
Activation of Autophagic Flux Maintains Mitochondrial Homeostasis during Cardiac Ischemia/Reperfusion Injury

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods and Materials
2.1. Experimental Animals
2.2. Generation of Inducible Cardiomyocyte-Specific ATG7 Knockout Mice
2.3. Mouse I/R Model
2.4. Primary Culture of Neonatal Rat Ventricular Myocytes
2.5. Adult Mouse Cardiomyocyte (AMVM) Isolation
2.6. Simulated I/R in Cultured Cells
2.7. Reagents
2.8. Fluorescent Microscopy
2.9. Isolation of DNA and RNA, Measurement of mtDNA Copy Number, and mtDNA Damage
2.10. Assays of Cell Death
2.11. Electron Microscope (EM)
2.12. ATP Concentration Measurement in AMVMs
2.13. Cellular Bioenergetics
2.14. Statistical Methods
3. Results
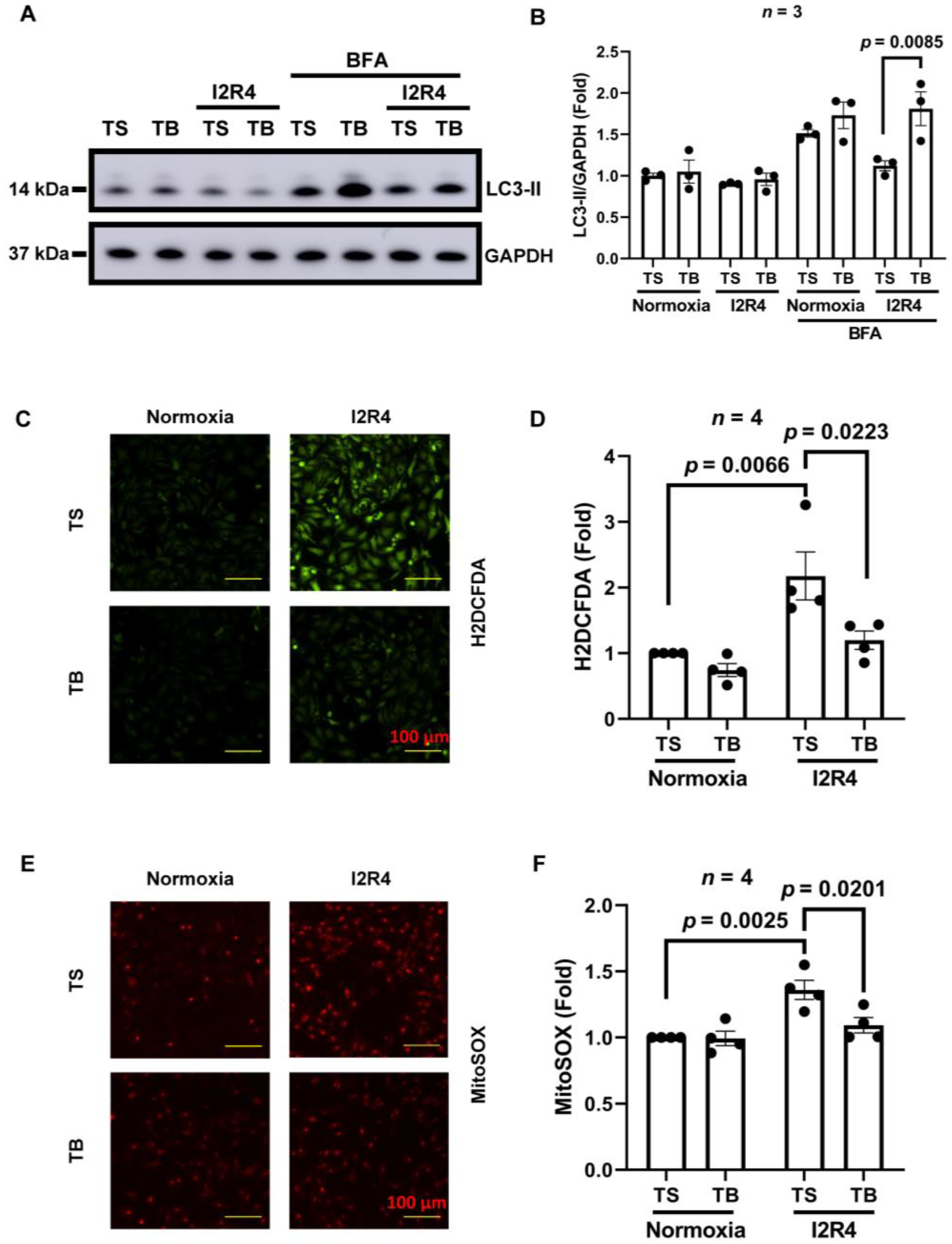
3.1. Tat-Beclin 1 Induces Autophagy and Reduces Oxidative Stress in Neonatal Rat Ventricular Cardiomyocytes (NRVMs) Simulated I/R Injury
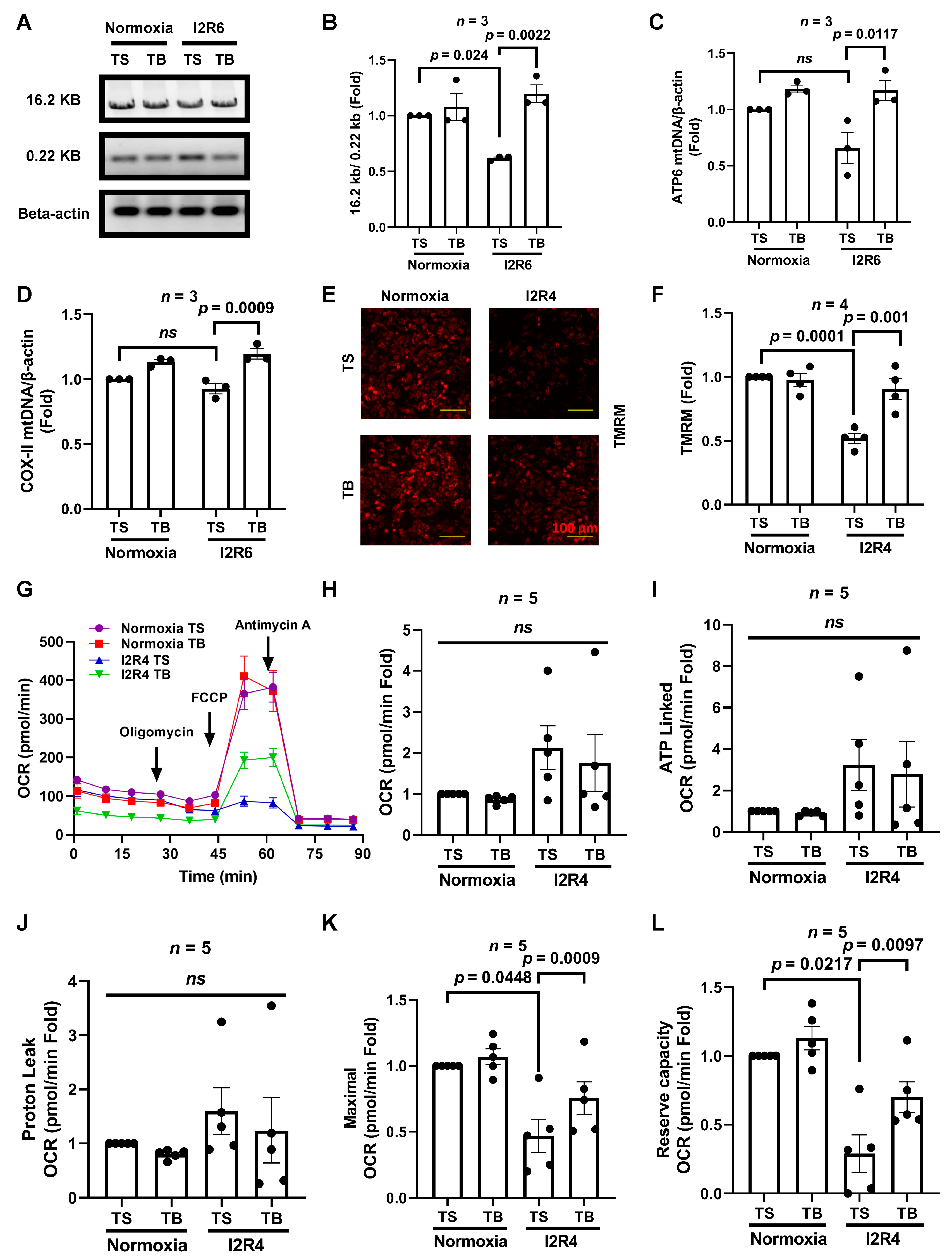
3.2. Tat-Beclin 1 Treatment Increases Mitochondrial DNA Levels, Partially Maintains Mitochondrial Function, and Reduces Cell Death in Cardiomyocytes Subjected to Simulated I/R Injury
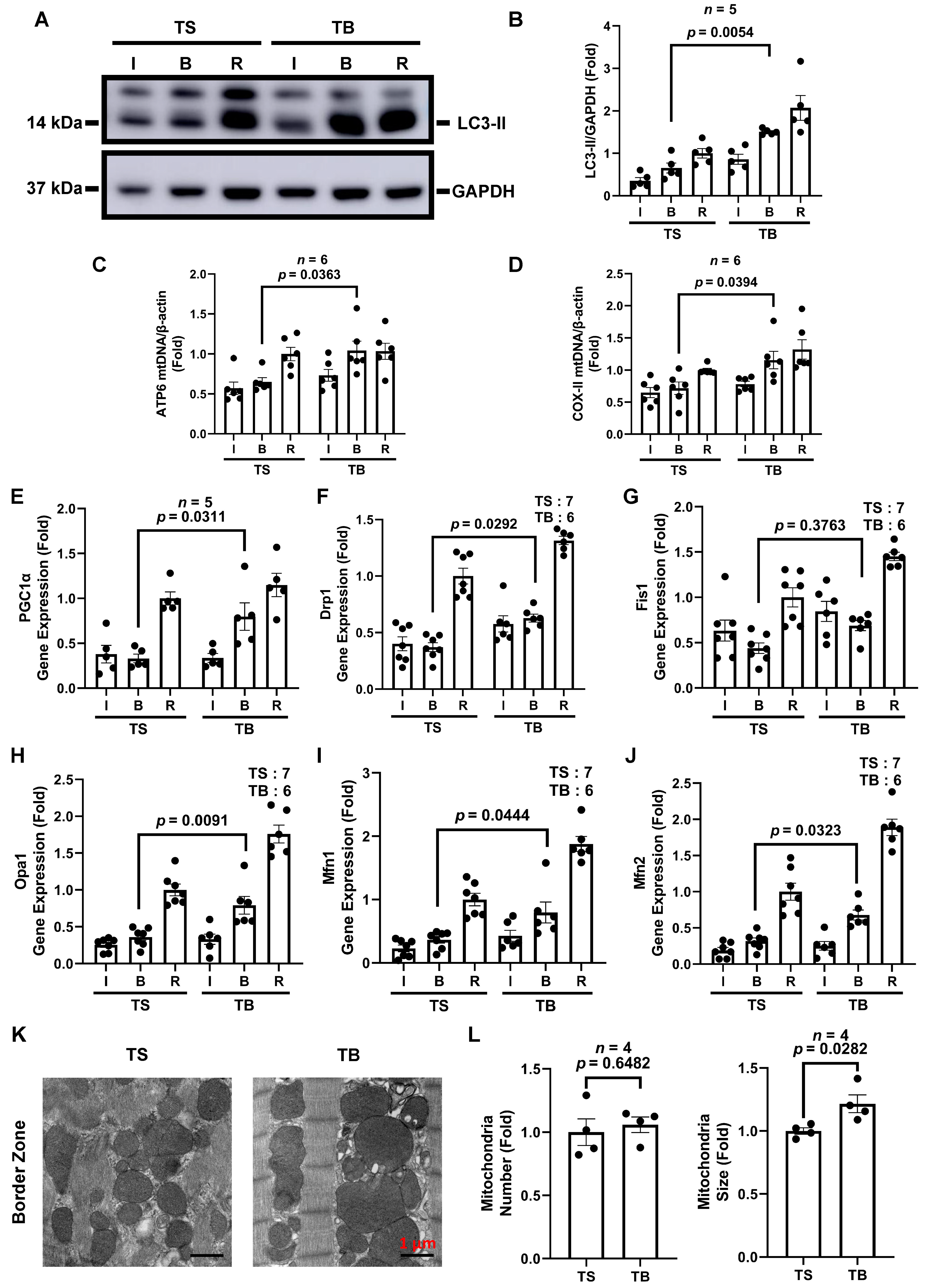
3.3. Tat-Beclin 1 Treatment Induces Autophagy and Maintains Mitochondrial Homeostasis in Mouse Myocardium Treated with I/R Injury
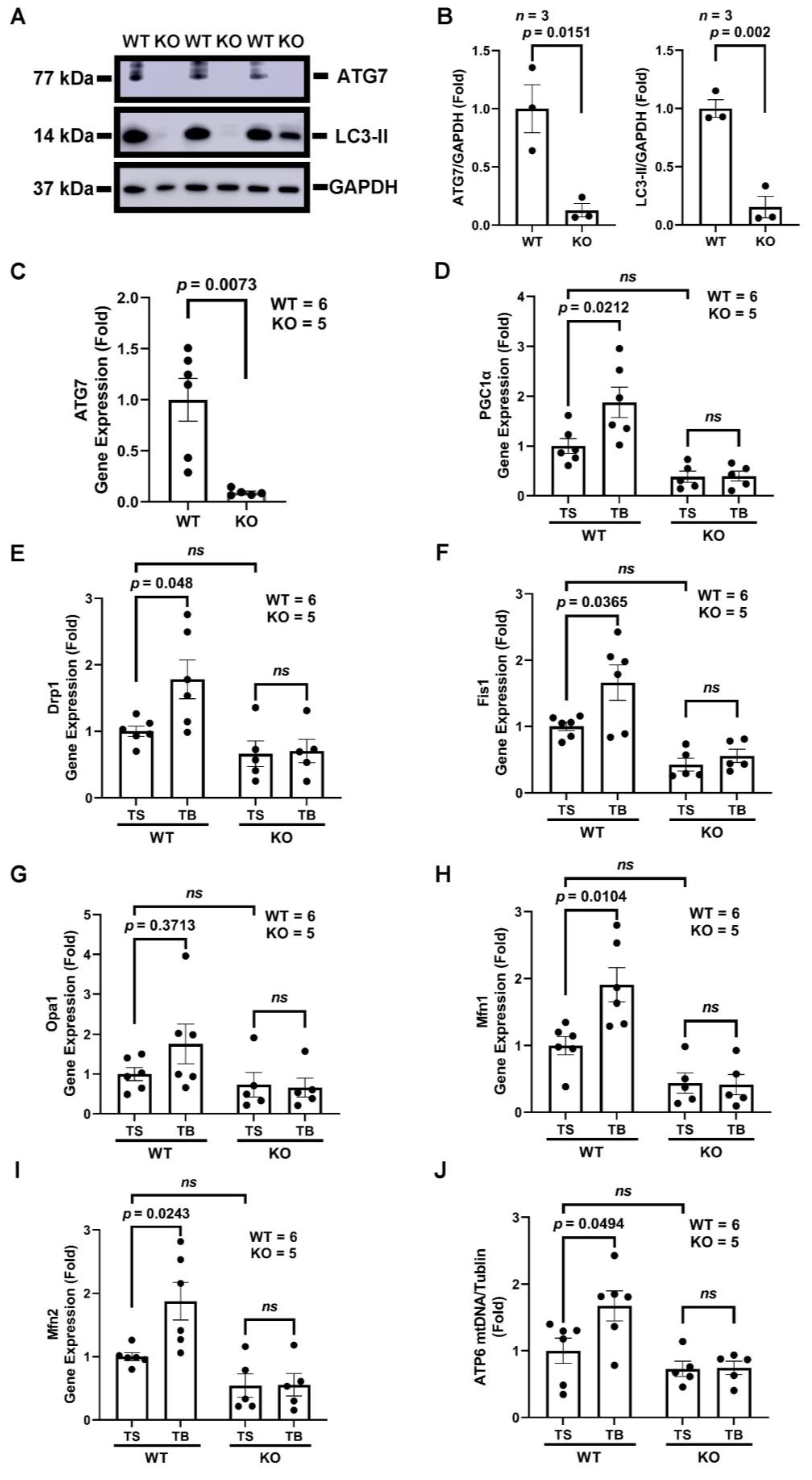
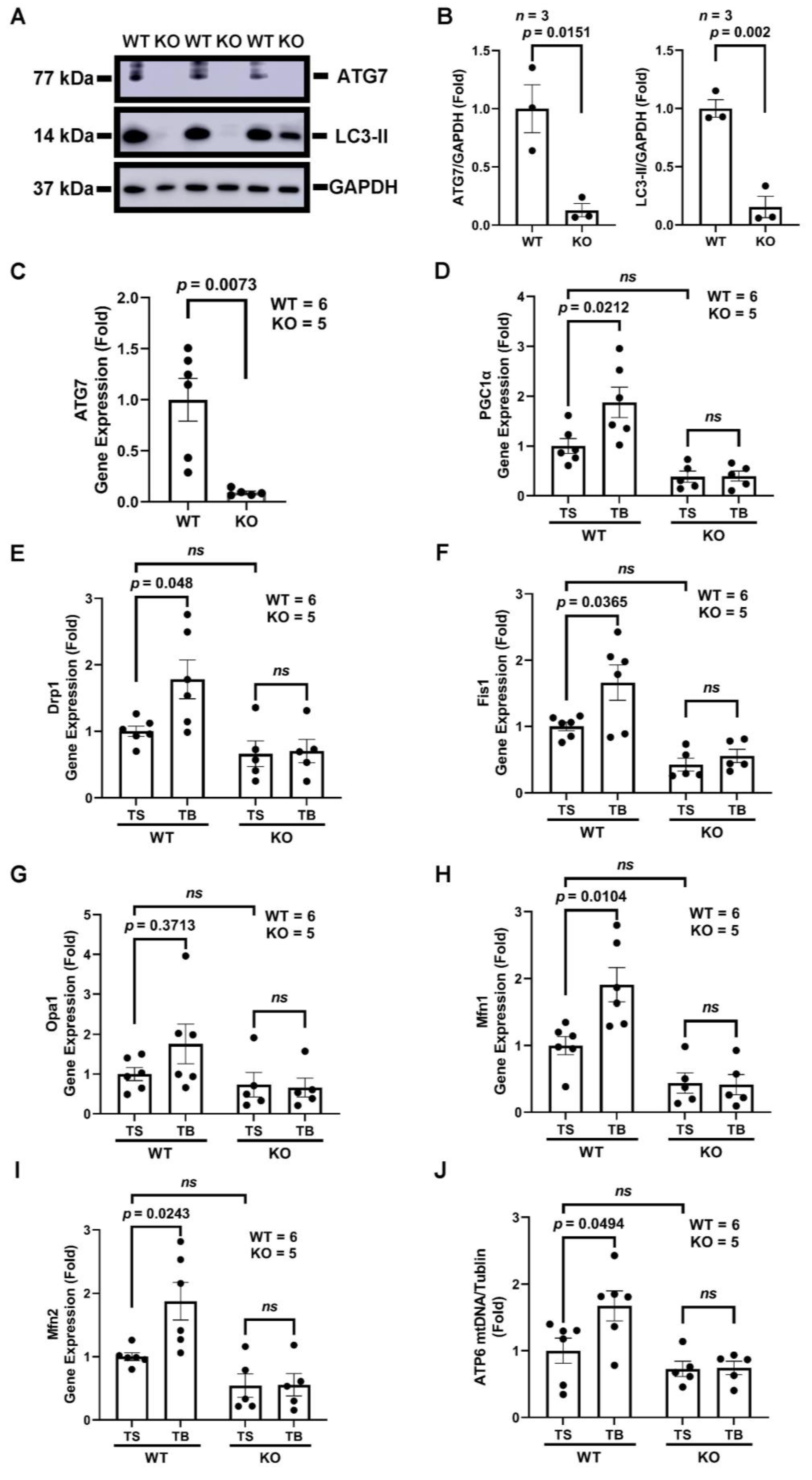
3.4. Tat-Beclin 1 Increases Mitochondrial Biogenesis and Dynamics, Which Is Dependent on the Essential Autophagy Gene, ATG7, in Adult Mouse Ventricular Cardiomyocytes (AMVMs)
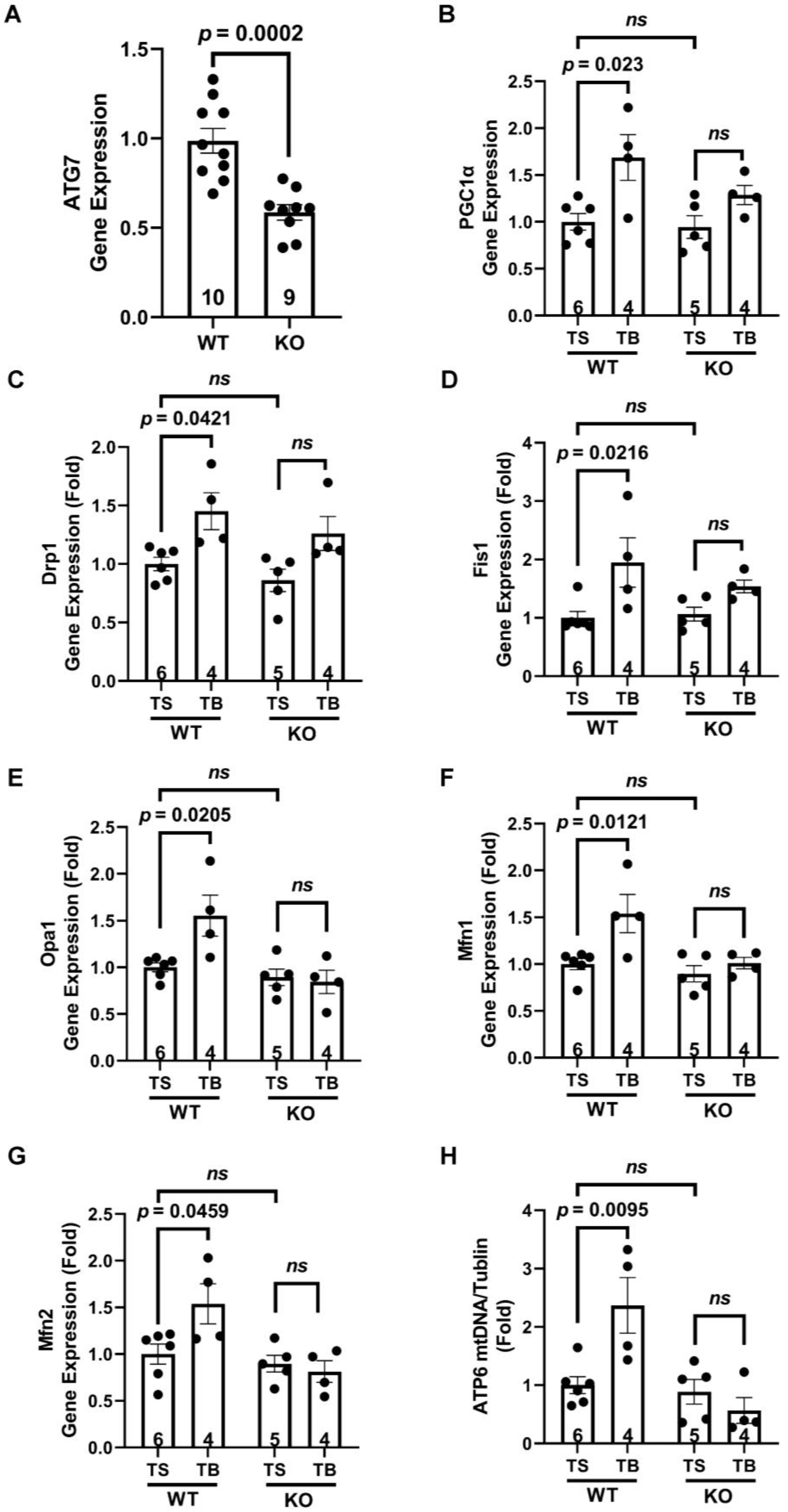
3.5. The Beneficial Effects of Tat-Beclin 1 on Mitochondria in Mouse Hearts Depend on Autophagy
4. Discussion
4.1. Mitochondrial Homeostasis Is a Possible Therapeutic Target for I/R Injury
4.2. Autophagic Removal of Damaged Mitochondria Becomes an Effective Mechanism to Prevent Cardiac Reperfusion Injury
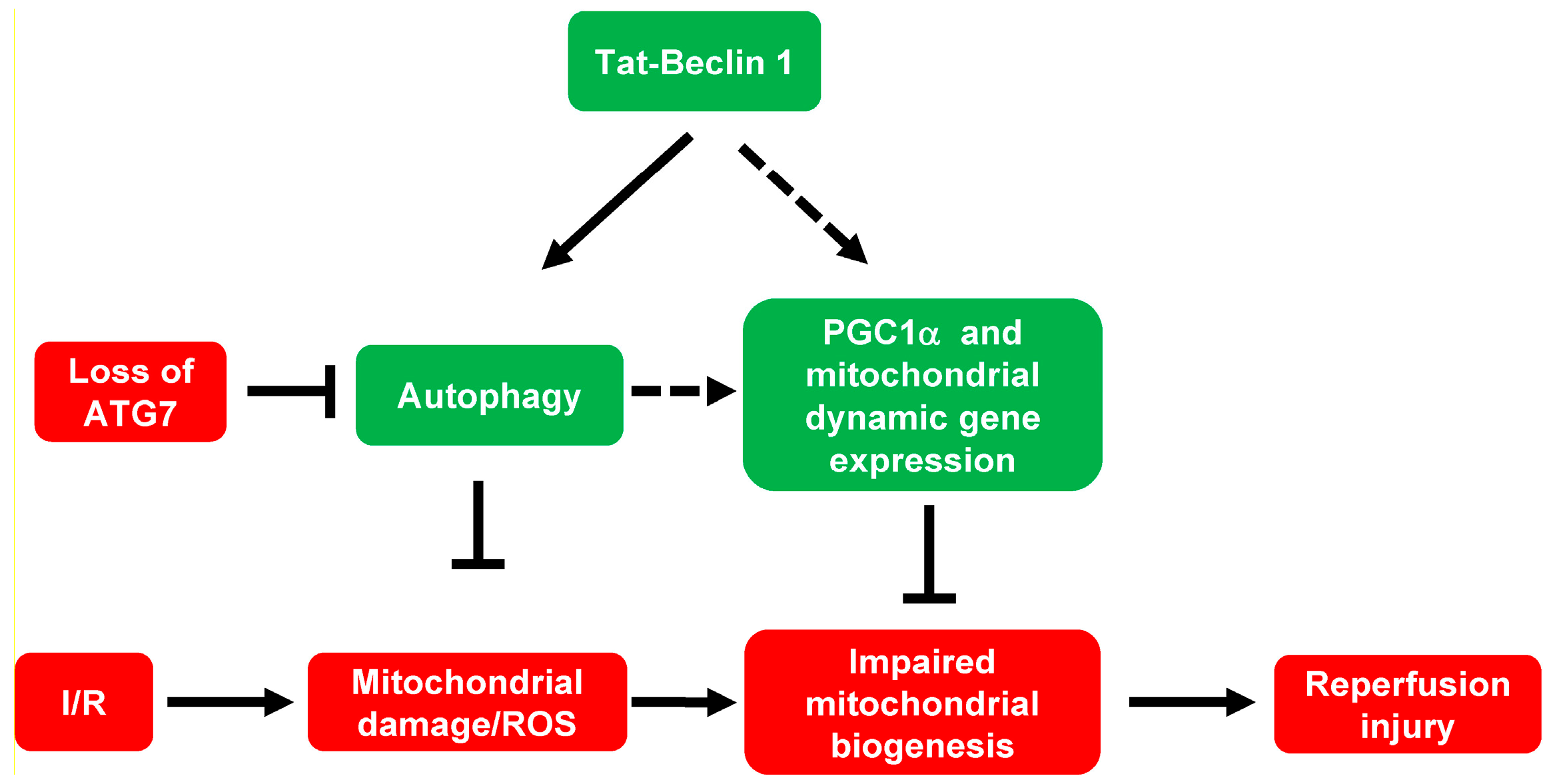
4.3. Tat-Beclin 1-Induced Autophagic Flux Maintains Mitochondrial Homeostasis and Possibly Dynamics during Cardiac I/R Injury
4.4. The Mechanism of Autophagy Induced PGC1α-Mediated Mitochondrial Biogenesis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef] [PubMed]
- Turer, A.T.; Hill, J.A. Pathogenesis of myocardial ischemia-reperfusion injury and rationale for therapy. Am. J. Cardiol. 2010, 106, 360–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloner, R.A.; Brown, D.A.; Csete, M.; Dai, W.; Downey, J.M.; Gottlieb, R.A.; Hale, S.L.; Shi, J. New and revisited approaches to preserving the reperfused myocardium. Nat. Rev. Cardiol. 2017, 14, 679–693. [Google Scholar] [CrossRef] [PubMed]
- Schwartz Longacre, L.; Kloner, R.A.; Arai, A.E.; Baines, C.P.; Bolli, R.; Braunwald, E.; Downey, J.; Gibbons, R.J.; Gottlieb, R.A.; Heusch, G.; et al. New horizons in cardioprotection: Recommendations from the 2010 National Heart, Lung, and Blood Institute Workshop. Circulation 2011, 124, 1172–1179. [Google Scholar] [CrossRef]
- Frohlich, G.M.; Meier, P.; White, S.K.; Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury: Looking beyond primary PCI. Eur. Heart J. 2013, 34, 1714–1722. [Google Scholar] [CrossRef] [Green Version]
- Xie, M.; Kong, Y.; Tan, W.; May, H.; Battiprolu, P.K.; Pedrozo, Z.; Wang, Z.V.; Morales, C.; Luo, X.; Cho, G.; et al. Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation 2014, 129, 1139–1151. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; He, J.; Ismail, M.; Tweeten, S.; Zeng, F.; Gao, L.; Ballinger, S.; Young, M.; Prabhu, S.D.; Rowe, G.C.; et al. HDAC inhibition induces autophagy and mitochondrial biogenesis to maintain mitochondrial homeostasis during cardiac ischemia/reperfusion injury. J. Mol. Cell Cardiol. 2019, 130, 36–48. [Google Scholar] [CrossRef]
- McKinsey, T.A. The biology and therapeutic implications of HDACs in the heart. Handb. Exp. Pharmacol. 2011, 206, 57–78. [Google Scholar] [CrossRef]
- Shoji-Kawata, S.; Sumpter, R.; Leveno, M.; Campbell, G.R.; Zou, Z.; Kinch, L.; Wilkins, A.D.; Sun, Q.; Pallauf, K.; MacDuff, D.; et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature 2013, 494, 201–206. [Google Scholar] [CrossRef] [Green Version]
- Xie, M.; Cho, G.W.; Kong, Y.; Li, D.L.; Altamirano, F.; Luo, X.; Morales, C.R.; Jiang, N.; Schiattarella, G.G.; May, H.I.; et al. Activation of Autophagic Flux Blunts Cardiac Ischemia/Reperfusion Injury. Circ. Res. 2021, 129, 435–450. [Google Scholar] [CrossRef]
- Ju, J.S.; Jeon, S.I.; Park, J.Y.; Lee, J.Y.; Lee, S.C.; Cho, K.J.; Jeong, J.M. Autophagy plays a role in skeletal muscle mitochondrial biogenesis in an endurance exercise-trained condition. J. Physiol. Sci. 2016, 66, 417–430. [Google Scholar] [CrossRef] [PubMed]
- McKinsey, T.A. Therapeutic potential for HDAC inhibitors in the heart. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 303–319. [Google Scholar] [CrossRef] [PubMed]
- Morales, C.R.; Li, D.L.; Pedrozo, Z.; May, H.I.; Jiang, N.; Kyrychenko, V.; Cho, G.W.; Kim, S.Y.; Wang, Z.V.; Rotter, D.; et al. Inhibition of class I histone deacetylases blunts cardiac hypertrophy through TSC2-dependent mTOR repression. Sci. Signal. 2016, 9, ra34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herr, D.J.; Baarine, M.; Aune, S.E.; Li, X.; Ball, L.E.; Lemasters, J.J.; Beeson, C.C.; Chou, J.C.; Menick, D.R. HDAC1 localizes to the mitochondria of cardiac myocytes and contributes to early cardiac reperfusion injury. J. Mol. Cell Cardiol. 2018, 114, 309–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu, M.; Waguri, S.; Ueno, T.; Iwata, J.; Murata, S.; Tanida, I.; Ezaki, J.; Mizushima, N.; Ohsumi, Y.; Uchiyama, Y.; et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 2005, 169, 425–434. [Google Scholar] [CrossRef]
- Sohal, D.S.; Nghiem, M.; Crackower, M.A.; Witt, S.A.; Kimball, T.R.; Tymitz, K.M.; Penninger, J.M.; Molkentin, J.D. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ. Res. 2001, 89, 20–25. [Google Scholar] [CrossRef]
- Louch, W.E.; Sheehan, K.A.; Wolska, B.M. Methods in cardiomyocyte isolation, culture, and gene transfer. J. Mol. Cell Cardiol. 2011, 51, 288–298. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Wu, J.; Bai, Y.; Zhao, X.; Liu, L. Isolation and culture of adult mouse cardiomyocytes for cell signaling and in vitro cardiac hypertrophy. J. Vis. Exp. 2014, 87, e51357. [Google Scholar] [CrossRef] [Green Version]
- Yitzhaki, S.; Huang, C.; Liu, W.; Lee, Y.; Gustafsson, A.B.; Mentzer, R.M., Jr.; Gottlieb, R.A. Autophagy is required for preconditioning by the adenosine A1 receptor-selective agonist CCPA. Basic Res. Cardiol. 2009, 104, 157–167. [Google Scholar] [CrossRef] [Green Version]
- Ballinger, S.W.; Patterson, C.; Knight-Lozano, C.A.; Burow, D.L.; Conklin, C.A.; Hu, Z.; Reuf, J.; Horaist, C.; Lebovitz, R.; Hunter, G.C.; et al. Mitochondrial integrity and function in atherogenesis. Circulation 2002, 106, 544–549. [Google Scholar] [CrossRef]
- Yang, N.C.; Ho, W.M.; Chen, Y.H.; Hu, M.L. A convenient one-step extraction of cellular ATP using boiling water for the luciferin-luciferase assay of ATP. Anal. Biochem. 2002, 306, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Dranka, B.P.; Benavides, G.A.; Diers, A.R.; Giordano, S.; Zelickson, B.R.; Reily, C.; Zou, L.; Chatham, J.C.; Hill, B.G.; Zhang, J.; et al. Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free Radic. Biol. Med. 2011, 51, 1621–1635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redmann, M.; Benavides, G.A.; Wani, W.Y.; Berryhill, T.F.; Ouyang, X.; Johnson, M.S.; Ravi, S.; Mitra, K.; Barnes, S.; Darley-Usmar, V.M.; et al. Methods for assessing mitochondrial quality control mechanisms and cellular consequences in cell culture. Redox Biol. 2018, 17, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Morales, C.R.; Lavandero, S.; Hill, J.A. Tuning flux: Autophagy as a target of heart disease therapy. Curr. Opin. Cardiol. 2011, 26, 216–222. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Gu, S.; Li, X.; Tan, J.; Liu, S.; Jiang, Y.; Zhang, C.; Gao, L.; Yang, H.-T. Berbamine postconditioning protects the heart from ischemia/reperfusion injury through modulation of autophagy. Cell Death Dis. 2017, 8, e2577. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Yan, C.; Tian, X.; Zhu, N.; Li, Y.; Liu, D.; Liu, Y.; Liu, M.; Peng, C.; Zhang, Q.; et al. CREG protects from myocardial ischemia/reperfusion injury by regulating myocardial autophagy and apoptosis. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1893–1903. [Google Scholar] [CrossRef]
- Murphy, E.; Steenbergen, C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol. Rev. 2008, 88, 581–609. [Google Scholar] [CrossRef] [Green Version]
- Hendgen-Cotta, U.B.; Merx, M.W.; Shiva, S.; Schmitz, J.; Becher, S.; Klare, J.P.; Steinhoff, H.J.; Goedecke, A.; Schrader, J.; Gladwin, M.T.; et al. Nitrite reductase activity of myoglobin regulates respiration and cellular viability in myocardial ischemia-reperfusion injury. Proc. Natl. Acad. Sci. USA 2008, 105, 10256–10261. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.S. Role of mitochondrial function in cell death and body metabolism. Front. Biosci. (Landmark Ed.) 2016, 21, 1233–1244. [Google Scholar]
- Nazari, A.; Sadr, S.S.; Faghihi, M.; Azizi, Y.; Hosseini, M.J.; Mobarra, N.; Tavakoli, A.; Imani, A. Vasopressin attenuates ischemia-reperfusion injury via reduction of oxidative stress and inhibition of mitochondrial permeability transition pore opening in rat hearts. Eur. J. Pharmacol. 2015, 760, 96–102. [Google Scholar] [CrossRef]
- Saito, T.; Sadoshima, J. Molecular mechanisms of mitochondrial autophagy/mitophagy in the heart. Circ. Res. 2015, 116, 1477–1490. [Google Scholar] [CrossRef] [PubMed]
- Hill, B.G.; Dranka, B.P.; Zou, L.; Chatham, J.C.; Darley-Usmar, V.M. Importance of the bioenergetic reserve capacity in response to cardiomyocyte stress induced by 4-hydroxynonenal. Biochem. J. 2009, 424, 99–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamuri, S.; Sperling, J.A.; Sure, V.N.; Dholakia, M.H.; Peterson, N.R.; Rutkai, I.; Mahalingam, P.S.; Satou, R.; Katakam, P.V.G. Measurement of respiratory function in isolated cardiac mitochondria using Seahorse XFe24 Analyzer: Applications for aging research. Geroscience 2018, 40, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W., 2nd; Vega, R.B.; Kelly, D.P. Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev. 2015, 29, 1981–1991. [Google Scholar] [CrossRef] [Green Version]
- Vainshtein, A.; Tryon, L.D.; Pauly, M.; Hood, D.A. Role of PGC-1α during acute exercise-induced autophagy and mitophagy in skeletal muscle. Am. J. Physiol. Cell Physiol. 2015, 308, C710–C719. [Google Scholar] [CrossRef] [Green Version]
- Halling, J.F.; Ringholm, S.; Nielsen, M.M.; Overby, P.; Pilegaard, H. PGC-1α promotes exercise-induced autophagy in mouse skeletal muscle. Physiol. Rep. 2016, 4, e12698. [Google Scholar] [CrossRef] [Green Version]
- Grootaert, M.O.; da Costa Martins, P.A.; Bitsch, N.; Pintelon, I.; De Meyer, G.R.; Martinet, W.; Schrijvers, D.M. Defective autophagy in vascular smooth muscle cells accelerates senescence and promotes neointima formation and atherogenesis. Autophagy 2015, 11, 2014–2032. [Google Scholar] [CrossRef]
- Shin, J.H.; Ko, H.S.; Kang, H.; Lee, Y.; Lee, Y.I.; Pletinkova, O.; Troconso, J.C.; Dawson, V.L.; Dawson, T.M. PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson’s disease. Cell 2011, 144, 689–702. [Google Scholar] [CrossRef] [Green Version]
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef]
- Xue, R.Q.; Xu, M.; Yu, X.J.; Liu, L.Z.; Zang, W.J. Recent progress of mitochondrial quality control in ischemic heart disease and its role in cardio-protection of vagal nerve. Acta Physiol. Sin. 2017, 69, 579–586. [Google Scholar]
- Bliksoen, M.; Baysa, A.; Eide, L.; Bjoras, M.; Suganthan, R.; Vaage, J.; Stenslokken, K.O.; Valen, G. Mitochondrial DNA damage and repair during ischemia-reperfusion injury of the heart. J. Mol. Cell Cardiol. 2015, 78, 9–22. [Google Scholar] [CrossRef]
- Valero, T. Mitochondrial biogenesis: Pharmacological approaches. Curr. Pharm. Des. 2014, 20, 5507–5509. [Google Scholar] [CrossRef]
- Onyango, I.G.; Lu, J.; Rodova, M.; Lezi, E.; Crafter, A.B.; Swerdlow, R.H. Regulation of neuron mitochondrial biogenesis and relevance to brain health. Biochim. Biophys. Acta 2010, 1802, 228–234. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.C.; Wei, Y.H. Mitochondrial biogenesis and mitochondrial DNA maintenance of mammalian cells under oxidative stress. Int. J. Biochem. Cell Biol. 2005, 37, 822–834. [Google Scholar] [CrossRef]
- Song, M.; Franco, A.; Fleischer, J.A.; Zhang, L.; Dorn, G.W., 2nd. Abrogating Mitochondrial Dynamics in Mouse Hearts Accelerates Mitochondrial Senescence. Cell Metab. 2017, 26, 872–883. [Google Scholar] [CrossRef] [Green Version]
- Stevens, D.A.; Lee, Y.; Kang, H.C.; Lee, B.D.; Lee, Y.I.; Bower, A.; Jiang, H.; Kang, S.U.; Andrabi, S.A.; Dawson, V.L.; et al. Parkin loss leads to PARIS-dependent declines in mitochondrial mass and respiration. Proc. Natl. Acad. Sci. USA 2015, 112, 11696–11701. [Google Scholar] [CrossRef] [Green Version]
- Tao, L.; Bei, Y.; Lin, S.; Zhang, H.; Zhou, Y.; Jiang, J.; Chen, P.; Shen, S.; Xiao, J.; Li, X. Exercise Training Protects Against Acute Myocardial Infarction via Improving Myocardial Energy Metabolism and Mitochondrial Biogenesis. Cell Physiol. Biochem. 2015, 37, 162–175. [Google Scholar] [CrossRef]
- Ma, X.; Liu, H.; Foyil, S.R.; Godar, R.J.; Weinheimer, C.J.; Diwan, A. Autophagy is impaired in cardiac ischemia-reperfusion injury. Autophagy 2012, 8, 1394–1396. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Nadtochiy, S.M.; Urciuoli, W.R.; Brookes, P.S. The cardioprotective compound cloxyquin uncouples mitochondria and induces autophagy. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H29–H38. [Google Scholar] [CrossRef] [Green Version]
- Di Lisa, F.; Bernardi, P. Modulation of Mitochondrial Permeability Transition in Ischemia-Reperfusion Injury of the Heart. Advantages and Limitations. Curr. Med. Chem. 2015, 22, 2480–2487. [Google Scholar] [CrossRef]
- Dutta, D.; Xu, J.; Kim, J.S.; Dunn, W.A., Jr.; Leeuwenburgh, C. Upregulated autophagy protects cardiomyocytes from oxidative stress-induced toxicity. Autophagy 2013, 9, 328–344. [Google Scholar] [CrossRef] [Green Version]
- Chouchani, E.T.; Methner, C.; Nadtochiy, S.M.; Logan, A.; Pell, V.R.; Ding, S.; James, A.M.; Cocheme, H.M.; Reinhold, J.; Lilley, K.S.; et al. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat. Med. 2013, 19, 753–759. [Google Scholar] [CrossRef] [Green Version]
- Chinda, K.; Sanit, J.; Chattipakorn, S.; Chattipakorn, N. Dipeptidyl peptidase-4 inhibitor reduces infarct size and preserves cardiac function via mitochondrial protection in ischaemia-reperfusion rat heart. Diab. Vasc. Dis. Res. 2014, 11, 75–83. [Google Scholar] [CrossRef] [Green Version]
- Andres, A.M.; Hernandez, G.; Lee, P.; Huang, C.; Ratliff, E.P.; Sin, J.; Thornton, C.A.; Damasco, M.V.; Gottlieb, R.A. Mitophagy is required for acute cardioprotection by simvastatin. Antioxid. Redox Signal. 2014, 21, 1960–1973. [Google Scholar] [CrossRef]
- Delbridge, L.M.; Mellor, K.M.; Taylor, D.J.; Gottlieb, R.A. Myocardial autophagic energy stress responses--macroautophagy, mitophagy, and glycophagy. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H1194–H1204. [Google Scholar] [CrossRef] [Green Version]
- Scarffe, L.A.; Stevens, D.A.; Dawson, V.L.; Dawson, T.M. Parkin and PINK1: Much more than mitophagy. Trends Neurosci. 2014, 37, 315–324. [Google Scholar] [CrossRef] [Green Version]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Murphy, E.; Ardehali, H.; Balaban, R.S.; DiLisa, F.; Dorn, G.W., 2nd; Kitsis, R.N.; Otsu, K.; Ping, P.; Rizzuto, R.; Sack, M.N.; et al. Mitochondrial Function, Biology, and Role in Disease: A Scientific Statement From the American Heart Association. Circ. Res. 2016, 118, 1960–1991. [Google Scholar] [CrossRef]
- Schiattarella, G.G.; Hill, J.A. Therapeutic targeting of autophagy in cardiovascular disease. J. Mol. Cell Cardiol. 2016, 95, 86–93. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Wu, Y.; Zhang, P.; Sha, H.; Jia, J.; Hu, Y.; Zhu, J. Exercise induces mitochondrial biogenesis after brain ischemia in rats. Neuroscience 2012, 205, 10–17. [Google Scholar] [CrossRef]
- Funk, J.A.; Schnellmann, R.G. Accelerated recovery of renal mitochondrial and tubule homeostasis with SIRT1/PGC-1alpha activation following ischemia-reperfusion injury. Toxicol. Appl. Pharmacol. 2013, 273, 345–354. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.D.; Yang, D.I.; Lin, T.K.; Shaw, F.Z.; Liou, C.W.; Chuang, Y.C. Roles of oxidative stress, apoptosis, PGC-1alpha and mitochondrial biogenesis in cerebral ischemia. Int. J. Mol. Sci. 2011, 12, 7199–7215. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Zhang, Q.; Pires, K.; Abel, E.D. Abstract 12606: Autophagy is Required for Mitochondrial Biogenesis in the Heart. Circulation 2014, 130, A12606. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, L.; Chu, Y.; Yang, J.; He, J.; Hua, Y.; Chen, Y.; Benavides, G.; Rowe, G.C.; Zhou, L.; Ballinger, S.; et al. Activation of Autophagic Flux Maintains Mitochondrial Homeostasis during Cardiac Ischemia/Reperfusion Injury. Cells 2022, 11, 2111. https://doi.org/10.3390/cells11132111
He L, Chu Y, Yang J, He J, Hua Y, Chen Y, Benavides G, Rowe GC, Zhou L, Ballinger S, et al. Activation of Autophagic Flux Maintains Mitochondrial Homeostasis during Cardiac Ischemia/Reperfusion Injury. Cells. 2022; 11(13):2111. https://doi.org/10.3390/cells11132111
Chicago/Turabian StyleHe, Lihao, Yuxin Chu, Jing Yang, Jin He, Yutao Hua, Yunxi Chen, Gloria Benavides, Glenn C. Rowe, Lufang Zhou, Scott Ballinger, and et al. 2022. "Activation of Autophagic Flux Maintains Mitochondrial Homeostasis during Cardiac Ischemia/Reperfusion Injury" Cells 11, no. 13: 2111. https://doi.org/10.3390/cells11132111
APA StyleHe, L., Chu, Y., Yang, J., He, J., Hua, Y., Chen, Y., Benavides, G., Rowe, G. C., Zhou, L., Ballinger, S., Darley-Usmar, V., Young, M. E., Prabhu, S. D., Sethu, P., Zhou, Y., Zhang, C., & Xie, M. (2022). Activation of Autophagic Flux Maintains Mitochondrial Homeostasis during Cardiac Ischemia/Reperfusion Injury. Cells, 11(13), 2111. https://doi.org/10.3390/cells11132111