci. 2020, 16, 447–459. [Google Scholar] [CrossRef]
- Vermeulen, L.; Todaro, M.; De Sousa Mello, F.; Sprick, M.R.; Kemper, K.; Perez Alea, M.; Richel, D.J.; Stassi, G.; Medema, J.P. Single-cell cloning of colon cancer stem cells reveals a multi-lineage differentiation capacity. Proc. Natl. Acad. Sci. USA 2008, 105, 13427–13432. [Google Scholar] [CrossRef]
- Medeiros, M.; Zheng, X.; Novak, P.; Wnek, S.M.; Chyan, V.; Escudero-Lourdes, C.; Gandolfi, A.J. Global gene expression changes in human urothelial cells exposed to low-level monomethylarsonous acid. Toxicology 2012, 291, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Johnen, G.; Rozynek, P.; von der Gathen, Y.; Bryk, O.; Zdrenka, R.; Johannes, C.; Weber, D.G.; Igwilo-Okuefuna, O.; Raiko, I.; Hippler, J.; et al. Cross-contamination of a UROtsa stock with T24 cells--molecular comparison of different cell lines and stocks. PLoS ONE 2013, 8, e64139. [Google Scholar] [CrossRef] [PubMed]
- Petrick, J.S.; Jagadish, B.; Mash, E.A.; Aposhian, H.V. Monomethylarsonous acid (MMA(III)) and arsenite: LD(50) in hamsters and in vitro inhibition of pyruvate dehydrogenase. Chem. Res. Toxicol. 2001, 14, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Zeng, M.; Zhu, H.; Chen, X.; Weng, Z.; Li, S. Emerging role of Hippo signalling pathway in bladder cancer. J. Cell. Mol. Med. 2018, 22, 4–15. [Google Scholar] [CrossRef]
- Khan, K.; Quispe, C.; Javed, Z.; Iqbal, M.J.; Sadia, H.; Raza, S.; Irshad, A.; Salehi, B.; Reiner, Ž.; Sharifi-Rad, J. Resveratrol, curcumin, paclitaxel and miRNAs mediated regulation of PI3K/Akt/mTOR pathway: Go four better to treat bladder cancer. Cancer Cell Int. 2020, 20, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Dangle, P.; Zaharieva, B.; Jia, H.; Pohar, K. Ras-MAPK pathway as a therapeutic target in cancer—Emphasis on bladder cancer. Recent Pat. Anticancer. Drug Discov. 2009, 4, 125–136. [Google Scholar] [CrossRef]
- Stueckle, T.A.; Lu, Y.; Davis, M.E.; Wang, L.; Jiang, B.H.; Holaskova, I.; Schafer, R.; Barnett, J.B.; Rojanasakul, Y. Chronic occupational exposure to arsenic induces carcinogenic gene signaling networks and neoplastic transformation in human lung epithelial cells. Toxicol. Appl. Pharmacol. 2012, 261, 204–216. [Google Scholar] [CrossRef]
- Gao, N.; Shen, L.; Zhang, Z.; Leonard, S.S.; He, H.; Zhang, X.G.; Shi, X.; Jiang, B.H. Arsenite induces HIF-1alpha and VEGF through PI3K, Akt and reactive oxygen species in DU145 human prostate carcinoma cells. Mol. Cell. Biochem. 2004, 255, 33–45. [Google Scholar] [CrossRef]
- Lau, A.; Zheng, Y.; Tao, S.; Wang, H.; Whitman, S.A.; White, E.; Zhang, D.D. Arsenic inhibits autophagic flux, activating the Nrf2-Keap1 pathway in a p62-dependent manner. Mol. Cell. Biol. 2013, 33, 2436–2446. [Google Scholar] [CrossRef]
- Zhang, T.; Qi, Y.; Liao, M.; Xu, M.; Bower, K.A.; Frank, J.A.; Shen, H.M.; Luo, J.; Shi, X.; Chen, G. Autophagy is a cell self-protective mechanism against arsenic-induced cell transformation. Toxicol. Sci. 2012, 130, 298–308. [Google Scholar] [CrossRef]
- Tanaka, Y.; Kanda, M.; Sugimoto, H.; Shimizu, D.; Sueoka, S.; Takami, H.; Ezaka, K.; Hashimoto, R.; Okamura, Y.; Iwata, N.; et al. Translational implication of Kallmann syndrome-1 gene expression in hepatocellular carcinoma. Int. J. Oncol. 2015, 46, 2546–2554. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.L.; Gui, S.L.; Ma, J.G. Aberrant methylation of CDH11 predicts a poor outcome for patients with bladder cancer. Oncol. Lett. 2015, 10, 647–652. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ikeda, K.; Iyama, K.I.; Ishikawa, N.; Egami, H.; Nakao, M.; Sado, Y.; Ninomiya, Y.; Baba, H. Loss of expression of type IV collagen alpha5 and alpha6 chains in colorectal cancer associated with the hypermethylation of their promoter region. Am. J. Pathol. 2006, 168, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.J.; Yoshioka, N.; Tambe, Y.; Kushima, R.; Okada, Y.; Inoue, H. Periostin is down-regulated in high grade human bladder cancers and suppresses in vitro cell invasiveness and in vivo metastasis of cancer cells. Int. J. Cancer 2005, 117, 51–58. [Google Scholar] [CrossRef]
- Yamakawa, T.; Kurosawa, N.; Kadomatsu, K.; Matsui, T.; Itoh, K.; Maeda, N.; Noda, M.; Muramatsu, T. Levels of expression of pleiotrophin and protein tyrosine phosphatase zeta are decreased in human colorectal cancers. Cancer Lett. 1999, 135, 91–96. [Google Scholar] [CrossRef]
- Foltz, G.; Ryu, G.Y.; Yoon, J.G.; Nelson, T.; Fahey, J.; Frakes, A.; Lee, H.; Field, L.; Zander, K.; Sibenaller, Z.; et al. Genome-wide analysis of epigenetic silencing identifies BEX1 and BEX2 as candidate tumor suppressor genes in malignant glioma. Cancer Res. 2006, 66, 6665–6674. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Pathway | FDR | Class |
|---|---|---|---|
| Upregulated Genes | |||
| ALDH1A1 | EMT | 9.75 × 10−9 | Activation |
| CDH1 | RTK | 0.006827 | Activation |
| GAL3ST1 | PI3K/AKT | 0.047569 | Activation |
| HNF1B | RTK | 0.000231 | Activation |
| PTPRD | EMT | 1.77 × 10−15 | Activation |
| MAL | Hormone AR | 0.000131 | Activation |
| NR1H4 | Hormone AR | 8.8 × 10−5 | Activation |
| PREX2 | Hormone AR | 0.01951 | Activation |
| HAVCR1 | Hormone AR | 0.000703 | Activation |
| Downregulated Genes | |||
| SULF1 | Apoptosis | 0.006361 | Activation |
| ANOS1 | Hormone AR | 0.002647 | Inhibition |
| CDH11 | Hormone AR | 0.000904 | Inhibition |
| COL3A1 | Hormone AR | 0.000184 | Inhibition |
| COL4A5 | Hormone ER | 1.69 × 10−7 | Inhibition |
| ITGA11 | PI3K/AKT | 0.018696 | Inhibition |
| ITGA11 | Hormone AR | 0.006147 | Inhibition |
| POSTN | Hormone AR | 2.13 × 10−6 | Inhibition |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shukla, V.; Chandrasekaran, B.; Tyagi, A.; Navin, A.K.; Saran, U.; Adam, R.M.; Damodaran, C. A Comprehensive Transcriptomic Analysis of Arsenic-Induced Bladder Carcinogenesis. Cells 2022, 11, 2435. https://doi.org/10.3390/cells11152435
Shukla V, Chandrasekaran B, Tyagi A, Navin AK, Saran U, Adam RM, Damodaran C. A Comprehensive Transcriptomic Analysis of Arsenic-Induced Bladder Carcinogenesis. Cells. 2022; 11(15):2435. https://doi.org/10.3390/cells11152435
Chicago/Turabian StyleShukla, Vaibhav, Balaji Chandrasekaran, Ashish Tyagi, Ajit Kumar Navin, Uttara Saran, Rosalyn M. Adam, and Chendil Damodaran. 2022. "A Comprehensive Transcriptomic Analysis of Arsenic-Induced Bladder Carcinogenesis" Cells 11, no. 15: 2435. https://doi.org/10.3390/cells11152435
APA StyleShukla, V., Chandrasekaran, B., Tyagi, A., Navin, A. K., Saran, U., Adam, R. M., & Damodaran, C. (2022). A Comprehensive Transcriptomic Analysis of Arsenic-Induced Bladder Carcinogenesis. Cells, 11(15), 2435. https://doi.org/10.3390/cells11152435