
Promises and Challenges of Cell-Based Therapies to Promote Lung Regeneration in Idiopathic Pulmonary Fibrosis


Abstract
1. Introduction
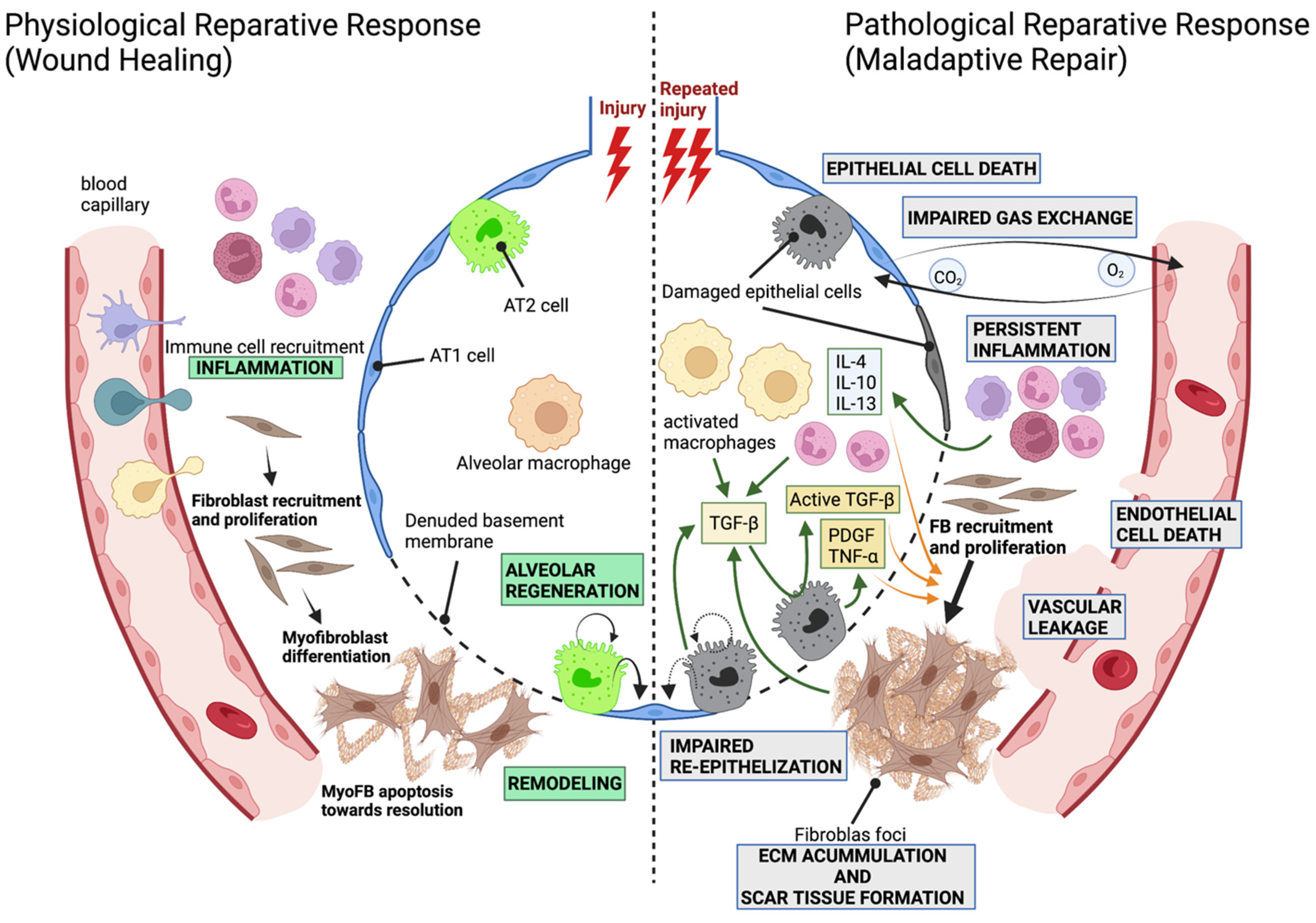
1.1. The Alveolar Compartment and the Development of Pathological Fibrosis
1.2. Modeling Lung Fibrosis
2. Regeneration and Stem/Progenitor Cells
2.1. Epithelial Stem and Progenitor Cells Are Contributing to Regenerate the Alveolar Epithelium
2.2. Mechanisms of Alveolar Regeneration
2.3. Alveolar Regeneration in Human Lungs
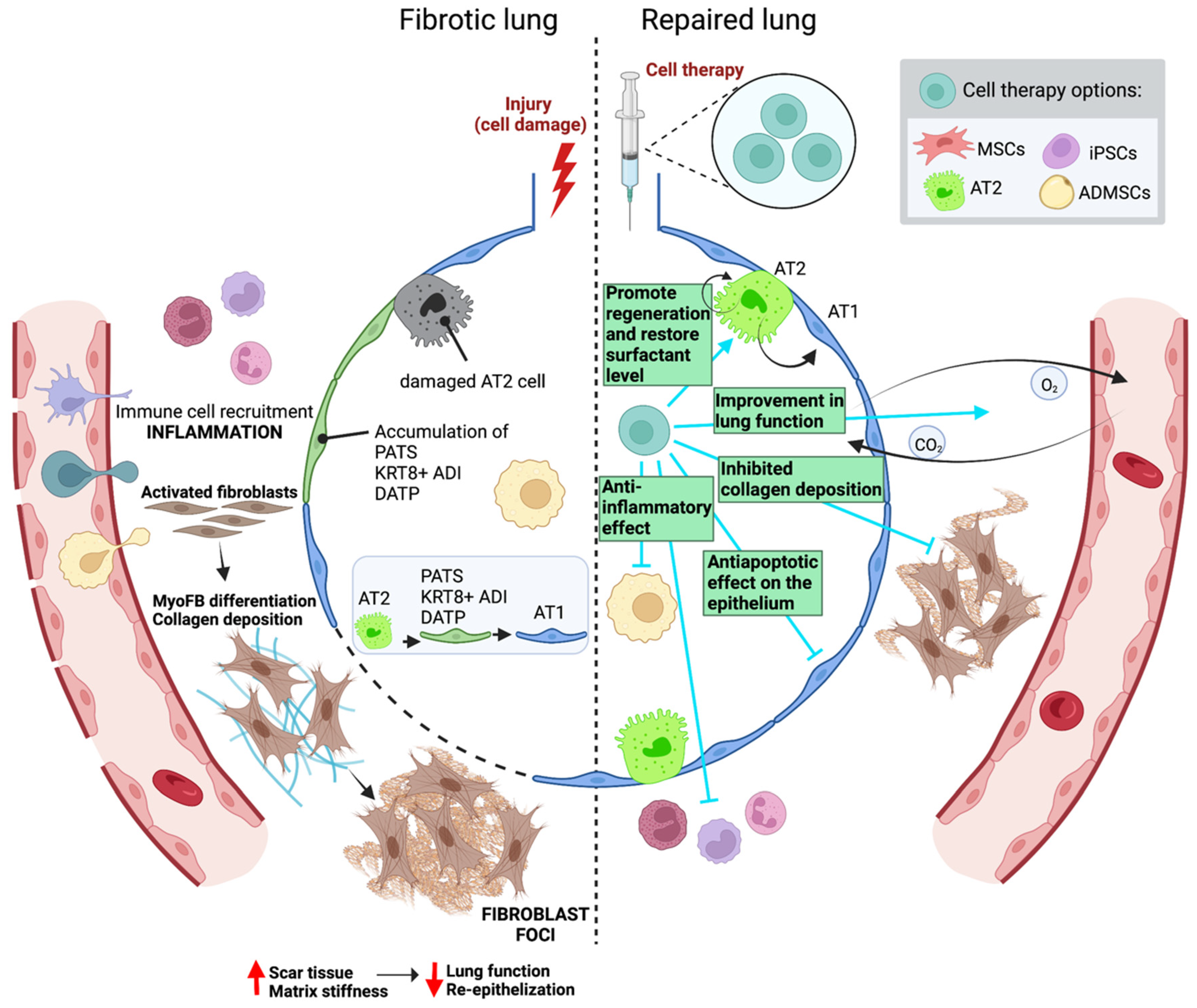
3. Cell Therapy in Lung Fibrosis
3.1. Preclinical Mouse Studies
3.1.1. Epithelial Cells: Alveolar Type 2 Cells
3.1.2. Adult Mesenchymal Stromal/Stem Cells
3.1.3. Induced Pluripotent Stem Cells
3.2. Clinical Human Studies

{kind=link}
{kind=link}
{kind=link}
| Type of Study | Cell Source | Cell Delivery Route, Dose and Time of Administration | Time of Readouts and Results | Ref |
|---|---|---|---|---|
| Preclinical mouse studies | AT2 cells | Intratracheal route. A dose of 2.5 × 106 cells/rat 14 days after a single intratracheal bleomycin administration | The animals were euthanized 21 days after bleomycin challenge. Treated rats after bleomycin instillation showed a reduction in the degree of fibrosis and a complete recovery to normal levels of surfactant proteins | [145] |
| AT2 cells | Intratracheal route. A dose of 2.5 × 106 cells/rat 3, 7 or 15 days after a single intratracheal bleomycin administration | The animals were euthanized 21 days after bleomycin challenge. Treated rats after bleomycin instillation showed reduced collagen deposition and reduction in the severity of pulmonary fibrosis (regardless the time point of AT2 cell treatment) | [146] | |
| AT2 cells | Intratracheal route. A dose of 2.5 × 106 cells/rat 3 or 7 days after a single intratracheal bleomycin administration | The animals were euthanized 7 or 14 days after bleomycin challenge. Treated rats 7 days after bleomycin instillation showed an improvement in lung performance, structure and surfactant ultrastructure in bleomycin-induced lung fibrosis, while those treated 3 days after bleomycin instillation were only able to slightly recover the volume of AT2 and volume fraction of lamellar bodies in AT2 | [147] | |
| Adult lung spheroid cells (LSCs) | Intravenous route. A dose of either 5 × 106 syngeneic or allogeneic LSCs/rat 24 h after a single intratracheal bleomycin administration | The animals were euthanized 14 days after bleomycin challenge. Treated rats with allogeneic/syngeneic LSCs show an attenuation in the progression and severity of pulmonary fibrosis, decreasing apoptosis, protecting alveolar structures and increasing angiogenesis. Safety and efficacy of allogeneic LSCs treatment is demonstrated | [148] | |
| Human BM-MSCs | Intravenous route. A dose of 5 × 10⁵ cells/humanized mouse 2 days after a single intratracheal bleomycin administration | The animals were euthanized 7 or 21 days after bleomycin challenge. Treated humanized mice with human MSCs showed an attenuation of pulmonary fibrosis development. MSCs are suggested to suppress T-cell overactivation via PD-1 and PD-L1 interaction. Human MSCs have a therapeutic effect only in the early phase of pulmonary fibrosis | [151] | |
| Human BM-MSCs | Intravenous route. A dose of 0.5 × 106 modified * or nonmodified cells/mouse 7 days after a single intratracheal bleomycin administration. * Cell modification refers to their prior transduction of miRNAs (let-7d or miR-154) using lentiviral vectors | The animals were euthanized 14 days after bleomycin challenge. Treated mice with human modified (let-7d) MSCs revealed shifts in animal weight loss, collagen activity after treatment and decrease in CD45+ cells, partially reducing the effects of bleomycin-induced lung injury. This study suggests the use of miRNA-modified BM-MSCs as a potential therapeutic strategy | [152] | |
| BM-MSCs | Intravenous route. A dose of 5 × 10⁵ cells/mouse immediately after or 7 days after a single intratracheal bleomycin administration | The animals were euthanized 14 days after bleomycin challenge. Immediately after bleomycin instillation, treated mice showed an amelioration in the fibrotic injuries, while those treated 7 days after bleomycin instillation, even though engraftment was not inhibited, the ability of the cells to alter the course of disease progression was eliminated | [155] | |
| BM-MSCs | Intravenous route. A dose of 2.5 × 106 cells/rat immediately after or 7 days after a single intratracheal bleomycin administration | The animals were euthanized 7, 14 or 28 days after bleomycin challenge. The present study demonstrates that when MSCs were administered after bleomycin challenge, exogenous MSCs were immediately detected in lung tissues from rats sacrificed at different time points and the number of MSCs in the lung tissue increased over time, while this did not happen to the group treated after 7 days of bleomycin instillation | [156] | |
| BM-MSCs | Intravenous route. Two doses of 0.5 × 106 cells/mouse. The first one was administered after a single oropharyngeal bleomycin administration and the second dose, 3 days after the first dose | The animals were euthanized 14 days after bleomycin challenge. This study demonstrates that BM-MSCs expressing keratinocyte growth factor via an inducible lentivirus protects against bleomycin-induced lung fibrosis | [157] | |
| Human BM-MSCs | Intravenous route. A dose of 5 × 10⁵/mouse 24 h after a single intratracheal bleomycin administration | The animals were euthanized 14 days after bleomycin challenge. In this study, the authors show that MSCs can correct the inadequate communication between epithelial and mesenchymal cells through STC1 (Stanniocalcin-1) secretion after bleomycin instillation | [158] | |
| BM-MSCs | Intratracheal route. A dose of either 5 × 10⁵ hypoxia-preconditioned or control cells/mouse 3 days after a single intratracheal bleomycin administration | The animals were euthanized 7 or 21 days after bleomycin challenge. This study reports that hypoxia-preconditioned BM-MSCs improve pulmonary functions and reduce inflammatory and fibrotic mediators after bleomycin-induced lung fibrosis | [159] | |
| Oncostatin M (OSM)-preconditioned BM-MSCs | Intratracheal route. A dose of either 2 × 10⁵ oncostatin M (OSM)-preconditioned or control cells/mouse 3 days after a single intratracheal bleomycin administration | The animals were euthanized 7 or 21 days after bleomycin challenge. Transplantation of OSM-preconditioned MSCs significantly improved pulmonary respiratory functions and downregulated expression of inflammatory factors and fibrotic factors after bleomycin instillation | [160] | |
| BM-MSCs | Intravenous route. A dose of 1 × 106 cells/mL/rat 14 days after a single intratracheal bleomycin administration | The animals were euthanized 28 days after bleomycin challenge. Animals treated with BM-MSCs showed a significant decrease in the alveolar wall thickening, in the inflammatory infiltrate and in the collagen fiber deposition. The conclusion of the study was that the therapeutic pulmonary anti-fibrotic activity of BM-MSCs is mediated through their anti-inflammatory properties and inhibition of SMAD-3/TGFβ expression | [161] | |
| Resident lung MSCs (luMSCs) | Intravenous route. A dose of either 0.15 × 106 or 0.25 × 106 cells/mouse immediately after a single intratracheal bleomycin administration | The animals were euthanized 14 or 35 days after bleomycin challenge. Treated animals showed a decrease in numbers of lymphocytes and granulocytes in bronchoalveolar fluid and display reduced collagen deposition. Also, treatment with luMSCs significantly decreased weight loss associated with bleomycin and increased survival from 50% at 14 days with bleomycin alone to 80% when mice had been treated with luMSCs | [162] | |
| BM-MSCs | Intravenous route. A dose of 5 × 10⁴ allogeneic cells/g/mouse 6–8 h or 9 days after a single intranasal bleomycin administration | The animals were euthanized 28 days after bleomycin challenge. Early treatment with allogeneic MSCs protected the lung architecture and significantly reduced fibrosis, apoptosis and IL1-production, while delayed MSC treatment failed to protect the mice from bleomycin-induced lung fibrosis. Of note, this is the first study to definitively show the importance of naturally derived HFG in MSC protection in the bleomycin model | [164] | |
| Amnion-MSCs vs. BM-MSCs vs. human amniotic epithelial cells (hAECs) | Intravenous route. A dose of 1 × 106 cells/mouse 3 days after introducing the second bleomycin injury (bleomycin administration was done intra-nasally, and the second dose was given 7 days after the first one) | The animals were euthanized 17 or 31 days after bleomycin challenge. This study concluded that amnion-MSCs may be more effective than BM-MSCs and hAECs in reducing injury following delayed injection in the setting of repeated lung injury | [165] | |
| ADSCs | Intravenous route. A dose of either 5 × 10⁵ young-donor or old-donor cells/mouse 24 h after a single intratracheal bleomycin administration | The animals were euthanized 21 days after bleomycin challenge. Treated old mice with young ADSC displayed a greater reduction in fibrosis, oxidative stress, MMP-2 activity and apoptosis markers than mice treated with old ADSCs | [168] | |
| ADSCs | Intravenous route. A dose of either 2.5 × 10⁴ or 2.5 × 10⁵ cells/mouse immediately after subcutaneous bleomycin administration for 7 days | The animals were euthanized 7 or 21 days after bleomycin challenge. ADCSs accumulated in the pulmonary interstitium and inhibited both inflammation and fibrosis in the lung. Treated mice showed decreased lung fibrosis and inflammation in a dose-dependent manner | [169] | |
| ADSCs (human) | Intraperitoneal route. During the latter 2 months of bleomycin exposure * 3 × 10⁵ human cells were administered repeatedly at the same time as bleomycin. * Bleomycin was injected intratracheally in eight biweekly doses | The animals were euthanized 14 days after bleomycin challenge. Treated mice showed decreased lung fibrosis, inflammatory cell infiltration, epithelial hyperplasia, TGFβ expression and epithelial apoptosis | [170] | |
| ADSCs | Intravenous route. A dose of 5 × 10⁵ cells/mouse 24 h after a single intratracheal bleomycin administration | Mice treated with ADSCs showed attenuated bleomycin-induced lung and skin fibrosis and accelerated wound healing. This study suggests that ADSCs may prime injured tissues and prevent end-organ fibrosis | [171] | |
| ADSCs (human) | Intravenous route. A dose of 40 × 106/kg body weight/mouse 3, 6 and 9 days after a single intratracheal bleomycin administration | The animals were euthanized 24 days after bleomycin challenge. Mice treated with ADSCs showed a higher increase in survivability, organ weight reduction and collagen deposition when compared to those treated with pirfenidone. Also, ADSCs potently suppressed profibrotic genes induced by bleomycin and also inhibited pro-inflammatory related transcripts | [172] | |
| Human Placental MSCs of fetal origins (hfPMSCs) | Intravenous route. A dose of 1 × 10⁵ cells/mouse 3 days after a single intratracheal bleomycin administration | The animals were euthanized 0, 7 and 28 days after bleomycin challenge. Treatment with hfPMSCs showed that these cells can attenuate bleomycin-induced lung inflammation and fibrosis in mice, in part through a mechanism by attenuating MyD88-mediated inflammation | [173] | |
| Amniotic fluid stem cells (AFSCs) | Intravenous route. A dose of 1 × 106 cells/mouse either 2 h or 14 days after a single intratracheal bleomycin administration | The animals were euthanized 3, 14, 28 days after bleomycin challenge, depending on the group. Treated mice at both time points showed inhibition in the changes in lung function associated with bleomycin-induced lung injury and decreased collagen deposition | [174] | |
| iPSCs | Intravenous route. A dose of 2 × 106 cells/mouse 24 h after a single intratracheal bleomycin administration | The animals were euthanized 21 days after bleomycin challenge. Treated mice after bleomycin showed an inhibition of EMT, inflammatory response and TGF-β1/Smad2/3 signaling pathway | [175] | |
| iPSCs | Intravenous route. A dose of 2 × 106 cells/mouse (cells either lacking c-Myc or in condition medium) 24 h after a single intratracheal bleomycin administration | The animals were euthanized 3, 7, 14 or 21 days after bleomycin challenge. Treated mice, after bleomycin instillation, showed an attenuation in collagen content, diminished neutrophil accumulation and rescued pulmonary function and recipient survival after bleomycin-induced lung injury | [176] | |
| Mouse iPSCs-derived AT2 cells | Intravenous route. A dose of 5 × 10⁵ cells/mouse 24 h after a single intratracheal bleomycin administration | The animals were euthanized 13 days after bleomycin challenge. Treated mice after bleomycin have decreased collagen deposition and lung inflammation | [179] | |
| Human iPSCs-derived AT2 cells | Intratracheal route. A dose of 3 × 106 cells/rat 15 days after a single intratracheal bleomycin administration | The animals were sacrificed 21 days after bleomycin administration. Transplanted lungs showed no inflammation, no edema, no epithelial damage and reduced fibrosis | [182] | |
| AT2, AT1 and Club cells derived from human embryonic stem cells (hESCs) | Intratracheal route. A dose of 1 × 10⁵ differentiated hESCs/mouse 7 days after a single intratracheal bleomycin administration and immediately after sublethal irradiation to avoid graft rejection | The animals were euthanized 14 days after bleomycin challenge. Treated mice, after bleomycin instillation, showed an increase in progenitor number in the airways and reduced collagen content | [180] | |
| Clinical human studies | AT2 cells (heterologous) | Intratracheal route. A total of 16 IPF patients. Four doses of 1000–1.200 × 106 cells/patient | Enrolled patients were monitored for 1 year. Administered AT2 cells were both safe and well tolerated. There was no deterioration in pulmonary function, respiratory symptoms or disease extent after 12 months of follow-up. This study lacks a control group due to ethical issues | [142] |
| SOX9 + BCs (autologous) | Endobronchial route. A total of 2 bronchiectasis patients. A dose of 1 × 106 cells/kg body weight/patient | This study was the first autologous SOX9 + BCs transplantation clinical trial. Lung tissue repair and pulmonary function enhancement was observed in patients 3–12 months after cell transplantation | [186] | |
| SOX9 + BCs (autologous) | Endobronchial route. A total of 7 bronchiectasis. A dose of 1 × 106 cells/kg body weight/patient | Enrolled patients were monitored for 1 year. Transplantation of autologous SOX9 + BCs had positive effects and is safe for patients with bronchiectasis | [187] | |
| BM-MSCs (allogeneic) | Intravenous route. A total of 20 patients with usual interstitial pneumonia and a history of lung function decline over the last 12 months, among other characteristics. Two doses of 200 × 106 cells/patient, every 3 months | Enrolled patients were monitored for 1 year. This study concluded that therapy with high doses of allogeneic MSCs is a safe and promising method to reduce disease progression in IPF patients with rapid pulmonary function decline | [188] | |
| ADSC-SVF (stromal vascular function) | Endobronchial route. A total of 14 IPF patients. A dose of 0.5 × 106 cells/kg body weight/patient/month (a total of 3 months) | Enrolled patients were monitored for 1 year. There was no formation of ectopic tissues and no difference in adverse events compared to placebo effect. Treatment was safe for IPF patients | [189] | |
| ADSC-SVF (stromal vascular function) | Endobronchial route. A total of 14 IPF patients. A dose of 0.5 × 106 cells/kg body weight/patient/month (a total of 3 months) | This study is the follow-up of the study above. They saw a significant functional decline was observed at 24 months after the first administration and highlighted the need of further clinical trials using these cells | [190] | |
| BM-MSCs (allogeneic) | Intravenous route. A total of 9 IPF patients. A dose of either 20 × 106, 100 × 106 or 200 × 106 cells/patient | Safety was assessed for 15 months in total. No treatment-emergent serious adverse events were reported in this study. This trial (called AETHER) was the first clinical trial conducted for 15 months to assess the safety of a single intravenous infusion of BM-MSCs | [191] | |
| Placental MSCs (allogeneic) | Intravenous route. A total of 8 IPF patients. A dose of either 1 × 106 or 2 × 106 cells/kg body weight/patient | Enrolled patients were followed for 6 months. Intravenous administration of these cells was proven to be feasible and to have a good short-term safety profile in patients with moderately severe IPF | [192] | |
| BM-MSCs (allogeneic) | Intravenous route. A total of 9 IPF patients. A dose of either 20 × 106, 100 × 106 or 200 × 106 cells/patient | This study is a follow-up of the AETHER trial. The subjects receiving the higher dose demonstrated better results when compared to those receiving the lowest dose | [193] |
4. Current Challenges
5. Future Perspectives and Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wijsenbeek, M.; Suzuki, A.; Maher, T.M. Interstitial lung diseases. Lancet 2022. [Google Scholar] [CrossRef]
- Confalonieri, P.; Volpe, M.C.; Jacob, J.; Maiocchi, S.; Salton, F.; Ruaro, B.; Confalonieri, M.; Braga, L. Regeneration or Repair? The Role of Alveolar Epithelial Cells in the Pathogenesis of Idiopathic Pulmonary Fibrosis (IPF). Cells 2022, 11, 2095. [Google Scholar] [CrossRef] [PubMed]
- Sauleda, J.; Núñez, B.; Sala, E.; Soriano, J.B. Idiopathic Pulmonary Fibrosis: Epidemiology, Natural History, Phenotypes. Med. Sci. 2018, 6, 110. [Google Scholar] [CrossRef] [PubMed]
- Salton, F.; Ruaro, B.; Confalonieri, P.; Confalonieri, M. Epithelial–Mesenchymal Transition: A Major Pathogenic Driver in Idiopathic Pulmonary Fibrosis? Medicina 2020, 56, 608. [Google Scholar] [CrossRef] [PubMed]
- Maher, T.M.; Bendstrup, E.; Dron, L.; Langley, J.; Smith, G.; Khalid, J.M.; Patel, H.; Kreuter, M. Global incidence and prevalence of idiopathic pulmonary fibrosis. Respir. Res. 2021, 22, 197. [Google Scholar] [CrossRef] [PubMed]
- Wakwaya, Y.; Brown, K.K. Idiopathic Pulmonary Fibrosis: Epidemiology, Diagnosis and Outcomes. Am. J. Med Sci. 2019, 357, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Lederer, D.J.; Martinez, F.J. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef]
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic pulmonary fibrosis. Lancet 2017, 389, 1941–1952. [Google Scholar] [CrossRef]
- Torrisi, S.E.; Kahn, N.; Vancheri, C.; Kreuter, M. Evolution and treatment of idiopathic pulmonary fibrosis. Presse Med. 2020, 49, 104025. [Google Scholar] [CrossRef]
- Yanagihara, T.; Scallan, C.; Ask, K.; Kolb, M.R.J. Emerging therapeutic targets for idiopathic pulmonary fibrosis: Preclinical progress and therapeutic implications. Expert Opin. Ther. Targets 2021, 25, 939–948. [Google Scholar] [CrossRef]
- Amor, M.S.; Rosengarten, D.; Shitenberg, D.; Pertzov, B.; Shostak, Y.; Kramer, M.R. Lung Transplantation in Idiopathic Pulmonary Fibrosis: Risk Factors and Outcome. Isr. Med. Assoc. J. 2020, 22, 741–746. [Google Scholar] [PubMed]
- Barkauskas, C.E.; Noble, P.W. Cellular Mechanisms of Tissue Fibrosis. 7. New insights into the cellular mechanisms of pulmonary fibrosis. Am. J. Physiol. Physiol. 2014, 306, C987–C996. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.Y.; Lagares, D.; Tager, A.M.; Kapoor, M. Fibrosis—A lethal component of systemic sclerosis. Nat. Rev. Rheumatol. 2014, 10, 390–402. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro-Gabazza, C.N.; Kobayashi, T.; Yasuma, T.; Toda, M.; Kim, H.; Fujimoto, H.; Hataji, O.; Takeshita, A.; Nishihama, K.; Okano, T.; et al. A Staphylococcus pro-apoptotic peptide induces acute exacerbation of pulmonary fibrosis. Nat. Commun. 2020, 11, 1539. [Google Scholar] [CrossRef]
- Fabbrizzi, A.; Nannini, G.; Lavorini, F.; Tomassetti, S.; Amedei, A. Microbiota and IPF: Hidden and detected relationships. Sarcoidosis Vasc. Diffus. Lung. Dis. 2021, 38, e2021028. [Google Scholar] [CrossRef]
- Wendisch, D.; Dietrich, O.; Mari, T.; von Stillfried, S.; Ibarra, I.L.; Mittermaier, M.; Mache, C.; Chua, R.L.; Knoll, R.; Timm, S.; et al. SARS-CoV-2 infection triggers profibrotic macrophage responses and lung fibrosis. Cell 2021, 184, 6243–6261.e27. [Google Scholar] [CrossRef]
- Guo, C.; Lv, S.; Liu, Y.; Li, Y. Biomarkers for the adverse effects on respiratory system health associated with atmospheric particulate matter exposure. J. Hazard. Mater. 2022, 421, 126760. [Google Scholar] [CrossRef]
- Meiners, S.; Eickelberg, O.; Königshoff, M. Hallmarks of the ageing lung. Eur. Respir. J. 2015, 45, 807–827. [Google Scholar] [CrossRef]
- Horowitz, J.C.; Thannickal, V.J. Mechanisms for the Resolution of Organ Fibrosis. Physiology 2019, 34, 43–55. [Google Scholar] [CrossRef]
- Jun, J.-I.; Lau, L.F. Resolution of organ fibrosis. J. Clin. Investig. 2018, 128, 97–107. [Google Scholar] [CrossRef]
- Winters, N.I.; Burman, A.; Kropski, J.A.; Blackwell, T.S. Epithelial Injury and Dysfunction in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Am. J. Med. Sci. 2019, 357, 374–378. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.B.; Penkala, I.J.; Zepp, J.A.; Sivakumar, A.; Linares-Saldana, R.; Zacharias, W.J.; Stolz, K.G.; Pankin, J.; Lu, M.; Wang, Q.; et al. Early lineage specification defines alveolar epithelial ontogeny in the murine lung. Proc. Natl. Acad. Sci. USA 2019, 116, 4362–4371. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Perl, A.-K.; Li, R.; Bell, S.M.; Sajti, E.; Kalinichenko, V.V.; Kalin, T.V.; Misra, R.S.; Deshmukh, H.; Clair, G.; et al. A census of the lung: CellCards from LungMAP. Dev. Cell 2022, 57, 112–145.e2. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Du, Y.; Gokey, J.; Ray, S.; Bell, S.M.; Adam, M.; Sudha, P.; Perl, A.K.; Deshmukh, H.; Potter, S.S.; et al. Single cell RNA analysis identifies cellular heterogeneity and adaptive responses of the lung at birth. Nat. Commun. 2019, 10, 37. [Google Scholar] [CrossRef]
- Travaglini, K.J.; Nabhan, A.N.; Penland, L.; Sinha, R.; Gillich, A.; Sit, R.V.; Chang, S.; Conley, S.D.; Mori, Y.; Seita, J.; et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 2020, 587, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Braga, F.A.V.; Kar, G.; Berg, M.; Carpaij, O.A.; Polański, K.; Simon, L.M.; Brouwer, S.; Gomes, T.; Hesse, L.; Jiang, J.; et al. A cellular census of human lungs identifies novel cell states in health and in asthma. Nat. Med. 2019, 25, 1153–1163. [Google Scholar] [CrossRef] [PubMed]
- Ciccimarra, R.; Bolognesi, M.M.; Zoboli, M.; Cattoretti, G.; Stellari, F.F.; Ravanetti, F. The normal and fibrotic mouse lung classified by spatial proteomic analysis. Sci. Rep. 2022, 12, 8742. [Google Scholar] [CrossRef]
- Aspal, M.; Zemans, R.L. Mechanisms of ATII-to-ATI Cell Differentiation during Lung Regeneration. Int. J. Mol. Sci. 2020, 21, 3188. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Transforming growth factor–β in tissue fibrosis. J. Exp. Med. 2020, 217, e20190103. [Google Scholar] [CrossRef]
- Pardo, A.; Selman, M. Lung Fibroblasts, Aging, and Idiopathic Pulmonary Fibrosis. Ann. Am. Thorac. Soc. 2016, 13, S417–S421. [Google Scholar] [CrossRef]
- Hinz, B.; Lagares, D. Myofibroblasts; Springer: New York, NY, USA, 2021. [Google Scholar]
- Fernandez, I.E.; Eickelberg, O. New cellular and molecular mechanisms of lung injury and fibrosis in idiopathic pulmonary fibrosis. Lancet 2012, 380, 680–688. [Google Scholar] [CrossRef]
- Carrington, R.; Jordan, S.; Pitchford, S.; Page, C. Use of animal models in IPF research. Pulm. Pharmacol. Ther. 2018, 51, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Shi, J.; Tang, H. Animal models of drug-induced pulmonary fibrosis: An overview of molecular mechanisms and characteristics. Cell Biol. Toxicol. 2021, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Oglesby, I.K.; Schweikert, A.; Fox, B.; Redmond, C.; Donnelly, S.C.; Hurley, K. Lung organoids and other preclinical models of pulmonary fibrosis. QJM Int. J. Med. 2021, 114, 167–173. [Google Scholar] [CrossRef]
- Li, B.; Mu, M.; Sun, Q.; Cao, H.; Liu, Q.; Liu, J.; Zhang, J.; Xu, K.; Hu, D.; Tao, X.; et al. A suitable silicosis mouse model was constructed by repeated inhalation of silica dust via nose. Toxicol. Lett. 2021, 353, 1–12. [Google Scholar] [CrossRef]
- Cheresh, P.; Morales-Nebreda, L.; Kim, S.-J.; Yeldandi, A.; Williams, D.B.; Cheng, Y.; Mutlu, G.M.; Budinger, G.R.S.; Ridge, K.; Schumacker, P.T.; et al. Asbestos-Induced Pulmonary Fibrosis Is Augmented in 8-Oxoguanine DNA Glycosylase Knockout Mice. Am. J. Respir. Cell Mol. Biol. 2015, 52, 25–36. [Google Scholar] [CrossRef]
- Yee, M.; Gelein, R.; Mariani, T.J.; Lawrence, B.P.; O’Reilly, M.A. The Oxygen Environment at Birth Specifies the Population of Alveolar Epithelial Stem Cells in the Adult Lung. Stem Cells 2016, 34, 1396–1406. [Google Scholar] [CrossRef]
- Marinova, M.; Solopov, P.; Dimitropoulou, C.; Biancatelli, R.M.L.C.; Catravas, J.D. Acute exposure of mice to hydrochloric acid leads to the development of chronic lung injury and pulmonary fibrosis. Inhal. Toxicol. 2019, 31, 147–160. [Google Scholar] [CrossRef]
- Lee, C.G.; Kang, H.-R.; Homer, R.J.; Chupp, G.; Elias, J.A. Transgenic Modeling of Transforming Growth Factor- 1: Role of Apoptosis in Fibrosis and Alveolar Remodeling. Proc. Am. Thorac. Soc. 2006, 3, 418–423. [Google Scholar] [CrossRef]
- Roberts, S.N.; Howie, S.E.M.; Wallace, W.A.H.; Brown, D.M.; Lamb, D.; Ramage, E.A.; Donaldson, K. A novel model for human interstitial lung disease: Hapten-driven lung fibrosis in rodents. J. Pathol. 1995, 176, 309–318. [Google Scholar] [CrossRef]
- Karvonen, R.L.; Fernandez-Madrid, F.; Maughan, R.L.; Palmer, K.C.; Fernandez-Madrid, I. An Animal Model of Pulmonary Radiation Fibrosis with Biochemical, Physiologic, Immunologic and Morphologic Observations. Radiat. Res. 1987, 111, 68. [Google Scholar] [CrossRef] [PubMed]
- Naikawadi, R.P.; Disayabutr, S.; Mallavia, B.; Donne, M.L.; Green, G.; La, J.L.; Rock, J.R.; Looney, M.R.; Wolters, P.J. Telomere dysfunction in alveolar epithelial cells causes lung remodeling and fibrosis. JCI Insight 2016, 1, e86704. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, G.; Moore, B.B.; Chambers, R.C.; Eickelberg, O.; Königshoff, M.; Kolb, M.; Laurent, G.J.; Nanthakumar, C.B.; Olman, M.A.; Pardo, A.; et al. An Official American Thoracic Society Workshop Report: Use of Animal Models for the Preclinical Assessment of Potential Therapies for Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2017, 56, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Degryse, A.L.; Tanjore, H.; Xu, X.C.; Polosukhin, V.V.; Jones, B.R.; McMahon, F.B.; Gleaves, L.A.; Blackwell, T.S.; Lawson, W.E. Repetitive intratracheal bleomycin models several features of idiopathic pulmonary fibrosis. Am. J. Physiol. Cell. Mol. Physiol. 2010, 299, L442–L452. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Lis, R.; Ginsberg, M.; Chavez, D.; Shido, K.; Rabbany, S.Y.; Fong, G.-H.; Sakmar, T.; Rafii, S.; Ding, B.-S. Targeting of the pulmonary capillary vascular niche promotes lung alveolar repair and ameliorates fibrosis. Nat. Med. 2016, 22, 154–162. [Google Scholar] [CrossRef]
- Liu, T.; de Los Santos, F.G.; Phan, S.H. The Bleomycin Model of Pulmonary Fibrosis. Methods Mol. Biol. 2017, 1627, 27–42. [Google Scholar] [CrossRef]
- Moore, B.B.; Lawson, W.E.; Oury, T.D.; Sisson, T.H.; Raghavendran, K.; Hogaboam, C.M. Animal Models of Fibrotic Lung Disease. Am. J. Respir. Cell Mol. Biol. 2013, 49, 167–179. [Google Scholar] [CrossRef]
- Hirota, J.A.; Hiebert, P.R.; Gold, M.; Wu, D.; Graydon, C.; Smith, J.A.; Ask, K.; McNagny, K.; Granville, D.J.; Knight, D.A. Granzyme B Deficiency Exacerbates Lung Inflammation in Mice after Acute Lung Injury. Am. J. Respir. Cell Mol. Biol. 2013, 49, 453–462. [Google Scholar] [CrossRef]
- Li, Y.J.; Azuma, A.; Usuki, J.; Abe, S.; Matsuda, K.; Sunazuka, T.; Shimizu, T.; Hirata, Y.; Inagaki, H.; Kawada, T.; et al. EM703 improves bleomycin-induced pulmonary fibrosis in mice by the inhibition of TGF-β signaling in lung fibroblasts. Respir. Res. 2006, 7, 16. [Google Scholar] [CrossRef]
- Babin, A.L.; Cannet, C.; Gérard, C.; Saint-Mezard, P.; Page, C.P.; Sparrer, H.; Matsuguchi, T.; Beckmann, N. Bleomycin-induced lung injury in mice investigated by MRI: Model assessment for target analysis. Magn. Reson. Med. 2012, 67, 499–509. [Google Scholar] [CrossRef]
- Le, T.-T.T.; Karmouty-Quintana, H.; Melicoff, E.; Le, T.-T.T.; Weng, T.; Chen, N.-Y.; Pedroza, M.; Zhou, Y.; Davies, J.; Philip, K.; et al. Blockade of IL-6 Trans Signaling Attenuates Pulmonary Fibrosis. J. Immunol. 2014, 193, 3755–3768. [Google Scholar] [CrossRef] [PubMed]
- Egger, C.; Cannet, C.; Gérard, C.; Jarman, E.; Jarai, G.; Feige, A.; Suply, T.; Micard, A.; Dunbar, A.; Tigani, B.; et al. Administration of Bleomycin via the Oropharyngeal Aspiration Route Leads to Sustained Lung Fibrosis in Mice and Rats as Quantified by UTE-MRI and Histology. PLoS ONE 2013, 8, e63432. [Google Scholar] [CrossRef] [PubMed]
- Oku, H.; Shimizu, T.; Kawabata, T.; Nagira, M.; Hikita, I.; Ueyama, A.; Matsushima, S.; Torii, M.; Arimura, A. Antifibrotic action of pirfenidone and prednisolone: Different effects on pulmonary cytokines and growth factors in bleomycin-induced murine pulmonary fibrosis. Eur. J. Pharmacol. 2008, 590, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Tashiro, J.; Rubio, G.A.; Limper, A.H.; Williams, K.; Elliot, S.J.; Ninou, I.; Aidinis, V.; Tzouvelekis, A.; Glassberg, M.K. Exploring Animal Models That Resemble Idiopathic Pulmonary Fibrosis. Front. Med. 2017, 4, 118. [Google Scholar] [CrossRef]
- Chen, I.-T.; Huang, L.-T.; Chen, C.-C.; Chen, C.-M. Molecular mechanisms underlying hyperoxia-induced lung fibrosis. Pediatr. Neonatol. 2022, 63, 109–116. [Google Scholar] [CrossRef]
- Bozyk, P.D.; Bentley, J.K.; Popova, A.P.; Anyanwu, A.C.; Linn, M.D.; Goldsmith, A.M.; Pryhuber, G.S.; Moore, B.B.; Hershenson, M.B. Neonatal Periostin Knockout Mice Are Protected from Hyperoxia-Induced Alveolar Simplication. PLoS ONE 2012, 7, e31336. [Google Scholar] [CrossRef]
- Alsafadi, H.N.; Staab-Weijnitz, C.; Lehmann, M.; Lindner, M.; Peschel, B.; Königshoff, M.; Wagner, D.E. An ex vivo model to induce early fibrosis-like changes in human precision-cut lung slices. Am. J. Physiol. Cell. Mol. Physiol. 2017, 312, L896–L902. [Google Scholar] [CrossRef]
- Kim, S.Y.; Mongey, R.; Wang, P.; Rothery, S.; Gaboriau, D.C.; Hind, M.; Griffiths, M.; Dean, C.H. The acid injury and repair (AIR) model: A novel ex-vivo tool to understand lung repair. Biomaterials 2021, 267, 120480. [Google Scholar] [CrossRef]
- Strikoudis, A.; Cieślak, A.; Loffredo, L.; Chen, Y.-W.; Patel, N.; Saqi, A.; Lederer, D.J.; Snoeck, H.-W. Modeling of Fibrotic Lung Disease Using 3D Organoids Derived from Human Pluripotent Stem Cells. Cell Rep. 2019, 27, 3709–3723.e5. [Google Scholar] [CrossRef]
- Mejías, J.C.; Nelson, M.R.; Liseth, O.; Roy, K. A 96-well format microvascularized human lung-on-a-chip platform for microphysiological modeling of fibrotic diseases. Lab Chip 2020, 20, 3601–3611. [Google Scholar] [CrossRef]
- Varone, A.; Nguyen, J.K.; Leng, L.; Barrile, R.; Sliz, J.; Lucchesi, C.; Wen, N.; Gravanis, A.; Hamilton, G.A.; Karalis, K.; et al. A novel organ-chip system emulates three-dimensional architecture of the human epithelia and the mechanical forces acting on it. Biomaterials 2021, 275, 120957. [Google Scholar] [CrossRef]
- Chen, H.; Qu, J.; Huang, X.; Kurundkar, A.; Zhu, L.; Yang, N.; Venado, A.; Ding, Q.; Liu, G.; Antony, V.B.; et al. Mechanosensing by the α6-integrin confers an invasive fibroblast phenotype and mediates lung fibrosis. Nat. Commun. 2016, 7, 12564. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Armendariz, A.I.; Barroso, M.M.; El Agha, E.; Herold, S. 3D In Vitro Models: Novel Insights into Idiopathic Pulmonary Fibrosis Pathophysiology and Drug Screening. Cells 2022, 11, 1526. [Google Scholar] [CrossRef] [PubMed]
- Bailey, K.E.; Pino, C.; Lennon, M.L.; Lyons, A.; Jacot, J.G.; Lammers, S.R.; Königshoff, M.; Magin, C.M. Embedding of Precision-Cut Lung Slices in Engineered Hydrogel Biomaterials Supports Extended Ex Vivo Culture. Am. J. Respir. Cell Mol. Biol. 2020, 62, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Hogan, B.L.; Barkauskas, C.E.; Chapman, H.A.; Epstein, J.A.; Jain, R.; Hsia, C.C.; Niklason, L.; Calle, E.; Le, A.; Randell, S.H.; et al. Repair and Regeneration of the Respiratory System: Complexity, Plasticity, and Mechanisms of Lung Stem Cell Function. Cell Stem Cell 2014, 15, 123–138. [Google Scholar] [CrossRef]
- Lee, J.-H.; Rawlins, E.L. Developmental mechanisms and adult stem cells for therapeutic lung regeneration. Dev. Biol. 2018, 433, 166–176. [Google Scholar] [CrossRef]
- Tata, P.R.; Rajagopal, J. Plasticity in the lung: Making and breaking cell identity. Development 2017, 144, 755–766. [Google Scholar] [CrossRef]
- Konkimalla, A.; Tata, A.; Tata, P.R. Lung Regeneration: Cells, Models, and Mechanisms. Cold Spring Harb. Perspect. Biol. 2021, a040873. [Google Scholar] [CrossRef]
- Basil, M.C.; Katzen, J.; Engler, A.E.; Guo, M.; Herriges, M.J.; Kathiriya, J.J.; Windmueller, R.; Ysasi, A.B.; Zacharias, W.J.; Chapman, H.A.; et al. The Cellular and Physiological Basis for Lung Repair and Regeneration: Past, Present and Future. Cell Stem Cell 2020, 26, 482–502. [Google Scholar] [CrossRef]
- Wu, H.; Tang, N. Stem cells in pulmonary alveolar regeneration. Development 2021, 148, dev193458. [Google Scholar] [CrossRef]
- Leach, J.P.; Morrisey, E.E. Repairing the lungs one breath at a time: How dedicated or facultative are you? Genes Dev. 2018, 32, 1461–1471. [Google Scholar] [CrossRef]
- Xie, T.; Lynn, H.; Parks, W.C.; Stripp, B.; Chen, P.; Jiang, D.; Noble, P.W. Abnormal respiratory progenitors in fibrotic lung injury. Stem Cell Res. Ther. 2022, 13, 64. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.J.; Bils, R.F. Identification of Cells Labeled with Tritiated Thymidine in the Pulmonary Alveolar Walls of the Mouse. Am. Rev. Respir. Dis. 1969, 100, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.J.; Dekker, N.P.; Cabral-Anderson, L.J.; Freeman, G. Quantitation of Damage to the Alveolar Epithelium by Means of Type 2 Cell Proliferation. Am. Rev. Respir. Dis. 1978, 118, 787–790. [Google Scholar] [CrossRef]
- Evans, M.J.; Cabral, L.J.; Stephens, R.J.; Freeman, G. Renewal of alveolar epithelium in the rat following exposure to NO2. Am. J. Pathol. 1973, 70, 175–198. [Google Scholar] [PubMed]
- Chapman, H.A.; Li, X.; Alexander, J.P.; Brumwell, A.; Lorizio, W.; Tan, K.; Sonnenberg, A.; Wei, Y.; Vu, T.H. Integrin α6β4 identifies an adult distal lung epithelial population with regenerative potential in mice. J. Clin. Investig. 2011, 121, 2855–2862. [Google Scholar] [CrossRef] [PubMed]
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.J.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L.M. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Investig. 2013, 123, 3025–3036. [Google Scholar] [CrossRef]
- Desai, T.J.; Brownfield, D.; Krasnow, M.A. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature 2014, 507, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Katsura, H.; Kobayashi, Y.; Tata, P.R.; Hogan, B.L. IL-1 and TNFα Contribute to the Inflammatory Niche to Enhance Alveolar Regeneration. Stem Cell Rep. 2019, 12, 657–666. [Google Scholar] [CrossRef]
- Khatri, A.; Kraft, B.D.; Tata, P.R.; Randell, S.H.; Piantadosi, C.A.; Pendergast, A.M. ABL kinase inhibition promotes lung regeneration through expansion of an SCGB1A1+ SPC+ cell population following bacterial pneumonia. Proc. Natl. Acad. Sci. USA 2019, 116, 1603–1612. [Google Scholar] [CrossRef]
- Liu, Z.; Wu, H.; Jiang, K.; Wang, Y.; Zhang, W.; Chu, Q.; Li, J.; Huang, H.; Cai, T.; Ji, H.; et al. MAPK-Mediated YAP Activation Controls Mechanical-Tension-Induced Pulmonary Alveolar Regeneration. Cell Rep. 2016, 16, 1810–1819. [Google Scholar] [CrossRef] [PubMed]
- Rock, J.R.; Barkauskas, C.E.; Cronce, M.J.; Xue, Y.; Harris, J.R.; Liang, J.; Noble, P.W.; Hogan, B.L. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc. Natl. Acad. Sci. USA 2011, 108, E1475–E1483. [Google Scholar] [CrossRef] [PubMed]
- Basil, M.C.; Morrisey, E.E. Lung regeneration: A tale of mice and men. Semin. Cell Dev. Biol. 2020, 100, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Yu, Y.; Huang, H.; Hu, Y.; Fu, S.; Wang, Z.; Shi, M.; Zhao, X.; Yuan, J.; Li, J.; et al. Progressive Pulmonary Fibrosis Is Caused by Elevated Mechanical Tension on Alveolar Stem Cells. Cell 2020, 180, 107–121.e17. [Google Scholar] [CrossRef]
- Chen, Q.; Kumar, V.S.; Finn, J.; Jiang, D.; Liang, J.; Zhao, Y.-Y.; Liu, Y. CD44high alveolar type II cells show stem cell properties during steady-state alveolar homeostasis. Am. J. Physiol. Cell. Mol. Physiol. 2017, 313, L41–L51. [Google Scholar] [CrossRef]
- Nabhan, A.N.; Brownfield, D.G.; Harbury, P.B.; Krasnow, M.A.; Desai, T.J. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science 2018, 359, 1118–1123. [Google Scholar] [CrossRef]
- Zacharias, W.; Frank, D.B.; Zepp, J.A.; Morley, M.P.; Alkhaleel, F.A.; Kong, J.; Zhou, S.; Cantu, E.; Morrisey, E.E. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature 2018, 555, 251–255. [Google Scholar] [CrossRef]
- Xi, Y.; Kim, T.; Brumwell, A.N.; Driver, I.H.; Wei, Y.; Tan, V.; Jackson, J.R.; Xu, J.; Lee, D.-K.; Gotts, J.E.; et al. Local lung hypoxia determines epithelial fate decisions during alveolar regeneration. Nature 2017, 19, 904–914. [Google Scholar] [CrossRef]
- Giangreco, A.; Reynolds, S.D.; Stripp, B.R. Terminal Bronchioles Harbor a Unique Airway Stem Cell Population That Localizes to the Bronchoalveolar Duct Junction. Am. J. Pathol. 2002, 161, 173–182. [Google Scholar] [CrossRef]
- Kim, C.F.B.; Jackson, E.L.; Woolfenden, A.E.; Lawrence, S.; Babar, I.; Vogel, S.; Crowley, D.; Bronson, R.T.; Jacks, T. Identification of Bronchioalveolar Stem Cells in Normal Lung and Lung Cancer. Cell 2005, 121, 823–835. [Google Scholar] [CrossRef]
- Salwig, I.; Spitznagel, B.; Vazquez-Armendariz, A.I.; Khalooghi, K.; Guenther, S.; Herold, S.; Szibor, M.; Braun, T. Bronchioalveolar stem cells are a main source for regeneration of distal lung epithelia in vivo. EMBO J. 2019, 38, e102099. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Liu, K.; Cui, G.; Huang, X.; Yao, S.; Guo, W.; Qin, Z.; Li, Y.; Yang, R.; Pu, W.; et al. Lung regeneration by multipotent stem cells residing at the bronchioalveolar-duct junction. Nat. Genet. 2019, 51, 728–738. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Tang, M.; Liu, Q.; Han, X.; Jin, H.; Zhu, H.; Li, Y.; He, L.; Ji, H.; Zhou, B. Bi-directional differentiation of single bronchioalveolar stem cells during lung repair. Cell Discov. 2020, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Van Winkle, L.S.; Buckpitt, A.R.; Nishio, S.J.; Isaac, J.M.; Plopper, C.G. Cellular response in naphthalene-induced Clara cell injury and bronchiolar epithelial repair in mice. Am. J. Physiol. Cell. Mol. Physiol. 1995, 269, L800–L818. [Google Scholar] [CrossRef]
- Reynolds, S.D.; Reynolds, P.R.; Pryhuber, G.S.; Finder, J.D.; Stripp, B.R. Secretoglobins SCGB3A1 and SCGB3A2 Define Secretory Cell Subsets in Mouse and Human Airways. Am. J. Respir. Crit. Care Med. 2002, 166, 1498–1509. [Google Scholar] [CrossRef] [PubMed]
- Rawlins, E.L.; Okubo, T.; Xue, Y.; Brass, D.M.; Auten, R.L.; Hasegawa, H.; Wang, F.; Hogan, B.L. The Role of Scgb1a1+ Clara Cells in the Long-Term Maintenance and Repair of Lung Airway, but Not Alveolar, Epithelium. Cell Stem Cell 2009, 4, 525–534. [Google Scholar] [CrossRef]
- McQualter, J.L.; Yuen, K.; Williams, B.; Bertoncello, I. Evidence of an epithelial stem/progenitor cell hierarchy in the adult mouse lung. Proc. Natl. Acad. Sci. USA 2010, 107, 1414–1419. [Google Scholar] [CrossRef]
- Kumar, P.A.; Hu, Y.; Yamamoto, Y.; Hoe, N.B.; Wei, T.S.; Mu, D.; Sun, Y.; Joo, L.S.; Dagher, R.; Zielonka, E.M.; et al. Distal Airway Stem Cells Yield Alveoli In Vitro and during Lung Regeneration following H1N1 Influenza Infection. Cell 2011, 147, 525–538. [Google Scholar] [CrossRef]
- Lee, J.-H.; Bhang, D.H.; Beede, A.; Huang, T.L.; Stripp, B.R.; Bloch, K.D.; Wagers, A.J.; Tseng, Y.-H.; Ryeom, S.; Kim, C.F. Lung Stem Cell Differentiation in Mice Directed by Endothelial Cells via a BMP4-NFATc1-Thrombospondin-1 Axis. Cell 2014, 156, 440–455. [Google Scholar] [CrossRef]
- Vaughan, A.E.; Brumwell, A.N.; Xi, Y.; Gotts, J.E.; Brownfield, D.G.; Treutlein, B.; Tan, K.; Tan, V.; Liu, F.C.; Looney, M.R.; et al. Lineage-negative progenitors mobilize to regenerate lung epithelium after major injury. Nature 2015, 517, 621–625. [Google Scholar] [CrossRef]
- Zuo, W.; Zhang, T.; Wu, D.Z.; Guan, S.P.; Liew, A.-A.; Yamamoto, Y.; Wang, X.; Lim, S.J.; Vincent, M.; Lessard, M.; et al. p63+ Krt5+ distal airway stem cells are essential for lung regeneration. Nature 2015, 517, 616–620. [Google Scholar] [CrossRef] [PubMed]
- McConnell, A.; Yao, C.; Yeckes, A.R.; Wang, Y.; Selvaggio, A.S.; Tang, J.; Kirsch, D.G.; Stripp, B.R. p53 Regulates Progenitor Cell Quiescence and Differentiation in the Airway. Cell Rep. 2016, 17, 2173–2182. [Google Scholar] [CrossRef] [PubMed]
- Guha, A.; Deshpande, A.; Jain, A.; Sebastiani, P.; Cardoso, W.V. Uroplakin 3a+ Cells Are a Distinctive Population of Epithelial Progenitors that Contribute to Airway Maintenance and Post-injury Repair. Cell Rep. 2017, 19, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Riccio, P.; Schotsaert, M.; Mori, M.; Lu, J.; Lee, D.-K.; García-Sastre, A.; Xu, J.; Cardoso, W.V. Spatial-Temporal Lineage Restrictions of Embryonic p63+ Progenitors Establish Distinct Stem Cell Pools in Adult Airways. Dev. Cell 2018, 44, 752–761.e4. [Google Scholar] [CrossRef] [PubMed]
- Kathiriya, J.J.; Brumwell, A.N.; Jackson, J.R.; Tang, X.; Chapman, H.A. Distinct Airway Epithelial Stem Cells Hide among Club Cells but Mobilize to Promote Alveolar Regeneration. Cell. Stem. Cell. 2020, 26, 346–358.e4. [Google Scholar] [CrossRef] [PubMed]
- Strunz, M.; Simon, L.M.; Ansari, M.; Kathiriya, J.J.; Angelidis, I.; Mayr, C.H.; Tsidiridis, G.; Lange, M.; Mattner, L.F.; Yee, M.; et al. Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat. Commun. 2020, 11, 3559. [Google Scholar] [CrossRef]
- Jain, R.; Barkauskas, C.E.; Takeda, N.; Bowie, E.; Aghajanian, H.; Wang, Q.; Padmanabhan, A.; Manderfield, L.J.; Gupta, M.; Li, D.; et al. Plasticity of Hopx+ type I alveolar cells to regenerate type II cells in the lung. Nat. Commun. 2015, 6, 6727. [Google Scholar] [CrossRef]
- Penkala, I.J.; Liberti, D.C.; Pankin, J.; Sivakumar, A.; Kremp, M.M.; Jayachandran, S.; Katzen, J.; Leach, J.P.; Windmueller, R.; Stolz, K.; et al. Age-dependent alveolar epithelial plasticity orchestrates lung homeostasis and regeneration. Cell Stem Cell 2021, 28, 1775–1789.e5. [Google Scholar] [CrossRef]
- Pardo-Saganta, A.; Tata, P.R.; Law, B.M.; Saez, B.; Chow, R.; Prabhu, M.; Gridley, T.; Rajagopal, J. Parent stem cells can serve as niches for their daughter cells. Nature 2015, 523, 597–601. [Google Scholar] [CrossRef]
- D’Alessio, F.R.; Tsushima, K.; Aggarwal, N.R.; West, E.E.; Willett, M.H.; Britos, M.F.; Pipeling, M.R.; Brower, R.G.; Tuder, R.M.; McDyer, J.F.; et al. CD4+ CD25+ Foxp3+ Tregs resolve experimental lung injury in mice and are present in humans with acute lung injury. J. Clin. Investig. 2009, 119, 2898–2913. [Google Scholar] [CrossRef]
- Flozak, A.S.; Lam, A.P.; Russell, S.; Jain, M.; Peled, O.N.; Sheppard, K.A.; Beri, R.; Mutlu, G.M.; Budinger, G.S.; Gottardi, C.J. β-Catenin/T-cell Factor Signaling Is Activated during Lung Injury and Promotes the Survival and Migration of Alveolar Epithelial Cells*. J. Biol. Chem. 2010, 285, 3157–3167. [Google Scholar] [CrossRef] [PubMed]
- Al Alam, D.; Green, M.; Irani, R.T.; Parsa, S.; Danopoulos, S.; Sala, F.G.; Branch, J.; El Agha, E.; Tiozzo, C.; Voswinckel, R.; et al. Contrasting Expression of Canonical Wnt Signaling Reporters TOPGAL, BATGAL and Axin2LacZ during Murine Lung Development and Repair. PLoS ONE 2011, 6, e23139. [Google Scholar] [CrossRef] [PubMed]
- Aumiller, V.; Balsara, N.; Wilhelm, J.; Günther, A.; Königshoff, M. WNT/β-Catenin Signaling Induces IL-1β Expression by Alveolar Epithelial Cells in Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2013, 49, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Lange, A.W.; Sridharan, A.; Xu, Y.; Stripp, B.R.; Perl, A.-K.; Whitsett, J.A. Hippo/Yap signaling controls epithelial progenitor cell proliferation and differentiation in the embryonic and adult lung. J. Mol. Cell Biol. 2015, 7, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, Z.; Chu, Q.; Jiang, K.; Li, J.; Tang, N. The Strength of Mechanical Forces Determines the Differentiation of Alveolar Epithelial Cells. Dev. Cell 2018, 44, 297–312.e5. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.-I.; Bujnis, M.; Barkauskas, C.E.; Kobayashi, Y.; Hogan, B.L.M. Niche-mediated BMP/SMAD signaling regulates lung alveolar stem cell proliferation and differentiation. Development 2018, 145, dev163014. [Google Scholar] [CrossRef]
- Lacanna, R.; Liccardo, D.; Zhang, P.; Tragesser, L.; Wang, Y.; Cao, T.; Chapman, H.A.; Morrisey, E.E.; Shen, H.; Koch, W.J.; et al. Yap/Taz regulate alveolar regeneration and resolution of lung inflammation. J. Clin. Investig. 2019, 129, 2107–2122. [Google Scholar] [CrossRef]
- Finn, J.; Sottoriva, K.; Pajcini, K.V.; Kitajewski, J.K.; Chen, C.; Zhang, W.; Malik, A.B.; Liu, Y. Dlk1-Mediated Temporal Regulation of Notch Signaling Is Required for Differentiation of Alveolar Type II to Type I Cells during Repair. Cell Rep. 2019, 26, 2942–2954.e5. [Google Scholar] [CrossRef]
- Zepp, J.A.; Morrisey, E.E. Cellular crosstalk in the development and regeneration of the respiratory system. Nat. Rev. Mol. Cell Biol. 2019, 20, 551–566. [Google Scholar] [CrossRef]
- Zepp, J.A.; Zacharias, W.J.; Frank, D.B.; Cavanaugh, C.A.; Zhou, S.; Morley, M.P.; Morrisey, E.E. Distinct Mesenchymal Lineages and Niches Promote Epithelial Self-Renewal and Myofibrogenesis in the Lung. Cell 2017, 170, 1134–1148.e10. [Google Scholar] [CrossRef]
- Zhou, Y.; Horowitz, J.C.; Naba, A.; Ambalavanan, N.; Atabai, K.; Balestrini, J.; Bitterman, P.B.; Corley, R.A.; Ding, B.-S.; Engler, A.J.; et al. Extracellular matrix in lung development, homeostasis and disease. Matrix Biol. 2018, 73, 77–104. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Tata, A.; Konkimalla, A.; Katsura, H.; Lee, R.F.; Ou, J.; Banovich, N.E.; Kropski, J.A.; Tata, P.R. Persistence of a regeneration-associated, transitional alveolar epithelial cell state in pulmonary fibrosis. Nature 2020, 22, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Park, J.-E.; Tsagkogeorga, G.; Yanagita, M.; Koo, B.-K.; Han, N.; Lee, J.-H. Inflammatory Signals Induce AT2 Cell-Derived Damage-Associated Transient Progenitors that Mediate Alveolar Regeneration. Cell Stem Cell 2020, 27, 366–382.e7. [Google Scholar] [CrossRef] [PubMed]
- Riemondy, K.A.; Jansing, N.L.; Jiang, P.; Redente, E.F.; Gillen, A.E.; Fu, R.; Miller, A.J.; Spence, J.R.; Gerber, A.N.; Hesselberth, J.R.; et al. Single-cell RNA sequencing identifies TGF-β as a key regenerative cue following LPS-induced lung injury. JCI Insight 2019, 4, e123637. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Gil de Rubio, R.; Hrycaj, S.M.; Gurczynski, S.J.; Riemondy, K.A.; Moore, B.B.; Omary, M.B.; Ridge, K.M.; Zemans, R.L. Ineffectual Type 2-to-Type 1 Alveolar Epithelial Cell Differentiation in Idiopathic Pulmonary Fibrosis: Persistence of the KRT8hi Transitional State. Am. J. Respir. Crit. Care Med. 2020, 201, 1443–1447. [Google Scholar] [CrossRef]
- Yao, C.; Guan, X.; Carraro, G.; Parimon, T.; Liu, X.; Huang, G.; Mulay, A.; Soukiasian, H.J.; David, G.; Weigt, S.S.; et al. Senescence of Alveolar Type 2 Cells Drives Progressive Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2021, 203, 707–717. [Google Scholar] [CrossRef]
- Murthy, P.K.L.; Sontake, V.; Tata, A.; Kobayashi, Y.; Macadlo, L.; Okuda, K.; Conchola, A.S.; Nakano, S.; Gregory, S.; Miller, L.A.; et al. Human distal lung maps and lineage hierarchies reveal a bipotent progenitor. Nature 2022, 604, 111–119. [Google Scholar] [CrossRef]
- Basil, M.C.; Cardenas-Diaz, F.L.; Kathiriya, J.J.; Morley, M.P.; Carl, J.; Brumwell, A.N.; Katzen, J.; Slovik, K.J.; Babu, A.; Zhou, S.; et al. Human distal airways contain a multipotent secretory cell that can regenerate alveoli. Nature 2022, 604, 120–126. [Google Scholar] [CrossRef]
- Schupp, J.C.; Adams, T.S.; Ahangari, F.; McDonough, J.E.; DeIuliis, G.; Poli, S.; Rosas, I.O.; Yan, X.; Kaminski, N. Single cell RNA velocity analysis of aberrant basaloid cells in pulmonary fibrosis reveals trajectory towards an alveolar type i like cell state. In TP112. TP112 Proteomics/Genomics/Metabolomics in Lung Disease; American Thoracic Society: New York, NY, USA, 2021; p. A4350. [Google Scholar]
- Reyfman, P.A.; Walter, J.M.; Joshi, N.; Anekalla, K.R.; McQuattie-Pimentel, A.C.; Chiu, S.; Fernandez, R.; Akbarpour, M.; Chen, C.-I.; Ren, Z.; et al. Single-Cell Transcriptomic Analysis of Human Lung Provides Insights into the Pathobiology of Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1517–1536. [Google Scholar] [CrossRef]
- Adams, T.S.; Schupp, J.C.; Poli, S.; Ayaub, E.A.; Neumark, N.; Ahangari, F.; Chu, S.G.; Raby, B.A.; DeIuliis, G.; Januszyk, M.; et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1983. [Google Scholar] [CrossRef]
- Habermann, A.C.; Gutierrez, A.J.; Bui, L.T.; Yahn, S.L.; Winters, N.I.; Calvi, C.L.; Peter, L.; Chung, M.-I.; Taylor, C.J.; Jetter, C.; et al. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1972. [Google Scholar] [CrossRef] [PubMed]
- Calle, E.A.; Ghaedi, M.; Sundaram, S.; Sivarapatna, A.; Tseng, M.K.; Niklason, L.E. Strategies for Whole Lung Tissue Engineering. IEEE Trans. Biomed. Eng. 2014, 61, 1482–1496. [Google Scholar] [CrossRef] [PubMed]
- Weiss, D.J. Concise Review: Current Status of Stem Cells and Regenerative Medicine in Lung Biology and Diseases. Stem Cells 2014, 32, 16–25. [Google Scholar] [CrossRef]
- Rosen, C.; Shezen, E.; Aronovich, A.; Klionsky, Y.Z.; Yaakov, Y.; Assayag, M.; Biton, I.E.; Tal, O.; Shakhar, G.; Ben-Hur, H.; et al. Preconditioning allows engraftment of mouse and human embryonic lung cells, enabling lung repair in mice. Nat. Med. 2015, 21, 869–879. [Google Scholar] [CrossRef]
- Nikolić, M.Z.; Caritg, O.; Jeng, Q.; Johnson, J.-A.; Sun, D.; Howell, K.J.; Brady, J.L.; Laresgoiti, U.; Allen, G.; Butler, R.; et al. Human embryonic lung epithelial tips are multipotent progenitors that can be expanded in vitro as long-term self-renewing organoids. eLife 2017, 6, e26575. [Google Scholar] [CrossRef] [PubMed]
- King, T.E., Jr.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A Phase 3 Trial of Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis. New Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef]
- Richeldi, L.; Du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and Safety of Nintedanib in Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; El-Hashash, A.H.K. Cell-based therapy for idiopathic pulmonary fibrosis. Stem Cell Investig. 2019, 6, 22. [Google Scholar] [CrossRef]
- Schlötzer-Schrehardt, U.; Freudenberg, U.; Kruse, F.E. Zukunftstechnologie Tissue-Engineering. Ophthalmologe 2017, 114, 327–340. [Google Scholar] [CrossRef]
- Serrano-Mollar, A.; Gay-Jordi, G.; Guillamat-Prats, R.; Closa, D.; Hernandez-Gonzalez, F.; Marin, P.; Burgos, F.; Martorell, J.; Sánchez, M.; Arguis, P.; et al. Safety and Tolerability of Alveolar Type II Cell Transplantation in Idiopathic Pulmonary Fibrosis. Chest 2016, 150, 533–543. [Google Scholar] [CrossRef]
- Friedenstein, A.J.; Petrakova, K.V.; Kurolesova, A.I.; Frolova, G.P. Heterotopic of bone marrow. Analysis of precursor cells for osteogenic and hematopoietic tissues. Transplantation 1968, 6, 230–247. [Google Scholar] [CrossRef] [PubMed]
- Ruaro, B.; Salton, F.; Braga, L.; Wade, B.; Confalonieri, P.; Volpe, M.C.; Baratella, E.; Maiocchi, S.; Confalonieri, M. The History and Mystery of Alveolar Epithelial Type II Cells: Focus on Their Physiologic and Pathologic Role in Lung. Int. J. Mol. Sci. 2021, 22, 2566. [Google Scholar] [CrossRef] [PubMed]
- Guillamat-Prats, R.; Gay-Jordi, G.; Xaubet, A.; Peinado, V.I.; Serrano-Mollar, A. Alveolar Type II cell transplantation restores pulmonary surfactant protein levels in lung fibrosis. J. Hear. Lung Transplant. 2014, 33, 758–765. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Mollar, A.; Nacher, M.; Gay-Jordi, G.; Closa, D.; Xaubet, A.; Bulbena, O. Intratracheal Transplantation of Alveolar Type II Cells Reverses Bleomycin-induced Lung Fibrosis. Am. J. Respir. Crit. Care Med. 2007, 176, 1261–1268. [Google Scholar] [CrossRef]
- Lopez-Rodriguez, E.; Gay-Jordi, G.; Knudsen, L.; Ochs, M.; Serrano-Mollar, A. Improved Alveolar Dynamics and Structure After Alveolar Epithelial Type II Cell Transplantation in Bleomycin Induced Lung Fibrosis. Front. Med. 2021, 8, 640020. [Google Scholar] [CrossRef]
- Cores, J.; Hensley, M.T.; Kinlaw, K.; Rikard, S.M.; Dinh, P.-U.; Paudel, D.; Tang, J.; Vandergriff, A.C.; Allen, T.A.; Li, Y.; et al. Safety and Efficacy of Allogeneic Lung Spheroid Cells in a Mismatched Rat Model of Pulmonary Fibrosis. Stem Cells Transl. Med. 2017, 6, 1905–1916. [Google Scholar] [CrossRef]
- Fitzsimmons, R.E.B.; Mazurek, M.S.; Soos, A.; Simmons, C.A. Mesenchymal Stromal/Stem Cells in Regenerative Medicine and Tissue Engineering. Stem Cells Int. 2018, 2018, 8031718. [Google Scholar] [CrossRef]
- Keating, A. Mesenchymal Stromal Cells: New Directions. Cell Stem Cell 2012, 10, 709–716. [Google Scholar] [CrossRef]
- Ni, K.; Liu, M.; Zheng, J.; Wen, L.; Chen, Q.; Xiang, Z.; Lam, K.-T.; Liu, Y.; Chan, G.C.-F.; Lau, Y.-L.; et al. PD-1/PD-L1 Pathway Mediates the Alleviation of Pulmonary Fibrosis by Human Mesenchymal Stem Cells in Humanized Mice. Am. J. Respir. Cell Mol. Biol. 2018, 58, 684–695. [Google Scholar] [CrossRef]
- Huleihel, L.; Sellares, J.; Cardenes, N.; Álvarez, D.; Faner, R.; Sakamoto, K.; Yu, G.; Kapetanaki, M.G.; Kaminski, N.; Rojas, M. Modified mesenchymal stem cells using miRNA transduction alter lung injury in a bleomycin model. Am. J. Physiol. Cell. Mol. Physiol. 2017, 313, L92–L103. [Google Scholar] [CrossRef]
- Yan, X.; Liu, Y.; Han, Q.; Jia, M.; Liao, L.; Qi, M.; Zhao, R.C. Injured microenvironment directly guides the differentiation of engrafted Flk-1+ mesenchymal stem cell in lung. Exp. Hematol. 2007, 35, 1466–1475. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, A. Mesenchymal Stem Cell Delivery Routes and Fate. Int. J. Stem Cells 2008, 1, 1158861. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, L.A.; Gambelli, F.; McBride, C.; Gaupp, D.; Baddoo, M.; Kaminski, N.; Phinney, D.G. Mesenchymal stem cell engraftment in lung is enhanced in response to bleomycin exposure and ameliorates its fibrotic effects. Proc. Natl. Acad. Sci. USA 2003, 100, 8407–8411. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Kang, X.; Wang, X.; Wu, S.; Xiao, J.; Li, Z.; Wu, X.; Zhang, W. Conversion of bone marrow mesenchymal stem cells into type II alveolar epithelial cells reduces pulmonary fibrosis by decreasing oxidative stress in rats. Mol. Med. Rep. 2015, 11, 1685–1692. [Google Scholar] [CrossRef]
- Aguilar, S.; Scotton, C.J.; McNulty, K.; Nye, E.; Stamp, G.; Laurent, G.; Bonnet, D.; Janes, S.M. Bone Marrow Stem Cells Expressing Keratinocyte Growth Factor via an Inducible Lentivirus Protects against Bleomycin-Induced Pulmonary Fibrosis. PLoS ONE 2009, 4, e8013. [Google Scholar] [CrossRef]
- Ono, M.; Ohkouchi, S.; Kanehira, M.; Tode, N.; Kobayashi, M.; Ebina, M.; Nukiwa, T.; Irokawa, T.; Ogawa, H.; Akaike, T.; et al. Mesenchymal Stem Cells Correct Inappropriate Epithelial–mesenchyme Relation in Pulmonary Fibrosis Using Stanniocalcin-1. Mol. Ther. 2015, 23, 549–560. [Google Scholar] [CrossRef]
- Lan, Y.-W.; Choo, K.-B.; Chen, C.-M.; Hsing, T.H.; Chen, Y.-B.; Hsieh, C.-H.; Kuo, H.-P.; Chong, K.-Y. Hypoxia-preconditioned mesenchymal stem cells attenuate bleomycin-induced pulmonary fibrosis. Stem Cell Res. Ther. 2015, 6, 97. [Google Scholar] [CrossRef]
- Lan, Y.-W.; Theng, S.-M.; Huang, T.-T.; Choo, K.-B.; Chen, C.-M.; Kuo, H.-P.; Chong, K.-Y. Oncostatin M-Preconditioned Mesenchymal Stem Cells Alleviate Bleomycin-Induced Pulmonary Fibrosis Through Paracrine Effects of the Hepatocyte Growth Factor. Stem Cells Transl. Med. 2017, 6, 1006–1017. [Google Scholar] [CrossRef]
- Gad, E.S.; Salama, A.A.A.; El-Shafie, M.F.; Arafa, H.M.M.; Abdelsalam, R.M.; Khattab, M. The Anti-fibrotic and Anti-inflammatory Potential of Bone Marrow–Derived Mesenchymal Stem Cells and Nintedanib in Bleomycin-Induced Lung Fibrosis in Rats. Inflammation 2020, 43, 123–134. [Google Scholar] [CrossRef]
- Jun, D.; Garat, C.; West, J.; Thorn, N.; Chow, K.; Cleaver, T.; Sullivan, T.; Torchia, E.C.; Childs, C.; Shade, T.; et al. The Pathology of Bleomycin-Induced Fibrosis Is Associated with Loss of Resident Lung Mesenchymal Stem Cells That Regulate Effector T-cell Proliferation. Stem Cells 2011, 29, 725–735. [Google Scholar] [CrossRef]
- Barczyk, M.; Schmidt, M.; Mattoli, S. Stem Cell-Based Therapy in Idiopathic Pulmonary Fibrosis. Stem Cell Rev. Rep. 2015, 11, 598–620. [Google Scholar] [CrossRef] [PubMed]
- Cahill, E.F.; Kennelly, H.; Carty, F.; Mahon, B.P.; English, K. Hepatocyte Growth Factor Is Required for Mesenchymal Stromal Cell Protection against Bleomycin-Induced Pulmonary Fibrosis. Stem Cells Transl. Med. 2016, 5, 1307–1318. [Google Scholar] [CrossRef] [PubMed]
- Moodley, Y.; Vaghjiani, V.; Chan, J.; Baltic, S.; Ryan, M.; Tchongue, J.; Samuel, C.S.; Murthi, P.; Parolini, O.; Manuelpillai, U. Anti-Inflammatory Effects of Adult Stem Cells in Sustained Lung Injury: A Comparative Study. PLoS ONE 2013, 8, e69299. [Google Scholar] [CrossRef] [PubMed]
- Tiono, J.; Surate Solaligue, D.E.; Mižíková, I.; Nardiello, C.; Vadász, I.; Böttcher-Friebertshäuser, E.; Ehrhardt, H.; Herold, S.; Seeger, W.; Morty, R.E. Mouse genetic background impacts susceptibility to hyperoxia-driven perturbations to lung maturation. Pediatr. Pulmonol. 2019, 54, 1060–1077. [Google Scholar] [CrossRef]
- Srour, N.; Thébaud, B. Mesenchymal Stromal Cells in Animal Bleomycin Pulmonary Fibrosis Models: A Systematic Review. Stem Cells Transl. Med. 2015, 4, 1500–1510. [Google Scholar] [CrossRef]
- Tashiro, J.; Elliot, S.J.; Gerth, D.J.; Xia, X.; Pereira-Simon, S.; Choi, R.; Catanuto, P.; Shahzeidi, S.; Toonkel, R.L.; Shah, R.H.; et al. Therapeutic benefits of young, but not old, adipose-derived mesenchymal stem cells in a chronic mouse model of bleomycin-induced pulmonary fibrosis. Transl. Res. 2015, 166, 554–567. [Google Scholar] [CrossRef]
- Kotani, T.; Masutani, R.; Suzuka, T.; Oda, K.; Makino, S.; Ii, M. Anti-inflammatory and anti-fibrotic effects of intravenous adipose-derived stem cell transplantation in a mouse model of bleomycin-induced interstitial pneumonia. Sci. Rep. 2017, 7, 14608. [Google Scholar] [CrossRef]
- Lee, S.H.; Lee, E.J.; Kim, J.H.; Shim, J.J.; Shin, C.; In, K.H.; Kang, K.H.; Uhm, C.S.; Kim, H.-K.; Yang, K.-S.; et al. The effect of adipose stem cell therapy on pulmonary fibrosis induced by repetitive intratracheal bleomycin in mice. Exp. Lung Res. 2014, 40, 117–125. [Google Scholar] [CrossRef]
- Rubio, G.A.; Elliot, S.J.; Wikramanayake, T.C.; Xia, X.; Pereira-Simon, S.; Thaller, S.R.; Glinos, G.D.; Jozic, I.; Hirt, P.; Pastar, I.; et al. Mesenchymal stromal cells prevent bleomycin-induced lung and skin fibrosis in aged mice and restore wound healing. J. Cell. Physiol. 2018, 233, 5503–5512. [Google Scholar] [CrossRef]
- Reddy, M.; Fonseca, L.; Gowda, S.; Chougule, B.; Hari, A.; Totey, S. Human Adipose-derived Mesenchymal Stem Cells Attenuate Early Stage of Bleomycin Induced Pulmonary Fibrosis: Comparison with Pirfenidone. Int. J. Stem Cells 2016, 9, 192–206. [Google Scholar] [CrossRef]
- Li, F.; Han, F.; Li, H.; Zhang, J.; Qiao, X.; Shi, J.; Yang, L.; Dong, J.; Luo, M.; Wei, J.; et al. Human placental mesenchymal stem cells of fetal origins-alleviated inflammation and fibrosis by attenuating MyD88 signaling in bleomycin-induced pulmonary fibrosis mice. Mol. Immunol. 2017, 90, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Garcia, O.; Carraro, G.; Turcatel, G.; Hall, M.; Sedrakyan, S.; Roche, T.; Buckley, S.; Driscoll, B.; Perin, L.; Warburton, D. Amniotic Fluid Stem Cells Inhibit the Progression of Bleomycin-Induced Pulmonary Fibrosis via CCL2 Modulation in Bronchoalveolar Lavage. PLoS ONE 2013, 8, e71679. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; He, Z.; Gao, Y.; Zheng, R.; Zhang, X.; Zhao, L.; Tan, M. Induced Pluripotent Stem Cells Inhibit Bleomycin-Induced Pulmonary Fibrosis in Mice through Suppressing TGF-β1/Smad-Mediated Epithelial to Mesenchymal Transition. Front. Pharmacol. 2016, 7, 430. [Google Scholar] [CrossRef] [PubMed]
- How, C.-K.; Chien, Y.; Yang, K.-Y.; Shih, H.-C.; Juan, C.-C.; Yang, Y.-P.; Chiou, G.-Y.; Huang, P.-I.; Chang, Y.-L.; Chen, L.-K.; et al. Induced Pluripotent Stem Cells Mediate the Release of Interferon Gamma–Induced Protein 10 and Alleviate Bleomycin-Induced Lung Inflammation and Fibrosis. Shock 2013, 39, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Geoghegan, E.; Byrnes, L. Mouse induced pluripotent stem cells. Int. J. Dev. Biol. 2008, 52, 1015–1022. [Google Scholar] [CrossRef]
- Zhou, Q.; Ye, X.; Sun, R.; Matsumoto, Y.; Moriyama, M.; Asano, Y.; Ajioka, Y.; Saijo, Y. Differentiation of Mouse Induced Pluripotent Stem Cells into Alveolar Epithelial Cells In Vitro for Use In Vivo. Stem Cells Transl. Med. 2014, 3, 675–685. [Google Scholar] [CrossRef]
- Banerjee, E.R.; Laflamme, M.; Papayannopoulou, T.; Kahn, M.; Murry, C.E.; Henderson, W.R., Jr. Human Embryonic Stem Cells Differentiated to Lung Lineage-Specific Cells Ameliorate Pulmonary Fibrosis in a Xenograft Transplant Mouse Model. PLoS ONE 2012, 7, e33165. [Google Scholar] [CrossRef]
- Jacob, A.; Morley, M.; Hawkins, F.; McCauley, K.B.; Jean, J.; Heins, H.; Na, C.-L.; Weaver, T.E.; Vedaie, M.; Hurley, K.; et al. Differentiation of Human Pluripotent Stem Cells into Functional Lung Alveolar Epithelial Cells. Cell Stem Cell 2017, 21, 472–488.e10. [Google Scholar] [CrossRef]
- Alvarez-Palomo, B.; Sanchez-Lopez, L.I.; Moodley, Y.; Edel, M.J.; Serrano-Mollar, A. Induced pluripotent stem cell-derived lung alveolar epithelial type II cells reduce damage in bleomycin-induced lung fibrosis. Stem Cell Res. Ther. 2020, 11, 213. [Google Scholar] [CrossRef]
- Qiao, Y.; Agboola, O.S.; Hu, X.; Wu, Y.; Lei, L. Tumorigenic and Immunogenic Properties of Induced Pluripotent Stem Cells: A Promising Cancer Vaccine. Stem Cell Rev. Rep. 2020, 16, 1049–1061. [Google Scholar] [CrossRef]
- Hogan, B.; Tata, P.R. Cellular organization and biology of the respiratory system. Nat. Cell Biol. 2019, 1. [Google Scholar] [CrossRef]
- Rock, J.R.; Onaitis, M.W.; Rawlins, E.L.; Lu, Y.; Clark, C.P.; Xue, Y.; Randell, S.H.; Hogan, B.L.M. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc. Natl. Acad. Sci. USA 2009, 106, 12771–12775. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Ma, Y.; Dai, X.; Ren, T.; Fu, Y.; Liu, W.; Han, Y.; Wu, Y.; Cheng, Y.; Zhang, T.; et al. Regeneration of functional alveoli by adult human SOX9+ airway basal cell transplantation. Protein Cell 2018, 9, 267–282. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Cheng, L.; Guo, H.; Sun, Y.; Ma, Y.; Wang, Y.; Feng, W.; Yuan, Q.; Dai, X. Application of autologous SOX9+ airway basal cells in patients with bronchiectasis. Clin. Respir. J. 2020, 14, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Averyanov, A.; Koroleva, I.; Konoplyannikov, M.; Revkova, V.; Lesnyak, V.; Kalsin, V.; Danilevskaya, O.; Nikitin, A.; Sotnikova, A.; Kotova, S.; et al. First-in-human high-cumulative-dose stem cell therapy in idiopathic pulmonary fibrosis with rapid lung function decline. Stem Cells Transl. Med. 2020, 9, 6–16. [Google Scholar] [CrossRef]
- Tzouvelekis, A.; Paspaliaris, V.; Koliakos, G.; Ntolios, P.; Bouros, E.; Oikonomou, A.; Zissimopoulos, A.; Boussios, N.; Dardzinski, B.; Gritzalis, D.; et al. A prospective, non-randomized, no placebo-controlled, phase Ib clinical trial to study the safety of the adipose derived stromal cells-stromal vascular fraction in idiopathic pulmonary fibrosis. J. Transl. Med. 2013, 11, 171. [Google Scholar] [CrossRef]
- Ntolios, P.; Manoloudi, E.; Tzouvelekis, A.; Bouros, E.; Steiropoulos, P.; Anevlavis, S.; Bouros, D.; Froudarakis, M.E. Longitudinal outcomes of patients enrolled in a phase Ib clinical trial of the adipose-derived stromal cells-stromal vascular fraction in idiopathic pulmonary fibrosis. Clin. Respir. J. 2018, 12, 2084–2089. [Google Scholar] [CrossRef]
- Glassberg, M.K.; Minkiewicz, J.; Toonkel, R.L.; Simonet, E.S.; Rubio, G.A.; DiFede, D.; Shafazand, S.; Khan, A.; Pujol, M.V.; LaRussa, V.F.; et al. Allogeneic Human Mesenchymal Stem Cells in Patients with Idiopathic Pulmonary Fibrosis via Intravenous Delivery (AETHER): A Phase I Safety Clinical Trial. Chest 2017, 151, 971–981. [Google Scholar] [CrossRef]
- Chambers, D.C.; Enever, D.; Ilic, N.; Sparks, L.; Whitelaw, K.; Ayres, J.; Yerkovich, S.; Khalil, D.; Atkinson, K.M.; Hopkins, P.M. A phase 1b study of placenta-derived mesenchymal stromal cells in patients with idiopathic pulmonary fibrosis. Respirology 2014, 19, 1013–1018. [Google Scholar] [CrossRef]
- Fishman, J.E.; Kim, G.-H.J.; Kyeong, N.-Y.; Goldin, J.G.; Glassber, M.K. Intravenous stem cell dose and changes in quantitative lung fibrosis and DLCO in the AETHER trial: A pilot study. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 7568–7572. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Jia, Z. Cell-based therapy in lung regenerative medicine. Regen. Med. Res. 2014, 2, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Reicherzer, T.; Häffner, S.; Shahzad, T.; Gronbach, J.; Mysliwietz, J.; Hübener, C.; Hasbargen, U.; Gertheiss, J.; Schulze, A.; Bellusci, S.; et al. Activation of the NF-κB pathway alters the phenotype of MSCs in the tracheal aspirates of preterm infants with severe BPD. Am. J. Physiol. Cell. Mol. Physiol. 2018, 315, L87–L101. [Google Scholar] [CrossRef] [PubMed]
- Gronbach, J.; Shahzad, T.; Radajewski, S.; Chao, C.-M.; Bellusci, S.; Morty, R.E.; Reicherzer, T.; Ehrhardt, H. The Potentials and Caveats of Mesenchymal Stromal Cell-Based Therapies in the Preterm Infant. Stem Cells Int. 2018, 2018, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Adamson, I.Y.; Bowden, D.H. The pathogenesis of bleomycin-induced pulmonary fibrosis in mice. Am. J. Pathol. 1974, 77, 185–197. [Google Scholar] [PubMed]
- Zheng, Y.; Cai, W.; Zhou, S.; Xu, L.; Jiang, C. Protective effect of bone marrow derived mesenchymal stem cells in lipopolysaccharide-induced acute lung injury mediated by claudin-4 in a rat model. Am. J. Transl. Res. 2016, 8, 3769–3779. [Google Scholar]
- Islam, M.N.; Das, S.R.; Emin, M.T.; Wei, M.; Sun, L.; Westphalen, K.; Rowlands, D.J.; Quadri, S.K.; Bhattacharya, S.; Bhattacharya, J. Mitochondrial transfer from bone-marrow–derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 2012, 18, 759–765. [Google Scholar] [CrossRef]



| Murine Models | Main Pathological Features | Pros | Cons | Ref |
|---|---|---|---|---|
| Bleomycin (single dose (I.T, I.N, I.V) or repeated doses (I.T, I.N, I.P, O.A, I.V)) | Epithelial cell injury. Fibroblast foci. Macrophage oxidative stress. Fiber deposition | Some of the molecular signatures as well as some histopathological hallmarks at distinct stages of bleomycin-induced lung fibrosis resemble those encountered in human fibrotic lung diseases. Quick development of fibrosis. Relative ease of induction, reproducibility and versatility. Economical | Important role of inflammation in the development of fibrosis. Some reports show that the fibrotic lesions resolved naturally after day 21–28, while other recent studies indicated persistence of fibrosis, albeit with less inflammation as long as 6 months after a single or repetitive bleomycin treatment(s). However, the chronic model that uses several doses of bleomycin may overcome the natural-resolving fibrosis handicap | [45,47,48,49,50,51,52,53,54] |
| Silica | Fibrotic nodules develop around silica deposits and silica fibers are easily identified both by histology and polarization microscopy. Macrophage NALP3 inflammasome activation regulates disease development | Development of fibrotic nodules that resemble lesions that develop in humans following exposure to mineral fibers and particulate aerosols. Persistence of fibrotic lesions due to diminished clearance of silica particles from the lungs | Highly expensive and difficult delivery, prolonged waiting periods until fibrosis develops (4–16 weeks), lack of reproducibility of fibrotic pattern, absence of usual interstitial pneumonia (UIP)-like lesions | [48,55] |
| Asbestosis | Asbestos bodies embedded within the fibrous tissue, fewer myofibroblasts foci and bronchial wall fibrosis. In some cases, the pattern of UIP can be also present | Recapitulates asbestos exposure in human lung fibrosis | A single intratracheal administration elicits an uneven distribution of fibrosis between lungs which also tends to develop in the core of the lung rather than in the subpleura. The fibrosis developed from the inhalation model is more peripheral but requires at least a month for fibrosis to develop | [55] |
| Hyperoxia | Hypoalveolarization. Increased elastin and collagen-I deposition by α-actin-positive myofibroblasts. Increased periostin expression in the alveolar walls, particularly in areas of interstitial thickening | Allows the study of prolonged exposure to supplemental oxygen | Additional studies investigating controversial molecular mechanisms underlying hyperoxia-induced cell injury should be performed since these may be helpful in future pharmaceutical interventions | [56,57] |
| Acid instillation | Pattern of fibrosis involves interstitial rather than alveolar consolidation | Allows studies of hypoxemia, permeability injuries and effects of hyperoxia. It also models fibroproliferative changes seen with ALI and ARDS | Modifications (e.g., a fluid bolus, supplemental oxygen and careful monitoring to be assured of surviving the procedure) are imperative because without them the animals die of lung injury before the development of lung scarring | [48] |
| Cytokine overexpression | Epithelial apoptosis and myofibroblast accumulation. Airway and parenchymal fibrotic response | Ability to dissect downstream signaling events relevant to specific fibrotic-inducing cytokines. Fibrotic scarring tends to be more persistent in some models than those produced by bleomycin | Models limited to dissecting specific pathways. Highly variable and heterogeneous kinetics of injury regarding severity, lesions extension and lack of reproducibility | [55] |
| Fluorescent isothiocyanate (FITC) | AEC injury. Vascular leak | Relatively reproducible and persistent fibrotic phenotypes. Easily trackable fluorescence-labeled fibrotic tissues | Lack representative UIP and inflammatory infiltrates preceding fibrosis. Technical issues regarding FITC particles may compromise model robustness. Limited human relevance since this type of injurious stimulus has never been described in humans | [48,55] |
| Radiation-induced | AEC injury. Vascular remodeling. MSCs regulate repair responses | Results in fibrosis and can be local or systemic if other organs are not shielded | Fibrosis takes a long time to develop. Mainly dependent on inflammation and free-radical-mediated DNA damage and less on TFG-B | [48,55] |
| Familial models | Depends on the altered gene of study | Useful to study the disease genetic background | Mutations may produce a susceptible phenotype, requiring also a second hit from environmental origin to partially recapitulate the human phenotype | [55] |
| Humanized (NOD/SCID mice) | Immunodeficient mice | It allows for cell trafficking during different stages of fibrosis development and progression, offers insights into role of different fibroblast populations and dissects the contribution of epithelial-fibroblast crosstalk in the absence of immune cells | May not be representative of human disease where immune cells play a role. High cost and requires specialized housing. | [55] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Egea-Zorrilla, A.; Vera, L.; Saez, B.; Pardo-Saganta, A. Promises and Challenges of Cell-Based Therapies to Promote Lung Regeneration in Idiopathic Pulmonary Fibrosis. Cells 2022, 11, 2595. https://doi.org/10.3390/cells11162595
Egea-Zorrilla A, Vera L, Saez B, Pardo-Saganta A. Promises and Challenges of Cell-Based Therapies to Promote Lung Regeneration in Idiopathic Pulmonary Fibrosis. Cells. 2022; 11(16):2595. https://doi.org/10.3390/cells11162595
Chicago/Turabian StyleEgea-Zorrilla, Alejandro, Laura Vera, Borja Saez, and Ana Pardo-Saganta. 2022. "Promises and Challenges of Cell-Based Therapies to Promote Lung Regeneration in Idiopathic Pulmonary Fibrosis" Cells 11, no. 16: 2595. https://doi.org/10.3390/cells11162595
APA StyleEgea-Zorrilla, A., Vera, L., Saez, B., & Pardo-Saganta, A. (2022). Promises and Challenges of Cell-Based Therapies to Promote Lung Regeneration in Idiopathic Pulmonary Fibrosis. Cells, 11(16), 2595. https://doi.org/10.3390/cells11162595