
Current Opportunities for Targeting Dysregulated Neurodevelopmental Signaling Pathways in Glioblastoma

 , , and
, , and
Abstract
1. Glioblastoma
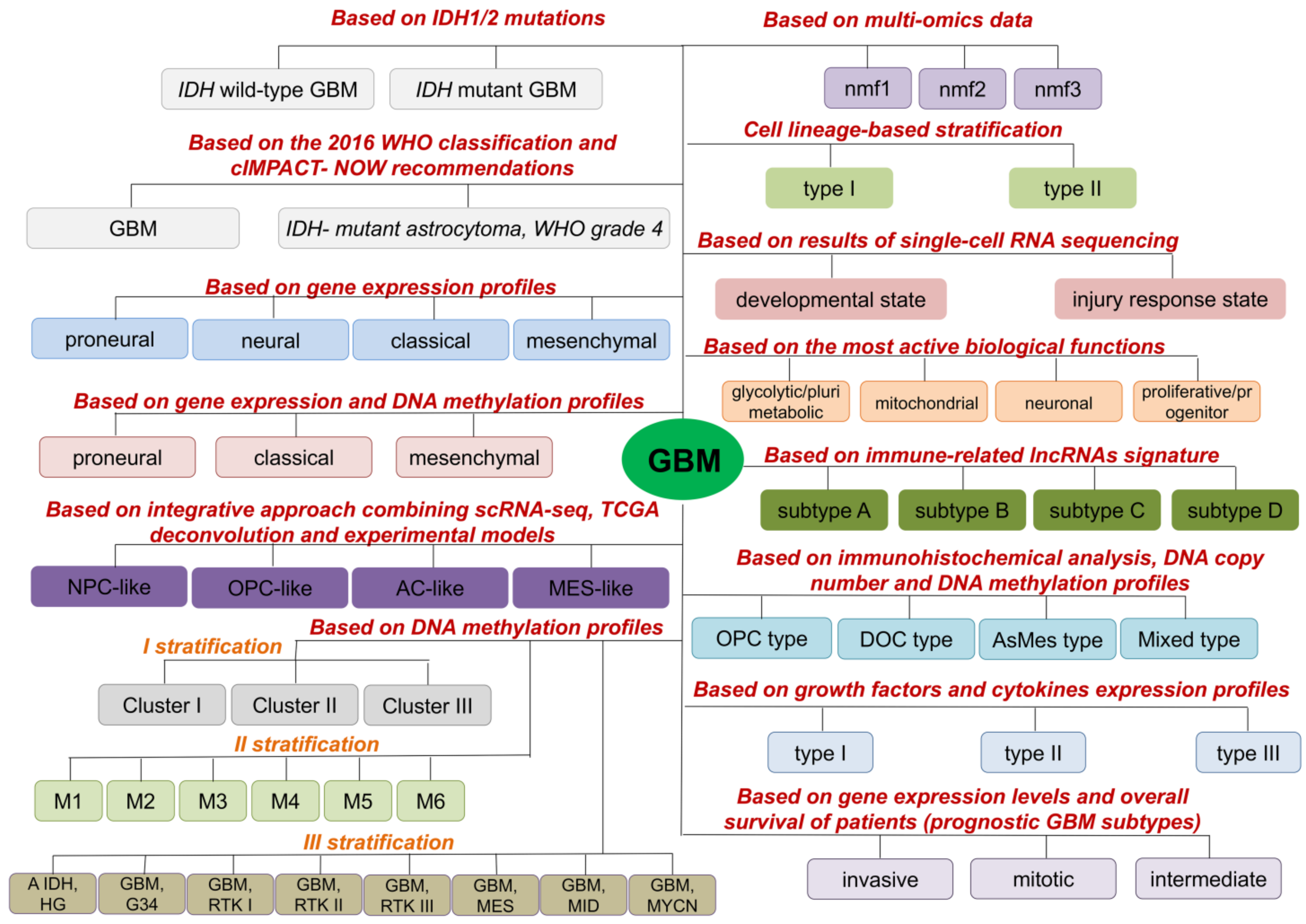
2. Molecular GBM Subtypes
3. Targeting Sonic Hedgehog Signaling Pathway in GBM
4. Targeting Canonical Wnt/β-Catenin Signaling Pathway in GBM


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Modulation of Wnt Signaling Activity | Effects on GBM Cells and GSCs Properties | Reference |
|---|---|---|---|
| A: Small molecules | |||
| ONC201 | inhibits expression of components of Wnt pathway and Wnt targets | induces apoptosis in GBM cells induces cytotoxicity in chemo- and radiation-resistant GBM patient samples inhibits the growth of GSCs in 3D neurospheres established from human GBM tumors inhibits tumor growth in GBM mouse models | [121,127] |
| SEN461 | induces AXIN stabilization | inhibits anchorage-independent growth of human GBM cell lines and patient-derived primary tumor cells reduces tumor growth in a mouse GBM xenograft model | [122] |
| XAV939 | antagonist of tankyrase-enzymes involved in the degradation of AXIN | decreases the survival and clonogenicity of GBM cells reduces GSC population increases radiosensitization in in vivo radiation model derived from a single human GBM specimen | [125] |
| LiCl SB216763 | inhibits GSK3β | induces the expression of differentiation markers in GBM cells depletes GSCs population reduces colony formation and induces cell death in GBM cell-lines | [126] |
| G007-LK | inhibitor of tankyrase-enzymes involved in the degradation of AXIN | decreases in vitro proliferation and sphere formation in primary GSC cultures reduction in GSC sphere formation in cotreatment with TMZ | [124] |
| IC261 | inhibitor of CK1 | inhibits growth of GBM cells and GSCs in vitro and induces growth inhibition of human GBM xenografts in mice | [129] |
| LGK974 | inhibitor of porcupine proteins that modulate Wnt ligands | acts synergistically with TMZ to inhibit growth of GBM cells | [128] |
| ICG-001 | CBP antagonist | reduces proliferation and survival of GBM cells | [123] |
| AZD2858 | inhibits GSK-3β | reduces proliferation and survival of GBM cells inhibits the invasion and migration of GBM cells | [123] |
| B: Natural agents | |||
| shikonin | inhibits β-catenin phosphorylation | inhibits proliferation, migration and invasion of GBM cells | [131] |
| Trichosanthin | inhibits expression of Wnt components | inhibits proliferation, invasion and migration and induces apoptosis of GBM cells | [132] |
| R. crenulata root extract | decreases nuclear localization of β-catenin | inhibits proliferation and tumorsphere formation and promotes differentiation of GBM cells | [133] |
| resveratrol | decreases expression of Wnt signaling components and Wnt targets | inhibits proliferation, motility and invasion of GSCs | [134] |
| carnosic acid | decreases expression of WISP1 | reduces GSC viability suppresses GSC tumorsphere formation inhibits the growth of GSC-derived xenografts | [135] |
| Indirubin | inhibitor of GSK-3β | reduces invasion of GBM and GSC-enriched neurospheres both in vitro and in vivo improves survival of intracranial glioma-bearing mice | [136] |
| DATS | decreases nuclear β-catenin level | inhibits cell growth, induces apoptosis and decreases migration and invasion in GBM cells | [137] |
| Sulforaphane | inhibits Wnt/β-catenin signaling | enhances TMZ-induced apoptosis | [138] |
| C: Repurposed drugs | |||
| NSAIDs diclofenac celecoxib aspirin | reduces phosphorylation of GSK3β inhibits expression of Wnt targets | inhibits proliferation, colony formation and migration of GBM cells inhibits proliferation and invasion and induces apoptosis of GBM cells | [139] [140] |
| Niclosamide | decreases concentration of β-catenin in the nucleus | decreases cell viability, exerts antimigratory effects and inhibits the malignant potential of primary GBMs combined treatment with TMZ inhibits viability, stemness, and invasive properties of human GBM tumorspheres and decreases tumor growth in mouse xenograft models | [141] [142] |
| QUE | decreases phosphorylation of GSK3β | suppresses GSCs-initiated tumor growth in mouse models of gliomas acts synergistically with TMZ to suppress growth of TMZ-resistant tumors originated from GSCs | [143] |
| Pioglitazone | inhibits β-catenin expression | reduces cell viability, suppresses invasion and induces apoptosis of GBM cells induces decrease in cell viability and proliferation of GSC lines isolated from GBM patients | [144] [145] |
5. Targeting Canonical Notch Signaling Pathway in GBM
6. Targeting TGFβ Signaling Pathway in GBM
7. Targeting BMP Signaling Pathway in GBM
8. Targeting Hippo Signaling Pathway in GBM
9. Targeting Retinoic Acid Signaling Pathway in GBM
10. Conclusions and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Parker, N.R.; Khong, P.; Parkinson, J.F.; Howell, V.M.; Wheeler, H.R. Molecular heterogeneity in glioblastoma: Potential clinical implications. Front. Oncol. 2015, 5, 55. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Platten, M. Understanding and Treating Glioblastoma. Neurol. Clin. 2018, 36, 485–499. [Google Scholar] [CrossRef] [PubMed]
- Soeda, A.; Hara, A.; Kunisada, T.; Yoshimura, S.; Iwama, T.; Park, D.M. The evidence of glioblastoma heterogeneity. Sci. Rep. 2015, 5, 7979. [Google Scholar] [CrossRef]
- Becker, A.P.; Sells, B.E.; Haque, S.J.; Chakravarti, A. Tumor Heterogeneity in Glioblastomas: From Light Microscopy to Molecular Pathology. Cancers 2021, 13, 761. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Gittleman, H.; Fulop, J.; Liu, M.; Blanda, R.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008–2012. Neuro-Oncology 2015, 17 (Suppl. 4), iv1–iv62. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Johnson, B.E.; Mazor, T.; Hong, C.; Barnes, M.; Aihara, K.; McLean, C.Y.; Fouse, S.D.; Yamamoto, S.; Ueda, H.; Tatsuno, K.; et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 2014, 343, 189–193. [Google Scholar] [CrossRef]
- Oronsky, B.; Reid, T.R.; Oronsky, A.; Sandhu, N.; Knox, S.J. A Review of Newly Diagnosed Glioblastoma. Front. Oncol. 2020, 10, 574012. [Google Scholar] [CrossRef]
- Birzu, C.; French, P.; Caccese, M.; Cerretti, G.; Idbaih, A.; Zagonel, V.; Lombardi, G. Recurrent Glioblastoma: From Molecular Landscape to New Treatment Perspectives. Cancers 2020, 13, 47. [Google Scholar] [CrossRef]
- Mao, H.; Lebrun, D.G.; Yang, J.; Zhu, V.F.; Li, M. Deregulated signaling pathways in glioblastoma multiforme: Molecular mechanisms and therapeutic targets. Cancer Investig. 2012, 30, 48–56. [Google Scholar] [CrossRef]
- Khabibov, M.; Garifullin, A.; Boumber, Y.; Khaddour, K.; Fernandez, M.; Khamitov, F.; Khalikova, L.; Kuznetsova, N.; Kit, O.; Kharin, L. Signaling pathways and therapeutic approaches in glioblastoma multiforme (Review). Int. J. Oncol. 2022, 60, 69. [Google Scholar] [CrossRef]
- Mehta, S.; Lo Cascio, C. Developmentally regulated signaling pathways in glioma invasion. Cell. Mol. Life Sci. 2018, 75, 385–402. [Google Scholar] [CrossRef]
- Lauko, A.; Lo, A.; Ahluwalia, M.S.; Lathia, J.D. Cancer cell heterogeneity & plasticity in glioblastoma and brain tumors. Semin Cancer Biol. 2022, 82, 162–175. [Google Scholar] [CrossRef]
- Ryskalin, L.; Gaglione, A.; Limanaqi, F.; Biagioni, F.; Familiari, P.; Frati, A.; Esposito, V.; Fornai, F. The Autophagy Status of Cancer Stem Cells in Gliobastoma Multiforme: From Cancer Promotion to Therapeutic Strategies. Int. J. Mol. Sci. 2019, 20, 3824. [Google Scholar] [CrossRef]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef]
- Stevanovic, M.; Kovacevic-Grujicic, N.; Mojsin, M.; Milivojevic, M.; Drakulic, D. SOX transcription factors and glioma stem cells: Choosing between stemness and differentiation. World J. Stem Cells 2021, 13, 1417–1445. [Google Scholar] [CrossRef]
- Prager, B.C.; Bhargava, S.; Mahadev, V.; Hubert, C.G.; Rich, J.N. Glioblastoma Stem Cells: Driving Resilience through Chaos. Trends Cancer 2020, 6, 223–235. [Google Scholar] [CrossRef]
- Cheng, L.; Wu, Q.; Guryanova, O.A.; Huang, Z.; Huang, Q.; Rich, J.N.; Bao, S. Elevated invasive potential of glioblastoma stem cells. Biochem. Biophys. Res. Commun. 2011, 406, 643–648. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef]
- Satterlee, A.B.; Dunn, D.E.; Lo, D.C.; Khagi, S.; Hingtgen, S. Tumoricidal stem cell therapy enables killing in novel hybrid models of heterogeneous glioblastoma. Neuro-Oncology 2019, 21, 1552–1564. [Google Scholar] [CrossRef]
- Tang, X.; Zuo, C.; Fang, P.; Liu, G.; Qiu, Y.; Huang, Y.; Tang, R. Targeting Glioblastoma Stem Cells: A Review on Biomarkers, Signal Pathways and Targeted Therapy. Front. Oncol. 2021, 11, 701291. [Google Scholar] [CrossRef]
- Nakada, M.; Kita, D.; Watanabe, T.; Hayashi, Y.; Teng, L.; Pyko, I.V.; Hamada, J. Aberrant signaling pathways in glioma. Cancers 2011, 3, 3242–3278. [Google Scholar] [CrossRef]
- Cruz Da Silva, E.; Mercier, M.C.; Etienne-Selloum, N.; Dontenwill, M.; Choulier, L. A Systematic Review of Glioblastoma-Targeted Therapies in Phases II, III, IV Clinical Trials. Cancers 2021, 13, 1795. [Google Scholar] [CrossRef]
- Fabro, F.; Lamfers, M.L.M.; Leenstra, S. Advancements, Challenges, and Future Directions in Tackling Glioblastoma Resistance to Small Kinase Inhibitors. Cancers 2022, 14, 600. [Google Scholar] [CrossRef]
- Liu, H.; Qiu, W.; Sun, T.; Wang, L.; Du, C.; Hu, Y.; Liu, W.; Feng, F.; Chen, Y.; Sun, H. Therapeutic strategies of glioblastoma (GBM): The current advances in the molecular targets and bioactive small molecule compounds. Acta Pharm. Sin. B 2022, 12, 1781–1804. [Google Scholar] [CrossRef]
- Zhang, G.L.; Wang, C.F.; Qian, C.; Ji, Y.X.; Wang, Y.Z. Role and mechanism of neural stem cells of the subventricular zone in glioblastoma. World J. Stem Cells 2021, 13, 877–893. [Google Scholar] [CrossRef]
- Matarredona, E.R.; Pastor, A.M. Neural Stem Cells of the Subventricular Zone as the Origin of Human Glioblastoma Stem Cells. Therapeutic Implications. Front. Oncol. 2019, 9, 779. [Google Scholar] [CrossRef]
- Couturier, C.P.; Ayyadhury, S.; Le, P.U.; Nadaf, J.; Monlong, J.; Riva, G.; Allache, R.; Baig, S.; Yan, X.; Bourgey, M.; et al. Single-cell RNA-seq reveals that glioblastoma recapitulates a normal neurodevelopmental hierarchy. Nat. Commun. 2020, 11, 3406. [Google Scholar] [CrossRef]
- Curry, R.N.; Glasgow, S.M. The Role of Neurodevelopmental Pathways in Brain Tumors. Front. Cell Dev. Biol. 2021, 9, 659055. [Google Scholar] [CrossRef]
- Bar, E.E.; Chaudhry, A.; Lin, A.; Fan, X.; Schreck, K.; Matsui, W.; Piccirillo, S.; Vescovi, A.L.; DiMeco, F.; Olivi, A.; et al. Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells 2007, 25, 2524–2533. [Google Scholar] [CrossRef] [PubMed]
- Clement, V.; Sanchez, P.; de Tribolet, N.; Radovanovic, I.; Ruiz i Altaba, A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 2007, 17, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.Y.; Zheng, M.H.; Cheng, G.; Li, L.; Liang, L.; Gao, F.; Wei, Y.N.; Fu, L.A.; Han, H. Notch signaling contributes to the maintenance of both normal neural stem cells and patient-derived glioma stem cells. BMC Cancer 2011, 11, 82. [Google Scholar] [CrossRef] [PubMed]
- Ikushima, H.; Todo, T.; Ino, Y.; Takahashi, M.; Miyazawa, K.; Miyazono, K. Autocrine TGF-beta signaling maintains tumorigenicity of glioma-initiating cells through Sry-related HMG-box factors. Cell Stem Cell 2009, 5, 504–514. [Google Scholar] [CrossRef]
- Piccirillo, S.G.; Reynolds, B.A.; Zanetti, N.; Lamorte, G.; Binda, E.; Broggi, G.; Brem, H.; Olivi, A.; Dimeco, F.; Vescovi, A.L. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature 2006, 444, 761–765. [Google Scholar] [CrossRef]
- Masui, K.; Mischel, P.S.; Reifenberger, G. Molecular classification of gliomas. Handb. Clin. Neurol. 2016, 134, 97–120. [Google Scholar] [CrossRef]
- Weller, M.; van den Bent, M.; Preusser, M.; Le Rhun, E.; Tonn, J.C.; Minniti, G.; Bendszus, M.; Balana, C.; Chinot, O.; Dirven, L.; et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat. Rev. Clin. Oncol. 2021, 18, 170–186. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Agnihotri, S.; Aldape, K.D.; Zadeh, G. Isocitrate dehydrogenase status and molecular subclasses of glioma and glioblastoma. Neurosurg. Focus 2014, 37, E13. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; de Carvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e46. [Google Scholar] [CrossRef]
- Gill, B.J.; Pisapia, D.J.; Malone, H.R.; Goldstein, H.; Lei, L.; Sonabend, A.; Yun, J.; Samanamud, J.; Sims, J.S.; Banu, M.; et al. MRI-localized biopsies reveal subtype-specific differences in molecular and cellular composition at the margins of glioblastoma. Proc. Natl. Acad. Sci. USA 2014, 111, 12550–12555. [Google Scholar] [CrossRef]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.A.; Jones, D.T.; Konermann, C.; Pfaff, E.; Tonjes, M.; Sill, M.; Bender, S.; et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef]
- Bhat, K.P.L.; Balasubramaniyan, V.; Vaillant, B.; Ezhilarasan, R.; Hummelink, K.; Hollingsworth, F.; Wani, K.; Heathcock, L.; James, J.D.; Goodman, L.D.; et al. Mesenchymal differentiation mediated by NF-kappaB promotes radiation resistance in glioblastoma. Cancer Cell 2013, 24, 331–346. [Google Scholar] [CrossRef]
- Huse, J.T.; Phillips, H.S.; Brennan, C.W. Molecular subclassification of diffuse gliomas: Seeing order in the chaos. Glia 2011, 59, 1190–1199. [Google Scholar] [CrossRef]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef]
- Zheng, S.; Chheda, M.G.; Verhaak, R.G. Studying a complex tumor: Potential and pitfalls. Cancer J. 2012, 18, 107–114. [Google Scholar] [CrossRef]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavare, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef]
- Ensenyat-Mendez, M.; Iniguez-Munoz, S.; Sese, B.; Marzese, D.M. iGlioSub: An integrative transcriptomic and epigenomic classifier for glioblastoma molecular subtypes. BioData Min. 2021, 14, 42. [Google Scholar] [CrossRef]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e821. [Google Scholar] [CrossRef]
- Ma, H.; Zhao, C.; Zhao, Z.; Hu, L.; Ye, F.; Wang, H.; Fang, Z.; Wu, Y.; Chen, X. Specific glioblastoma multiforme prognostic-subtype distinctions based on DNA methylation patterns. Cancer Gene 2020, 27, 702–714. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Capper, D.; Stichel, D.; Sahm, F.; Jones, D.T.W.; Schrimpf, D.; Sill, M.; Schmid, S.; Hovestadt, V.; Reuss, D.E.; Koelsche, C.; et al. Practical implementation of DNA methylation and copy-number-based CNS tumor diagnostics: The Heidelberg experience. Acta Neuropathol. 2018, 136, 181–210. [Google Scholar] [CrossRef]
- Wang, L.B.; Karpova, A.; Gritsenko, M.A.; Kyle, J.E.; Cao, S.; Li, Y.; Rykunov, D.; Colaprico, A.; Rothstein, J.H.; Hong, R.; et al. Proteogenomic and metabolomic characterization of human glioblastoma. Cancer Cell 2021, 39, 509–528.e520. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, D.; Chen, Y.J.; Xie, X.; Shi, Y.; Tabar, V.; Brennan, C.W.; Bale, T.A.; Jayewickreme, C.D.; Laks, D.R.; et al. Cell Lineage-Based Stratification for Glioblastoma. Cancer Cell 2020, 38, 366–379. [Google Scholar] [CrossRef]
- Hubert, C.G.; Lathia, J.D. Seeing the GBM diversity spectrum. Nat. Cancer 2021, 2, 135–137. [Google Scholar] [CrossRef]
- Richards, L.M.; Whitley, O.K.N.; MacLeod, G.; Cavalli, F.M.G.; Coutinho, F.J.; Jaramillo, J.E.; Svergun, N.; Riverin, M.; Croucher, D.C.; Kushida, M.; et al. Gradient of Developmental and Injury Response transcriptional states defines functional vulnerabilities underpinning glioblastoma heterogeneity. Nat. Cancer 2021, 2, 157–173. [Google Scholar] [CrossRef]
- Garofano, L.; Migliozzi, S.; Oh, Y.T.; D’Angelo, F.; Najac, R.D.; Ko, A.; Frangaj, B.; Caruso, F.P.; Yu, K.; Yuan, J.; et al. Pathway-based classification of glioblastoma uncovers a mitochondrial subtype with therapeutic vulnerabilities. Nat. Cancer 2021, 2, 141–156. [Google Scholar] [CrossRef]
- Yu, W.; Ma, Y.; Hou, W.; Wang, F.; Cheng, W.; Qiu, F.; Wu, P.; Zhang, G. Identification of Immune-Related lncRNA Prognostic Signature and Molecular Subtypes for Glioblastoma. Front. Immunol. 2021, 12, 706936. [Google Scholar] [CrossRef]
- Motomura, K.; Natsume, A.; Watanabe, R.; Ito, I.; Kato, Y.; Momota, H.; Nishikawa, R.; Mishima, K.; Nakasu, Y.; Abe, T.; et al. Immunohistochemical analysis-based proteomic subclassification of newly diagnosed glioblastomas. Cancer Sci. 2012, 103, 1871–1879. [Google Scholar] [CrossRef]
- Hu, B.; Ruan, Y.; Wei, F.; Qin, G.; Mo, X.; Wang, X.; Zou, D. Identification of three glioblastoma subtypes and a six-gene prognostic risk index based on the expression of growth factors and cytokines. Am. J. Transl. Res. 2020, 12, 4669–4682. [Google Scholar]
- Park, J.; Shim, J.K.; Yoon, S.J.; Kim, S.H.; Chang, J.H.; Kang, S.G. Transcriptome profiling-based identification of prognostic subtypes and multi-omics signatures of glioblastoma. Sci. Rep. 2019, 9, 10555. [Google Scholar] [CrossRef] [PubMed]
- Mao, P.; Joshi, K.; Li, J.; Kim, S.H.; Li, P.; Santana-Santos, L.; Luthra, S.; Chandran, U.R.; Benos, P.V.; Smith, L.; et al. Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc. Natl. Acad. Sci. USA 2013, 110, 8644–8649. [Google Scholar] [CrossRef] [PubMed]
- Piao, Y.; Liang, J.; Holmes, L.; Henry, V.; Sulman, E.; de Groot, J.F. Acquired resistance to anti-VEGF therapy in glioblastoma is associated with a mesenchymal transition. Clin. Cancer Res. 2013, 19, 4392–4403. [Google Scholar] [CrossRef] [PubMed]
- Singer, E.; Judkins, J.; Salomonis, N.; Matlaf, L.; Soteropoulos, P.; McAllister, S.; Soroceanu, L. Reactive oxygen species-mediated therapeutic response and resistance in glioblastoma. Cell Death Dis. 2015, 6, e1601. [Google Scholar] [CrossRef]
- Yin, J.; Oh, Y.T.; Kim, J.Y.; Kim, S.S.; Choi, E.; Kim, T.H.; Hong, J.H.; Chang, N.; Cho, H.J.; Sa, J.K.; et al. Transglutaminase 2 Inhibition Reverses Mesenchymal Transdifferentiation of Glioma Stem Cells by Regulating C/EBPbeta Signaling. Cancer Res. 2017, 77, 4973–4984. [Google Scholar] [CrossRef]
- Narayanan, A.; Gagliardi, F.; Gallotti, A.L.; Mazzoleni, S.; Cominelli, M.; Fagnocchi, L.; Pala, M.; Piras, I.S.; Zordan, P.; Moretta, N.; et al. The proneural gene ASCL1 governs the transcriptional subgroup affiliation in glioblastoma stem cells by directly repressing the mesenchymal gene NDRG1. Cell Death Differ. 2019, 26, 1813–1831. [Google Scholar] [CrossRef]
- Bhat, K.P.; Salazar, K.L.; Balasubramaniyan, V.; Wani, K.; Heathcock, L.; Hollingsworth, F.; James, J.D.; Gumin, J.; Diefes, K.L.; Kim, S.H.; et al. The transcriptional coactivator TAZ regulates mesenchymal differentiation in malignant glioma. Genes Dev. 2011, 25, 2594–2609. [Google Scholar] [CrossRef]
- Beachy, P.A.; Karhadkar, S.S.; Berman, D.M. Tissue repair and stem cell renewal in carcinogenesis. Nature 2004, 432, 324–331. [Google Scholar] [CrossRef]
- Huangfu, D.; Anderson, K.V. Signaling from Smo to Ci/Gli: Conservation and divergence of Hedgehog pathways from Drosophila to vertebrates. Development 2006, 133, 3–14. [Google Scholar] [CrossRef]
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087. [Google Scholar] [CrossRef]
- Ericson, J.; Morton, S.; Kawakami, A.; Roelink, H.; Jessell, T.M. Two critical periods of Sonic Hedgehog signaling required for the specification of motor neuron identity. Cell 1996, 87, 661–673. [Google Scholar] [CrossRef]
- Ruiz i Altaba, A. Gli proteins and Hedgehog signaling: Development and cancer. Trends Genet. 1999, 15, 418–425. [Google Scholar] [CrossRef]
- Wechsler-Reya, R.J.; Scott, M.P. Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron 1999, 22, 103–114. [Google Scholar] [CrossRef]
- Stone, D.M.; Hynes, M.; Armanini, M.; Swanson, T.A.; Gu, Q.; Johnson, R.L.; Scott, M.P.; Pennica, D.; Goddard, A.; Phillips, H.; et al. The tumour-suppressor gene patched encodes a candidate receptor for Sonic hedgehog. Nature 1996, 384, 129–134. [Google Scholar] [CrossRef]
- Hooper, J.E.; Scott, M.P. Communicating with Hedgehogs. Nat. Rev. Mol. Cell Biol. 2005, 6, 306–317. [Google Scholar] [CrossRef]
- Villavicencio, E.H.; Walterhouse, D.O.; Iannaccone, P.M. The sonic hedgehog-patched-gli pathway in human development and disease. Am. J. Hum. Genet. 2000, 67, 1047–1054. [Google Scholar] [CrossRef]
- Carballo, G.B.; Honorato, J.R.; de Lopes, G.P.F.; Spohr, T. A highlight on Sonic hedgehog pathway. Cell Commun. Signal. 2018, 16, 11. [Google Scholar] [CrossRef]
- Gupta, S.; Takebe, N.; Lorusso, P. Targeting the Hedgehog pathway in cancer. Adv. Med. Oncol. 2010, 2, 237–250. [Google Scholar] [CrossRef]
- Chandra, V.; Das, T.; Gulati, P.; Biswas, N.K.; Rote, S.; Chatterjee, U.; Ghosh, S.N.; Deb, S.; Saha, S.K.; Chowdhury, A.K.; et al. Hedgehog signaling pathway is active in GBM with GLI1 mRNA expression showing a single continuous distribution rather than discrete high/low clusters. PLoS ONE 2015, 10, e0116390. [Google Scholar] [CrossRef]
- Yan, G.N.; Yang, L.; Lv, Y.F.; Shi, Y.; Shen, L.L.; Yao, X.H.; Guo, Q.N.; Zhang, P.; Cui, Y.H.; Zhang, X.; et al. Endothelial cells promote stem-like phenotype of glioma cells through activating the Hedgehog pathway. J. Pathol. 2014, 234, 11–22. [Google Scholar] [CrossRef]
- Hung, H.C.; Liu, C.C.; Chuang, J.Y.; Su, C.L.; Gean, P.W. Inhibition of Sonic Hedgehog Signaling Suppresses Glioma Stem-Like Cells Likely Through Inducing Autophagic Cell Death. Front. Oncol. 2020, 10, 1233. [Google Scholar] [CrossRef]
- Lee, S.T.; Welch, K.D.; Panter, K.E.; Gardner, D.R.; Garrossian, M.; Chang, C.W. Cyclopamine: From cyclops lambs to cancer treatment. J. Agric. Food Chem. 2014, 62, 7355–7362. [Google Scholar] [CrossRef]
- Carballo, G.B.; Matias, D.; Ribeiro, J.H.; Pessoa, L.S.; Arrais-Neto, A.M.; Spohr, T. Cyclopamine sensitizes glioblastoma cells to temozolomide treatment through Sonic hedgehog pathway. Life Sci. 2020, 257, 118027. [Google Scholar] [CrossRef]
- Xie, H.; Paradise, B.D.; Ma, W.W.; Fernandez-Zapico, M.E. Recent Advances in the Clinical Targeting of Hedgehog/GLI Signaling in Cancer. Cells 2019, 8, 394. [Google Scholar] [CrossRef]
- Bureta, C.; Saitoh, Y.; Tokumoto, H.; Sasaki, H.; Maeda, S.; Nagano, S.; Komiya, S.; Taniguchi, N.; Setoguchi, T. Synergistic effect of arsenic trioxide, vismodegib and temozolomide on glioblastoma. Oncol. Rep. 2019, 41, 3404–3412. [Google Scholar] [CrossRef]
- Rudin, C.M.; Hann, C.L.; Laterra, J.; Yauch, R.L.; Callahan, C.A.; Fu, L.; Holcomb, T.; Stinson, J.; Gould, S.E.; Coleman, B.; et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N. Engl. J. Med. 2009, 361, 1173–1178. [Google Scholar] [CrossRef]
- Sloan, A.E.; Nock, C.J.; Kerstetter, A.; Supko, J.; Ye, X.; Barnholtz-Sloan, J.S.; Miller, R.; Rich, J.; Takebe, N.; Prados, M.; et al. Targeting glioma stem cells (GSC): A biomarker and phase ii study of gdc-0449 in patients with recurrent glioblastoma multiforme (GBM). Neuro-Oncology 2012, 14, vi101–vi105. [Google Scholar] [CrossRef]
- Kieran, M.W.; Chisholm, J.; Casanova, M.; Brandes, A.A.; Aerts, I.; Bouffet, E.; Bailey, S.; Leary, S.; MacDonald, T.J.; Mechinaud, F.; et al. Phase I study of oral sonidegib (LDE225) in pediatric brain and solid tumors and a phase II study in children and adults with relapsed medulloblastoma. Neuro-Oncology 2017, 19, 1542–1552. [Google Scholar] [CrossRef]
- Thomas, X.; Heiblig, M. An evaluation of glasdegib for the treatment of acute myelogenous leukemia. Expert Opin. Pharm. 2020, 21, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Aftab, B.T.; Dobromilskaya, I.; Liu, J.O.; Rudin, C.M. Itraconazole inhibits angiogenesis and tumor growth in non-small cell lung cancer. Cancer Res. 2011, 71, 6764–6772. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Heath, E.I.; Smith, D.C.; Rathkopf, D.; Blackford, A.L.; Danila, D.C.; King, S.; Frost, A.; Ajiboye, A.S.; Zhao, M.; et al. Repurposing itraconazole as a treatment for advanced prostate cancer: A noncomparative randomized phase II trial in men with metastatic castration-resistant prostate cancer. Oncologist 2013, 18, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Ip, K.H.; McKerrow, K. Itraconazole in the treatment of basal cell carcinoma: A case-based review of the literature. Australas J. Derm. 2021, 62, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Brahmer, J.R.; Juergens, R.A.; Hann, C.L.; Ettinger, D.S.; Sebree, R.; Smith, R.; Aftab, B.T.; Huang, P.; Liu, J.O. Phase 2 study of pemetrexed and itraconazole as second-line therapy for metastatic nonsquamous non-small-cell lung cancer. J. Thorac. Oncol. 2013, 8, 619–623. [Google Scholar] [CrossRef]
- Beauchamp, E.M.; Ringer, L.; Bulut, G.; Sajwan, K.P.; Hall, M.D.; Lee, Y.C.; Peaceman, D.; Ozdemirli, M.; Rodriguez, O.; Macdonald, T.J.; et al. Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/GLI pathway. J. Clin. Investig. 2011, 121, 148–160. [Google Scholar] [CrossRef]
- Kumthekar, P.; Grimm, S.; Chandler, J.; Mehta, M.; Marymont, M.; Levy, R.; Muro, K.; Helenowski, I.; McCarthy, K.; Fountas, L.; et al. A phase II trial of arsenic trioxide and temozolomide in combination with radiation therapy for patients with malignant gliomas. J. Neurooncol. 2017, 133, 589–594. [Google Scholar] [CrossRef]
- Shen, H.; He, D.; Wang, S.; Ding, P.; Wang, J.; Ju, J. Preparation, characterization, and pharmacokinetics study of a novel genistein-loaded mixed micelles system. Drug Dev. Ind. Pharm. 2018, 44, 1536–1542. [Google Scholar] [CrossRef]
- Honorato, J.R.; Hauser-Davis, R.A.; Saggioro, E.M.; Correia, F.V.; Sales-Junior, S.F.; Soares, L.O.S.; Lima, L.D.R.; Moura-Neto, V.; Lopes, G.P.F.; Spohr, T. Role of Sonic hedgehog signaling in cell cycle, oxidative stress, and autophagy of temozolomide resistant glioblastoma. J. Cell Physiol. 2020, 235, 3798–3814. [Google Scholar] [CrossRef]
- Li, J.; Cai, J.; Zhao, S.; Yao, K.; Sun, Y.; Li, Y.; Chen, L.; Li, R.; Zhai, X.; Zhang, J.; et al. GANT61, a GLI inhibitor, sensitizes glioma cells to the temozolomide treatment. J. Exp. Clin. Cancer Res. 2016, 35, 184. [Google Scholar] [CrossRef]
- Krishnamurthy, N.; Kurzrock, R. Targeting the Wnt/beta-catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treat. Rev. 2018, 62, 50–60. [Google Scholar] [CrossRef]
- Nusse, R.; Clevers, H. Wnt/beta-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef]
- Croce, J.C.; McClay, D.R. Evolution of the Wnt pathways. Methods Mol. Biol. 2008, 469, 3–18. [Google Scholar] [CrossRef]
- Gordon, M.D.; Nusse, R. Wnt signaling: Multiple pathways, multiple receptors, and multiple transcription factors. J. Biol. Chem 2006, 281, 22429–22433. [Google Scholar] [CrossRef]
- Logan, C.Y.; Nusse, R. The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 2004, 20, 781–810. [Google Scholar] [CrossRef]
- Bilic, J.; Huang, Y.L.; Davidson, G.; Zimmermann, T.; Cruciat, C.M.; Bienz, M.; Niehrs, C. Wnt induces LRP6 signalosomes and promotes dishevelled-dependent LRP6 phosphorylation. Science 2007, 316, 1619–1622. [Google Scholar] [CrossRef]
- He, X.; Semenov, M.; Tamai, K.; Zeng, X. LDL receptor-related proteins 5 and 6 in Wnt/beta-catenin signaling: Arrows point the way. Development 2004, 131, 1663–1677. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef]
- Cheng, X.; Xu, X.; Chen, D.; Zhao, F.; Wang, W. Therapeutic potential of targeting the Wnt/beta-catenin signaling pathway in colorectal cancer. Biomed. Pharm. 2019, 110, 473–481. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, L.; Qu, Y. Targeting the beta-catenin signaling for cancer therapy. Pharm. Res. 2020, 160, 104794. [Google Scholar] [CrossRef]
- Latour, M.; Her, N.G.; Kesari, S.; Nurmemmedov, E. WNT Signaling as a Therapeutic Target for Glioblastoma. Int. J. Mol. Sci. 2021, 22, 8428. [Google Scholar] [CrossRef]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef]
- Pulvirenti, T.; Van Der Heijden, M.; Droms, L.A.; Huse, J.T.; Tabar, V.; Hall, A. Dishevelled 2 signaling promotes self-renewal and tumorigenicity in human gliomas. Cancer Res. 2011, 71, 7280–7290. [Google Scholar] [CrossRef]
- Tang, C.; Guo, J.; Chen, H.; Yao, C.J.; Zhuang, D.X.; Wang, Y.; Tang, W.J.; Ren, G.; Yao, Y.; Wu, J.S.; et al. Gene mutation profiling of primary glioblastoma through multiple tumor biopsy guided by 1H-magnetic resonance spectroscopy. Int. J. Clin. Exp. Pathol. 2015, 8, 5327–5335. [Google Scholar] [PubMed]
- Lorzadeh, S.; Kohan, L.; Ghavami, S.; Azarpira, N. Autophagy and the Wnt signaling pathway: A focus on Wnt/beta-catenin signaling. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118926. [Google Scholar] [CrossRef] [PubMed]
- Morris, L.G.; Kaufman, A.M.; Gong, Y.; Ramaswami, D.; Walsh, L.A.; Turcan, S.; Eng, S.; Kannan, K.; Zou, Y.; Peng, L.; et al. Recurrent somatic mutation of FAT1 in multiple human cancers leads to aberrant Wnt activation. Nat. Genet. 2013, 45, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Sareddy, G.R.; Pratap, U.P.; Viswanadhapalli, S.; Venkata, P.P.; Nair, B.C.; Krishnan, S.R.; Zheng, S.; Gilbert, A.R.; Brenner, A.J.; Brann, D.W.; et al. PELP1 promotes glioblastoma progression by enhancing Wnt/beta-catenin signaling. Neurooncol. Adv. 2019, 1, vdz042. [Google Scholar] [CrossRef]
- Shaji, S.K.; Sunilkumar, D.; Mahalakshmi, N.V.; Kumar, G.B.; Nair, B.G. Analysis of microarray data for identification of key microRNA signatures in glioblastoma multiforme. Oncol. Lett. 2019, 18, 1938–1948. [Google Scholar] [CrossRef]
- Vecera, M.; Sana, J.; Lipina, R.; Smrcka, M.; Slaby, O. Long Non-Coding RNAs in Gliomas: From Molecular Pathology to Diagnostic Biomarkers and Therapeutic Targets. Int. J. Mol. Sci. 2018, 19, 2754. [Google Scholar] [CrossRef]
- Wang, Y.N.; Chen, W.Q.; Shi, Y.X.; Yan, C.R.; Kong, Z.R.; Wang, Y.K.; Wang, Y.; Ma, W.B. Imposing Phase II and Phase III Clinical Trials of Targeted Drugs for Glioblastoma: Current Status and Progress. Front. Oncol. 2021, 11, 3611. [Google Scholar] [CrossRef]
- He, L.; Zhou, H.; Zeng, Z.; Yao, H.; Jiang, W.; Qu, H. Wnt/beta-catenin signaling cascade: A promising target for glioma therapy. J. Cell Physiol. 2019, 234, 2217–2228. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X. Targeting the Wnt/beta-catenin signaling pathway in cancer. J. Hematol. Oncol. 2020, 13, 165. [Google Scholar] [CrossRef]
- Allen, J.E.; Krigsfeld, G.; Mayes, P.A.; Patel, L.; Dicker, D.T.; Patel, A.S.; Dolloff, N.G.; Messaris, E.; Scata, K.A.; Wang, W.; et al. Dual inactivation of Akt and ERK by TIC10 signals Foxo3a nuclear translocation, TRAIL gene induction, and potent antitumor effects. Sci. Transl. Med. 2013, 5, 171ra117. [Google Scholar] [CrossRef]
- De Robertis, A.; Valensin, S.; Rossi, M.; Tunici, P.; Verani, M.; De Rosa, A.; Giordano, C.; Varrone, M.; Nencini, A.; Pratelli, C.; et al. Identification and characterization of a small-molecule inhibitor of Wnt signaling in glioblastoma cells. Mol. Cancer Ther. 2013, 12, 1180–1189. [Google Scholar] [CrossRef]
- Gao, L.; Chen, B.; Li, J.; Yang, F.; Cen, X.; Liao, Z.; Long, X. Wnt/beta-catenin signaling pathway inhibits the proliferation and apoptosis of U87 glioma cells via different mechanisms. PLoS ONE 2017, 12, e0181346. [Google Scholar] [CrossRef]
- Kierulf-Vieira, K.S.; Sandberg, C.J.; Waaler, J.; Lund, K.; Skaga, E.; Saberniak, B.M.; Panagopoulos, I.; Brandal, P.; Krauss, S.; Langmoen, I.A.; et al. A Small-Molecule Tankyrase Inhibitor Reduces Glioma Stem Cell Proliferation and Sphere Formation. Cancers 2020, 12, 1630. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, K.H.; Lee, J.; Lee, Y.A.; Kim, M.; Lee, S.J.; Park, K.; Yang, H.; Jin, J.; Joo, K.M.; et al. Wnt activation is implicated in glioblastoma radioresistance. Lab. Investig. 2012, 92, 466–473. [Google Scholar] [CrossRef]
- Korur, S.; Huber, R.M.; Sivasankaran, B.; Petrich, M.; Morin, P., Jr.; Hemmings, B.A.; Merlo, A.; Lino, M.M. GSK3beta regulates differentiation and growth arrest in glioblastoma. PLoS ONE 2009, 4, e7443. [Google Scholar] [CrossRef]
- Prabhu, V.V.; Lulla, A.R.; Madhukar, N.S.; Ralff, M.D.; Zhao, D.; Kline, C.L.B.; Van den Heuvel, A.P.J.; Lev, A.; Garnett, M.J.; McDermott, U.; et al. Cancer stem cell-related gene expression as a potential biomarker of response for first-in-class imipridone ONC201 in solid tumors. PLoS ONE 2017, 12, e0180541. [Google Scholar] [CrossRef]
- Suwala, A.K.; Koch, K.; Rios, D.H.; Aretz, P.; Uhlmann, C.; Ogorek, I.; Felsberg, J.; Reifenberger, G.; Kohrer, K.; Deenen, R.; et al. Inhibition of Wnt/beta-catenin signaling downregulates expression of aldehyde dehydrogenase isoform 3A1 (ALDH3A1) to reduce resistance against temozolomide in glioblastoma in vitro. Oncotarget 2018, 9, 22703–22716. [Google Scholar] [CrossRef]
- Varghese, R.T.; Young, S.; Pham, L.; Liang, Y.; Pridham, K.J.; Guo, S.; Murphy, S.; Kelly, D.F.; Sheng, Z. Casein Kinase 1 Epsilon Regulates Glioblastoma Cell Survival. Sci. Rep. 2018, 8, 13621. [Google Scholar] [CrossRef]
- Yu, W.K.; Xu, Z.Y.; Yuan, L.; Mo, S.; Xu, B.; Cheng, X.D.; Qin, J.J. Targeting beta-Catenin Signaling by Natural Products for Cancer Prevention and Therapy. Front. Pharm. 2020, 11, 984. [Google Scholar] [CrossRef]
- Zhang, F.Y.; Hu, Y.; Que, Z.Y.; Wang, P.; Liu, Y.H.; Wang, Z.H.; Xue, Y.X. Shikonin Inhibits the Migration and Invasion of Human Glioblastoma Cells by Targeting Phosphorylated beta-Catenin and Phosphorylated PI3K/Akt: A Potential Mechanism for the Anti-Glioma Efficacy of a Traditional Chinese Herbal Medicine. Int. J. Mol. Sci. 2015, 16, 23823–23848. [Google Scholar] [CrossRef]
- Miao, J.; Jiang, Y.; Wang, D.; Zhou, J.; Fan, C.; Jiao, F.; Liu, B.; Zhang, J.; Wang, Y.; Zhang, Q. Trichosanthin suppresses the proliferation of glioma cells by inhibiting LGR5 expression and the Wnt/beta-catenin signaling pathway. Oncol. Rep. 2015, 34, 2845–2852. [Google Scholar] [CrossRef]
- Mora, M.C.; Bassa, L.M.; Wong, K.E.; Tirabassi, M.V.; Arenas, R.B.; Schneider, S.S. Rhodiola crenulata inhibits Wnt/beta-catenin signaling in glioblastoma. J. Surg. Res. 2015, 197, 247–255. [Google Scholar] [CrossRef]
- Cilibrasi, C.; Riva, G.; Romano, G.; Cadamuro, M.; Bazzoni, R.; Butta, V.; Paoletta, L.; Dalpra, L.; Strazzabosco, M.; Lavitrano, M.; et al. Resveratrol Impairs Glioma Stem Cells Proliferation and Motility by Modulating the Wnt Signaling Pathway. PLoS ONE 2017, 12, e0169854. [Google Scholar] [CrossRef]
- Tao, W.; Chu, C.; Zhou, W.; Huang, Z.; Zhai, K.; Fang, X.; Huang, Q.; Zhang, A.; Wang, X.; Yu, X.; et al. Dual Role of WISP1 in maintaining glioma stem cells and tumor-supportive macrophages in glioblastoma. Nat. Commun. 2020, 11, 3015. [Google Scholar] [CrossRef]
- Williams, S.P.; Nowicki, M.O.; Liu, F.; Press, R.; Godlewski, J.; Abdel-Rasoul, M.; Kaur, B.; Fernandez, S.A.; Chiocca, E.A.; Lawler, S.E. Indirubins decrease glioma invasion by blocking migratory phenotypes in both the tumor and stromal endothelial cell compartments. Cancer Res. 2011, 71, 5374–5380. [Google Scholar] [CrossRef] [PubMed]
- Tao, Q.; Wu, C.; Xu, R.; Niu, L.; Qin, J.; Liu, N.; Zhang, P.; Wang, C. Diallyl trisulfide inhibits proliferation, invasion and angiogenesis of glioma cells by inactivating Wnt/beta-catenin signaling. Cell Tissue Res. 2017, 370, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Lan, F.; Pan, Q.; Yu, H.; Yue, X. Sulforaphane enhances temozolomide-induced apoptosis because of down-regulation of miR-21 via Wnt/beta-catenin signaling in glioblastoma. J. Neurochem. 2015, 134, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Sareddy, G.R.; Kesanakurti, D.; Kirti, P.B.; Babu, P.P. Nonsteroidal anti-inflammatory drugs diclofenac and celecoxib attenuates Wnt/beta-catenin/Tcf signaling pathway in human glioblastoma cells. Neurochem. Res. 2013, 38, 2313–2322. [Google Scholar] [CrossRef] [PubMed]
- Lan, F.; Yue, X.; Han, L.; Yuan, X.; Shi, Z.; Huang, K.; Yang, Y.; Zou, J.; Zhang, J.; Jiang, T.; et al. Antitumor effect of aspirin in glioblastoma cells by modulation of beta-catenin/T-cell factor-mediated transcriptional activity. J. Neurosurg. 2011, 115, 780–788. [Google Scholar] [CrossRef]
- Wieland, A.; Trageser, D.; Gogolok, S.; Reinartz, R.; Hofer, H.; Keller, M.; Leinhaas, A.; Schelle, R.; Normann, S.; Klaas, L.; et al. Anticancer effects of niclosamide in human glioblastoma. Clin. Cancer Res. 2013, 19, 4124–4136. [Google Scholar] [CrossRef]
- Oh, H.C.; Shim, J.K.; Park, J.; Lee, J.H.; Choi, R.J.; Kim, N.H.; Kim, H.S.; Moon, J.H.; Kim, E.H.; Chang, J.H.; et al. Combined effects of niclosamide and temozolomide against human glioblastoma tumorspheres. J. Cancer Res. Clin. Oncol. 2020, 146, 2817–2828. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, N.; Li, H.; Liu, S.; Chen, X.; Yu, S.; Wu, N.; Bian, X.W.; Shen, H.Y.; Li, C.; et al. Promoting oligodendroglial-oriented differentiation of glioma stem cell: A repurposing of quetiapine for the treatment of malignant glioma. Oncotarget 2017, 8, 37511–37524. [Google Scholar] [CrossRef]
- Wan, Z.; Shi, W.; Shao, B.; Shi, J.; Shen, A.; Ma, Y.; Chen, J.; Lan, Q. Peroxisome proliferator-activated receptor gamma agonist pioglitazone inhibits beta-catenin-mediated glioma cell growth and invasion. Mol. Cell Biochem 2011, 349, 1–10. [Google Scholar] [CrossRef]
- Cilibrasi, C.; Butta, V.; Riva, G.; Bentivegna, A. Pioglitazone Effect on Glioma Stem Cell Lines: Really a Promising Drug Therapy for Glioblastoma? PPAR Res. 2016, 2016, 7175067. [Google Scholar] [CrossRef]
- Jin, X.; Jeon, H.Y.; Joo, K.M.; Kim, J.K.; Jin, J.; Kim, S.H.; Kang, B.G.; Beck, S.; Lee, S.J.; Kim, J.K.; et al. Frizzled 4 regulates stemness and invasiveness of migrating glioma cells established by serial intracranial transplantation. Cancer Res. 2011, 71, 3066–3075. [Google Scholar] [CrossRef]
- Zeppernick, F.; Ahmadi, R.; Campos, B.; Dictus, C.; Helmke, B.M.; Becker, N.; Lichter, P.; Unterberg, A.; Radlwimmer, B.; Herold-Mende, C.C. Stem cell marker CD133 affects clinical outcome in glioma patients. Clin. Cancer Res. 2008, 14, 123–129. [Google Scholar] [CrossRef]
- Arrillaga-Romany, I.; Kurz, S.; Tarapore, R.S.; Sumrall, A.; Butowski, N.; Harrison, R.; de Groot, J.; Chi, A.; Shonka, N.; Umemura, Y.; et al. Efficacy of Onc201 in Patients with Onc201 for Recurrent H3 K27m-Mutant Diffuse Midline Glioma. Neuro-Oncology 2020, 22, 50–51. [Google Scholar] [CrossRef]
- Le, P.N.; McDermott, J.D.; Jimeno, A. Targeting the Wnt pathway in human cancers: Therapeutic targeting with a focus on OMP-54F28. Pharmacol. Ther. 2015, 146, 1–11. [Google Scholar] [CrossRef]
- Gurney, A.; Axelrod, F.; Bond, C.J.; Cain, J.; Chartier, C.; Donigan, L.; Fischer, M.; Chaudhari, A.; Ji, M.; Kapoun, A.M.; et al. Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 11717–11722. [Google Scholar] [CrossRef]
- Pavlovic, Z.; Adams, J.J.; Blazer, L.L.; Gakhal, A.K.; Jarvik, N.; Steinhart, Z.; Robitaille, M.; Mascall, K.; Pan, J.; Angers, S.; et al. A synthetic anti-Frizzled antibody engineered for broadened specificity exhibits enhanced anti-tumor properties. Mabs 2018, 10, 1157–1167. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Qian, B.; Nag, S.A.; Su, Y.; Voruganti, S.; Qin, J.J.; Zhang, R.; Cho, W.C. miRNAs in cancer prevention and treatment and as molecular targets for natural product anticancer agents. Curr. Cancer Drug Targets 2013, 13, 519–541. [Google Scholar] [CrossRef]
- Qin, J.; Wang, W.; Zhang, R. Novel natural product therapeutics targeting both inflammation and cancer. Chin. J. Nat. Med. 2017, 15, 401–416. [Google Scholar] [CrossRef]
- Penas-Prado, M.; Hess, K.R.; Fisch, M.J.; Lagrone, L.W.; Groves, M.D.; Levin, V.A.; De Groot, J.F.; Puduvalli, V.K.; Colman, H.; Volas-Redd, G.; et al. Randomized phase II adjuvant factorial study of dose-dense temozolomide alone and in combination with isotretinoin, celecoxib, and/or thalidomide for glioblastoma. Neuro-Oncology 2015, 17, 266–273. [Google Scholar] [CrossRef]
- Huang, M.; Zhang, D.; Wu, J.Y.; Xing, K.; Yeo, E.; Li, C.; Zhang, L.; Holland, E.; Yao, L.; Qin, L.; et al. Wnt-mediated endothelial transformation into mesenchymal stem cell-like cells induces chemoresistance in glioblastoma. Sci. Transl. Med. 2020, 12, eaay7522. [Google Scholar] [CrossRef]
- Seano, G.; Jain, R.K. Vessel co-option in glioblastoma: Emerging insights and opportunities. Angiogenesis 2020, 23, 9–16. [Google Scholar] [CrossRef]
- Jain, R.K. Antiangiogenesis strategies revisited: From starving tumors to alleviating hypoxia. Cancer Cell 2014, 26, 605–622. [Google Scholar] [CrossRef]
- Griveau, A.; Seano, G.; Shelton, S.J.; Kupp, R.; Jahangiri, A.; Obernier, K.; Krishnan, S.; Lindberg, O.R.; Yuen, T.J.; Tien, A.C.; et al. A Glial Signature and Wnt7 Signaling Regulate Glioma-Vascular Interactions and Tumor Microenvironment. Cancer Cell 2018, 33, 874–889.e877. [Google Scholar] [CrossRef]
- Watkins, S.; Robel, S.; Kimbrough, I.F.; Robert, S.M.; Ellis-Davies, G.; Sontheimer, H. Disruption of astrocyte-vascular coupling and the blood-brain barrier by invading glioma cells. Nat. Commun. 2014, 5, 4196. [Google Scholar] [CrossRef]
- Baker, G.J.; Yadav, V.N.; Motsch, S.; Koschmann, C.; Calinescu, A.A.; Mineharu, Y.; Camelo-Piragua, S.I.; Orringer, D.; Bannykh, S.; Nichols, W.S.; et al. Mechanisms of glioma formation: Iterative perivascular glioma growth and invasion leads to tumor progression, VEGF-independent vascularization, and resistance to antiangiogenic therapy. Neoplasia 2014, 16, 543–561. [Google Scholar] [CrossRef] [PubMed]
- Di Tomaso, E.; Snuderl, M.; Kamoun, W.S.; Duda, D.G.; Auluck, P.K.; Fazlollahi, L.; Andronesi, O.C.; Frosch, M.P.; Wen, P.Y.; Plotkin, S.R.; et al. Glioblastoma recurrence after cediranib therapy in patients: Lack of “rebound” revascularization as mode of escape. Cancer Res. 2011, 71, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Voutouri, C.; Kirkpatrick, N.D.; Chung, E.; Mpekris, F.; Baish, J.W.; Munn, L.L.; Fukumura, D.; Stylianopoulos, T.; Jain, R.K. Experimental and computational analyses reveal dynamics of tumor vessel cooption and optimal treatment strategies. Proc. Natl. Acad. Sci. USA 2019, 116, 2662–2671. [Google Scholar] [CrossRef] [PubMed]
- Whelan, R.; Hargaden, G.C.; Knox, A.J.S. Modulating the Blood-Brain Barrier: A Comprehensive Review. Pharmaceutics 2021, 13, 1980. [Google Scholar] [CrossRef]
- Cho, C.; Smallwood, P.M.; Nathans, J. Reck and Gpr124 Are Essential Receptor Cofactors for Wnt7a/Wnt7b-Specific Signaling in Mammalian CNS Angiogenesis and Blood-Brain Barrier Regulation. Neuron 2017, 95, 1056–1073.e1055. [Google Scholar] [CrossRef]
- Vanhollebeke, B.; Stone, O.A.; Bostaille, N.; Cho, C.; Zhou, Y.; Maquet, E.; Gauquier, A.; Cabochette, P.; Fukuhara, S.; Mochizuki, N.; et al. Tip cell-specific requirement for an atypical Gpr124- and Reck-dependent Wnt/beta-catenin pathway during brain angiogenesis. eLife 2015, 4, e06489. [Google Scholar] [CrossRef]
- Martin, M.; Vermeiren, S.; Bostaille, N.; Eubelen, M.; Spitzer, D.; Vermeersch, M.; Profaci, C.P.; Pozuelo, E.; Toussay, X.; Raman-Nair, J.; et al. Engineered Wnt ligands enable blood-brain barrier repair in neurological disorders. Science 2022, 375, eabm4459. [Google Scholar] [CrossRef]
- Chavali, M.; Ulloa-Navas, M.J.; Perez-Borreda, P.; Garcia-Verdugo, J.M.; McQuillen, P.S.; Huang, E.J.; Rowitch, D.H. Wnt-Dependent Oligodendroglial-Endothelial Interactions Regulate White Matter Vascularization and Attenuate Injury. Neuron 2020, 108, 1130–1145.e1135. [Google Scholar] [CrossRef]
- Reis, M.; Czupalla, C.J.; Ziegler, N.; Devraj, K.; Zinke, J.; Seidel, S.; Heck, R.; Thom, S.; Macas, J.; Bockamp, E.; et al. Endothelial Wnt/beta-catenin signaling inhibits glioma angiogenesis and normalizes tumor blood vessels by inducing PDGF-B expression. J. Exp. Med. 2012, 209, 1611–1627. [Google Scholar] [CrossRef]
- Andersson, E.R.; Sandberg, R.; Lendahl, U. Notch signaling: Simplicity in design, versatility in function. Development 2011, 138, 3593–3612. [Google Scholar] [CrossRef]
- Liu, J.; Sato, C.; Cerletti, M.; Wagers, A. Notch signaling in the regulation of stem cell self-renewal and differentiation. Curr. Top. Dev. Biol. 2010, 92, 367–409. [Google Scholar] [CrossRef]
- Imayoshi, I.; Sakamoto, M.; Yamaguchi, M.; Mori, K.; Kageyama, R. Essential roles of Notch signaling in maintenance of neural stem cells in developing and adult brains. J. Neurosci. 2010, 30, 3489–3498. [Google Scholar] [CrossRef]
- Pierfelice, T.; Alberi, L.; Gaiano, N. Notch in the vertebrate nervous system: An old dog with new tricks. Neuron 2011, 69, 840–855. [Google Scholar] [CrossRef]
- Bray, S.J. Notch signalling: A simple pathway becomes complex. Nat. Rev. Mol. Cell Biol. 2006, 7, 678–689. [Google Scholar] [CrossRef]
- Kopan, R.; Ilagan, M.X. The canonical Notch signaling pathway: Unfolding the activation mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef]
- Cheng, H.T.; Kim, M.; Valerius, M.T.; Surendran, K.; Schuster-Gossler, K.; Gossler, A.; McMahon, A.P.; Kopan, R. Notch2, but not Notch1, is required for proximal fate acquisition in the mammalian nephron. Development 2007, 134, 801–811. [Google Scholar] [CrossRef]
- Krebs, L.T.; Iwai, N.; Nonaka, S.; Welsh, I.C.; Lan, Y.; Jiang, R.; Saijoh, Y.; O’Brien, T.P.; Hamada, H.; Gridley, T. Notch signaling regulates left-right asymmetry determination by inducing Nodal expression. Genes Dev. 2003, 17, 1207–1212. [Google Scholar] [CrossRef]
- Matsuura, A.; Ito, M.; Sakaidani, Y.; Kondo, T.; Murakami, K.; Furukawa, K.; Nadano, D.; Matsuda, T.; Okajima, T. O-linked N-acetylglucosamine is present on the extracellular domain of notch receptors. J. Biol. Chem 2008, 283, 35486–35495. [Google Scholar] [CrossRef]
- Moloney, D.J.; Shair, L.H.; Lu, F.M.; Xia, J.; Locke, R.; Matta, K.L.; Haltiwanger, R.S. Mammalian Notch1 is modified with two unusual forms of O-linked glycosylation found on epidermal growth factor-like modules. J. Biol. Chem 2000, 275, 9604–9611. [Google Scholar] [CrossRef]
- Blaumueller, C.M.; Qi, H.; Zagouras, P.; Artavanis-Tsakonas, S. Intracellular cleavage of Notch leads to a heterodimeric receptor on the plasma membrane. Cell 1997, 90, 281–291. [Google Scholar] [CrossRef]
- Sanchez-Irizarry, C.; Carpenter, A.C.; Weng, A.P.; Pear, W.S.; Aster, J.C.; Blacklow, S.C. Notch subunit heterodimerization and prevention of ligand-independent proteolytic activation depend, respectively, on a novel domain and the LNR repeats. Mol. Cell Biol. 2004, 24, 9265–9273. [Google Scholar] [CrossRef] [PubMed]
- Brou, C.; Logeat, F.; Gupta, N.; Bessia, C.; LeBail, O.; Doedens, J.R.; Cumano, A.; Roux, P.; Black, R.A.; Israel, A. A novel proteolytic cleavage involved in Notch signaling: The role of the disintegrin-metalloprotease TACE. Mol. Cell 2000, 5, 207–216. [Google Scholar] [CrossRef]
- Van Tetering, G.; van Diest, P.; Verlaan, I.; van der Wall, E.; Kopan, R.; Vooijs, M. Metalloprotease ADAM10 is required for Notch1 site 2 cleavage. J. Biol. Chem. 2009, 284, 31018–31027. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B.; Annaert, W.; Cupers, P.; Saftig, P.; Craessaerts, K.; Mumm, J.S.; Schroeter, E.H.; Schrijvers, V.; Wolfe, M.S.; Ray, W.J.; et al. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature 1999, 398, 518–522. [Google Scholar] [CrossRef]
- Mumm, J.S.; Schroeter, E.H.; Saxena, M.T.; Griesemer, A.; Tian, X.; Pan, D.J.; Ray, W.J.; Kopan, R. A ligand-induced extracellular cleavage regulates gamma-secretase-like proteolytic activation of Notch1. Mol. Cell 2000, 5, 197–206. [Google Scholar] [CrossRef]
- Christensen, S.; Kodoyianni, V.; Bosenberg, M.; Friedman, L.; Kimble, J. lag-1, a gene required for lin-12 and glp-1 signaling in Caenorhabditis elegans, is homologous to human CBF1 and Drosophila Su(H). Development 1996, 122, 1373–1383. [Google Scholar] [CrossRef]
- Fortini, M.E.; Artavanis-Tsakonas, S. The suppressor of hairless protein participates in notch receptor signaling. Cell 1994, 79, 273–282. [Google Scholar] [CrossRef]
- Tamura, K.; Taniguchi, Y.; Minoguchi, S.; Sakai, T.; Tun, T.; Furukawa, T.; Honjo, T. Physical interaction between a novel domain of the receptor Notch and the transcription factor RBP-J kappa/Su(H). Curr. Biol. 1995, 5, 1416–1423. [Google Scholar] [CrossRef]
- Wu, L.; Sun, T.; Kobayashi, K.; Gao, P.; Griffin, J.D. Identification of a family of mastermind-like transcriptional coactivators for mammalian notch receptors. Mol. Cell Biol. 2002, 22, 7688–7700. [Google Scholar] [CrossRef]
- Kovall, R.A. More complicated than it looks: Assembly of Notch pathway transcription complexes. Oncogene 2008, 27, 5099–5109. [Google Scholar] [CrossRef]
- Nam, Y.; Sliz, P.; Song, L.; Aster, J.C.; Blacklow, S.C. Structural basis for cooperativity in recruitment of MAML coactivators to Notch transcription complexes. Cell 2006, 124, 973–983. [Google Scholar] [CrossRef]
- Wilson, J.J.; Kovall, R.A. Crystal structure of the CSL-Notch-Mastermind ternary complex bound to DNA. Cell 2006, 124, 985–996. [Google Scholar] [CrossRef]
- Kadam, S.; Emerson, B.M. Transcriptional specificity of human SWI/SNF BRG1 and BRM chromatin remodeling complexes. Mol. Cell 2003, 11, 377–389. [Google Scholar] [CrossRef]
- Wallberg, A.E.; Pedersen, K.; Lendahl, U.; Roeder, R.G. p300 and PCAF act cooperatively to mediate transcriptional activation from chromatin templates by notch intracellular domains in vitro. Mol. Cell Biol. 2002, 22, 7812–7819. [Google Scholar] [CrossRef]
- Iso, T.; Sartorelli, V.; Chung, G.; Shichinohe, T.; Kedes, L.; Hamamori, Y. HERP, a new primary target of Notch regulated by ligand binding. Mol. Cell Biol. 2001, 21, 6071–6079. [Google Scholar] [CrossRef]
- Jarriault, S.; Brou, C.; Logeat, F.; Schroeter, E.H.; Kopan, R.; Israel, A. Signalling downstream of activated mammalian Notch. Nature 1995, 377, 355–358. [Google Scholar] [CrossRef]
- El Hindy, N.; Keyvani, K.; Pagenstecher, A.; Dammann, P.; Sandalcioglu, I.E.; Sure, U.; Zhu, Y. Implications of Dll4-Notch signaling activation in primary glioblastoma multiforme. Neuro-Oncology 2013, 15, 1366–1378. [Google Scholar] [CrossRef]
- Purow, B.W.; Haque, R.M.; Noel, M.W.; Su, Q.; Burdick, M.J.; Lee, J.; Sundaresan, T.; Pastorino, S.; Park, J.K.; Mikolaenko, I.; et al. Expression of Notch-1 and its ligands, Delta-like-1 and Jagged-1, is critical for glioma cell survival and proliferation. Cancer Res. 2005, 65, 2353–2363. [Google Scholar] [CrossRef]
- Shih, A.H.; Holland, E.C. Notch signaling enhances nestin expression in gliomas. Neoplasia 2006, 8, 1072–1082. [Google Scholar] [CrossRef]
- Parmigiani, E.; Taylor, V.; Giachino, C. Oncogenic and Tumor-Suppressive Functions of NOTCH Signaling in Glioma. Cells 2020, 9, 2304. [Google Scholar] [CrossRef]
- Giachino, C.; Boulay, J.L.; Ivanek, R.; Alvarado, A.; Tostado, C.; Lugert, S.; Tchorz, J.; Coban, M.; Mariani, L.; Bettler, B.; et al. A Tumor Suppressor Function for Notch Signaling in Forebrain Tumor Subtypes. Cancer Cell 2015, 28, 730–742. [Google Scholar] [CrossRef]
- Fan, X.; Khaki, L.; Zhu, T.S.; Soules, M.E.; Talsma, C.E.; Gul, N.; Koh, C.; Zhang, J.; Li, Y.M.; Maciaczyk, J.; et al. NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells 2010, 28, 5–16. [Google Scholar] [CrossRef]
- Zhu, T.S.; Costello, M.A.; Talsma, C.E.; Flack, C.G.; Crowley, J.G.; Hamm, L.L.; He, X.; Hervey-Jumper, S.L.; Heth, J.A.; Muraszko, K.M.; et al. Endothelial cells create a stem cell niche in glioblastoma by providing NOTCH ligands that nurture self-renewal of cancer stem-like cells. Cancer Res. 2011, 71, 6061–6072. [Google Scholar] [CrossRef]
- Wang, J.; Wakeman, T.P.; Lathia, J.D.; Hjelmeland, A.B.; Wang, X.F.; White, R.R.; Rich, J.N.; Sullenger, B.A. Notch promotes radioresistance of glioma stem cells. Stem Cells 2010, 28, 17–28. [Google Scholar] [CrossRef]
- Gilbert, C.A.; Daou, M.C.; Moser, R.P.; Ross, A.H. Gamma-secretase inhibitors enhance temozolomide treatment of human gliomas by inhibiting neurosphere repopulation and xenograft recurrence. Cancer Res. 2010, 70, 6870–6879. [Google Scholar] [CrossRef]
- Bazzoni, R.; Bentivegna, A. Role of Notch Signaling Pathway in Glioblastoma Pathogenesis. Cancers 2019, 11, 292. [Google Scholar] [CrossRef]
- Hovinga, K.E.; Shimizu, F.; Wang, R.; Panagiotakos, G.; Van Der Heijden, M.; Moayedpardazi, H.; Correia, A.S.; Soulet, D.; Major, T.; Menon, J.; et al. Inhibition of notch signaling in glioblastoma targets cancer stem cells via an endothelial cell intermediate. Stem Cells 2010, 28, 1019–1029. [Google Scholar] [CrossRef]
- Lin, J.; Zhang, X.M.; Yang, J.C.; Ye, Y.B.; Luo, S.Q. gamma-secretase inhibitor-I enhances radiosensitivity of glioblastoma cell lines by depleting CD133+ tumor cells. Arch. Med. Res. 2010, 41, 519–529. [Google Scholar] [CrossRef]
- Saito, N.; Fu, J.; Zheng, S.; Yao, J.; Wang, S.; Liu, D.D.; Yuan, Y.; Sulman, E.P.; Lang, F.F.; Colman, H.; et al. A high Notch pathway activation predicts response to gamma secretase inhibitors in proneural subtype of glioma tumor-initiating cells. Stem Cells 2014, 32, 301–312. [Google Scholar] [CrossRef]
- Xu, R.; Shimizu, F.; Hovinga, K.; Beal, K.; Karimi, S.; Droms, L.; Peck, K.K.; Gutin, P.; Iorgulescu, J.B.; Kaley, T.; et al. Molecular and Clinical Effects of Notch Inhibition in Glioma Patients: A Phase 0/I Trial. Clin. Cancer Res. 2016, 22, 4786–4796. [Google Scholar] [CrossRef]
- Pan, E.; Supko, J.G.; Kaley, T.J.; Butowski, N.A.; Cloughesy, T.; Jung, J.; Desideri, S.; Grossman, S.; Ye, X.; Park, D.M. Phase I study of RO4929097 with bevacizumab in patients with recurrent malignant glioma. J. Neurooncol. 2016, 130, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Peereboom, D.M.; Ye, X.; Mikkelsen, T.; Lesser, G.J.; Lieberman, F.S.; Robins, H.I.; Ahluwalia, M.S.; Sloan, A.E.; Grossman, S.A. A Phase II and Pharmacodynamic Trial of RO4929097 for Patients With Recurrent/Progressive Glioblastoma. Neurosurgery 2021, 88, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W.; Messersmith, W.A.; Mikulski, S.M.; Papadopoulos, K.P.; Kwak, E.L.; Gibbon, D.G.; Patnaik, A.; Falchook, G.S.; Dasari, A.; Shapiro, G.I.; et al. Phase I study of RO4929097, a gamma secretase inhibitor of Notch signaling, in patients with refractory metastatic or locally advanced solid tumors. J. Clin. Oncol. 2012, 30, 2348–2353. [Google Scholar] [CrossRef] [PubMed]
- Fouladi, M.; Stewart, C.F.; Olson, J.; Wagner, L.M.; Onar-Thomas, A.; Kocak, M.; Packer, R.J.; Goldman, S.; Gururangan, S.; Gajjar, A.; et al. Phase I trial of MK-0752 in children with refractory CNS malignancies: A pediatric brain tumor consortium study. J. Clin. Oncol. 2011, 29, 3529–3534. [Google Scholar] [CrossRef] [PubMed]
- Krop, I.; Demuth, T.; Guthrie, T.; Wen, P.Y.; Mason, W.P.; Chinnaiyan, P.; Butowski, N.; Groves, M.D.; Kesari, S.; Freedman, S.J.; et al. Phase I pharmacologic and pharmacodynamic study of the gamma secretase (Notch) inhibitor MK-0752 in adult patients with advanced solid tumors. J. Clin. Oncol. 2012, 30, 2307–2313. [Google Scholar] [CrossRef]
- Milano, J.; McKay, J.; Dagenais, C.; Foster-Brown, L.; Pognan, F.; Gadient, R.; Jacobs, R.T.; Zacco, A.; Greenberg, B.; Ciaccio, P.J. Modulation of notch processing by gamma-secretase inhibitors causes intestinal goblet cell metaplasia and induction of genes known to specify gut secretory lineage differentiation. Toxicol. Sci. 2004, 82, 341–358. [Google Scholar] [CrossRef]
- Riccio, O.; van Gijn, M.E.; Bezdek, A.C.; Pellegrinet, L.; van Es, J.H.; Zimber-Strobl, U.; Strobl, L.J.; Honjo, T.; Clevers, H.; Radtke, F. Loss of intestinal crypt progenitor cells owing to inactivation of both Notch1 and Notch2 is accompanied by derepression of CDK inhibitors p27Kip1 and p57Kip2. EMBO Rep. 2008, 9, 377–383. [Google Scholar] [CrossRef]
- Beel, A.J.; Sanders, C.R. Substrate specificity of gamma-secretase and other intramembrane proteases. Cell Mol. Life Sci. 2008, 65, 1311–1334. [Google Scholar] [CrossRef]
- Hemming, M.L.; Elias, J.E.; Gygi, S.P.; Selkoe, D.J. Proteomic profiling of gamma-secretase substrates and mapping of substrate requirements. PLoS Biol. 2008, 6, e257. [Google Scholar] [CrossRef]
- Aste-Amezaga, M.; Zhang, N.; Lineberger, J.E.; Arnold, B.A.; Toner, T.J.; Gu, M.; Huang, L.; Vitelli, S.; Vo, K.T.; Haytko, P.; et al. Characterization of Notch1 antibodies that inhibit signaling of both normal and mutated Notch1 receptors. PLoS ONE 2010, 5, e9094. [Google Scholar] [CrossRef]
- Wu, Y.; Cain-Hom, C.; Choy, L.; Hagenbeek, T.J.; de Leon, G.P.; Chen, Y.; Finkle, D.; Venook, R.; Wu, X.; Ridgway, J.; et al. Therapeutic antibody targeting of individual Notch receptors. Nature 2010, 464, 1052–1057. [Google Scholar] [CrossRef]
- Li, Y.; Hickson, J.A.; Ambrosi, D.J.; Haasch, D.L.; Foster-Duke, K.D.; Eaton, L.J.; DiGiammarino, E.L.; Panchal, S.C.; Jiang, F.; Mudd, S.R.; et al. ABT-165, a Dual Variable Domain Immunoglobulin (DVD-Ig) Targeting DLL4 and VEGF, Demonstrates Superior Efficacy and Favorable Safety Profiles in Preclinical Models. Mol. Cancer 2018, 17, 1039–1050. [Google Scholar] [CrossRef]
- Parakh, S.; Nicolazzo, J.; Scott, A.M.; Gan, H.K. Antibody Drug Conjugates in Glioblastoma—Is There a Future for Them? Front. Oncol. 2021, 11, 718590. [Google Scholar] [CrossRef]
- Spino, M.; Kurz, S.C.; Chiriboga, L.; Serrano, J.; Zeck, B.; Sen, N.; Patel, S.; Shen, G.; Vasudevaraja, V.; Tsirigos, A.; et al. Cell Surface Notch Ligand DLL3 is a Therapeutic Target in Isocitrate Dehydrogenase-mutant Glioma. Clin. Cancer Res. 2019, 25, 1261–1271. [Google Scholar] [CrossRef]
- Rudin, C.M.; Pietanza, M.C.; Bauer, T.M.; Ready, N.; Morgensztern, D.; Glisson, B.S.; Byers, L.A.; Johnson, M.L.; Burris, H.A., 3rd; Robert, F.; et al. Rovalpituzumab tesirine, a DLL3-targeted antibody-drug conjugate, in recurrent small-cell lung cancer: A first-in-human, first-in-class, open-label, phase 1 study. Lancet Oncol. 2017, 18, 42–51. [Google Scholar] [CrossRef]
- Blackhall, F.; Jao, K.; Greillier, L.; Cho, B.C.; Penkov, K.; Reguart, N.; Majem, M.; Nackaerts, K.; Syrigos, K.; Hansen, K.; et al. Efficacy and Safety of Rovalpituzumab Tesirine Compared With Topotecan as Second-Line Therapy in DLL3-High SCLC: Results From the Phase 3 TAHOE Study. J. Thorac. Oncol. 2021, 16, 1547–1558. [Google Scholar] [CrossRef]
- Johnson, M.L.; Zvirbule, Z.; Laktionov, K.; Helland, A.; Cho, B.C.; Gutierrez, V.; Colinet, B.; Lena, H.; Wolf, M.; Gottfried, M.; et al. Rovalpituzumab Tesirine as a Maintenance Therapy After First-Line Platinum-Based Chemotherapy in Patients With Extensive-Stage-SCLC: Results From the Phase 3 MERU Study. J. Thorac. Oncol. 2021, 16, 1570–1581. [Google Scholar] [CrossRef]
- Chen, J.; Kesari, S.; Rooney, C.; Strack, P.R.; Chen, J.; Shen, H.; Wu, L.; Griffin, J.D. Inhibition of notch signaling blocks growth of glioblastoma cell lines and tumor neurospheres. Genes Cancer 2010, 1, 822–835. [Google Scholar] [CrossRef]
- Opacak-Bernardi, T.; Ryu, J.S.; Raucher, D. Effects of cell penetrating Notch inhibitory peptide conjugated to elastin-like polypeptide on glioblastoma cells. J. Drug Target. 2017, 25, 523–531. [Google Scholar] [CrossRef]
- Alvarez-Trotta, A.; Guerrant, W.; Astudillo, L.; Lahiry, M.; Diluvio, G.; Shersher, E.; Kaneku, H.; Robbins, D.J.; Orton, D.; Capobianco, A.J. Pharmacological Disruption of the Notch1 Transcriptional Complex Inhibits Tumor Growth by Selectively Targeting Cancer Stem Cells. Cancer Res. 2021, 81, 3347–3357. [Google Scholar] [CrossRef]
- Astudillo, L.; Da Silva, T.G.; Wang, Z.; Han, X.; Jin, K.; VanWye, J.; Zhu, X.; Weaver, K.; Oashi, T.; Lopes, P.E.; et al. The Small Molecule IMR-1 Inhibits the Notch Transcriptional Activation Complex to Suppress Tumorigenesis. Cancer Res. 2016, 76, 3593–3603. [Google Scholar] [CrossRef]
- Lehal, R.; Zaric, J.; Vigolo, M.; Urech, C.; Frismantas, V.; Zangger, N.; Cao, L.; Berger, A.; Chicote, I.; Loubery, S.; et al. Pharmacological disruption of the Notch transcription factor complex. Proc. Natl. Acad. Sci. USA 2020, 117, 16292–16301. [Google Scholar] [CrossRef]
- Perron, A.; Nishikawa, Y.; Iwata, J.; Shimojo, H.; Takaya, J.; Kobayashi, K.; Imayoshi, I.; Mbenza, N.M.; Takenoya, M.; Kageyama, R.; et al. Small-molecule screening yields a compound that inhibits the cancer-associated transcription factor Hes1 via the PHB2 chaperone. J. Biol. Chem. 2018, 293, 8285–8294. [Google Scholar] [CrossRef]
- Floyd, D.H.; Kefas, B.; Seleverstov, O.; Mykhaylyk, O.; Dominguez, C.; Comeau, L.; Plank, C.; Purow, B. Alpha-secretase inhibition reduces human glioblastoma stem cell growth in vitro and in vivo by inhibiting Notch. Neuro-Oncology 2012, 14, 1215–1226. [Google Scholar] [CrossRef]
- Roti, G.; Carlton, A.; Ross, K.N.; Markstein, M.; Pajcini, K.; Su, A.H.; Perrimon, N.; Pear, W.S.; Kung, A.L.; Blacklow, S.C.; et al. Complementary genomic screens identify SERCA as a therapeutic target in NOTCH1 mutated cancer. Cancer Cell 2013, 23, 390–405. [Google Scholar] [CrossRef]
- Piccioni, D.; Juarez, T.; Brown, B.; Rose, L.; Allgood, V.; Kesari, S. Atct-18phase Ii Study of Mipsagargin (G-202), a Psma-Activated Prodrug Targeting the Tumor Endothelium, in Adult Patients with Recurrent or Progressive Glioblastoma. Neuro-Oncology 2015, 17, v5. [Google Scholar] [CrossRef][Green Version]
- Zhang, H.; Liu, L.; Liu, C.; Pan, J.; Lu, G.; Zhou, Z.; Chen, Z.; Qian, C. Notch3 overexpression enhances progression and chemoresistance of urothelial carcinoma. Oncotarget 2017, 8, 34362–34373. [Google Scholar] [CrossRef]
- Granit, R.Z.; Masury, H.; Condiotti, R.; Fixler, Y.; Gabai, Y.; Glikman, T.; Dalin, S.; Winter, E.; Nevo, Y.; Carmon, E.; et al. Regulation of Cellular Heterogeneity and Rates of Symmetric and Asymmetric Divisions in Triple-Negative Breast Cancer. Cell Rep. 2018, 24, 3237–3250. [Google Scholar] [CrossRef]
- Ding, W.; Zeng, T.; Tao, W.; Ge, W.; Deng, J.; Lei, H.; Xiao, Y.; Liao, F. Effect of lenalidomide on the human gastric cancer cell line SGC7901/vincristine Notch signaling. J. Cancer Res. 2018, 14, S237–S242. [Google Scholar] [CrossRef]
- Ponnurangam, S.; Dandawate, P.R.; Dhar, A.; Tawfik, O.W.; Parab, R.R.; Mishra, P.D.; Ranadive, P.; Sharma, R.; Mahajan, G.; Umar, S.; et al. Quinomycin A targets Notch signaling pathway in pancreatic cancer stem cells. Oncotarget 2016, 7, 3217–3232. [Google Scholar] [CrossRef]
- Hanashima, Y.; Sano, E.; Sumi, K.; Ozawa, Y.; Yagi, C.; Tatsuoka, J.; Yoshimura, S.; Yamamuro, S.; Ueda, T.; Nakayama, T.; et al. Antitumor effect of lenalidomide in malignant glioma cell lines. Oncol. Rep. 2020, 43, 1580–1590. [Google Scholar] [CrossRef] [PubMed]
- Kader, B.A.; Distefano, R.; West, K.L.; West, A.G. EZH2 inhibition in glioblastoma stem cells increases the expression of neuronal genes and the neuronal developmental regulators ZIC2, ZNF423 and MAFB. bioRxiv 2021, 20211122469535. [Google Scholar] [CrossRef]
- Yin, D.; Ong, J.M.; Hu, J.; Desmond, J.C.; Kawamata, N.; Konda, B.M.; Black, K.L.; Koeffler, H.P. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor: Effects on gene expression and growth of glioma cells in vitro and in vivo. Clin. Cancer Res. 2007, 13, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Y.; Zhao, S.; Li, Q.; Li, Y.; Kawamoto, K. Arsenic trioxide-mediated Notch pathway inhibition depletes the cancer stem-like cell population in gliomas. Cancer Lett. 2010, 292, 64–72. [Google Scholar] [CrossRef]
- Wu, J.; Ji, Z.; Liu, H.; Liu, Y.; Han, D.; Shi, C.; Shi, C.; Wang, C.; Yang, G.; Chen, X.; et al. Arsenic trioxide depletes cancer stem-like cells and inhibits repopulation of neurosphere derived from glioblastoma by downregulation of Notch pathway. Toxicol. Lett. 2013, 220, 61–69. [Google Scholar] [CrossRef]
- Schwalfenberg, G.K. N-Acetylcysteine: A Review of Clinical Usefulness (an Old Drug with New Tricks). J. Nutr. Metab. 2021, 2021, 9949453. [Google Scholar] [CrossRef]
- Deng, J.; Liu, A.D.; Hou, G.Q.; Zhang, X.; Ren, K.; Chen, X.Z.; Li, S.S.C.; Wu, Y.S.; Cao, X. N-acetylcysteine decreases malignant characteristics of glioblastoma cells by inhibiting Notch2 signaling. J. Exp. Clin. Cancer Res. 2019, 38, 2. [Google Scholar] [CrossRef]
- Majumder, S.; Crabtree, J.S.; Golde, T.E.; Minter, L.M.; Osborne, B.A.; Miele, L. Targeting Notch in oncology: The path forward. Nat. Rev. Drug Discov. 2021, 20, 125–144. [Google Scholar] [CrossRef]
- Zhdanovskaya, N.; Firrincieli, M.; Lazzari, S.; Pace, E.; Scribani Rossi, P.; Felli, M.P.; Talora, C.; Screpanti, I.; Palermo, R. Targeting Notch to Maximize Chemotherapeutic Benefits: Rationale, Advanced Strategies, and Future Perspectives. Cancers 2021, 13, 5106. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Z.; Jin, J.; Zhu, S.X.; He, G.Q.; Li, S.H.; Wang, J.; Cai, Y. Quercetin pretreatment enhances the radiosensitivity of colon cancer cells by targeting Notch-1 pathway. Biochem. Biophys. Res. Commun. 2020, 523, 947–953. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, J.; Ma, D.; Zhang, L.; Si, M.; Yin, H.; Li, J. Curcumin inhibits proliferation and invasion of osteosarcoma cells through inactivation of Notch-1 signaling. FEBS J. 2012, 279, 2247–2259. [Google Scholar] [CrossRef]
- Mori, M.; Tottone, L.; Quaglio, D.; Zhdanovskaya, N.; Ingallina, C.; Fusto, M.; Ghirga, F.; Peruzzi, G.; Crestoni, M.E.; Simeoni, F.; et al. Identification of a novel chalcone derivative that inhibits Notch signaling in T-cell acute lymphoblastic leukemia. Sci. Rep. 2017, 7, 2213. [Google Scholar] [CrossRef]
- Lai, I.C.; Shih, P.H.; Yao, C.J.; Yeh, C.T.; Wang-Peng, J.; Lui, T.N.; Chuang, S.E.; Hu, T.S.; Lai, T.Y.; Lai, G.M. Elimination of cancer stem-like cells and potentiation of temozolomide sensitivity by Honokiol in glioblastoma multiforme cells. PLoS ONE 2015, 10, e0114830. [Google Scholar] [CrossRef]
- Lubecka, K.; Kurzava, L.; Flower, K.; Buvala, H.; Zhang, H.; Teegarden, D.; Camarillo, I.; Suderman, M.; Kuang, S.; Andrisani, O.; et al. Stilbenoids remodel the DNA methylation patterns in breast cancer cells and inhibit oncogenic NOTCH signaling through epigenetic regulation of MAML2 transcriptional activity. Carcinogenesis 2016, 37, 656–668. [Google Scholar] [CrossRef]
- Sun, Z.; Zhou, C.; Liu, F.; Zhang, W.; Chen, J.; Pan, Y.; Ma, L.; Liu, Q.; Du, Y.; Yang, J.; et al. Inhibition of breast cancer cell survival by Xanthohumol via modulation of the Notch signaling pathway in vivo and in vitro. Oncol. Lett. 2018, 15, 908–916. [Google Scholar] [CrossRef]
- Kang, M.S.; Baek, S.H.; Chun, Y.S.; Moore, A.Z.; Landman, N.; Berman, D.; Yang, H.O.; Morishima-Kawashima, M.; Osawa, S.; Funamoto, S.; et al. Modulation of lipid kinase PI4KIIalpha activity and lipid raft association of presenilin 1 underlies gamma-secretase inhibition by ginsenoside (20S)-Rg3. J. Biol. Chem. 2013, 288, 20868–20882. [Google Scholar] [CrossRef]
- Kiesel, V.A.; Stan, S.D. Diallyl trisulfide, a chemopreventive agent from Allium vegetables, inhibits alpha-secretases in breast cancer cells. Biochem. Biophys. Res. Commun. 2017, 484, 833–838. [Google Scholar] [CrossRef]
- Ohtaka, M.; Itoh, M.; Tohda, S. BMI1 Inhibitors Down-regulate NOTCH Signaling and Suppress Proliferation of Acute Leukemia Cells. Anticancer Res. 2017, 37, 6047–6053. [Google Scholar] [CrossRef]
- Sun, D.W.; Zhang, H.D.; Mao, L.; Mao, C.F.; Chen, W.; Cui, M.; Ma, R.; Cao, H.X.; Jing, C.W.; Wang, Z.; et al. Luteolin Inhibits Breast Cancer Development and Progression In Vitro and In Vivo by Suppressing Notch Signaling and Regulating MiRNAs. Cell Physiol. Biochem. 2015, 37, 1693–1711. [Google Scholar] [CrossRef]
- Giuli, M.V.; Diluvio, G.; Giuliani, E.; Franciosa, G.; Di Magno, L.; Pignataro, M.G.; Tottone, L.; Nicoletti, C.; Besharat, Z.M.; Peruzzi, G.; et al. Notch3 contributes to T-cell leukemia growth via regulation of the unfolded protein response. Oncogenesis 2020, 9, 93. [Google Scholar] [CrossRef]
- Koduru, S.; Kumar, R.; Srinivasan, S.; Evers, M.B.; Damodaran, C. Notch-1 inhibition by Withaferin-A: A therapeutic target against colon carcinogenesis. Mol. Cancer 2010, 9, 202–210. [Google Scholar] [CrossRef]
- Dandawate, P.; Subramaniam, D.; Panovich, P.; Standing, D.; Krishnamachary, B.; Kaushik, G.; Thomas, S.M.; Dhar, A.; Weir, S.J.; Jensen, R.A.; et al. Cucurbitacin B and I inhibits colon cancer growth by targeting the Notch signaling pathway. Sci. Rep. 2020, 10, 1290. [Google Scholar] [CrossRef]
- Kubiczkova, L.; Sedlarikova, L.; Hajek, R.; Sevcikova, S. TGF-beta—An excellent servant but a bad master. J. Transl. Med. 2012, 10, 183. [Google Scholar] [CrossRef]
- Xu, X.; Zheng, L.; Yuan, Q.; Zhen, G.; Crane, J.L.; Zhou, X.; Cao, X. Transforming growth factor-beta in stem cells and tissue homeostasis. Bone Res. 2018, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Meyers, E.A.; Kessler, J.A. TGF-beta Family Signaling in Neural and Neuronal Differentiation, Development, and Function. Cold Spring Harb. Perspect. Biol. 2017, 9, a022244. [Google Scholar] [CrossRef]
- Falk, S.; Wurdak, H.; Ittner, L.M.; Ille, F.; Sumara, G.; Schmid, M.T.; Draganova, K.; Lang, K.S.; Paratore, C.; Leveen, P.; et al. Brain area-specific effect of TGF-beta signaling on Wnt-dependent neural stem cell expansion. Cell Stem Cell 2008, 2, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Baba, A.B.; Rah, B.; Bhat, G.R.; Mushtaq, I.; Parveen, S.; Hassan, R.; Hameed Zargar, M.; Afroze, D. Transforming Growth Factor-Beta (TGF-beta) Signaling in Cancer-A Betrayal Within. Front. Pharm. 2022, 13, 791272. [Google Scholar] [CrossRef]
- Proetzel, G.; Pawlowski, S.A.; Wiles, M.V.; Yin, M.; Boivin, G.P.; Howles, P.N.; Ding, J.; Ferguson, M.W.; Doetschman, T. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat. Genet. 1995, 11, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Letterio, J.J.; Roberts, A.B. Transforming growth factor-beta1-deficient mice: Identification of isoform-specific activities in vivo. J. Leukoc. Biol. 1996, 59, 769–774. [Google Scholar] [CrossRef] [PubMed]
- Sanford, L.P.; Ormsby, I.; Gittenberger-de Groot, A.C.; Sariola, H.; Friedman, R.; Boivin, G.P.; Cardell, E.L.; Doetschman, T. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development 1997, 124, 2659–2670. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Alvarez-Breckenridge, C.A.; Wang, Q.E.; Yu, J. TGF-beta signaling and its targeting for glioma treatment. Am. J. Cancer Res. 2015, 5, 945–955. [Google Scholar]
- Nakao, A.; Afrakhte, M.; Moren, A.; Nakayama, T.; Christian, J.L.; Heuchel, R.; Itoh, S.; Kawabata, M.; Heldin, N.E.; Heldin, C.H.; et al. Identification of Smad7, a TGFbeta-in.nducible antagonist of TGF-beta signalling. Nature 1997, 389, 631–635. [Google Scholar] [CrossRef]
- Moustakas, A.; Heldin, C.H. Non-Smad TGF-beta signals. J. Cell Sci. 2005, 118, 3573–3584. [Google Scholar] [CrossRef]
- Teixeira, A.F.; Ten Dijke, P.; Zhu, H.J. On-Target Anti-TGF-beta Therapies Are Not Succeeding in Clinical Cancer Treatments: What Are Remaining Challenges? Front. Cell Dev. Biol. 2020, 8, 605. [Google Scholar] [CrossRef]
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef]
- Lindholm, D.; Castren, E.; Kiefer, R.; Zafra, F.; Thoenen, H. Transforming growth factor-beta 1 in the rat brain: Increase after injury and inhibition of astrocyte proliferation. J. Cell Biol. 1992, 117, 395–400. [Google Scholar] [CrossRef]
- Lebrun, J.J. The Dual Role of TGFbeta in Human Cancer: From Tumor Suppression to Cancer Metastasis. ISRN Mol. Biol. 2012, 2012, 381428. [Google Scholar] [CrossRef]
- Kjellman, C.; Olofsson, S.P.; Hansson, O.; Von Schantz, T.; Lindvall, M.; Nilsson, I.; Salford, L.G.; Sjogren, H.O.; Widegren, B. Expression of TGF-beta isoforms, TGF-beta receptors, and SMAD molecules at different stages of human glioma. Int. J. Cancer 2000, 89, 251–258. [Google Scholar] [CrossRef]
- Bruna, A.; Darken, R.S.; Rojo, F.; Ocana, A.; Penuelas, S.; Arias, A.; Paris, R.; Tortosa, A.; Mora, J.; Baselga, J.; et al. High TGFbeta-Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell 2007, 11, 147–160. [Google Scholar] [CrossRef]
- Alexandrow, M.G.; Moses, H.L. Transforming growth factor beta and cell cycle regulation. Cancer Res. 1995, 55, 1452–1457. [Google Scholar]
- Wesolowska, A.; Kwiatkowska, A.; Slomnicki, L.; Dembinski, M.; Master, A.; Sliwa, M.; Franciszkiewicz, K.; Chouaib, S.; Kaminska, B. Microglia-derived TGF-beta as an important regulator of glioblastoma invasion--an inhibition of TGF-beta-dependent effects by shRNA against human TGF-beta type II receptor. Oncogene 2008, 27, 918–930. [Google Scholar] [CrossRef] [PubMed]
- Nana, A.W.; Yang, P.M.; Lin, H.Y. Overview of Transforming Growth Factor beta Superfamily Involvement in Glioblastoma Initiation and Progression. Asian Pac. J. Cancer Prev. 2015, 16, 6813–6823. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Katz, L.H.; Li, Y.; Chen, J.S.; Munoz, N.M.; Majumdar, A.; Chen, J.; Mishra, L. Targeting TGF-beta signaling in cancer. Expert Opin Targets 2013, 17, 743–760. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Alexander, P.B.; Wang, X.F. TGF-beta Family Signaling in the Control of Cell Proliferation and Survival. Cold Spring Harb. Perspect. Biol. 2017, 9, a022145. [Google Scholar] [CrossRef]
- Batlle, E.; Massague, J. Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Gu, S.; Feng, X.H. TGF-beta signaling in cancer. Acta Biochim. Biophys. Sin. 2018, 50, 941–949. [Google Scholar] [CrossRef]
- Gieryng, A.; Pszczolkowska, D.; Walentynowicz, K.A.; Rajan, W.D.; Kaminska, B. Immune microenvironment of gliomas. Lab. Investig. 2017, 97, 498–518. [Google Scholar] [CrossRef]
- Li, W.; Graeber, M.B. The molecular profile of microglia under the influence of glioma. Neuro-Oncology 2012, 14, 958–978. [Google Scholar] [CrossRef]
- Xue, V.W.; Chung, J.Y.; Cordoba, C.A.G.; Cheung, A.H.; Kang, W.; Lam, E.W.; Leung, K.T.; To, K.F.; Lan, H.Y.; Tang, P.M. Transforming Growth Factor-beta: A Multifunctional Regulator of Cancer Immunity. Cancers 2020, 12, 3099. [Google Scholar] [CrossRef]
- Friese, M.A.; Wischhusen, J.; Wick, W.; Weiler, M.; Eisele, G.; Steinle, A.; Weller, M. RNA interference targeting transforming growth factor-beta enhances NKG2D-mediated antiglioma immune response, inhibits glioma cell migration and invasiveness, and abrogates tumorigenicity in vivo. Cancer Res. 2004, 64, 7596–7603. [Google Scholar] [CrossRef]
- Schlingensiepen, K.H.; Schlingensiepen, R.; Steinbrecher, A.; Hau, P.; Bogdahn, U.; Fischer-Blass, B.; Jachimczak, P. Targeted tumor therapy with the TGF-beta 2 antisense compound AP 12009. Cytokine Growth Factor Rev. 2006, 17, 129–139. [Google Scholar] [CrossRef]
- Jaschinski, F.; Rothhammer, T.; Jachimczak, P.; Seitz, C.; Schneider, A.; Schlingensiepen, K.H. The antisense oligonucleotide trabedersen (AP 12009) for the targeted inhibition of TGF-beta2. Curr. Pharm. Biotechnol. 2011, 12, 2203–2213. [Google Scholar] [CrossRef]
- Hau, P.; Jachimczak, P.; Schlingensiepen, R.; Schulmeyer, F.; Jauch, T.; Steinbrecher, A.; Brawanski, A.; Proescholdt, M.; Schlaier, J.; Buchroithner, J.; et al. Inhibition of TGF-beta2 with AP 12009 in recurrent malignant gliomas: From preclinical to phase I/II studies. Oligonucleotides 2007, 17, 201–212. [Google Scholar] [CrossRef]
- Roth, P.; Silginer, M.; Goodman, S.L.; Hasenbach, K.; Thies, S.; Maurer, G.; Schraml, P.; Tabatabai, G.; Moch, H.; Tritschler, I.; et al. Integrin control of the transforming growth factor-beta pathway in glioblastoma. Brain 2013, 136, 564–576. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Gorlia, T.; Erridge, S.C.; Perry, J.; Hong, Y.-K.; Aldape, K.D.; Lhermitte, B.; Pietsch, T.; Grujicic, D.; et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 1100–1108. [Google Scholar] [CrossRef]
- Suzuki, E.; Kim, S.; Cheung, H.K.; Corbley, M.J.; Zhang, X.; Sun, L.; Shan, F.; Singh, J.; Lee, W.C.; Albelda, S.M.; et al. A novel small-molecule inhibitor of transforming growth factor beta type I receptor kinase (SM16) inhibits murine mesothelioma tumor growth in vivo and prevents tumor recurrence after surgical resection. Cancer Res. 2007, 67, 2351–2359. [Google Scholar] [CrossRef]
- Rausch, M.P.; Hahn, T.; Ramanathapuram, L.; Bradley-Dunlop, D.; Mahadevan, D.; Mercado-Pimentel, M.E.; Runyan, R.B.; Besselsen, D.G.; Zhang, X.; Cheung, H.K.; et al. An orally active small molecule TGF-beta receptor I antagonist inhibits the growth of metastatic murine breast cancer. Anticancer Res. 2009, 29, 2099–2109. [Google Scholar]
- Zhang, M.; Kleber, S.; Rohrich, M.; Timke, C.; Han, N.; Tuettenberg, J.; Martin-Villalba, A.; Debus, J.; Peschke, P.; Wirkner, U.; et al. Blockade of TGF-beta signaling by the TGFbetaR-I kinase inhibitor LY2109761 enhances radiation response and prolongs survival in glioblastoma. Cancer Res. 2011, 71, 7155–7167. [Google Scholar] [CrossRef]
- Zhang, M.; Herion, T.W.; Timke, C.; Han, N.; Hauser, K.; Weber, K.J.; Peschke, P.; Wirkner, U.; Lahn, M.; Huber, P.E. Trimodal glioblastoma treatment consisting of concurrent radiotherapy, temozolomide, and the novel TGF-beta receptor I kinase inhibitor LY2109761. Neoplasia 2011, 13, 537–549. [Google Scholar] [CrossRef]
- Yingling, J.M.; McMillen, W.T.; Yan, L.; Huang, H.; Sawyer, J.S.; Graff, J.; Clawson, D.K.; Britt, K.S.; Anderson, B.D.; Beight, D.W.; et al. Preclinical assessment of galunisertib (LY2157299 monohydrate), a first-in-class transforming growth factor-beta receptor type I inhibitor. Oncotarget 2018, 9, 6659–6677. [Google Scholar] [CrossRef]
- Wick, A.; Desjardins, A.; Suarez, C.; Forsyth, P.; Gueorguieva, I.; Burkholder, T.; Cleverly, A.L.; Estrem, S.T.; Wang, S.; Lahn, M.M.; et al. Phase 1b/2a study of galunisertib, a small molecule inhibitor of transforming growth factor-beta receptor I, in combination with standard temozolomide-based radiochemotherapy in patients with newly diagnosed malignant glioma. Investig. New Drugs 2020, 38, 1570–1579. [Google Scholar] [CrossRef]
- Brandes, A.A.; Carpentier, A.F.; Kesari, S.; Sepulveda-Sanchez, J.M.; Wheeler, H.R.; Chinot, O.; Cher, L.; Steinbach, J.P.; Capper, D.; Specenier, P.; et al. A Phase II randomized study of galunisertib monotherapy or galunisertib plus lomustine compared with lomustine monotherapy in patients with recurrent glioblastoma. Neuro-Oncology 2016, 18, 1146–1156. [Google Scholar] [CrossRef]
- Spender, L.C.; Ferguson, G.J.; Hughes, G.D.; Davies, B.R.; Goldberg, F.W.; Herrera, B.; Taylor, R.G.; Strathearn, L.S.; Sansom, O.J.; Barry, S.T.; et al. Preclinical Evaluation of AZ12601011 and AZ12799734, Inhibitors of Transforming Growth Factor beta Superfamily Type 1 Receptors. Mol. Pharm. 2019, 95, 222–234. [Google Scholar] [CrossRef]
- Ueda, R.; Fujita, M.; Zhu, X.; Sasaki, K.; Kastenhuber, E.R.; Kohanbash, G.; McDonald, H.A.; Harper, J.; Lonning, S.; Okada, H. Systemic inhibition of transforming growth factor-beta in glioma-bearing mice improves the therapeutic efficacy of glioma-associated antigen peptide vaccines. Clin. Cancer Res. 2009, 15, 6551–6559. [Google Scholar] [CrossRef]
- Hulper, P.; Schulz-Schaeffer, W.; Dullin, C.; Hoffmann, P.; Harper, J.; Kurtzberg, L.; Lonning, S.; Kugler, W.; Lakomek, M.; Erdlenbruch, B. Tumor localization of an anti-TGF-beta antibody and its effects on gliomas. Int. J. Oncol. 2011, 38, 51–59. [Google Scholar]
- Den Hollander, M.W.; Bensch, F.; Glaudemans, A.W.; Oude Munnink, T.H.; Enting, R.H.; den Dunnen, W.F.; Heesters, M.A.; Kruyt, F.A.; Lub-de Hooge, M.N.; Cees de Groot, J.; et al. TGF-beta Antibody Uptake in Recurrent High-Grade Glioma Imaged with 89Zr-Fresolimumab PET. J. Nucl. Med. 2015, 56, 1310–1314. [Google Scholar] [CrossRef]
- Shaim, H.; Shanley, M.; Basar, R.; Daher, M.; Gumin, J.; Zamler, D.B.; Uprety, N.; Wang, F.; Huang, Y.; Gabrusiewicz, K.; et al. Targeting the alphav integrin/TGF-beta axis improves natural killer cell function against glioblastoma stem cells. J. Clin. Investig. 2021, 131, e142116. [Google Scholar] [CrossRef]
- Niedbala, M.; Malarz, K.; Sharma, G.; Kramer-Marek, G.; Kaspera, W. Glioblastoma: Pitfalls and Opportunities of Immunotherapeutic Combinations. OncoTargets Ther. 2022, 15, 437–468. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Najafi, M.; Orouei, S.; Zabolian, A.; Saleki, H.; Azami, N.; Sharifi, N.; Hushmandi, K.; Zarrabi, A.; Ahn, K.S. Resveratrol Modulates Transforming Growth Factor-Beta (TGF-beta) Signaling Pathway for Disease Therapy: A New Insight into Its Pharmacological Activities. Biomedicines 2020, 8, 261. [Google Scholar] [CrossRef]
- Kabel, A.M.; Atef, A.; Estfanous, R.S. Ameliorative potential of sitagliptin and/or resveratrol on experimentally-induced clear cell renal cell carcinoma. Biomed. Pharm. 2018, 97, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, S.; Yang, Y.; Liu, T. Resveratrol induces immunogenic cell death of human and murine ovarian carcinoma cells. Infect. Agent Cancer 2019, 14, 27. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Chen, Y.; Li, Y.; Lyu, X.; Cui, J.; Cheng, Y.; Zheng, T.; Zhao, L.; Zhao, G. Resveratrol Suppresses Epithelial-Mesenchymal Transition in GBM by Regulating Smad-Dependent Signaling. Biomed. Res. Int. 2019, 2019, 1321973. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.; Luukko, K.; Kettunen, P. BMP signalling in craniofacial development. Int. J. Dev. Biol. 2006, 50, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Bond, A.M.; Bhalala, O.G.; Kessler, J.A. The dynamic role of bone morphogenetic proteins in neural stem cell fate and maturation. Dev. Neurobiol 2012, 72, 1068–1084. [Google Scholar] [CrossRef]
- Chen, H.L.; Panchision, D.M. Concise review: Bone morphogenetic protein pleiotropism in neural stem cells and their derivatives--alternative pathways, convergent signals. Stem Cells 2007, 25, 63–68. [Google Scholar] [CrossRef]
- Gamez, B.; Rodriguez-Carballo, E.; Ventura, F. BMP signaling in telencephalic neural cell specification and maturation. Front. Cell Neurosci. 2013, 7, 87. [Google Scholar] [CrossRef]
- Katagiri, T.; Watabe, T. Bone Morphogenetic Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a021899. [Google Scholar] [CrossRef]
- Bond, A.M.; Peng, C.Y.; Meyers, E.A.; McGuire, T.; Ewaleifoh, O.; Kessler, J.A. BMP signaling regulates the tempo of adult hippocampal progenitor maturation at multiple stages of the lineage. Stem Cells 2014, 32, 2201–2214. [Google Scholar] [CrossRef]
- Choe, Y.; Pleasure, S.J.; Mira, H. Control of Adult Neurogenesis by Short-Range Morphogenic-Signaling Molecules. Cold Spring Harb. Perspect. Biol. 2015, 8, a018887. [Google Scholar] [CrossRef]
- Panchision, D.M.; McKay, R.D. The control of neural stem cells by morphogenic signals. Curr. Opin. Genet. Dev. 2002, 12, 478–487. [Google Scholar] [CrossRef]
- Wang, R.N.; Green, J.; Wang, Z.; Deng, Y.; Qiao, M.; Peabody, M.; Zhang, Q.; Ye, J.; Yan, Z.; Denduluri, S.; et al. Bone Morphogenetic Protein (BMP) signaling in development and human diseases. Genes Dis 2014, 1, 87–105. [Google Scholar] [CrossRef]
- Choi, S.; Yu, J.; Park, A.; Dubon, M.J.; Do, J.; Kim, Y.; Nam, D.; Noh, J.; Park, K.S. BMP-4 enhances epithelial mesenchymal transition and cancer stem cell properties of breast cancer cells via Notch signaling. Sci. Rep. 2019, 9, 11724. [Google Scholar] [CrossRef]
- Ye, L.; Jiang, W.G. Bone morphogenetic proteins in tumour associated angiogenesis and implication in cancer therapies. Cancer Lett 2016, 380, 586–597. [Google Scholar] [CrossRef]
- Guyot, B.; Lefort, S.; Voeltzel, T.; Pecheur, E.I.; Maguer-Satta, V. Altered BMP2/4 Signaling in Stem Cells and Their Niche: Different Cancers but Similar Mechanisms, the Example of Myeloid Leukemia and Breast Cancer. Front. Cell Dev. Biol. 2021, 9, 787989. [Google Scholar] [CrossRef]
- Kallioniemi, A. Bone morphogenetic protein 4-a fascinating regulator of cancer cell behavior. Cancer Genet. 2012, 205, 267–277. [Google Scholar] [CrossRef]
- Lowery, J.W.; Brookshire, B.; Rosen, V. A Survey of Strategies to Modulate the Bone Morphogenetic Protein Signaling Pathway: Current and Future Perspectives. Stem Cells Int. 2016, 2016, 7290686. [Google Scholar] [CrossRef]
- Bach, D.H.; Park, H.J.; Lee, S.K. The Dual Role of Bone Morphogenetic Proteins in Cancer. Mol. Oncolytics 2018, 8, 1–13. [Google Scholar] [CrossRef]
- Zhang, L.; Ye, Y.; Long, X.; Xiao, P.; Ren, X.; Yu, J. BMP signaling and its paradoxical effects in tumorigenesis and dissemination. Oncotarget 2016, 7, 78206–78218. [Google Scholar] [CrossRef]
- Bao, Z.; Zhang, C.; Yan, W.; Liu, Y.; Li, M.; Zhang, W.; Jiang, T. BMP4, a strong better prognosis predictor, has a subtype preference and cell development association in gliomas. J. Transl. Med. 2013, 11, 100. [Google Scholar] [CrossRef]
- Wu, Q.; Yao, J. BMP4, a new prognostic factor for glioma. World J. Surg. Oncol. 2013, 11, 264. [Google Scholar] [CrossRef]
- Liu, C.; Tian, G.; Tu, Y.; Fu, J.; Lan, C.; Wu, N. Expression pattern and clinical prognostic relevance of bone morphogenetic protein-2 in human gliomas. Jpn J. Clin. Oncol. 2009, 39, 625–631. [Google Scholar] [CrossRef]
- Chirasani, S.R.; Sternjak, A.; Wend, P.; Momma, S.; Campos, B.; Herrmann, I.M.; Graf, D.; Mitsiadis, T.; Herold-Mende, C.; Besser, D.; et al. Bone morphogenetic protein-7 release from endogenous neural precursor cells suppresses the tumourigenicity of stem-like glioblastoma cells. Brain 2010, 133, 1961–1972. [Google Scholar] [CrossRef]
- Tate, C.M.; Pallini, R.; Ricci-Vitiani, L.; Dowless, M.M.; Shiyanova, T.; D’Alessandris, G.Q.; Morgante, L.; Giannetti, S.; Larocca, L.M.; di Martino, S.; et al. A BMP7 variant inhibits the tumorigenic potential of glioblastoma stem-like cells. Cell Death Differ. 2012, 19, 1644–1654. [Google Scholar] [CrossRef]
- Gonzalez-Gomez, P.; Crecente-Campo, J.; Zahonero, C.; de la Fuente, M.; Hernandez-Lain, A.; Mira, H.; Sanchez-Gomez, P.; Garcia-Fuentes, M. Controlled release microspheres loaded with BMP7 suppress primary tumors from human glioblastoma. Oncotarget 2015, 6, 10950–10963. [Google Scholar] [CrossRef]
- Zhou, Z.; Sun, L.; Wang, Y.; Wu, Z.; Geng, J.; Miu, W.; Pu, Y.; You, Y.; Yang, Z.; Liu, N. Bone morphogenetic protein 4 inhibits cell proliferation and induces apoptosis in glioma stem cells. Cancer Biother. Radiopharm. 2011, 26, 77–83. [Google Scholar] [CrossRef]
- Liu, B.; Tian, D.; Yi, W.; Wu, L.; Cai, Q.; Dong, H.; Shen, H.; Ji, B.; Wang, L.; Zhang, S.; et al. Effect of bone morphogenetic protein 4 in the human brain glioma cell line U251. Cell Biochem. Biophys. 2010, 58, 91–96. [Google Scholar] [CrossRef]
- Liu, B.; Chen, Q.; Tian, D.; Wu, L.; Dong, H.; Wang, J.; Ji, B.; Zhu, X.; Cai, Q.; Wang, L.; et al. BMP4 reverses multidrug resistance through modulation of BCL-2 and GDNF in glioblastoma. Brain Res. 2013, 1507, 115–124. [Google Scholar] [CrossRef]
- Persano, L.; Pistollato, F.; Rampazzo, E.; Della Puppa, A.; Abbadi, S.; Frasson, C.; Volpin, F.; Indraccolo, S.; Scienza, R.; Basso, G. BMP2 sensitizes glioblastoma stem-like cells to Temozolomide by affecting HIF-1alpha stability and MGMT expression. Cell Death Dis. 2012, 3, e412. [Google Scholar] [CrossRef]
- Yan, K.; Wu, Q.; Yan, D.H.; Lee, C.H.; Rahim, N.; Tritschler, I.; DeVecchio, J.; Kalady, M.F.; Hjelmeland, A.B.; Rich, J.N. Glioma cancer stem cells secrete Gremlin1 to promote their maintenance within the tumor hierarchy. Genes Dev. 2014, 28, 1085–1100. [Google Scholar] [CrossRef]
- Koguchi, M.; Nakahara, Y.; Ito, H.; Wakamiya, T.; Yoshioka, F.; Ogata, A.; Inoue, K.; Masuoka, J.; Izumi, H.; Abe, T. BMP4 induces asymmetric cell division in human glioma stem-like cells. Oncol. Lett. 2020, 19, 1247–1254. [Google Scholar] [CrossRef] [PubMed]
- Dalmo, E.; Johansson, P.; Niklasson, M.; Gustavsson, I.; Nelander, S.; Westermark, B. Growth-Inhibitory Activity of Bone Morphogenetic Protein 4 in Human Glioblastoma Cell Lines Is Heterogeneous and Dependent on Reduced SOX2 Expression. Mol. Cancer Res. 2020, 18, 981–991. [Google Scholar] [CrossRef] [PubMed]
- Kaye, J.; Mondal, A.; Foty, R.; Jia, D.; Langenfeld, J. Bone morphogenetic protein receptor inhibitors suppress the growth of glioblastoma cells. Mol. Cell Biochem. 2022, 477, 1583–1595. [Google Scholar] [CrossRef] [PubMed]
- Lasorella, A.; Benezra, R.; Iavarone, A. The ID proteins: Master regulators of cancer stem cells and tumour aggressiveness. Nat. Rev. Cancer 2014, 14, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Rampazzo, E.; Dettin, M.; Maule, F.; Scabello, A.; Calvanese, L.; D’Auria, G.; Falcigno, L.; Porcu, E.; Zamuner, A.; Della Puppa, A.; et al. A synthetic BMP-2 mimicking peptide induces glioblastoma stem cell differentiation. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 2282–2292. [Google Scholar] [CrossRef]
- Lowery, J.W.; Rosen, V. Bone Morphogenetic Protein-Based Therapeutic Approaches. Cold Spring Harb. Perspect. Biol. 2018, 10, a022327. [Google Scholar] [CrossRef]
- Huang, J.; Wu, S.; Barrera, J.; Matthews, K.; Pan, D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell 2005, 122, 421–434. [Google Scholar] [CrossRef]
- Halder, G.; Johnson, R.L. Hippo signaling: Growth control and beyond. Development 2011, 138, 9–22. [Google Scholar] [CrossRef]
- Bae, S.J.; Luo, X. Activation mechanisms of the Hippo kinase signaling cascade. Biosci. Rep. 2018, 38, BSR20171469. [Google Scholar] [CrossRef]
- Nishio, M.; Goto, H.; Suzuki, M.; Fujimoto, A.; Mimori, K.; Suzuki, A. The Hippo Signaling Pathway: A Candidate New Drug Target for Malignant Tumors. In Innovative Medicine; Nakao, K., Minato, N., Uemoto, S., Eds.; Springer: Tokyo, Japan, 2015. [Google Scholar]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L.; et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007, 21, 2747–2761. [Google Scholar] [CrossRef]
- Pobbati, A.V.; Hong, W. Emerging roles of TEAD transcription factors and its coactivators in cancers. Cancer Biol. 2013, 14, 390–398. [Google Scholar] [CrossRef]
- Meng, Z.; Moroishi, T.; Guan, K.L. Mechanisms of Hippo pathway regulation. Genes Dev. 2016, 30, 1–17. [Google Scholar] [CrossRef]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef]
- Johnson, R.; Halder, G. The two faces of Hippo: Targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat. Rev. Drug Discov. 2014, 13, 63–79. [Google Scholar] [CrossRef]
- Orr, B.A.; Bai, H.; Odia, Y.; Jain, D.; Anders, R.A.; Eberhart, C.G. Yes-associated protein 1 is widely expressed in human brain tumors and promotes glioblastoma growth. J. Neuropathol. Exp. Neurol. 2011, 70, 568–577. [Google Scholar] [CrossRef]
- Castellan, M.; Guarnieri, A.; Fujimura, A.; Zanconato, F.; Battilana, G.; Panciera, T.; Sladitschek, H.L.; Contessotto, P.; Citron, A.; Grilli, A.; et al. Single-cell analyses reveal YAP/TAZ as regulators of stemness and cell plasticity in Glioblastoma. Nat. Cancer 2021, 2, 174–188. [Google Scholar] [CrossRef]
- Fernandez, L.A.; Squatrito, M.; Northcott, P.; Awan, A.; Holland, E.C.; Taylor, M.D.; Nahle, Z.; Kenney, A.M. Oncogenic YAP promotes radioresistance and genomic instability in medulloblastoma through IGF2-mediated Akt activation. Oncogene 2012, 31, 1923–1937. [Google Scholar] [CrossRef]
- Tian, T.; Li, A.; Lu, H.; Luo, R.; Zhang, M.; Li, Z. TAZ promotes temozolomide resistance by upregulating MCL-1 in human glioma cells. Biochem. Biophys. Res. Commun. 2015, 463, 638–643. [Google Scholar] [CrossRef]
- Liu-Chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.J.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012, 26, 1300–1305. [Google Scholar] [CrossRef]
- Hill, J.S.; Kaye, A.H.; Sawyer, W.H.; Morstyn, G.; Megison, P.D.; Stylli, S.S. Selective uptake of hematoporphyrin derivative into human cerebral glioma. Neurosurgery 1990, 26, 248–254. [Google Scholar] [CrossRef]
- Vigneswaran, K.; Boyd, N.H.; Oh, S.Y.; Lallani, S.; Boucher, A.; Neill, S.G.; Olson, J.J.; Read, R.D. YAP/TAZ Transcriptional Coactivators Create Therapeutic Vulnerability to Verteporfin in EGFR-mutant Glioblastoma. Clin. Cancer Res. 2021, 27, 1553–1569. [Google Scholar] [CrossRef]
- Han, W.; Guan, W. Valproic Acid: A Promising Therapeutic Agent in Glioma Treatment. Front. Oncol. 2021, 11, 687362. [Google Scholar] [CrossRef]
- Knupfer, M.M.; Hernaiz-Driever, P.; Poppenborg, H.; Wolff, J.E.; Cinatl, J. Valproic acid inhibits proliferation and changes expression of CD44 and CD56 of malignant glioma cells in vitro. Anticancer Res. 1998, 18, 3585–3589. [Google Scholar]
- Xu, Y.; Stamenkovic, I.; Yu, Q. CD44 attenuates activation of the hippo signaling pathway and is a prime therapeutic target for glioblastoma. Cancer Res. 2010, 70, 2455–2464. [Google Scholar] [CrossRef]
- Valiyaveettil, D.; Malik, M.; Joseph, D.M.; Ahmed, S.F.; Kothwal, S.A.; Vijayasaradhi, M. Effect of valproic acid on survival in glioblastoma: A prospective single-arm study. South. Asian J. Cancer 2018, 7, 159–162. [Google Scholar] [CrossRef]
- Yuan, Y.; Xiang, W.; Qing, M.; Yanhui, L.; Jiewen, L.; Yunhe, M. Survival analysis for valproic acid use in adult glioblastoma multiforme: A meta-analysis of individual patient data and a systematic review. Seizure 2014, 23, 830–835. [Google Scholar] [CrossRef]
- Liu, Z.; Wei, Y.; Zhang, L.; Yee, P.P.; Johnson, M.; Zhang, X.; Gulley, M.; Atkinson, J.M.; Trebak, M.; Wang, H.G.; et al. Induction of store-operated calcium entry (SOCE) suppresses glioblastoma growth by inhibiting the Hippo pathway transcriptional coactivators YAP/TAZ. Oncogene 2019, 38, 120–139. [Google Scholar] [CrossRef]
- Miller, P.D.; Chines, A.A.; Christiansen, C.; Hoeck, H.C.; Kendler, D.L.; Lewiecki, E.M.; Woodson, G.; Levine, A.B.; Constantine, G.; Delmas, P.D. Effects of bazedoxifene on BMD and bone turnover in postmenopausal women: 2-yr results of a randomized, double-blind, placebo-, and active-controlled study. J. Bone Min. Res. 2008, 23, 525–535. [Google Scholar] [CrossRef]
- Wu, X.; Cao, Y.; Xiao, H.; Li, C.; Lin, J. Bazedoxifene as a Novel GP130 Inhibitor for Pancreatic Cancer Therapy. Mol. Cancer 2016, 15, 2609–2619. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Zhao, P.; Li, H.; Fu, H.; Liu, X.; Liu, Y.; Wu, J.; Fu, W. Bazedoxifene enhances paclitaxel efficacy to suppress glioblastoma via altering Hippo/YAP pathway. J. Cancer 2020, 11, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Wightman, S.M.; Alban, T.J.; Chen, X.; Lathia, J.D.; Wang, Y.; Stark, G.R. Bazedoxifene inhibits sustained STAT3 activation and increases survival in GBM. Transl. Oncol. 2021, 14, 101192. [Google Scholar] [CrossRef] [PubMed]
- Xiao, A.; Brenneman, B.; Floyd, D.; Comeau, L.; Spurio, K.; Olmez, I.; Lee, J.; Nakano, I.; Godlewski, J.; Bronisz, A.; et al. Statins affect human glioblastoma and other cancers through TGF-beta inhibition. Oncotarget 2019, 10, 1716–1728. [Google Scholar] [CrossRef]
- Oku, Y.; Nishiya, N.; Shito, T.; Yamamoto, R.; Yamamoto, Y.; Oyama, C.; Uehara, Y. Small molecules inhibiting the nuclear localization of YAP/TAZ for chemotherapeutics and chemosensitizers against breast cancers. FEBS Open Bio 2015, 5, 542–549. [Google Scholar] [CrossRef]
- Xie, Y.; Lu, Q.; Lenahan, C.; Yang, S.; Zhou, D.; Qi, X. Whether statin use improves the survival of patients with glioblastoma?: A meta-analysis. Medicine 2020, 99, e18997. [Google Scholar] [CrossRef]
- Cunningham, T.J.; Duester, G. Mechanisms of retinoic acid signalling and its roles in organ and limb development. Nat. Rev. Mol. Cell Biol. 2015, 16, 110–123. [Google Scholar] [CrossRef]
- Wiesinger, A.; Boink, G.J.J.; Christoffels, V.M.; Devalla, H.D. Retinoic acid signaling in heart development: Application in the differentiation of cardiovascular lineages from human pluripotent stem cells. Stem Cell Rep. 2021, 16, 2589–2606. [Google Scholar] [CrossRef]
- Dubey, A.; Rose, R.E.; Jones, D.R.; Saint-Jeannet, J.P. Generating retinoic acid gradients by local degradation during craniofacial development: One cell’s cue is another cell’s poison. Genesis 2018, 56, e23091. [Google Scholar] [CrossRef]
- Ni, X.; Hu, G.; Cai, X. The success and the challenge of all-trans retinoic acid in the treatment of cancer. Crit. Rev. Food Sci. Nutr. 2019, 59, S71–S80. [Google Scholar] [CrossRef]
- Ozgun, G.; Senturk, S.; Erkek-Ozhan, S. Retinoic acid signaling and bladder cancer: Epigenetic deregulation, therapy and beyond. Int. J. Cancer 2020, 148, 2364–2374. [Google Scholar] [CrossRef]
- Thompson, B.; Katsanis, N.; Apostolopoulos, N.; Thompson, D.C.; Nebert, D.W.; Vasiliou, V. Genetics and functions of the retinoic acid pathway, with special emphasis on the eye. Hum. Genom. 2019, 13, 61. [Google Scholar] [CrossRef]
- Fex, G.; Johannesson, G. Retinol transfer across and between phospholipid bilayer membranes. Biochim. Biophys. Acta 1988, 944, 249–255. [Google Scholar] [CrossRef]
- Luo, T.; Wagner, E.; Drager, U.C. Integrating retinoic acid signaling with brain function. Dev. Psychol. 2009, 45, 139–150. [Google Scholar] [CrossRef]
- Hunsu, V.O.; Facey, C.O.B.; Fields, J.Z.; Boman, B.M. Retinoids as Chemo-Preventive and Molecular-Targeted Anti-Cancer Therapies. Int. J. Mol. Sci. 2021, 22, 7731. [Google Scholar] [CrossRef] [PubMed]
- Al Tanoury, Z.; Piskunov, A.; Rochette-Egly, C. Vitamin A and retinoid signaling: Genomic and nongenomic effects. J. Lipid Res. 2013, 54, 1761–1775. [Google Scholar] [CrossRef] [PubMed]
- Agnihotri, S.; Mansouri, S.; Burrell, K.; Li, M.; Mamatjan, Y.; Liu, J.; Nejad, R.; Kumar, S.; Jalali, S.; Singh, S.K.; et al. Ketoconazole and Posaconazole Selectively Target HK2-expressing Glioblastoma Cells. Clin. Cancer Res. 2019, 25, 844–855. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, N.; Butters, R.R.; Brown, E.M. Agonists of the retinoic acid- and retinoid X-receptors inhibit hepatocyte growth factor secretion and expression in U87 human astrocytoma cells. Brain Res. Mol. Brain Res. 2001, 87, 100–108. [Google Scholar] [CrossRef]
- Costa, S.L.; Paillaud, E.; Fages, C.; Rochette-Egly, C.; Plassat, J.L.; Jouault, H.; Perzelova, A.; Tardy, M. Effects of a novel synthetic retinoid on malignant glioma in vitro: Inhibition of cell proliferation, induction of apoptosis and differentiation. Eur. J. Cancer 2001, 37, 520–530. [Google Scholar] [CrossRef]
- Hacioglu, C.; Kar, F.; Kacar, S.; Sahinturk, V.; Kanbak, G. Bexarotene inhibits cell proliferation by inducing oxidative stress, DNA damage and apoptosis via PPARgamma/NF-kappaB signaling pathway in C6 glioma cells. Med. Oncol. 2021, 38, 31. [Google Scholar] [CrossRef] [PubMed]
- Lena, A.; Rechichi, M.; Salvetti, A.; Bartoli, B.; Vecchio, D.; Scarcelli, V.; Amoroso, R.; Benvenuti, L.; Gagliardi, R.; Gremigni, V.; et al. Drugs targeting the mitochondrial pore act as cytotoxic and cytostatic agents in temozolomide-resistant glioma cells. J. Transl. Med. 2009, 7, 13. [Google Scholar] [CrossRef]
- Yokosawa, M.; Sonoda, Y.; Sugiyama, S.; Saito, R.; Yamashita, Y.; Nishihara, M.; Satoh, T.; Kumabe, T.; Yokoyama, M.; Tominaga, T. Convection-enhanced delivery of a synthetic retinoid Am80, loaded into polymeric micelles, prolongs the survival of rats bearing intracranial glioblastoma xenografts. Tohoku J. Exp. Med. 2010, 221, 257–264. [Google Scholar] [CrossRef][Green Version]
- Di Masi, A.; Leboffe, L.; De Marinis, E.; Pagano, F.; Cicconi, L.; Rochette-Egly, C.; Lo-Coco, F.; Ascenzi, P.; Nervi, C. Retinoic acid receptors: From molecular mechanisms to cancer therapy. Mol. Asp. Med. 2015, 41, 1–115. [Google Scholar] [CrossRef]
- Li, M.; Sun, Y.; Guan, X.; Shu, X.; Li, C. Advanced progress on the relationship between RA and its receptors and malignant tumors. Crit. Rev. Oncol. Hematol. 2014, 91, 271–282. [Google Scholar] [CrossRef]
- Chou, A.P.; Chowdhury, R.; Li, S.; Chen, W.; Kim, A.J.; Piccioni, D.E.; Selfridge, J.M.; Mody, R.R.; Chang, S.; Lalezari, S.; et al. Identification of retinol binding protein 1 promoter hypermethylation in isocitrate dehydrogenase 1 and 2 mutant gliomas. J. Natl. Cancer Inst. 2012, 104, 1458–1469. [Google Scholar] [CrossRef]
- Campos, B.; Warta, R.; Chaisaingmongkol, J.; Geiselhart, L.; Popanda, O.; Hartmann, C.; von Deimling, A.; Unterberg, A.; Plass, C.; Schmezer, P.; et al. Epigenetically mediated downregulation of the differentiation-promoting chaperon protein CRABP2 in astrocytic gliomas. Int. J. Cancer 2012, 131, 1963–1968. [Google Scholar] [CrossRef]
- Sanders, S.; Herpai, D.M.; Rodriguez, A.; Huang, Y.; Chou, J.; Hsu, F.C.; Seals, D.; Mott, R.; Miller, L.D.; Debinski, W. The Presence and Potential Role of ALDH1A2 in the Glioblastoma Microenvironment. Cells 2021, 10, 2485. [Google Scholar] [CrossRef]
- Campos, B.; Weisang, S.; Osswald, F.; Ali, R.; Sedlmeier, G.; Bageritz, J.; Mallm, J.P.; Hartmann, C.; von Deimling, A.; Popanda, O.; et al. Retinoid resistance and multifaceted impairment of retinoic acid synthesis in glioblastoma. Glia 2015, 63, 1850–1859. [Google Scholar] [CrossRef]
- Barbus, S.; Tews, B.; Karra, D.; Hahn, M.; Radlwimmer, B.; Delhomme, N.; Hartmann, C.; Felsberg, J.; Krex, D.; Schackert, G.; et al. Differential retinoic acid signaling in tumors of long- and short-term glioblastoma survivors. J. Natl. Cancer Inst. 2011, 103, 598–606. [Google Scholar] [CrossRef]
- Piperi, C.; Themistocleous, M.S.; Papavassiliou, G.A.; Farmaki, E.; Levidou, G.; Korkolopoulou, P.; Adamopoulos, C.; Papavassiliou, A.G. High incidence of MGMT and RARbeta promoter methylation in primary glioblastomas: Association with histopathological characteristics, inflammatory mediators and clinical outcome. Mol. Med. 2010, 16, 1–9. [Google Scholar] [CrossRef]
- Bouterfa, H.; Picht, T.; Kess, D.; Herbold, C.; Noll, E.; Black, P.M.; Roosen, K.; Tonn, J.C. Retinoids inhibit human glioma cell proliferation and migration in primary cell cultures but not in established cell lines. Neurosurgery 2000, 46, 419–430. [Google Scholar] [CrossRef]
- Campos, B.; Wan, F.; Farhadi, M.; Ernst, A.; Zeppernick, F.; Tagscherer, K.E.; Ahmadi, R.; Lohr, J.; Dictus, C.; Gdynia, G.; et al. Differentiation therapy exerts antitumor effects on stem-like glioma cells. Clin. Cancer Res. 2010, 16, 2715–2728. [Google Scholar] [CrossRef]
- Rodriguez, V.; Bailey, R.; Larion, M.; Gilbert, M.R. Retinoid receptor turnover mediated by sumoylation, ubiquitination and the valosin-containing protein is disrupted in glioblastoma. Sci. Rep. 2019, 9, 16250. [Google Scholar] [CrossRef]
- Paillaud, E.; Costa, S.; Fages, C.; Plassat, J.L.; Rochette-Egly, C.; Monville, C.; Tardy, M. Retinoic acid increases proliferation rate of GL-15 glioma cells, involving activation of STAT-3 transcription factor. J. Neurosci. Res. 2002, 67, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.H.; Shih, C.M.; Chang, W.C.; Cheng, C.H.; Lin, C.W.; Ho, K.H.; Su, P.C.; Chen, K.C. MicroRNA-302b-inhibited E2F3 transcription factor is related to all trans retinoic acid-induced glioma cell apoptosis. J. Neurochem. 2014, 131, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Haque, A.; Das, A.; Hajiaghamohseni, L.M.; Younger, A.; Banik, N.L.; Ray, S.K. Induction of apoptosis and immune response by all-trans retinoic acid plus interferon-gamma in human malignant glioblastoma T98G and U87MG cells. Cancer Immunol. Immunother. 2007, 56, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Marjanović Vićentić, J.; Schwirtlich, M.; Kovačević-Grujičić, N.; Stevanović, M.; Drakulić, D. All-trans retinoic acid influences viability, migration and adhesion of U251 glioblastoma cells. Arch. Biol. Sci. 2017, 69, 699–706. [Google Scholar] [CrossRef]
- Zeng, Y.; Yang, Z.; Xu, J.G.; Yang, M.S.; Zeng, Z.X.; You, C. Differentially expressed genes from the glioblastoma cell line SHG-44 treated with all-trans retinoic acid in vitro. J. Clin. Neurosci. 2009, 16, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Liu, C. All-trans retinoic acid therapy induces asymmetric division of glioma stem cells from the U87MG cell line. Oncol. Lett. 2019, 18, 3646–3654. [Google Scholar] [CrossRef] [PubMed]
- Ling, G.Q.; Liu, Y.J.; Ke, Y.Q.; Chen, L.; Jiang, X.D.; Jiang, C.L.; Ye, W. All-trans retinoic acid impairs the vasculogenic mimicry formation ability of U87 stem-like cells through promoting differentiation. Mol. Med. Rep. 2015, 12, 165–172. [Google Scholar] [CrossRef]
- Ying, M.; Wang, S.; Sang, Y.; Sun, P.; Lal, B.; Goodwin, C.R.; Guerrero-Cazares, H.; Quinones-Hinojosa, A.; Laterra, J.; Xia, S. Regulation of glioblastoma stem cells by retinoic acid: Role for Notch pathway inhibition. Oncogene 2011, 30, 3454–3467. [Google Scholar] [CrossRef]
- Karsy, M.; Albert, L.; Tobias, M.E.; Murali, R.; Jhanwar-Uniyal, M. All-trans retinoic acid modulates cancer stem cells of glioblastoma multiforme in an MAPK-dependent manner. Anticancer Res. 2010, 30, 4915–4920. [Google Scholar]
- Niu, C.S.; Li, M.W.; Ni, Y.F.; Chen, J.M.; Mei, J.M.; Li, J.; Fu, X.M. Effect of all-trans retinoic acid on the proliferation and differentiation of brain tumor stem cells. J. Exp. Clin. Cancer Res. 2010, 29, 113. [Google Scholar] [CrossRef]
- Choschzick, I.; Hirseland, E.; Cramer, H.; Schultz, S.; Leppert, J.; Tronnier, V.; Zechel, C. Responsiveness of stem-like human glioma cells to all-trans retinoic acid and requirement of retinoic acid receptor isotypes alpha, beta and gamma. Neuroscience 2014, 279, 44–64. [Google Scholar] [CrossRef]
- Reboul, P.; George, P.; Louisot, P.; Broquet, P. Study of retinoic acid effect upon retinoic acid receptors beta (RAR-beta) in C6 cultured glioma cells. Biochem. Mol. Biol. Int. 1995, 36, 1097–1105. [Google Scholar]
- Rodts, G.E., Jr.; Black, K.L. Trans retinoic acid inhibits in vivo tumour growth of C6 glioma in rats: Effect negatively influenced by nerve growth factor. Neurol. Res 1994, 16, 184–186. [Google Scholar] [CrossRef]
- See, S.J.; Levin, V.A.; Yung, W.K.; Hess, K.R.; Groves, M.D. 13-cis-retinoic acid in the treatment of recurrent glioblastoma multiforme. Neuro-Oncology 2004, 6, 253–258. [Google Scholar] [CrossRef]
- Tang, K.; Cao, L.; Fan, S.Q.; Wu, M.H.; Huang, H.; Zhou, Y.H.; Zhou, M.; Tang, Y.L.; Wang, R.; Zeng, F.; et al. Effect of all-trans-retinoic acid on C6 glioma cell proliferation and differentiation. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2008, 33, 892–897. [Google Scholar]
- Milanovic, D.; Maier, P.; Lohr, F.; Wenz, F.; Herskind, C. Inhibition of 13-cis retinoic acid-induced gene expression of homeobox B7 by thalidomide. Int. J. Cancer 2007, 121, 1205–1211. [Google Scholar] [CrossRef]
- Heo, J.C.; Jung, T.H.; Lee, S.; Kim, H.Y.; Choi, G.; Jung, M.; Jung, D.; Lee, H.K.; Lee, J.O.; Park, J.H.; et al. Effect of bexarotene on differentiation of glioblastoma multiforme compared with ATRA. Clin. Exp. Metastasis 2016, 33, 417–429. [Google Scholar] [CrossRef]
- Shi, L.; Li, H.; Zhan, Y. All-trans retinoic acid enhances temozolomide-induced autophagy in human glioma cells U251 via targeting Keap1/Nrf2/ARE signaling pathway. Oncol. Lett. 2017, 14, 2709–2714. [Google Scholar] [CrossRef][Green Version]
- Karmakar, S.; Banik, N.L.; Patel, S.J.; Ray, S.K. Combination of all-trans retinoic acid and taxol regressed glioblastoma T98G xenografts in nude mice. Apoptosis 2007, 12, 2077–2087. [Google Scholar] [CrossRef]
- Karmakar, S.; Banik, N.L.; Ray, S.K. Combination of all-trans retinoic acid and paclitaxel-induced differentiation and apoptosis in human glioblastoma U87MG xenografts in nude mice. Cancer 2008, 112, 596–607. [Google Scholar] [CrossRef]
- Karmakar, S.; Choudhury, S.R.; Banik, N.L.; Ray, S.K. Activation of Multiple Molecular Mechanisms for Increasing Apoptosis in Human Glioblastoma T98G Xenograft. J. Cancer Sci. 2010, 2, 107–113. [Google Scholar] [CrossRef]
- Songthaveesin, C.; Sa-Nongdej, W.; Limboonreung, T.; Chongthammakun, S. Combination of metformin and 9-cis retinoic acid increases apoptosis in C6 glioma stem-like cells. Heliyon 2018, 4, e00638. [Google Scholar] [CrossRef]
- Milanovic, D.; Sticht, C.; Rohrich, M.; Maier, P.; Grosu, A.L.; Herskind, C. Inhibition of 13-cis retinoic acid-induced gene expression of reactive-resistance genes by thalidomide in glioblastoma tumours in vivo. Oncotarget 2015, 6, 28938–28948. [Google Scholar] [CrossRef][Green Version]
- Papi, A.; Tatenhorst, L.; Terwel, D.; Hermes, M.; Kummer, M.P.; Orlandi, M.; Heneka, M.T. PPARgamma and RXRgamma ligands act synergistically as potent antineoplastic agents in vitro and in vivo glioma models. J. Neurochem. 2009, 109, 1779–1790. [Google Scholar] [CrossRef] [PubMed]
- Pitz, M.W.; Lipson, M.; Hosseini, B.; Lambert, P.; Guilbert, K.; Lister, D.; Schroeder, G.; Jones, K.; Mihalicioiu, C.; Eisenstat, D.D. Extended adjuvant temozolomide with cis-retinoic acid for adult glioblastoma. Curr. Oncol. 2012, 19, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Butowski, N.; Prados, M.D.; Lamborn, K.R.; Larson, D.A.; Sneed, P.K.; Wara, W.M.; Malec, M.; Rabbitt, J.; Page, M.; Chang, S.M. A phase II study of concurrent temozolomide and cis-retinoic acid with radiation for adult patients with newly diagnosed supratentorial glioblastoma. Int. J. Radiat. Oncol. Biol. Phys. 2005, 61, 1454–1459. [Google Scholar] [CrossRef] [PubMed]
- Puduvalli, V.K.; Yung, W.K.; Hess, K.R.; Kuhn, J.G.; Groves, M.D.; Levin, V.A.; Zwiebel, J.; Chang, S.M.; Cloughesy, T.F.; Junck, L.; et al. Phase II study of fenretinide (NSC 374551) in adults with recurrent malignant gliomas: A North American Brain Tumor Consortium study. J. Clin. Oncol. 2004, 22, 4282–4289. [Google Scholar] [CrossRef]
- Levin, V.A.; Giglio, P.; Puduvalli, V.K.; Jochec, J.; Groves, M.D.; Yung, W.K.; Hess, K. Combination chemotherapy with 13-cis-retinoic acid and celecoxib in the treatment of glioblastoma multiforme. J. Neurooncol. 2006, 78, 85–90. [Google Scholar] [CrossRef]
- Jaeckle, K.A.; Hess, K.R.; Yung, W.K.; Greenberg, H.; Fine, H.; Schiff, D.; Pollack, I.F.; Kuhn, J.; Fink, K.; Mehta, M.; et al. Phase II evaluation of temozolomide and 13-cis-retinoic acid for the treatment of recurrent and progressive malignant glioma: A North American Brain Tumor Consortium study. J. Clin. Oncol. 2003, 21, 2305–2311. [Google Scholar] [CrossRef]
- Ferreira, R.; Napoli, J.; Enver, T.; Bernardino, L.; Ferreira, L. Advances and challenges in retinoid delivery systems in regenerative and therapeutic medicine. Nat. Commun. 2020, 11, 4265. [Google Scholar] [CrossRef]
- Jeong, Y.I.; Kim, S.H.; Jung, T.Y.; Kim, I.Y.; Kang, S.S.; Jin, Y.H.; Ryu, H.H.; Sun, H.S.; Jin, S.; Kim, K.K.; et al. Polyion complex micelles composed of all-trans retinoic acid and poly (ethylene glycol)-grafted-chitosan. J. Pharm. Sci. 2006, 95, 2348–2360. [Google Scholar] [CrossRef]
- Jones, T.; Zhang, B.; Major, S.; Webb, A. All-trans retinoic acid eluting poly(diol citrate) wafers for treatment of glioblastoma. J. Biomed. Mater. Res. B Appl. Biomater. 2020, 108, 619–628. [Google Scholar] [CrossRef]
- Reifenberger, G.; Liu, L.; Ichimura, K.; Schmidt, E.E.; Collins, V.P. Amplification and overexpression of the MDM2 gene in a subset of human malignant gliomas without p53 mutations. Cancer Res. 1993, 53, 2736–2739. [Google Scholar]
- Izumoto, S.; Arita, N.; Ohnishi, T.; Hiraga, S.; Taki, T.; Tomita, N.; Ohue, M.; Hayakawa, T. Microsatellite instability and mutated type II transforming growth factor-beta receptor gene in gliomas. Cancer Lett. 1997, 112, 251–256. [Google Scholar] [CrossRef]
- Suzuki, H.; Aoki, K.; Chiba, K.; Sato, Y.; Shiozawa, Y.; Shiraishi, Y.; Shimamura, T.; Niida, A.; Motomura, K.; Ohka, F.; et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nat. Genet. 2015, 47, 458–468. [Google Scholar] [CrossRef]
- Sakthikumar, S.; Roy, A.; Haseeb, L.; Pettersson, M.E.; Sundstrom, E.; Marinescu, V.D.; Lindblad-Toh, K.; Forsberg-Nilsson, K. Whole-genome sequencing of glioblastoma reveals enrichment of non-coding constraint mutations in known and novel genes. Genome Biol. 2020, 21, 127. [Google Scholar] [CrossRef]
- Madsen, H.; Hellwinkel, J.E.; Graner, M.W. Clinical Trials in Glioblastoma—Designs and Challenges. In Molecular Considerations and Evolving Surgical Management Issues in the Treatment of Patients with a Brain Tumor; Lichtor, T., Ed.; IntechOpen: Rijeka, Croatia, 2015. [Google Scholar] [CrossRef]
- Park, A.K.; Kim, P.; Ballester, L.Y.; Esquenazi, Y.; Zhao, Z. Subtype-specific signaling pathways and genomic aberrations associated with prognosis of glioblastoma. Neuro-Oncology 2019, 21, 59–70. [Google Scholar] [CrossRef]
- El-Sehemy, A.; Selvadurai, H.; Ortin-Martinez, A.; Pokrajac, N.; Mamatjan, Y.; Tachibana, N.; Rowland, K.; Lee, L.; Park, N.; Aldape, K.; et al. Norrin mediates tumor-promoting and -suppressive effects in glioblastoma via Notch and Wnt. J. Clin. Investig. 2020, 130, 3069–3086. [Google Scholar] [CrossRef]
- Lin, C.J.; Chen, T.L.; Tseng, Y.Y.; Wu, G.J.; Hsieh, M.H.; Lin, Y.W.; Chen, R.M. Honokiol induces autophagic cell death in malignant glioma through reactive oxygen species-mediated regulation of the p53/PI3K/Akt/mTOR signaling pathway. Toxicol. Appl. Pharm. 2016, 304, 59–69. [Google Scholar] [CrossRef]
- Jung, Y.; Park, H.; Zhao, H.Y.; Jeon, R.; Ryu, J.H.; Kim, W.Y. Systemic approaches identify a garlic-derived chemical, Z-ajoene, as a glioblastoma multiforme cancer stem cell-specific targeting agent. Mol. Cells 2014, 37, 547–553. [Google Scholar] [CrossRef]
- Ivkovic, I.; Novakovic, M.; Veljic, M.; Mojsin, M.; Stevanovic, M.; Marin, P.D.; Bukvicki, D. Bis-Bibenzyls from the Liverwort Pellia endiviifolia and Their Biological Activity. Plants 2021, 10, 1063. [Google Scholar] [CrossRef]
- Stojkovic, D.; Drakulic, D.; Dias, M.I.; Zengin, G.; Barros, L.; Ivanov, M.; Gasic, U.; Rajcevic, N.; Stevanovic, M.; Ferreira, I.; et al. Phlomis fruticosa L. exerts in vitro antineurodegenerative and antioxidant activities and induces prooxidant effect in glioblastoma cell line. EXCLI J. 2022, 21, 387–399. [Google Scholar] [CrossRef]
- Stojkovic, D.; Drakulic, D.; Gasic, U.; Zengin, G.; Stevanovic, M.; Rajcevic, N.; Sokovic, M. Ononis spinosa L., an edible and medicinal plant: UHPLC-LTQ-Orbitrap/MS chemical profiling and biological activities of the herbal extract. Food Funct. 2020, 11, 7138–7151. [Google Scholar] [CrossRef]
- Stojkovic, D.; Drakulic, D.; Schwirtlich, M.; Rajcevic, N.; Stevanovic, M.; Sokovic, M.D.; Gasic, U. Extract of Herba Anthrisci cerefolii: Chemical Profiling and Insights into Its Anti-Glioblastoma and Antimicrobial Mechanism of Actions. Pharmaceuticals 2021, 14, 55. [Google Scholar] [CrossRef]
- Mariappan, A.; Goranci-Buzhala, G.; Ricci-Vitiani, L.; Pallini, R.; Gopalakrishnan, J. Trends and challenges in modeling glioma using 3D human brain organoids. Cell Death Differ. 2021, 28, 15–23. [Google Scholar] [CrossRef]
- Goranci-Buzhala, G.; Mariappan, A.; Gabriel, E.; Ramani, A.; Ricci-Vitiani, L.; Buccarelli, M.; D’Alessandris, Q.G.; Pallini, R.; Gopalakrishnan, J. Rapid and Efficient Invasion Assay of Glioblastoma in Human Brain Organoids. Cell Rep. 2020, 31, 107738. [Google Scholar] [CrossRef]
- Chadwick, M.; Yang, C.; Liu, L.; Gamboa, C.M.; Jara, K.; Lee, H.; Sabaawy, H.E. Rapid Processing and Drug Evaluation in Glioblastoma Patient-Derived Organoid Models with 4D Bioprinted Arrays. iScience 2020, 23, 101365. [Google Scholar] [CrossRef]
- Stankovic, T.; Randelovic, T.; Dragoj, M.; Stojkovic Buric, S.; Fernandez, L.; Ochoa, I.; Perez-Garcia, V.M.; Pesic, M. In vitro biomimetic models for glioblastoma-a promising tool for drug response studies. Drug Resist. Updat. 2021, 55, 100753. [Google Scholar] [CrossRef]
- Alomari, S.; Zhang, I.; Hernandez, A.; Kraft, C.Y.; Raj, D.; Kedda, J.; Tyler, B. Drug Repurposing for Glioblastoma and Current Advances in Drug Delivery-A Comprehensive Review of the Literature. Biomolecules 2021, 11, 1870. [Google Scholar] [CrossRef]
- Aghabegi Moghanjoughi, A.; Khoshnevis, D.; Zarrabi, A. A concise review on smart polymers for controlled drug release. Drug Deliv. Transl. Res. 2016, 6, 333–340. [Google Scholar] [CrossRef]
- Nouri, Z.; Fakhri, S.; Nouri, K.; Wallace, C.E.; Farzaei, M.H.; Bishayee, A. Targeting Multiple Signaling Pathways in Cancer: The Rutin Therapeutic Approach. Cancers 2020, 12, 2276. [Google Scholar] [CrossRef] [PubMed]
- Fathi, M.; Sahandi Zangabad, P.; Majidi, S.; Barar, J.; Erfan-Niya, H.; Omidi, Y. Stimuli-responsive chitosan-based nanocarriers for cancer therapy. Bioimpacts 2017, 7, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Du, H.; Zhang, W.; Zhai, G. Internal stimuli-responsive nanocarriers for drug delivery: Design strategies and applications. Mater. Sci. Eng. C Mater. Biol. Appl. 2017, 71, 1267–1280. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Kazandjian, D.; Suzman, D.L.; Blumenthal, G.; Mushti, S.; He, K.; Libeg, M.; Keegan, P.; Pazdur, R. FDA Approval Summary: Nivolumab for the Treatment of Metastatic Non-Small Cell Lung Cancer with Progression on or after Platinum-Based Chemotherapy. Oncologist 2016, 21, 634–642. [Google Scholar] [CrossRef]
- Yu, M.W.; Quail, D.F. Immunotherapy for Glioblastoma: Current Progress and Challenges. Front. Immunol. 2021, 12, 676301. [Google Scholar] [CrossRef]
- Bausart, M.; Preat, V.; Malfanti, A. Immunotherapy for glioblastoma: The promise of combination strategies. J. Exp. Clin. Cancer Res. 2022, 41, 35. [Google Scholar] [CrossRef]
- Chan, H.Y.; Choi, J.; Jackson, C.; Lim, M. Combination immunotherapy strategies for glioblastoma. J. Neurooncol. 2021, 151, 375–391. [Google Scholar] [CrossRef]
- Yuan, B.; Wang, G.; Tang, X.; Tong, A.; Zhou, L. Immunotherapy of glioblastoma: Recent advances and future prospects. Hum. Vaccines Immunother. 2022, 18, 2055417. [Google Scholar] [CrossRef]
- Gonzalez Castro, L.N.; Tirosh, I.; Suva, M.L. Decoding Cancer Biology One Cell at a Time. Cancer Discov. 2021, 11, 960–970. [Google Scholar] [CrossRef]
- Tirosh, I.; Suva, M.L. Dissecting human gliomas by single-cell RNA sequencing. Neuro-Oncology 2018, 20, 37–43. [Google Scholar] [CrossRef]
- Suva, M.L.; Tirosh, I. The Glioma Stem Cell Model in the Era of Single-Cell Genomics. Cancer Cell 2020, 37, 630–636. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drakulic, D.; Schwirtlich, M.; Petrovic, I.; Mojsin, M.; Milivojevic, M.; Kovacevic-Grujicic, N.; Stevanovic, M. Current Opportunities for Targeting Dysregulated Neurodevelopmental Signaling Pathways in Glioblastoma. Cells 2022, 11, 2530. https://doi.org/10.3390/cells11162530
Drakulic D, Schwirtlich M, Petrovic I, Mojsin M, Milivojevic M, Kovacevic-Grujicic N, Stevanovic M. Current Opportunities for Targeting Dysregulated Neurodevelopmental Signaling Pathways in Glioblastoma. Cells. 2022; 11(16):2530. https://doi.org/10.3390/cells11162530
Chicago/Turabian StyleDrakulic, Danijela, Marija Schwirtlich, Isidora Petrovic, Marija Mojsin, Milena Milivojevic, Natasa Kovacevic-Grujicic, and Milena Stevanovic. 2022. "Current Opportunities for Targeting Dysregulated Neurodevelopmental Signaling Pathways in Glioblastoma" Cells 11, no. 16: 2530. https://doi.org/10.3390/cells11162530
APA StyleDrakulic, D., Schwirtlich, M., Petrovic, I., Mojsin, M., Milivojevic, M., Kovacevic-Grujicic, N., & Stevanovic, M. (2022). Current Opportunities for Targeting Dysregulated Neurodevelopmental Signaling Pathways in Glioblastoma. Cells, 11(16), 2530. https://doi.org/10.3390/cells11162530