
Varied Responses to a High m.3243A>G Mutation Load and Respiratory Chain Dysfunction in Patient-Derived Cardiomyocytes

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Cells and Induced Pluripotent Stem Cell Culture
2.2. Differentiation and Characterization of Cardiomyocytes
2.3. Flow Cytometry
2.4. Seahorse Analysis for Cellular Bioenergetics
2.5. Immunocytochemistry
2.6. Calcium Imaging
2.7. ROS Analysis
2.8. DNA Extraction
2.9. Quantitative PCR
2.10. Determination of the m.3243A>G Mutation Load
2.11. Western Blotting
2.12. ATP Concentration
2.13. Lactate Concentration
2.14. Statistical Analysis
3. Results
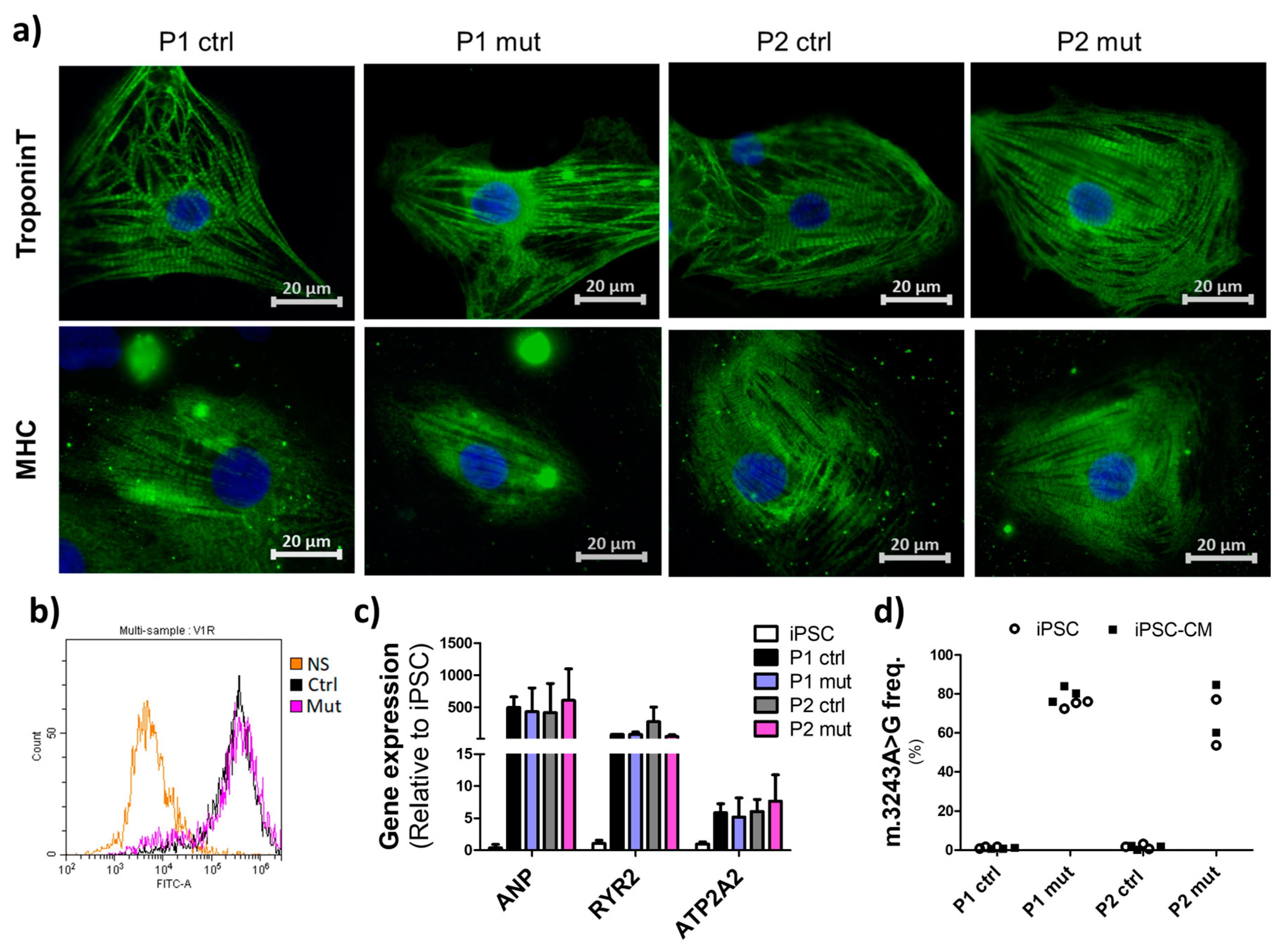
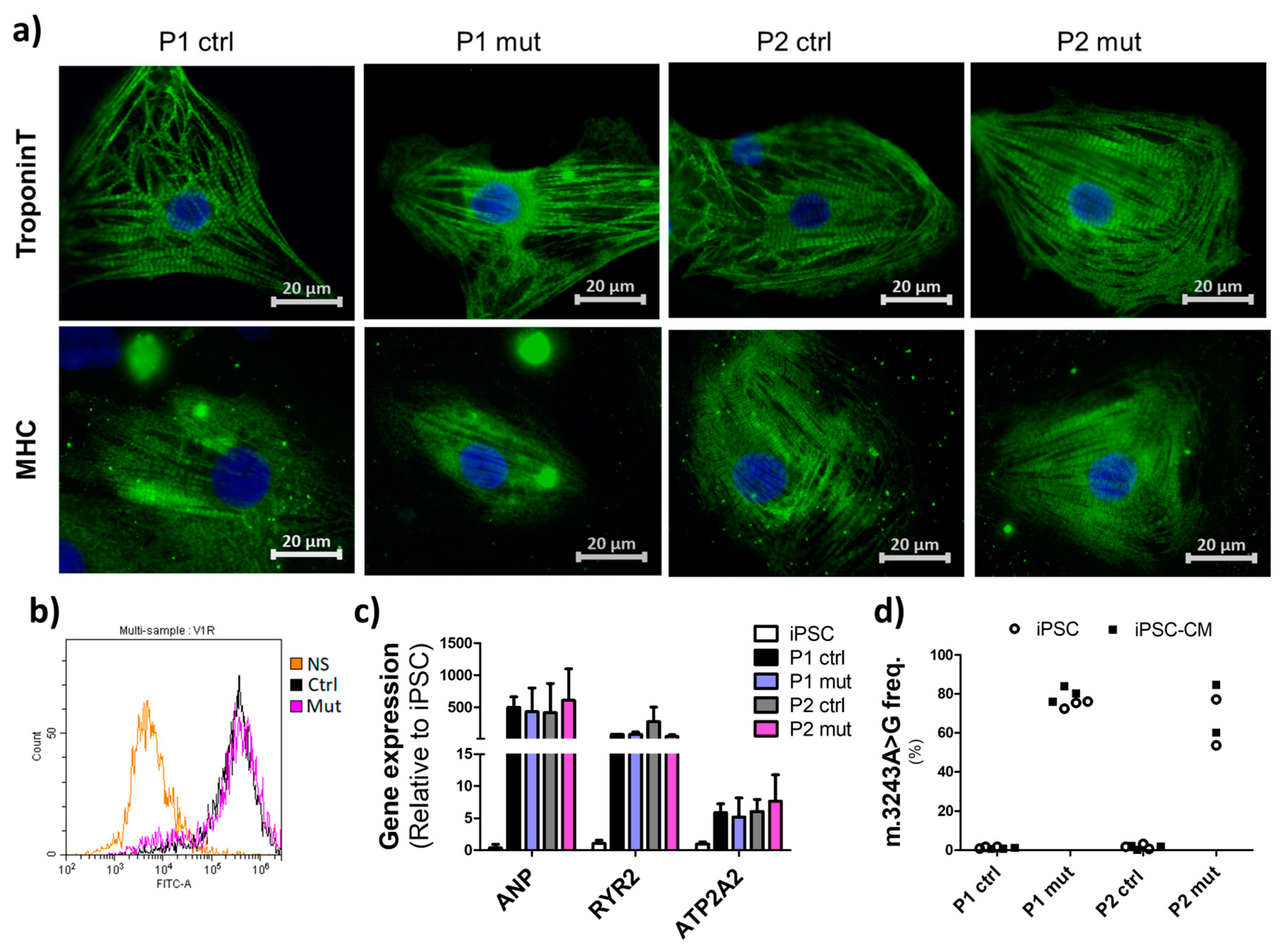
3.1. Cells with High mtDNA Mutation Load Show Normal Cardiac Differentiation Potential
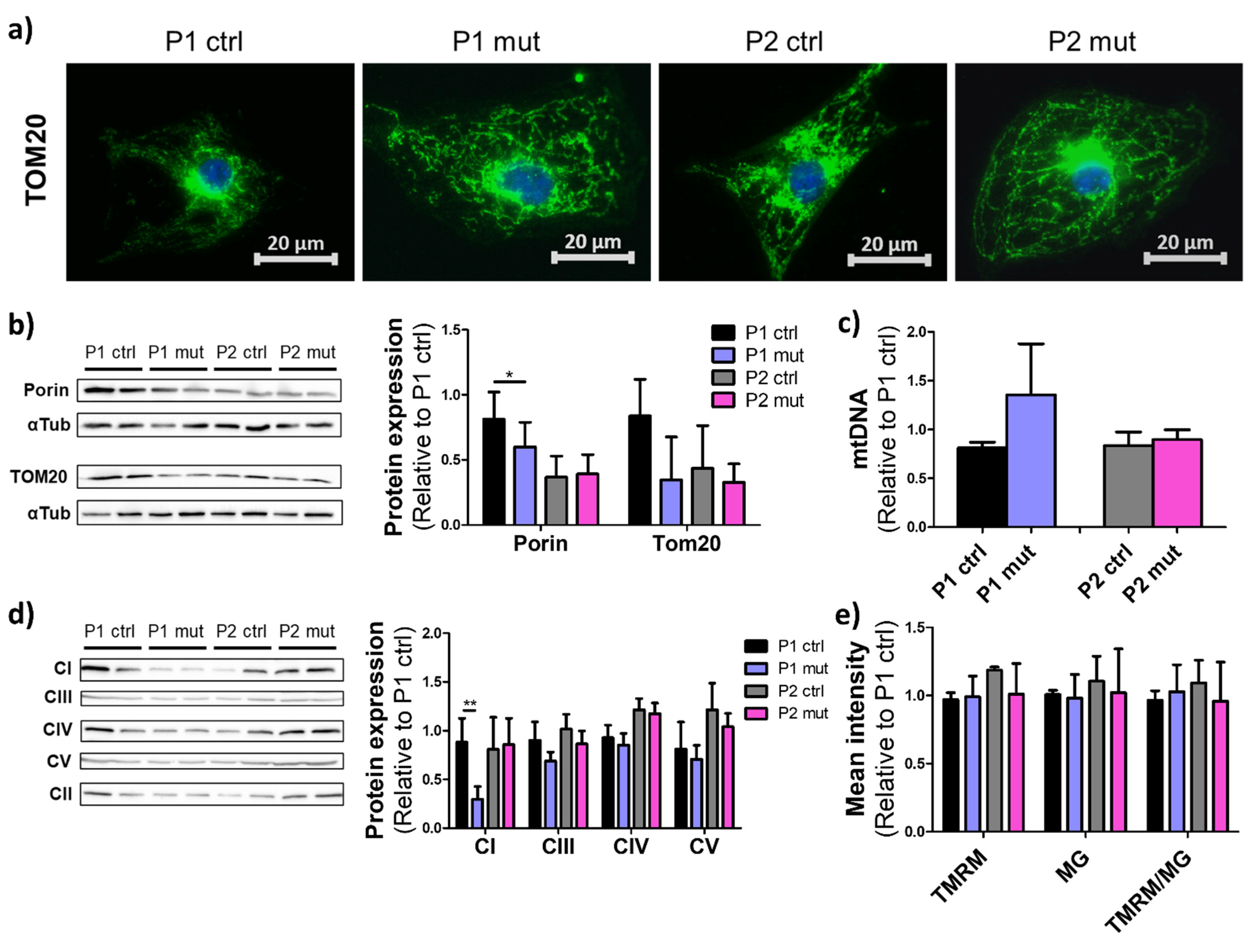
3.2. Mitochondrial Mutations Do Not Induce Drastic Effects on Mitochondrial Mass or Morphology in iPSC-CMs
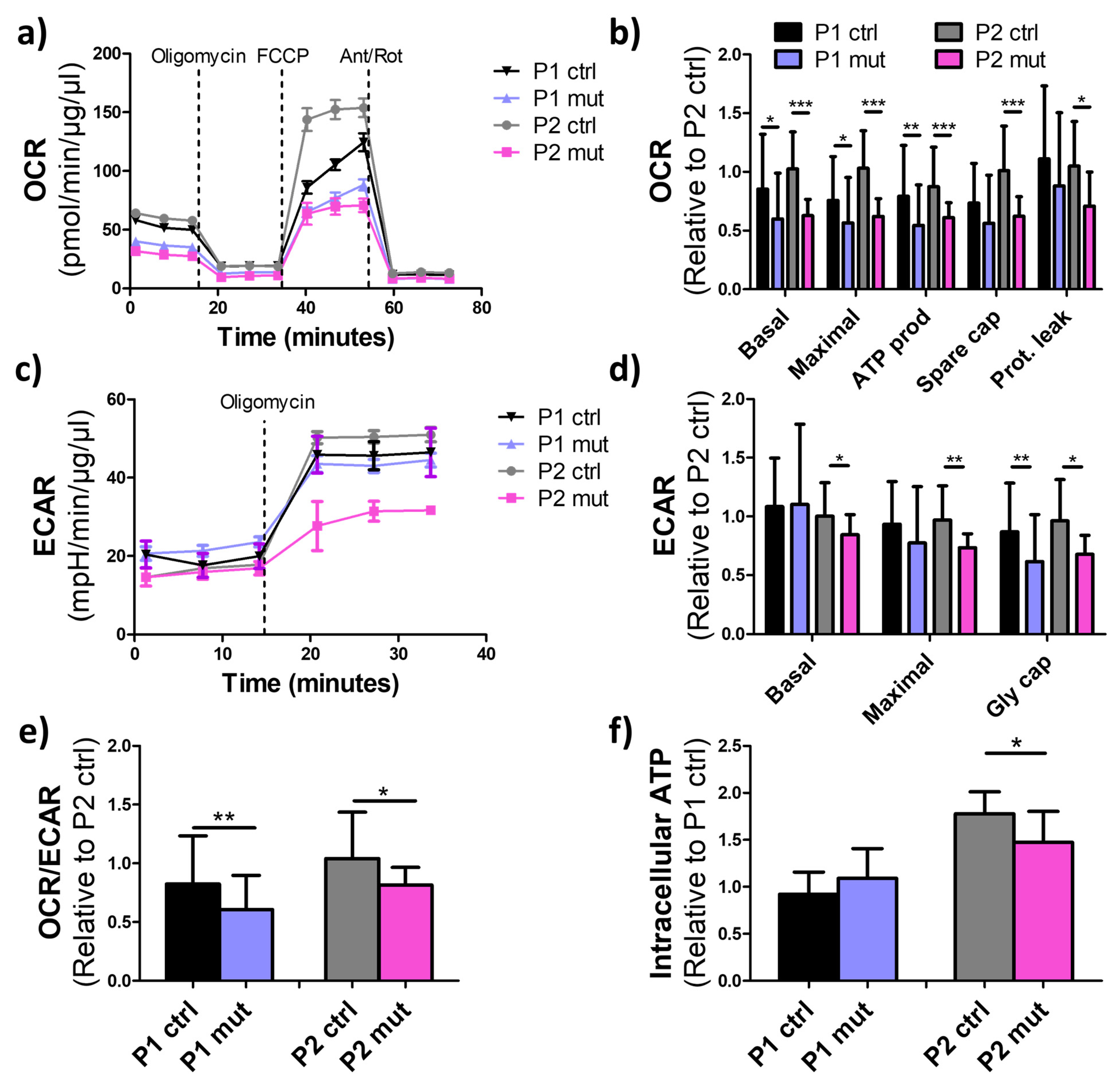
3.3. High m.3243A>G Mutation Load Impairs Oxidative Respiration in iPSC-CMs
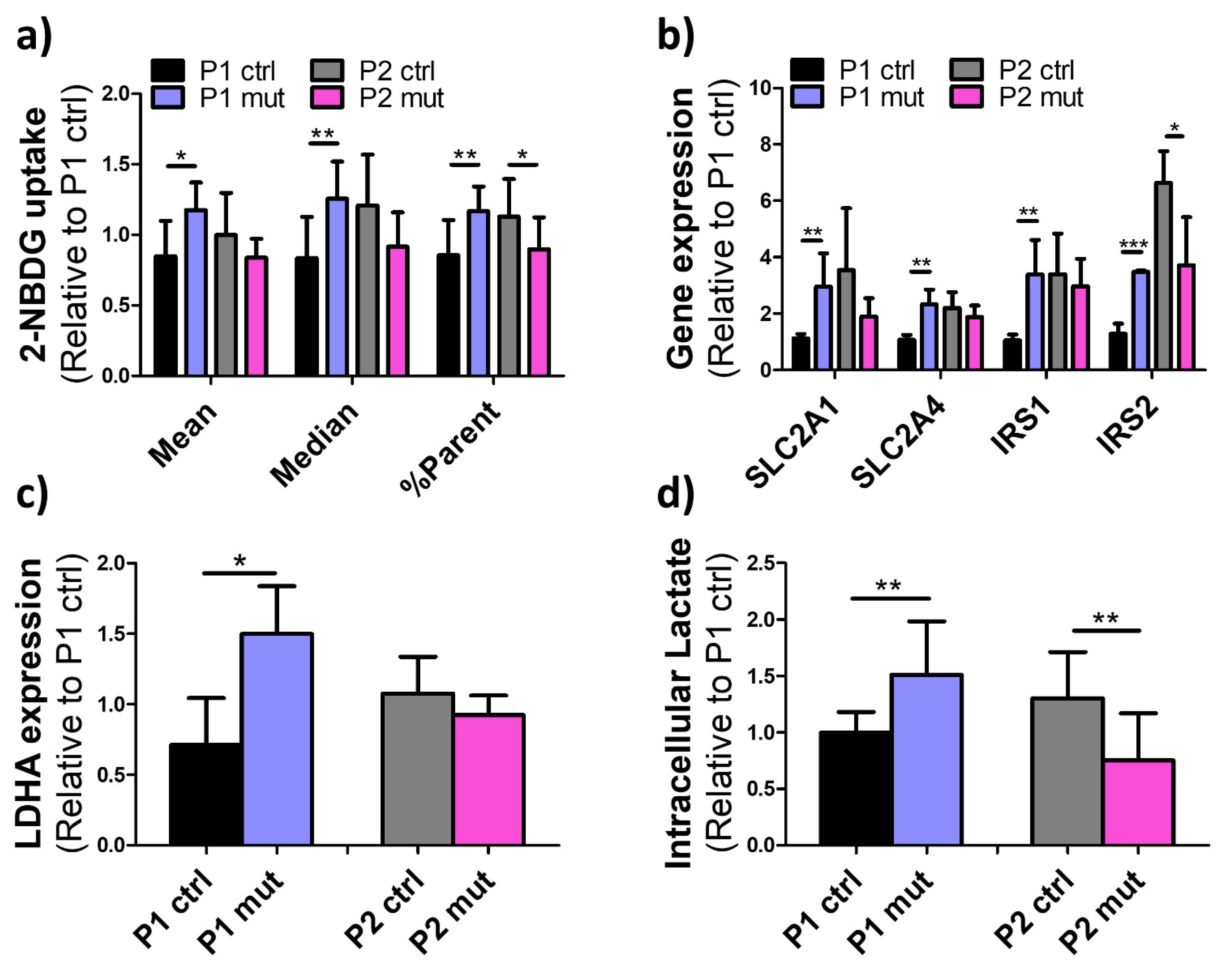
3.4. Compensatory Glucose Metabolism Is Upregulated in the MIDD Patient’s Cells upon High Mutation Load
3.5. High m.3243A>G Mutation Load Elevates ROS Production in the CMP Patient’s Cells
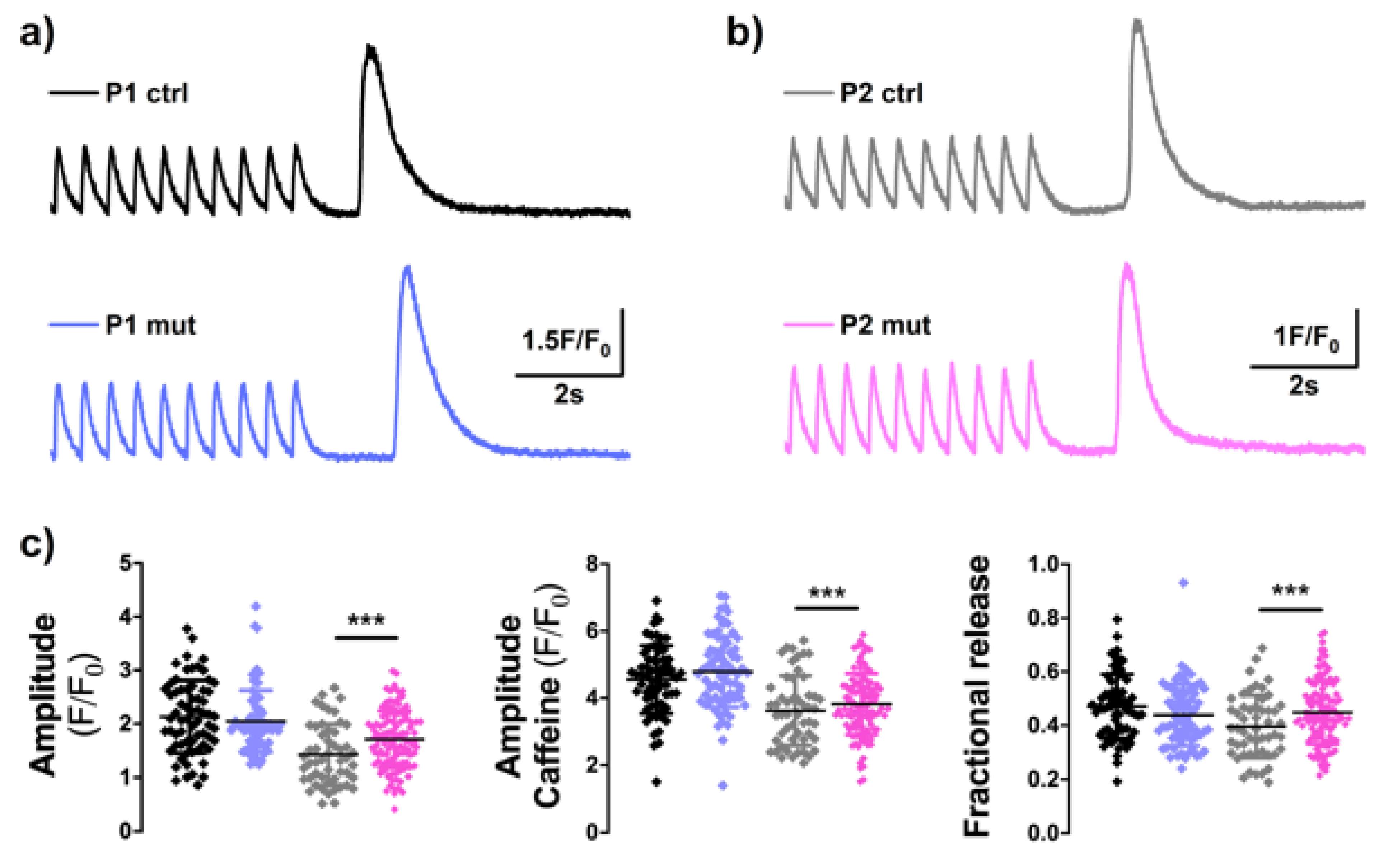
3.6. High m.3243A>G Mutation Load Induces Altererations in Calcium-Signaling in the CMP Patient’s Cells
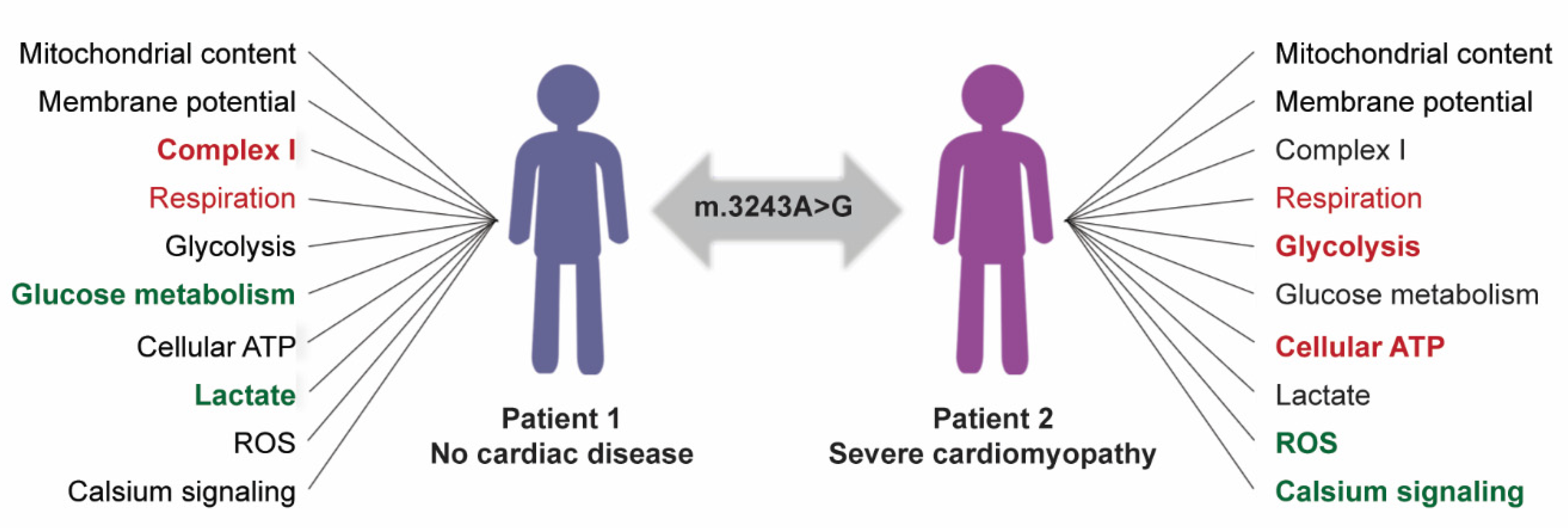
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Additional Methodological Details

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Antibody | Origin | Company (Product Code) | Application (Dilution) |
|---|---|---|---|---|
| Primary | Cardiac Troponin T | Goat | Abcam (ab64623) | IF (1:500) |
| TOM20 | Rabbit | Cell Signaling (42406) | IF (1:200) WB (1:500) | |
| Porin | Rabbit | Abcam (ab15895) | WB (1:1000) | |
| Total OXPHOS Human WB antibody | Mouse | Abcam (ab110411) | WB (1:1000) | |
| Myosin Heavy Chain | Mouse | Millipore (05-716) | IF (1:400) | |
| α-Tubulin | Mouse | Abcam (ab40742) | WB (1:2000) | |
| Secondary | Anti-mouse, Alexa fluor 488 | Donkey | Invitrogen (A21202) | IF (1:300) |
| Anti-goat, Alexa fluor 568 | Donkey | Invitrogen (A11057) | IF (1:300) | |
| Anti-goat, Alexa fluor 488 | Donkey | Invitrogen (A11055) | IF (1:300) | |
| Anti-rabbit, Alexa fluor 594 | Donkey | Invitrogen (A21207) | IF (1:300) | |
| Anti-mouse, biotinylated | Horse | Vector Laboratories (BA-2000) | WB (1:3000) | |
| Anti-rabbit, biotinylated | Goat | Vector Laboratories (BA-1000) | WB (1:5000) |
| Gene | Sequence 5′->3′ | Reference |
|---|---|---|
| APP forward | GCCTGCCTGATCCTCCAAAT | |
| APP reverse | AGGGTAGCGGATGATTCAGCC | |
| CYTB forward | TGTGTGCTCTCCCAGGTCTA | |
| CYTB reverse | CAGTTCTGGATGGTCACTGG | |
| ANP forward | GATCTGCCCTCCTAAAAAGCAA | |
| ANP reverse | ATCTCCGCAGGCTCCGA | |
| ATP2A2 forward | GCGGGCAATCTACAACAACA | |
| ATP2A2 reverse | GAGCAGCTGAACAGGAATCAAA | |
| RYR2 forward | AACTACTGTCCAAGCAGAGGAAAAG | |
| RYR2 reverse | CCGATGCCTTGGCAGATTAT | |
| SLC2A1 forward | GTCTGGCATCAACGCTGTCTT | |
| SLC2A1 reverse | CTGCTCGCTCCACCACAAAC | |
| SLC2A4 forward | CGACCAGCATCTTCGAGACAG | |
| SLC2A4 reverse | TCCACCAACAACACCGAGACC | |
| IRS1 forward | AACTGGACATCACAGCAGAATGAA | |
| IRS1 reverse | CAAACTGAAATGGATGCATCGTACC | |
| IRS2 forward | GCCACCATCGTGAAAGAGTGA | |
| IRS2 reverse | TTGCCTTGTTGGTGCCTCAT | |
| LDHA forward | AGCCCGATTCCGTTACCT | [44] |
| LDHA reverse | CACCAGCAACATTCATTCCA | [44] |
| GAPDH forward | AATGAAGGGGTCATTGATGG | |
| GAPDH reverse | AAGGTGAAGGTCGGAGTCAA | |
| TUBA3 forward | CATTGCCAATCTGGACACC | |
| TUBA3 reverse | GCAACAACCTCTCCTCTTCG | |
| HSF1 forward | TGAGAATGAGGCTCTGTGG | |
| HSF1 reverse | CTGAATGAGCTTGTTGACGA | |
| HSPB7 forward | AGCGAAGGTGTTCATGACG | |
| HSPB7 reverse | CCTATGAGTTTGCGGTGGAC | |
| NFE2L2 forward | GGTTGCCCACATTCCCAAATC | [45] |
| NFE2L2 reverse | CAAGTGACTGAAACGTAGCCG | [45] |
| HMOX1 forward | TTCAAGCAGCTCTACCGCTC | [45] |
| HMOX1 reverse | GAACGCAGTCTTGGCCTCTT | [45] |
| NQO1 forward | TGCAGCGGCTTTGAAGAAGAAAG | [46] |
| NQO1 reverse | TCGGCAGGATACTGAAAGTTCGC | [46] |
| GCLC forward | ATGGAGGTGCAATTAACAGAC | [46] |
| GCLC reverse | ACTGCATTGCCACCTTTGCA | [46] |
| GCLM forward | GCTGTATCAGTGGGCACAG | [46] |
| GCLM reverse | CGCTTGAATGTCAGGAATGC | [46] |
| Forward primer | 5′-CCACCCAAGAACAGGGTTTG-3’ |
| Reverse primer | 5′-GGAATTGAACCTCTGACTGTAAAGTTT-3’ |
| 3243A probe | VIC-AGATGGCAGAGCCCGGTA-MGB |
| 3243G probe | FAM-AGATGGCAGGGCCCGGTA-MGB |
References
- Ng, Y.S.; Turnbull, D. Mitochondrial Disease: Genetics and Management. J. Neurol. 2016, 263, 179–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunnari, J.; Suomalainen, A. Mitochondria: In Sickness and in Health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Hattab, A.W.; Scaglia, F. Mitochondrial Cardiomyopathies. Front. Cardiovasc. Med. 2016, 3, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorman, G.S.; Andrew, M.S.; Ng, Y.; Gomez, N.; Blakely, E.L.; Alston, C.L.; Feeney, C.; Horvath, R.; Yu-Wai-Man, P.; Chinnery, P.F.; et al. Prevalence of Nuclear and Mitochondrial DNA Mutations Related to Adult Mitochondrial Disease. Ann. Neurol. 2015, 77, 753–759. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Du, A. The Non-Syndromic Clinical Spectrums of MtDNA 3243A>G Mutation. Neurosciences 2021, 26, 128–133. [Google Scholar] [CrossRef]
- Majamaa, K.; Moilanen, J.S.; Uimonen, S.; Remes, A.M.; Salmela, P.I.; Kärppä, M.; Majamaa-Voltti, K.A.M.; Rusanen, H.; Sorri, M.; Peuhkurinen, K.J.; et al. Epidemiology of A3243G, the Mutation for Mitochondrial Encephalomyopathy, Lactic Acidosis, and Strokelike Episodes: Prevalence of the Mutation in an Adult Population. Am. J. Hum. Genet. 1998, 63, 447–454. [Google Scholar] [CrossRef] [Green Version]
- Manwaring, N.; Jones, M.M.; Wang, J.J.; Rochtchina, E.; Howard, C.; Mitchell, P.; Sue, C.M. Population Prevalence of the MELAS A3243G Mutation. Mitochondrion 2007, 7, 230–233. [Google Scholar] [CrossRef]
- Sproule, D.M.; Kaufmann, P. Mitochondrial Encephalopathy, Lactic Acidosis, and Strokelike Episodes: Basic Concepts, Clinical Phenotype, and Therapeutic Management of MELAS Syndrome. Ann. N. Y. Acad. Sci. 2008, 1142, 133–158. [Google Scholar] [CrossRef]
- Boggan, R.M.; Lim, A.; Taylor, R.W.; McFarland, R.; Pickett, S.J. Resolving Complexity in Mitochondrial Disease: Towards Precision Medicine. Mol. Genet. Metab. 2019, 128, 19–29. [Google Scholar] [CrossRef]
- Bates, M.G.; Newman, J.H.; Jakovljevic, D.G.; Hollingsworth, K.G.; Alston, C.L.; Zalewski, P.; Klawe, J.J.; Blamire, A.M.; MacGowan, G.A.; Keavney, B.D. Defining Cardiac Adaptations and Safety of Endurance Training in Patients with m.3243A>G-Related Mitochondrial Disease. Int. J. Cardiol. 2013, 168, 3599–3608. [Google Scholar] [CrossRef]
- Stewart, J.B.; Chinnery, P.F. The Dynamics of Mitochondrial DNA Heteroplasmy: Implications for Human Health and Disease. Nat. Rev. Genet. 2015, 16, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Lindroos, M.M.; Pärkkä, J.P.; Taittonen, M.T.; Iozzo, P.; Kärppä, M.; Hassinen, I.E.; Knuuti, J.; Nuutila, P.; Majamaa, K. Myocardial Glucose Uptake in Patients with the m.3243A > G Mutation in Mitochondrial DNA. J. Inherit. Metab. Dis. 2016, 39, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Saku, T.; Takashio, S.; Tsuruta, Y.; Otsuka, Y.; Takae, M.; Kiyama, T.; Yamamoto, E.; Kaikita, K.; Hotta, T.; Matsumoto, S.; et al. Comparison of Electron Microscopic Findings and Clinical Presentation in Three Patients with Mitochondrial Cardiomyopathy Caused by the Mitochondrial DNA Mutation m.3243A > G. Med. Mol. Morphol. 2021, 54, 181–186. [Google Scholar] [CrossRef]
- Scholle, L.M.; Zierz, S.; Mawrin, C.; Wickenhauser, C.; Urban, D.L. Heteroplasmy and Copy Number in the Common m.3243A>G Mutation-A Post-Mortem Genotype-Phenotype Analysis. Genes 2020, 11, 212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finsterer, J.; Zarrouk-Mahjoub, S. The Heart in m.3243A>G Carriers. Herz 2020, 45, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Hämäläinen, R.H.; Manninen, T.; Koivumäki, H.; Kislin, M.; Otonkoski, T.; Suomalainen, A. Tissue- and Cell-Type–Specific Manifestations of Heteroplasmic MtDNA 3243A>G Mutation in Human Induced Pluripotent Stem Cell-Derived Disease Model. Proc. Natl. Acad. Sci. USA 2013, 110, E3622–E3630. [Google Scholar] [CrossRef] [Green Version]
- Lian, X.; Zhang, J.; Azarin, S.M.; Zhu, K.; Hazeltine, L.B.; Bao, X.; Hsiao, C.; Kamp, T.J.; Palecek, S.P. Directed Cardiomyocyte Differentiation from Human Pluripotent Stem Cells by Modulating Wnt/β-Catenin Signaling under Fully Defined Conditions. Nat. Protoc. 2013, 8, 162–175. [Google Scholar] [CrossRef] [Green Version]
- Naumenko, N.; Mutikainen, M.; Holappa, L.; Ruas, J.L.; Tuomainen, T.; Tavi, P. PGC-1α Deficiency Reveals Sex-Specific Links between Cardiac Energy Metabolism and EC-Coupling during Development of Heart Failure in Mice. Cardiovasc. Res. 2021, 118, 1520–1534. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial Dynamics: Overview of Molecular Mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef] [Green Version]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial Membrane Potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W.; Adesina, A.M.; Jones, J.; Scaglia, F. MELAS Syndrome: Clinical Manifestations, Pathogenesis, and Treatment Options. Mol. Genet. Metab. 2015, 116, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.S.; Dighe, P.A.; Mezera, V.; Monternier, P.A.; Brand, M.D. Production of Superoxide and Hydrogen Peroxide from Specific Mitochondrial Sites under Different Bioenergetic Conditions. J. Biol. Chem. 2017, 292, 16804–16809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.M.; Maltagliati, A.J. Nrf2 at the Heart of Oxidative Stress and Cardiac Protection. Physiol. Genom. 2018, 50, 77–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Labbadia, J.; Morimoto, R. Rethinking HSF1 in Stress, Development and Organismal Health. Trends Cell Biol. 2017, 27, 895–905. [Google Scholar] [CrossRef]
- Landstrom, A.P.; Dobrev, D.; Wehrens, X.H.T. Calcium Signaling and Cardiac Arrhythmias. Circ. Res. 2017, 120, 1969–1993. [Google Scholar] [CrossRef]
- Brodehl, A.; Ebbinghaus, H.; Deutsch, M.-A.; Gummert, J.; Gärtner, A.; Ratnavadivel, S.; Milting, H. Human Induced Pluripotent Stem-Cell-Derived Cardiomyocytes as Models for Genetic Cardiomyopathies. Int. J. Mol. Sci. 2019, 20, 4381. [Google Scholar] [CrossRef] [Green Version]
- Modi, S.R.; Hämäläinen, R.H. IPSCs for Modeling MtDNA Diseases. In Recent Advances in IPSC Disease Modeling, 1st ed.; Birbrair, A., Ed.; Academic Press: Cambridge, MA, USA, 2020; Volume 1, pp. 47–70. [Google Scholar] [CrossRef]
- Yokota, M.; Hatakeyama, H.; Ono, Y.; Kanazawa, M.; Goto, Y. Mitochondrial Respiratory Dysfunction Disturbs Neuronal and Cardiac Lineage Commitment of Human IPSCs. Cell. Death Dis. 2017, 8, e2551. [Google Scholar] [CrossRef]
- Ma, H.; Folmes, C.D.L.; Wu, J.; Morey, R.; Mora-Castilla, S.; Ocampo, A.; Ma, L.; Poulton, J.; Wang, X.; Ahmed, R.; et al. Metabolic Rescue in Pluripotent Cells from Patients with MtDNA Disease. Nature 2015, 524, 234–238. [Google Scholar] [CrossRef]
- Chae, J.H.; Hwang, H.; Lim, B.C.; Cheong, H.I.; Hwang, Y.S.; Kim, K.J. Clinical Features of A3243G Mitochondrial TRNA Mutation. Brain Dev. 2004, 26, 459–462. [Google Scholar] [CrossRef]
- Eisner, D.A.; Caldwell, J.L.; Kistamás, K.; Trafford, A.W. Calcium and Excitation-Contraction Coupling in the Heart. Circ. Res. 2017, 121, 181195. [Google Scholar] [CrossRef] [PubMed]
- Drago, I.; De Stefani, D.; Rizzuto, R.; Pozzan, T. Mitochondrial Ca2+ Uptake Contributes to Buffering Cytoplasmic Ca2+ Peaks in Cardiomyocytes. Proc. Natl. Acad. Sci. USA 2012, 109, 12986–12991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.P. How Mitochondria Produce Reactive Oxygen Species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Seo, B.B.; Marella, M.; Yagi, T.; Matsuno-Yagi, A. The single subunit NADH dehydrogenase reduces generation of reactive oxygen species from complex I. FEBS Lett. 2006, 580, 6105–6108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, D.S.; Huang, Y.W.; Ho, C.S.; Hung, P.L.; Hsu, M.H.; Wang, T.J.; Wu, T.Y.; Lee, T.H.; Huang, Z.D.; Chang, P.C.; et al. Oxidative Insults and Mitochondrial DNA Mutation Promote Enhanced Autophagy and Mitophagy Compromising Cell Viability in Pluripotent Cell Model of Mitochondrial Disease. Cells 2019, 8, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heliö, T.M.; Götz, A.; Rapola, J.; Kiuru-Enari, S.; Kivistö, S.; Heikinheimo, T.; Suomalainen, A. Atrial Fibrillation Is Poorly Tolerated by Patients with Hypertrophic Concentric Cardiomyopathy Caused by Mitochondrial TRNALeu (UUR) Mutations. Cardiogenetics 2013, 3, e6. [Google Scholar] [CrossRef] [Green Version]
- Matsa, E.; Burridge, P.W.; Wu, J.C. Human Stem Cells for Modeling Heart Disease and for Drug Discovery. Sci. Transl. Med. 2014, 6, 239ps6. [Google Scholar] [CrossRef] [Green Version]
- Feric, N.T.; Radisic, M. Towards Adult-like Human Engineered Cardiac Tissue: Maturing Human Pluripotent Stem Cell-Derived Cardiomyocytes in Human Engineered Cardiac Tissues. Adv. Drug Deliv. Rev. 2016, 96, 110–134. [Google Scholar] [CrossRef] [Green Version]
- Thavandiran, N.; Dubois, N.; Mikryukov, A.; Massé, S.; Beca, B.; Simmons, C.A.; Deshpande, V.S.; McGarry, J.P.; Chen, C.S.; Nanthakumar, K.; et al. Design and Formulation of Functional Pluripotent Stem Cell-Derived Cardiac Microtissues. Proc. Natl. Acad. Sci. USA 2013, 110, E4698–E4707. [Google Scholar] [CrossRef] [Green Version]
- Giacomelli, E.; Bellin, M.; Sala, L.; van Meer, B.J.; Tertoolen, L.G.J.; Orlova, V.V.; Mummery, C.L. Three-Dimensional Cardiac Microtissues Composed of Cardiomyocytes and Endothelial Cells Co-Differentiated from Human Pluripotent Stem Cells. Development 2017, 144, 1008–1017. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Lee, S.; Baek, G.; Kim, A.; Kang, B.C.; Seo, H.; Kim, J.S. Mitochondrial DNA Editing in Mice with DddA-TALE Fusion Deaminases. Nat. Commun. 2021, 12, 1190. [Google Scholar] [CrossRef] [PubMed]
- Mok, B.Y.; de Moraes, M.H.; Zeng, J.; Bosch, D.E.; Kotrys, A.V.; Raguram, A.; Hsu, F.; Radey, M.C.; Peterson, S.B.; Mootha, V.K. A Bacterial Cytidine Deaminase Toxin Enables CRISPR-Free Mitochondrial Base Editing. Nature 2020, 583, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Liao, M.; Liu, R.; Chen, J.; Feng, H.; Fu, Z. Overexpression of lactate dehydrogenase-A in human intrahepatic cholangiocarcinoma: Its implication for treatment. World J. Surg. Oncol. 2014, 12, 78. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Zhong, H.; Wei, J.; Lin, S.; Zong, Z.; Gong, F.; Huang, X.; Sun, J.; Li, P.; Lin, H.; et al. Inhibition of Nrf2/HO-1 signaling leads to increased activation of the NLRP3 inflammasome in osteoarthritis. Arthritis Res. Ther. 2019, 21, 300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Zhou, L.; Yuen, J.; Birkner, N.; Leppert, V.; O’Day, P.A.; Forman, H.J. Delayed Nrf2-regulated antioxidant gene induction in response to silica nanoparticles. Free Radic. Biol. Med. 2017, 108, 311–319. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ryytty, S.; Modi, S.R.; Naumenko, N.; Shakirzyanova, A.; Rahman, M.O.; Vaara, M.; Suomalainen, A.; Tavi, P.; Hämäläinen, R.H. Varied Responses to a High m.3243A>G Mutation Load and Respiratory Chain Dysfunction in Patient-Derived Cardiomyocytes. Cells 2022, 11, 2593. https://doi.org/10.3390/cells11162593
Ryytty S, Modi SR, Naumenko N, Shakirzyanova A, Rahman MO, Vaara M, Suomalainen A, Tavi P, Hämäläinen RH. Varied Responses to a High m.3243A>G Mutation Load and Respiratory Chain Dysfunction in Patient-Derived Cardiomyocytes. Cells. 2022; 11(16):2593. https://doi.org/10.3390/cells11162593
Chicago/Turabian StyleRyytty, Sanna, Shalem R. Modi, Nikolay Naumenko, Anastasia Shakirzyanova, Muhammad Obaidur Rahman, Miia Vaara, Anu Suomalainen, Pasi Tavi, and Riikka H. Hämäläinen. 2022. "Varied Responses to a High m.3243A>G Mutation Load and Respiratory Chain Dysfunction in Patient-Derived Cardiomyocytes" Cells 11, no. 16: 2593. https://doi.org/10.3390/cells11162593
APA StyleRyytty, S., Modi, S. R., Naumenko, N., Shakirzyanova, A., Rahman, M. O., Vaara, M., Suomalainen, A., Tavi, P., & Hämäläinen, R. H. (2022). Varied Responses to a High m.3243A>G Mutation Load and Respiratory Chain Dysfunction in Patient-Derived Cardiomyocytes. Cells, 11(16), 2593. https://doi.org/10.3390/cells11162593