
How Azanucleosides Affect Myeloid Cell Fate


{kind=link}
{kind=link}
{kind=link}
{kind=link}
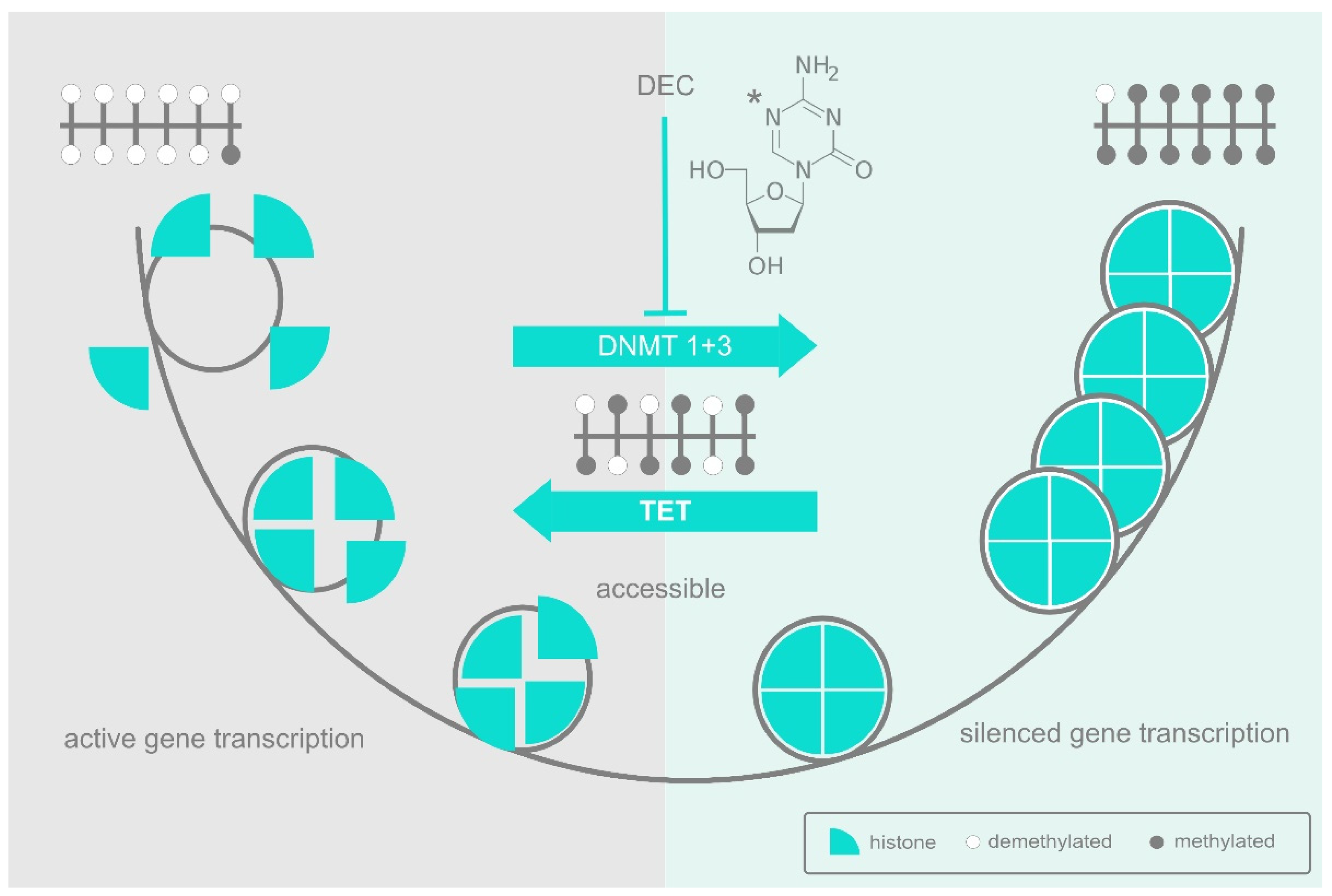
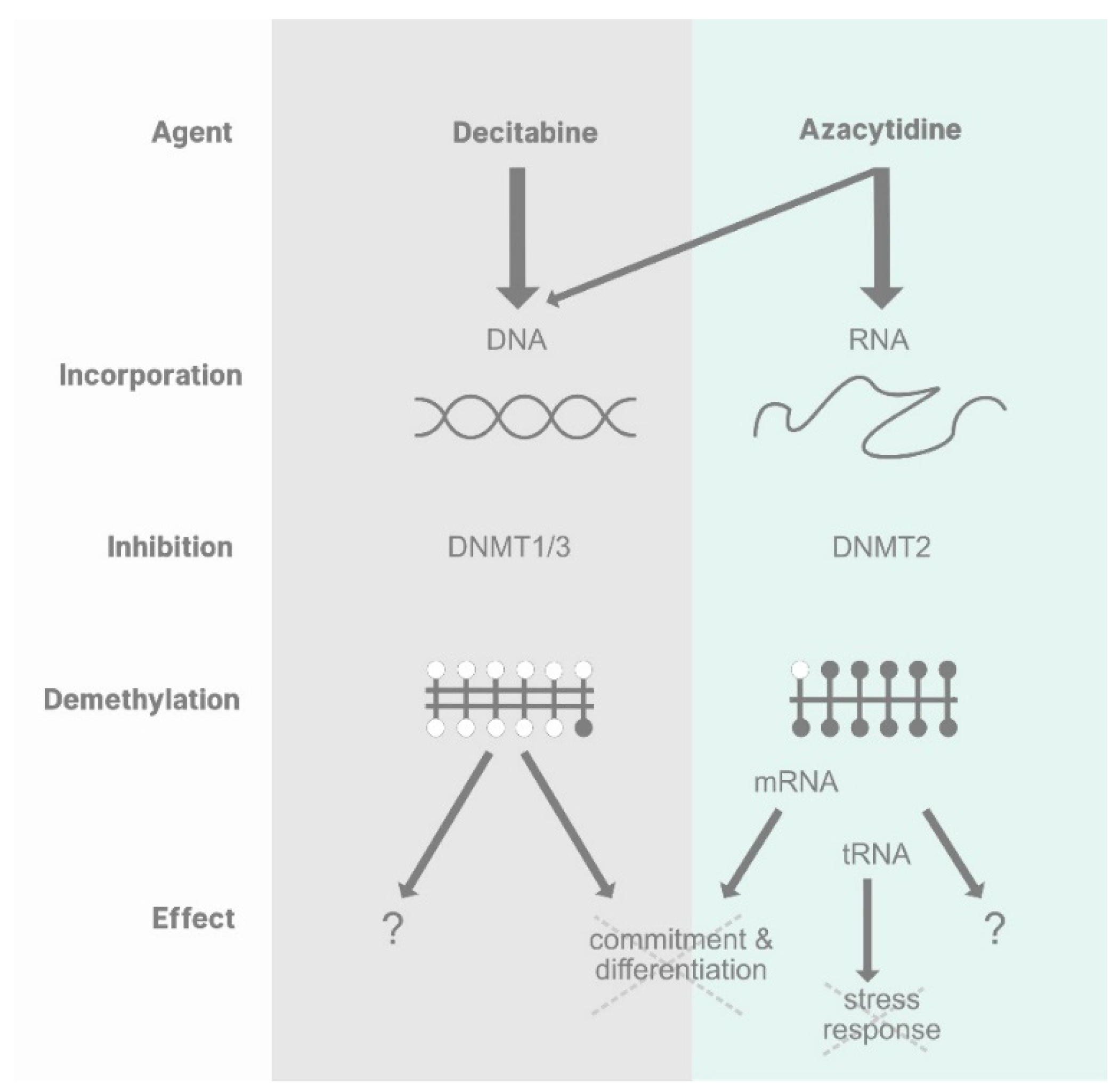{kind=link}
Abstract
:1. Introduction
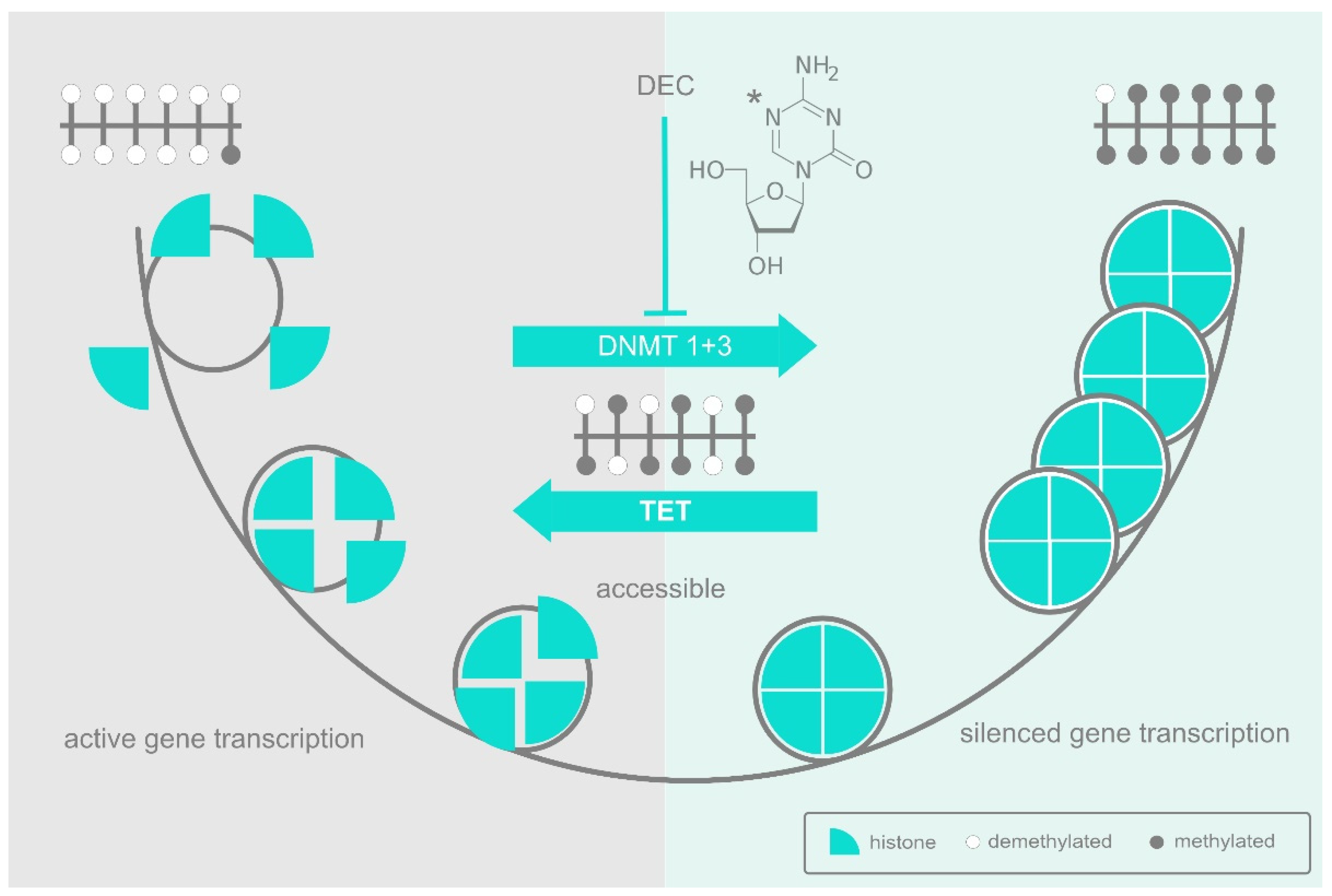
2. DNA Methylation and the DNA Methyltransferases
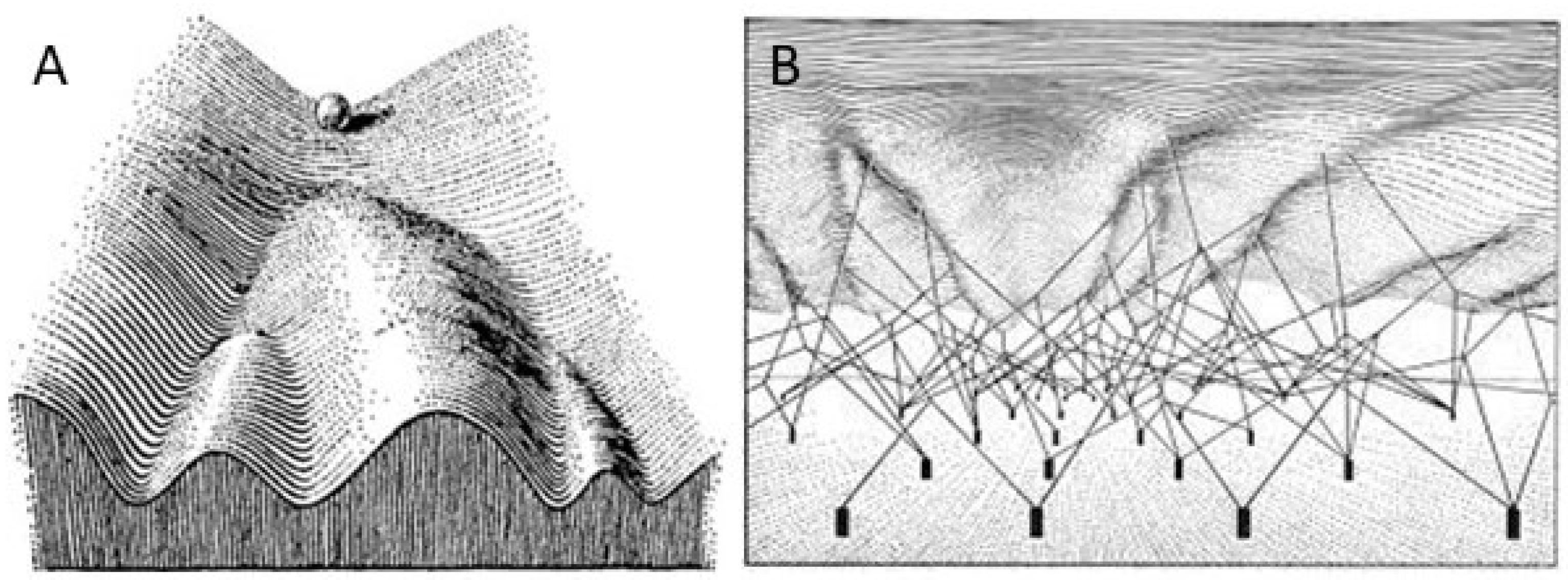
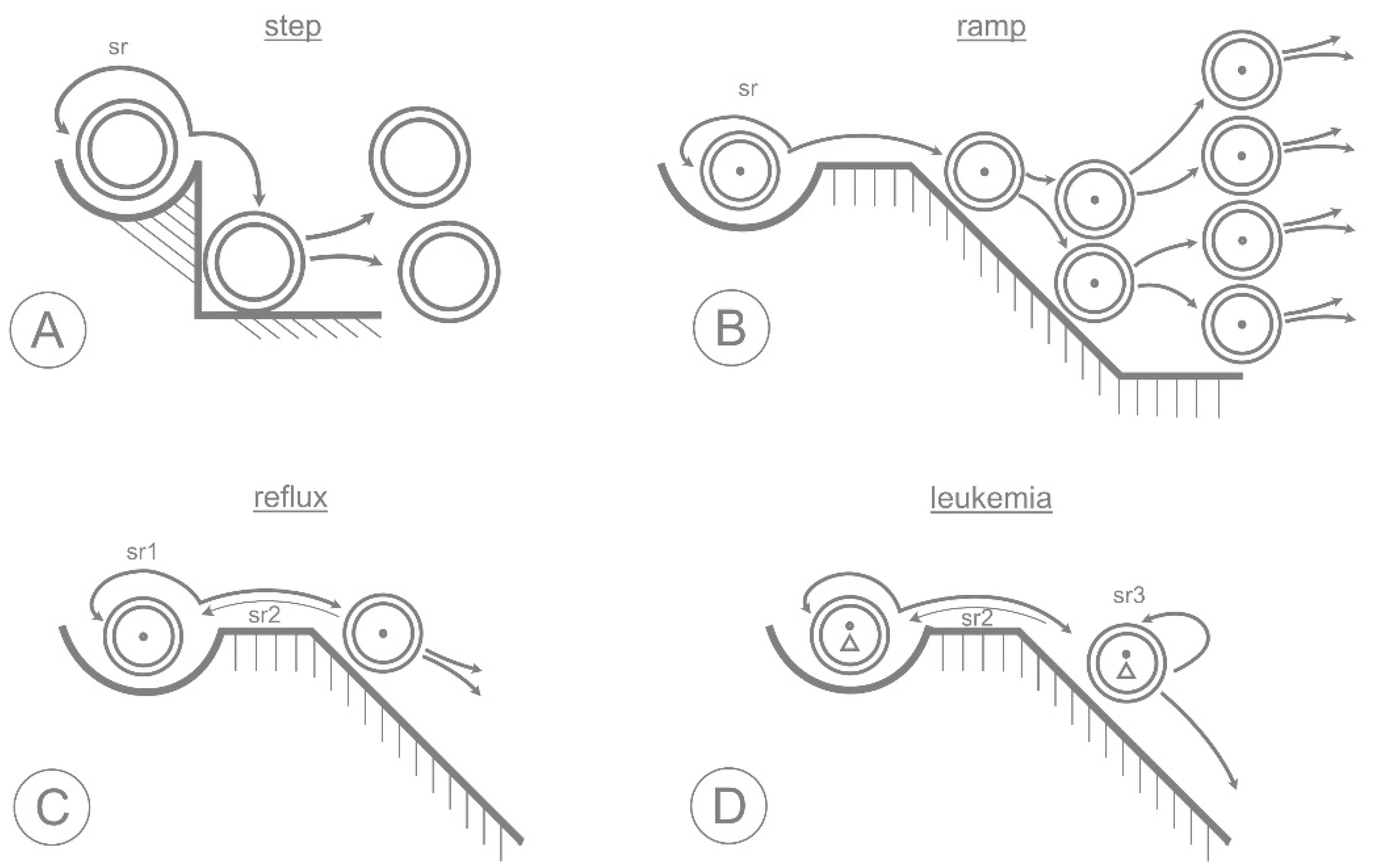
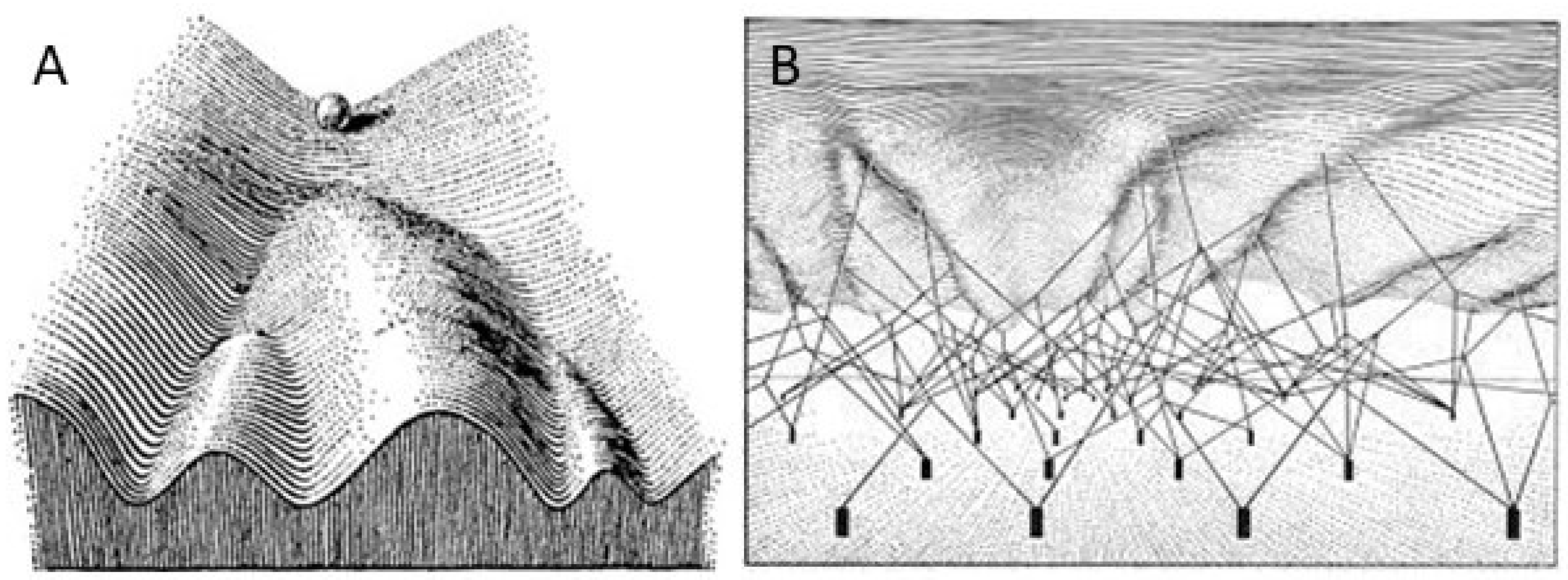
3. The Epigenetic Landscape
4. Lessons from Clonal Hematopoiesis
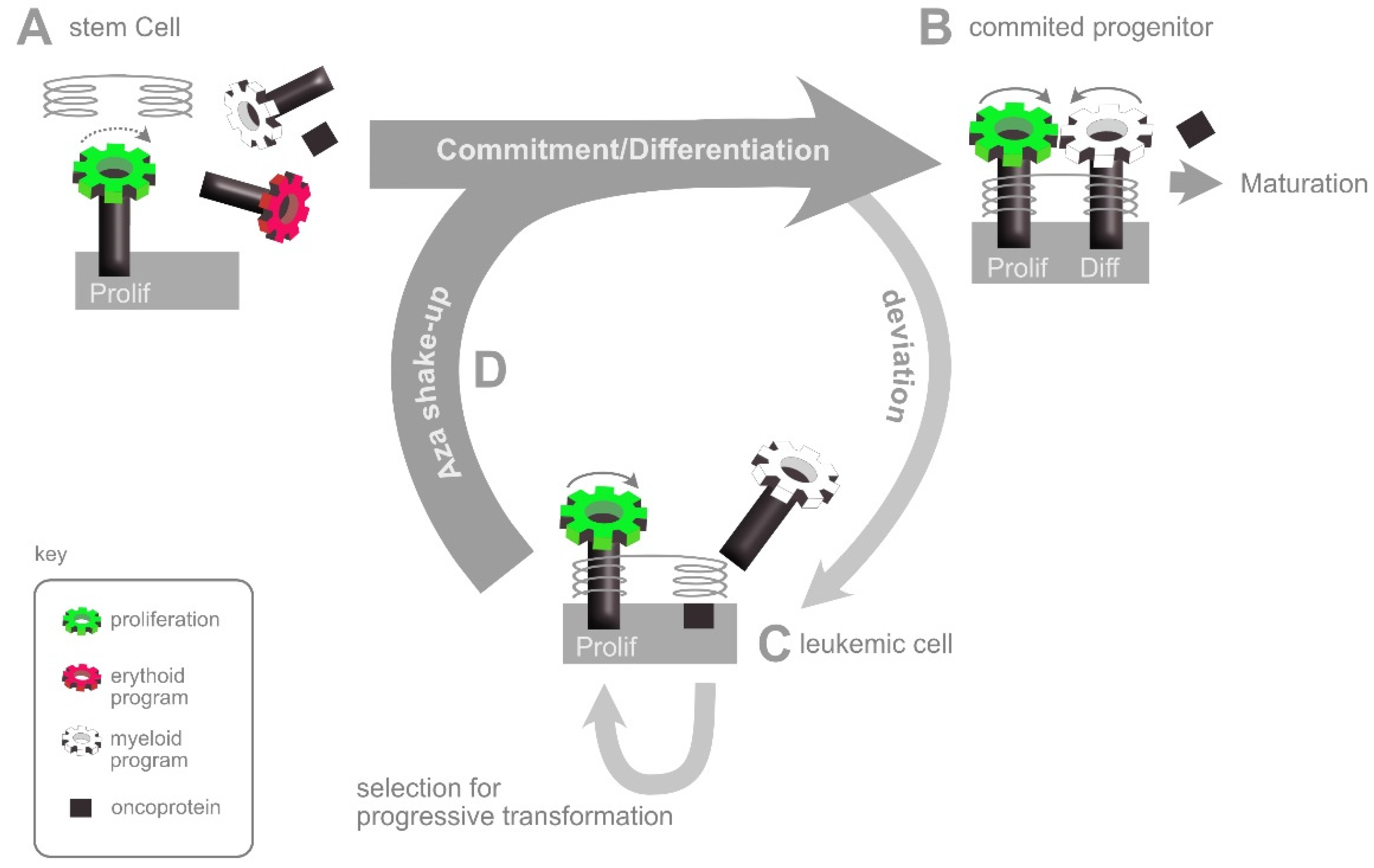
5. Neoplastic Transformation
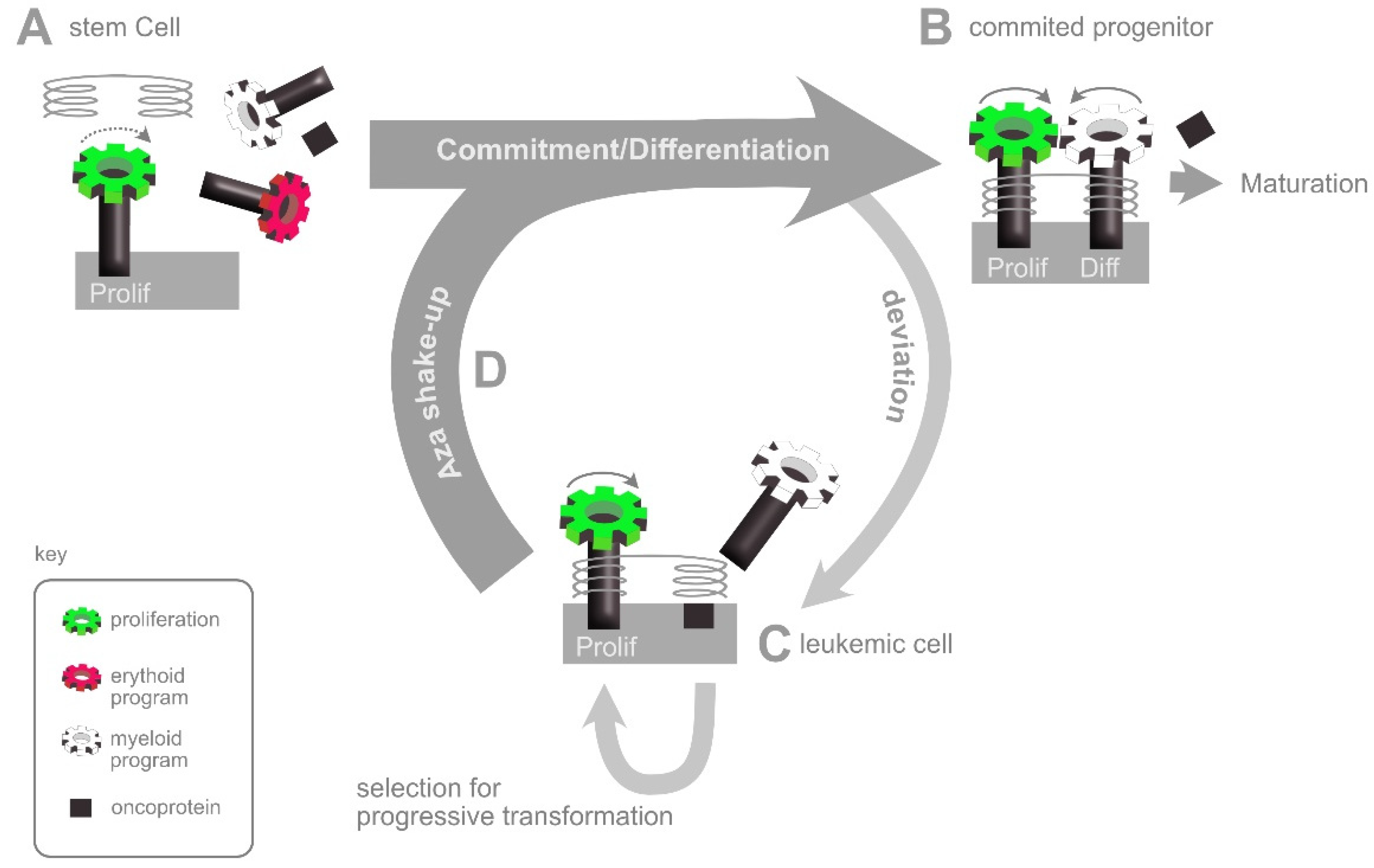
6. The AZA Shake-Up: An Interpretation of HMA Action
7. Strategies for Predicting and Improving Response
8. RNA Methylation
9. DNMT2 Methylation of mRNA
10. DNMT2 Methylation of tRNA
11. Summary and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- de Vos, D.; van Overveld, W. Decitabine: A historical review of the development of an epigenetic drug. Ann. Hematol. 2005, 84 (Suppl. S1), 3–8. [Google Scholar] [CrossRef] [PubMed]
- Issa, J.-P.J.; Garcia-Manero, G.; Giles, F.J.; Mannari, R.; Thomas, D.; Faderl, S.; Bayar, E.; Lyons, J.; Rosenfeld, C.S.; Cortes, J.; et al. Phase 1 study of low-dose prolonged exposure schedules of the hypomethylating agent 5-aza-2′-deoxycytidine (decitabine) in hematopoietic malignancies. Blood 2004, 103, 1635–1640. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Manero, G.; Fenaux, P. Hypomethylating agents and other novel strategies in myelodysplastic syndromes. J. Clin. Oncol. 2011, 29, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Stomper, J.; Rotondo, J.C.; Greve, G.; Lübbert, M. Hypomethylating agents (HMA) for the treatment of acute myeloid leukemia and myelodysplastic syndromes: Mechanisms of resistance and novel HMA-based therapies. Leukemia 2021, 35, 1873–1889. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Ge, Z. Comparison between Decitabine and Azacitidine for Patients with Acute Myeloid Leukemia and Higher-Risk Myelodysplastic Syndrome: A Systematic Review and Network Meta-Analysis. Front. Pharmacol. 2021, 12, 701690. [Google Scholar] [CrossRef] [PubMed]
- Hollenbach, P.W.; Nguyen, A.N.; Brady, H.; Williams, M.; Ning, Y.; Richard, N.; Krushel, L.; Aukerman, S.L.; Heise, C.; MacBeth, K.J. A comparison of azacitidine and decitabine activities in acute myeloid leukemia cell lines. PLoS ONE 2010, 5, e9001. [Google Scholar] [CrossRef]
- Flotho, C.; Claus, R.; Batz, C.; Schneider, M.; Sandrock, I.; Ihde, S.; Plass, C.; Niemeyer, C.M.; Lübbert, M. The DNA methyltransferase inhibitors azacitidine, decitabine and zebularine exert differential effects on cancer gene expression in acute myeloid leukemia cells. Leukemia 2009, 23, 1019–1028. [Google Scholar] [CrossRef]
- Jurkowski, T.P.; Jeltsch, A. On the evolutionary origin of eukaryotic DNA methyltransferases and Dnmt2. PLoS ONE 2011, 6, e28104. [Google Scholar] [CrossRef]
- Cross, M.A.; Enver, T. The lineage commitment of haemopoietic progenitor cells. Curr. Opin. Genet. Dev. 1997, 7, 609–613. [Google Scholar] [CrossRef]
- Akashi, K.; He, X.; Chen, J.; Iwasaki, H.; Niu, C.; Steenhard, B.; Zhang, J.; Haug, J.; Li, L. Transcriptional accessibility for genes of multiple tissues and hematopoietic lineages is hierarchically controlled during early hematopoiesis. Blood 2003, 101, 383–389. [Google Scholar] [CrossRef]
- Martin, E.W.; Krietsch, J.; Reggiardo, R.E.; Sousae, R.; Kim, D.H.; Forsberg, E.C. Chromatin accessibility maps provide evidence of multilineage gene priming in hematopoietic stem cells. Epigenet. Chromatin 2021, 14, 2. [Google Scholar] [CrossRef]
- Mizuno, S.; Chijiwa, T.; Okamura, T.; Akashi, K.; Fukumaki, Y.; Niho, Y.; Sasaki, H. Expression of DNA methyltransferases DNMT1, 3A, and 3B in normal hematopoiesis and in acute and chronic myelogenous leukemia. Blood 2001, 97, 1172–1179. [Google Scholar] [CrossRef] [PubMed]
- Holliday, R.; Grigg, G.W. DNA methylation and mutation. Mutat. Res./Fundam. Mol. Mech. Mutagenesis 1993, 285, 61–67. [Google Scholar] [CrossRef]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [PubMed]
- Waddington, C.H. The Strategy of the Genes; Allen and Unwin: London, UK, 1957. [Google Scholar]
- Kremsky, I.; Corces, V.G. Protection from DNA re-methylation by transcription factors in primordial germ cells and pre-implantation embryos can explain trans-generational epigenetic inheritance. Genome Biol. 2020, 21, 118. [Google Scholar] [CrossRef]
- Wei, J.-W.; Huang, K.; Yang, C.; Kang, C.-S. Non-coding RNAs as regulators in epigenetics (Review). Oncol. Rep. 2017, 37, 3–9. [Google Scholar] [CrossRef]
- Francis, N.J.; Sihou, D. Inheritance of Histone (H3/H4): A Binary Choice? Trends Biochem. Sci. 2021, 46, 5–14. [Google Scholar] [CrossRef]
- Huang, S. Genetic and non-genetic instability in tumor progression: Link between the fitness landscape and the epigenetic landscape of cancer cells. Cancer Metastasis Rev. 2013, 32, 423–448. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Ley, T.J.; Ding, L.; Walter, M.J.; McLellan, M.D.; Lamprecht, T.; Larson, D.E.; Kandoth, C.; Payton, J.E.; Baty, J.; Welch, J.; et al. DNMT3A mutations in acute myeloid leukemia. N. Engl. J. Med. 2010, 363, 2424–2433. [Google Scholar] [CrossRef]
- Abdel-Wahab, O.; Mullally, A.; Hedvat, C.; Garcia-Manero, G.; Patel, J.; Wadleigh, M.; Malinge, S.; Yao, J.; Kilpivaara, O.; Bhat, R.; et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood 2009, 114, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Young, A.L.; Challen, G.A.; Birmann, B.M.; Druley, T.E. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat. Commun. 2016, 7, 12484. [Google Scholar] [CrossRef]
- Midic, D.; Rinke, J.; Perner, F.; Müller, V.; Hinze, A.; Pester, F.; Landschulze, J.; Ernst, J.; Gruhn, B.; Matziolis, G.; et al. Prevalence and dynamics of clonal hematopoiesis caused by leukemia-associated mutations in elderly individuals without hematologic disorders. Leukemia 2020, 34, 2198–2205. [Google Scholar] [CrossRef] [PubMed]
- Hecker, J.S.; Hartmann, L.; Rivière, J.; Buck, M.C.; van der Garde, M.; Rothenberg-Thurley, M.; Fischer, L.; Winter, S.; Ksienzyk, B.; Ziemann, F.; et al. CHIP and hips: Clonal hematopoiesis is common in patients undergoing hip arthroplasty and is associated with autoimmune disease. Blood 2021, 138, 1727–1732. [Google Scholar] [CrossRef]
- Cobo, I.; Tanaka, T.; Glass, C.K.; Yeang, C. Clonal hematopoiesis driven by DNMT3A and TET2 mutations: Role in monocyte and macrophage biology and atherosclerotic cardiovascular disease. Curr. Opin. Hematol. 2022, 29, 1–7. [Google Scholar] [CrossRef]
- Winter, S.; Shoaie, S.; Kordasti, S.; Platzbecker, U. Integrating the “Immunome” in the Stratification of Myelodysplastic Syndromes and Future Clinical Trial Design. J. Clin. Oncol. 2020, 38, 1723–1735. [Google Scholar] [CrossRef]
- Challen, G.A.; Goodell, M.A. Clonal hematopoiesis: Mechanisms driving dominance of stem cell clones. Blood 2020, 136, 1590–1598. [Google Scholar] [CrossRef]
- Ostrander, E.L.; Kramer, A.C.; Mallaney, C.; Celik, H.; Koh, W.K.; Fairchild, J.; Haussler, E.; Zhang, C.R.C.; Challen, G.A. Divergent Effects of Dnmt3a and Tet2 Mutations on Hematopoietic Progenitor Cell Fitness. Stem Cell Rep. 2020, 14, 551–560. [Google Scholar] [CrossRef]
- Nerlov, C.; Querfurth, E.; Kulessa, H.; Graf, T. GATA-1 interacts with the myeloid PU.1 transcription factor and represses PU.1-dependent transcription. Blood 2000, 95, 2543–2551. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zhang, X.; Iwama, A.; Yu, C.; Smith, K.A.; Mueller, B.U.; Narravula, S.; Torbett, B.E.; Orkin, S.H.; Tenen, D.G.P. 1 inhibits GATA-1 function and erythroid differentiation by blocking GATA-1 DNA binding. Blood 2000, 96, 2641–2648. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.X.; Chen, L.; Li, Y.; Cloe, A.; Yue, M.; Wei, J.; Watanabe, K.A.; Shammo, J.M.; Anastasi, J.; Shen, Q.J.; et al. RNA cytosine methylation and methyltransferases mediate chromatin organization and 5-azacytidine response and resistance in leukaemia. Nat. Commun. 2018, 9, 1163. [Google Scholar] [CrossRef] [PubMed]
- Fabre, M.A.; de Almeida, J.G.; Fiorillo, E.; Mitchell, E.; Damaskou, A.; Rak, J.; Orrù, V.; Marongiu, M.; Chapman, M.S.; Vijayabaskar, M.S.; et al. The longitudinal dynamics and natural history of clonal haematopoiesis. Nature 2022, 606, 335–342. [Google Scholar] [CrossRef]
- Welch, J.S.; Petti, A.A.; Miller, C.A.; Fronick, C.C.; O’Laughlin, M.; Fulton, R.S.; Wilson, R.K.; Baty, J.D.; Duncavage, E.J.; Tandon, B.; et al. TP53 and Decitabine in Acute Myeloid Leukemia and Myelodysplastic Syndromes. N. Engl. J. Med. 2016, 375, 2023–2036. [Google Scholar] [CrossRef]
- Waddington, C.H. Canalization of development and the inheritance of acquired characteristics. Nature 1942, 150, 563–565. [Google Scholar] [CrossRef]
- Nerlov, C.; Graf, T.P. 1 induces myeloid lineage commitment in multipotent hematopoietic progenitors. Genes Dev. 1998, 12, 2403–2412. [Google Scholar] [CrossRef]
- Ruijtenberg, S.; van den Heuvel, S. Coordinating cell proliferation and differentiation: Antagonism between cell cycle regulators and cell type-specific gene expression. Cell Cycle 2016, 15, 196–212. [Google Scholar] [CrossRef]
- Wajed, S.A.; Laird, P.W.; DeMeester, T.R. DNA methylation: An alternative pathway to cancer. Ann. Surg. 2001, 234, 10–20. [Google Scholar] [CrossRef]
- Mund, C.; Brueckner, B.; Lyko, F. Reactivation of epigenetically silenced genes by DNA methyltransferase inhibitors: Basic concepts and clinical applications. Epigenetics 2006, 1, 7–13. [Google Scholar] [CrossRef]
- Hu, C.; Wang, X. Predictive and prognostic value of gene mutations in myelodysplastic syndrome treated with hypomethylating agents: A meta-analysis. Leuk. Lymphoma 2022, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Hunter, A.M.; Komrokji, R.S.; Yun, S.; Al Ali, N.; Chan, O.; Song, J.; Hussaini, M.; Talati, C.; Sweet, K.L.; Lancet, J.E.; et al. Baseline and serial molecular profiling predicts outcomes with hypomethylating agents in myelodysplastic syndromes. Blood Adv. 2021, 5, 1017–1028. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Wang, Q.; Li, S.; Wang, X. Resistance to Hypomethylating Agents in Myelodysplastic Syndrome and Acute Myeloid Leukemia from Clinical Data and Molecular Mechanism. Front. Oncol. 2021, 11, 706030. [Google Scholar] [CrossRef] [PubMed]
- Bejar, R.; Lord, A.; Stevenson, K.; Bar-Natan, M.; Pérez-Ladaga, A.; Zaneveld, J.; Wang, H.; Caughey, B.; Stojanov, P.; Getz, G.; et al. TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients. Blood 2014, 124, 2705–2712. [Google Scholar] [CrossRef] [PubMed]
- Nazha, A.; Sekeres, M.A.; Bejar, R.; Rauh, M.J.; Othus, M.; Komrokji, R.S.; Barnard, J.; Hilton, C.B.; Kerr, C.M.; Steensma, D.P.; et al. Genomic Biomarkers to Predict Resistance to Hypomethylating Agents in Patients with Myelodysplastic Syndromes Using Artificial Intelligence. JCO Precis. Oncol. 2019, 3. [Google Scholar] [CrossRef] [PubMed]
- Meldi, K.; Qin, T.; Buchi, F.; Droin, N.; Sotzen, J.; Micol, J.-B.; Selimoglu-Buet, D.; Masala, E.; Allione, B.; Gioia, D.; et al. Specific molecular signatures predict decitabine response in chronic myelomonocytic leukemia. J. Clin. Investig. 2015, 125, 1857–1872. [Google Scholar] [CrossRef]
- Cabezón, M.; Malinverni, R.; Bargay, J.; Xicoy, B.; Marcé, S.; Garrido, A.; Tormo, M.; Arenillas, L.; Coll, R.; Borras, J.; et al. Different methylation signatures at diagnosis in patients with high-risk myelodysplastic syndromes and secondary acute myeloid leukemia predict azacitidine response and longer survival. Clin. Epigenet 2021, 13, 9. [Google Scholar] [CrossRef]
- Wenk, C.; Garz, A.-K.; Grath, S.; Huberle, C.; Witham, D.; Weickert, M.; Malinverni, R.; Niggemeyer, J.; Kyncl, M.; Hecker, J.; et al. Direct modulation of the bone marrow mesenchymal stromal cell compartment by azacitidine enhances healthy hematopoiesis. Blood Adv. 2018, 2, 3447–3461. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Ohtani, H.; Chakravarthy, A.; de Carvalho, D.D. Epigenetic therapy in immune-oncology. Nat. Rev. Cancer 2019, 19, 151–161. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef]
- Daver, N.; Garcia-Manero, G.; Basu, S.; Boddu, P.C.; Alfayez, M.; Cortes, J.E.; Konopleva, M.; Ravandi-Kashani, F.; Jabbour, E.; Kadia, T.; et al. Efficacy, Safety, and Biomarkers of Response to Azacitidine and Nivolumab in Relapsed/Refractory Acute Myeloid Leukemia: A Nonrandomized, Open-Label, Phase II Study. Cancer Discov. 2019, 9, 370–383. [Google Scholar] [CrossRef]
- Sallman, D.A.; Asch, A.S.; Al Malki, M.M.; Lee, D.J.; Donnellan, W.B.; Marcucci, G.; Kambhampati, S.; Daver, N.G.; Garcia-Manero, G.; Komrokji, R.S.; et al. The First-in-Class Anti-CD47 Antibody Magrolimab (5F9) in Combination with Azacitidine Is Effective in MDS and AML Patients: Ongoing Phase 1b Results. Blood 2019, 134, 569. [Google Scholar] [CrossRef]
- Gallazzi, M.; Ucciero, M.A.M.; Faraci, D.G.; Mahmoud, A.M.; Al Essa, W.; Gaidano, G.; Mouhssine, S.; Crisà, E. New Frontiers in Monoclonal Antibodies for the Targeted Therapy of Acute Myeloid Leukemia and Myelodysplastic Syndromes. Int. J. Mol. Sci. 2022, 23, 7542. [Google Scholar] [CrossRef] [PubMed]
- Li, L.H.; Olin, E.J.; Buskirk, H.H.; Reineke, L.M. Cytotoxicity and mode of action of 5-azacytidine on L1210 leukemia. Cancer Res. 1970, 30, 2760–2769. [Google Scholar] [PubMed]
- Schaefer, M.; Hagemann, S.; Hanna, K.; Lyko, F. Azacytidine inhibits RNA methylation at DNMT2 target sites in human cancer cell lines. Cancer Res. 2009, 69, 8127–8132. [Google Scholar] [CrossRef]
- Suzuki, T. The expanding world of tRNA modifications and their disease relevance. Nat. Rev. Mol. Cell Biol. 2021, 22, 375–392. [Google Scholar] [CrossRef]
- Bohnsack, K.E.; Höbartner, C.; Bohnsack, M.T. Eukaryotic 5-methylcytosine (m⁵C) RNA Methyltransferases: Mechanisms, Cellular Functions, and Links to Disease. Genes 2019, 10, 102. [Google Scholar] [CrossRef]
- Goll, M.G.; Kirpekar, F.; Maggert, K.A.; Yoder, J.A.; Hsieh, C.-L.; Zhang, X.; Golic, K.G.; Jacobsen, S.E.; Bestor, T.H. Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science 2006, 311, 395–398. [Google Scholar] [CrossRef]
- Schaefer, M.; Pollex, T.; Hanna, K.; Tuorto, F.; Meusburger, M.; Helm, M.; Lyko, F. RNA methylation by Dnmt2 protects transfer RNAs against stress-induced cleavage. Genes Dev. 2010, 24, 1590–1595. [Google Scholar] [CrossRef]
- Tuorto, F.; Herbst, F.; Alerasool, N.; Bender, S.; Popp, O.; Federico, G.; Reitter, S.; Liebers, R.; Stoecklin, G.; Gröne, H.-J.; et al. The tRNA methyltransferase Dnmt2 is required for accurate polypeptide synthesis during haematopoiesis. EMBO J. 2015, 34, 2350–2362. [Google Scholar] [CrossRef]
- Diesch, J.; Le Pannérer, M.-M.; Winkler, R.; Casquero, R.; Muhar, M.; van der Garde, M.; Maher, M.; Herráez, C.M.; Bech-Serra, J.J.; Fellner, M.; et al. Inhibition of CBP synergizes with the RNA-dependent mechanisms of Azacitidine by limiting protein synthesis. Nat. Commun. 2021, 12, 6060. [Google Scholar] [CrossRef] [PubMed]
- Kubasch, A.S.; Platzbecker, U. Beyond the Edge of Hypomethylating Agents: Novel Combination Strategies for Older Adults with Advanced MDS and AML. Cancers 2018, 10, 158. [Google Scholar] [CrossRef]
- Shanmugam, R.; Fierer, J.; Kaiser, S.; Helm, M.; Jurkowski, T.P.; Jeltsch, A. Cytosine methylation of tRNA-Asp by DNMT2 has a role in translation of proteins containing poly-Asp sequences. Cell Discov. 2015, 1, 15010. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.; Müller, S.; Nellen, W.; Jurkowski, T.P.; Jeltsch, A.; Ehrenhofer-Murray, A.E. Pmt1, a Dnmt2 homolog in Schizosaccharomyces pombe, mediates tRNA methylation in response to nutrient signaling. Nucleic Acids Res. 2012, 40, 11648–11658. [Google Scholar] [CrossRef]
- Durdevic, Z.; Mobin, M.B.; Hanna, K.; Lyko, F.; Schaefer, M. The RNA methyltransferase Dnmt2 is required for efficient Dicer-2-dependent siRNA pathway activity in Drosophila. Cell Rep. 2013, 4, 931–937. [Google Scholar] [CrossRef]
- Ehrenhofer-Murray, A.E. Cross-Talk between Dnmt2-Dependent tRNA Methylation and Queuosine Modification. Biomolecules 2017, 7, 14. [Google Scholar] [CrossRef]
- Gingold, H.; Tehler, D.; Christoffersen, N.R.; Nielsen, M.M.; Asmar, F.; Kooistra, S.M.; Christophersen, N.S.; Christensen, L.L.; Borre, M.; Sørensen, K.D.; et al. A dual program for translation regulation in cellular proliferation and differentiation. Cell 2014, 158, 1281–1292. [Google Scholar] [CrossRef]
- Meier, F.; Suter, B.; Grosjean, H.; Keith, G.; Kubli, E. Queuosine modification of the wobble base in tRNAHis influences ‘in vivo’ decoding properties. EMBO J. 1985, 4, 823–827. [Google Scholar] [CrossRef]
- Ianevski, A.; Lahtela, J.; Javarappa, K.K.; Sergeev, P.; Ghimire, B.R.; Gautam, P.; Vähä-Koskela, M.; Turunen, L.; Linnavirta, N.; Kuusanmäki, H.; et al. Patient-tailored design for selective co-inhibition of leukemic cell subpopulations. Sci. Adv. 2021, 7, eabe4038. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stein, A.; Platzbecker, U.; Cross, M. How Azanucleosides Affect Myeloid Cell Fate. Cells 2022, 11, 2589. https://doi.org/10.3390/cells11162589
Stein A, Platzbecker U, Cross M. How Azanucleosides Affect Myeloid Cell Fate. Cells. 2022; 11(16):2589. https://doi.org/10.3390/cells11162589
Chicago/Turabian StyleStein, Anna, Uwe Platzbecker, and Michael Cross. 2022. "How Azanucleosides Affect Myeloid Cell Fate" Cells 11, no. 16: 2589. https://doi.org/10.3390/cells11162589
APA StyleStein, A., Platzbecker, U., & Cross, M. (2022). How Azanucleosides Affect Myeloid Cell Fate. Cells, 11(16), 2589. https://doi.org/10.3390/cells11162589