
The Interaction of Human Papillomavirus Infection and Prostaglandin E2 Signaling in Carcinogenesis: A Focus on Cervical Cancer Therapeutics

 ,
,  ,
,  and
and
Abstract
1. Introduction
2. Cervical Cancer and Human Papillomavirus
2.1. The Human Papillomavirus Life Cycle
2.2. Mechanisms Underlying HPV-Driven Carcinogenesis
3. Role of the Axis Cyclooxygenases/PGE2 and Its Receptors in Normal Physiology and Cancer
3.1. PGE2 Biosynthesis and Metabolism
3.2. PGE2 Receptors
3.3. Role of the COXs/PGE2/PTGERs Axis in Human Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type | COX-2/PGE2/PTGER1-4 | Tumorigenic Role | Factors and Associated Genes | References |
|---|---|---|---|---|
| Colorectal | COX-2/PGE2/PTGER2 | Angiogenesis | VEGF and Ang-2 | [126,127] |
| Colon | COX-2/PGE2/PTGER2 | Tumor microenvironment | CXCL1, IL6, WNT (2, 2B, 5A), MMP12 | [128] |
| Gastric | COX-2/PGE2/PTGER4 | Tumor microenvironment, metastasis | ADAM metalloproteases, EGFR ligands | [129] |
| PTGER2/PTGER4 | Cell growth inhibition | [130] | ||
| Cervical | PTGER2 | Prognostic marker of disease | [123] | |
| COX-2/PGE2/PTGER3 | Metastasis | uPAR | [125] | |
| COX-2/PGE2/PTGER4 | Carcinogenesis | [32] | ||
| Lung | COX-2 | Tumor microenvironment and inflammation | Cancer promoting cytokines | [131] |
| COX-2/PGE2/PTGER4 | Cell migration | [132] | ||
| COX-2/PGE2/PTGER1 | Cell proliferation and migration | ERK phosphorylation, β1 integrin activation | [133,134] | |
| COX-2/PGE2/PTGER3 | Cell migration | MMP 2-9 VEGF, TGFβ, p-Smad 2-3 | [135] | |
| Breast | COX-2 | Metastasis | MMP1 | [136] |
| Chemoresistance | MFGE8, KLK5, and KLK7 | [137] | ||
| PTGER3 | Prognostic factor for progression-free survival | [138] | ||
| COX-2/PGE2/PTGER2/ PTGER4 | Angiogenesis, cell proliferation and stemness | MMP 2-9 | [139,140] | |
| Nuclear PTGER1 | Good prognosis marker | [141] | ||
| Bladder | COX-2 | Stemness | Oct3/4, CD44v6 | [142] |
| Vulva | COX-2/PGE2/PTGER4 | Negative prognostic factor | [143] | |
| Bone | COX-2 | Cell migration | [144] | |
| Cell growth and progression, poor survival | [145] | |||
| Liver | COX-2 | Activation of AKT and mTOR oncogenic pathways | AKT, TET1, MTOR, LTBP1, ADCY5 and PRKCZ | [146] |
| Prostate | COX-2/PGE2/PTGER4 | Cell proliferation and migration | RANKL, RUNX2, MMP 2-9 | [147] |
| Oral squamous carcinoma | COX-2/PGE2 | Cell growth inhibition | [148] |
4. Crosstalk between HPV Infection and PGE2/PTGERs Signaling on Cancer Progression
4.1. Chronic Inflammation
4.2. Immune Response Evasion
5. Therapeutic Targeting of the COX/PGE2 Axis in Cancer
5.1. The Non-Selective NSAIDS as Antineoplastic Agents in Cervical Cancer
5.2. COX-2 Selective NSAIDs as Antineoplastic Agents in Cervical Cancer
5.3. Corticosteroids in Cervical Cancer
6. Targeting the Human Papillomavirus in Cancer
6.1. Preventing HPV Transmission Using Prophylactic Vaccines
6.2. Therapeutic Vaccines Targeting HPV Oncoproteins
6.2.1. HPV-Therapeutic Vaccines Designed with Bacterial Vectors
6.2.2. HPV-Therapeutic Vaccines Designed with Viral Vectors
6.2.3. Nucleic Acid-Based Vaccines against HPV
6.2.4. Peptide/Protein-Based Vaccines against HPV
7. Immunomodulators
8. Chemical Antivirals
8.1. Acyclic Nucleoside Phosphonates
8.2. Other Antivirals Targeting HPV Proteins Interaction
9. Therapeutic Strategy against Cervical Cancer Using Nanoparticles and Gene Therapy
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AA | Arachidonic acid |
| AC | Adenylate cyclase |
| AKT | Protein kinase B |
| APC | Antigen-presenting cells |
| ATR | Ataxia Telangiectasia and Rad3-related |
| cAMP | Cyclic adenosine monophosphate |
| CIN | Cervical intraepithelial neoplasia |
| COX | Cyclooxygenase |
| CRE | cAMP-responsive element |
| CREB | cAMP-response element binding protein |
| CTL | Cytotoxic T-lymphocytes |
| DAG | Diacylglycerol |
| DC | Dendritic cells |
| EGFR | Epidermal growth factor receptor |
| ERK | Extracellular signal-regulated kinase |
| HPV | Human papillomavirus |
| HSIL | High-grade squamous intraepithelial lesion |
| IC | Inflammatory cytokines |
| ICC | Invasive carcinoma of the cervix |
| IFNγ | Interferon-gamma |
| IL | Interleukin |
| IP3 | Inositol 1,4,5-trisphosphate |
| LCR | Long control region |
| LLO | Listeriolysin O |
| Lm | Listeria monocytogenes |
| LSIL | Low-grade squamous intraepithelial lesion |
| MDSCs | Myeloid-derived suppressor cells |
| MHC | Major histocompatibility complex |
| MMP | Matrix metallopeptidase |
| mTOR | Mammalian target of rapamycin |
| MVA | Modified Vaccinia Ankara |
| NF-κB | Nuclear factor-kappa B |
| NSAIDS | Non-steroidal anti-inflammatory drugs |
| PGE2 | Prostaglandin E2 |
| PGES | Prostaglandin E2 synthase |
| PI3K | Phosphatidylinositol 3-kinase |
| PIP2 | Phosphatidylinositol 4,5-bisphosphate |
| PKA | Protein kinase A |
| PKC | Protein kinase C |
| PLA2 | Phospholipase A2 |
| PLC | Phospholipase C |
| PTGER | Prostaglandin E2 receptor |
| pRB | Retinoblastoma protein |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| SLP | Synthetic long peptides |
| TERT | Telomerase reverse transcriptase |
| TGFβ | Transforming growth factor-beta |
| TLR | Toll-like receptor |
| TNFα | Tumor necrosis factor-alpha |
| TX | Thromboxane |
| URR | Upstream regulatory region |
| VEGF | Vascular endothelial growth factor |
| VIN | Vulval intraepithelial neoplasia |
| VLPs | Virus-like particles |
References
- Duesberg, P.; Li, R. Multistep carcinogenesis: A chain reaction of aneuploidizations. Cell Cycle 2003, 2, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Hsu, W.L.; Yang, H.I.; Lee, M.H.; Chen, H.C.; Chien, Y.C.; You, S.L. Epidemiology of virus infection and human cancer. Recent Results Cancer Res. 2014, 193, 11–32. [Google Scholar] [CrossRef] [PubMed]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Finetti, F.; Travelli, C.; Ercoli, J.; Colombo, G.; Buoso, E.; Trabalzini, L. Prostaglandin E2 and Cancer: Insight into Tumor Progression and Immunity. Biology 2020, 9, 434. [Google Scholar] [CrossRef] [PubMed]
- Arbyn, M.; Weiderpass, E.; Bruni, L.; de Sanjose, S.; Saraiya, M.; Ferlay, J.; Bray, F. Estimates of incidence and mortality of cervical cancer in 2018: A worldwide analysis. Lancet Glob. Health 2020, 8, e191–e203. [Google Scholar] [CrossRef]
- Schiffman, M.; Castle, P.E.; Jeronimo, J.; Rodriguez, A.C.; Wacholder, S. Human papillomavirus and cervical cancer. Lancet 2007, 370, 890–907. [Google Scholar] [CrossRef]
- Graham, S.V. Human papillomavirus: Gene expression, regulation and prospects for novel diagnostic methods and antiviral therapies. Future Microbiol. 2010, 5, 1493–1506. [Google Scholar] [CrossRef]
- Graham, S.V. Keratinocyte Differentiation-Dependent Human Papillomavirus Gene Regulation. Viruses 2017, 9, 245. [Google Scholar] [CrossRef]
- Shafti-Keramat, S.; Handisurya, A.; Kriehuber, E.; Meneguzzi, G.; Slupetzky, K.; Kirnbauer, R. Different heparan sulfate proteoglycans serve as cellular receptors for human papillomaviruses. J. Virol. 2003, 77, 13125–13135. [Google Scholar] [CrossRef]
- Culp, T.D.; Budgeon, L.R.; Marinkovich, M.P.; Meneguzzi, G.; Christensen, N.D. Keratinocyte-secreted laminin 5 can function as a transient receptor for human papillomaviruses by binding virions and transferring them to adjacent cells. J. Virol. 2006, 80, 8940–8950. [Google Scholar] [CrossRef]
- Yoon, C.S.; Kim, K.D.; Park, S.N.; Cheong, S.W. Alpha(6) Integrin is the main receptor of human papillomavirus type 16 VLP. Biochem. Biophys. Res. Commun. 2001, 283, 668–673. [Google Scholar] [CrossRef]
- Schelhaas, M.; Shah, B.; Holzer, M.; Blattmann, P.; Kuhling, L.; Day, P.M.; Schiller, J.T.; Helenius, A. Entry of human papillomavirus type 16 by actin-dependent, clathrin- and lipid raft-independent endocytosis. PLoS Pathog. 2012, 8, e1002657. [Google Scholar] [CrossRef]
- Popa, A.; Zhang, W.; Harrison, M.S.; Goodner, K.; Kazakov, T.; Goodwin, E.C.; Lipovsky, A.; Burd, C.G.; DiMaio, D. Direct binding of retromer to human papillomavirus type 16 minor capsid protein L2 mediates endosome exit during viral infection. PLoS Pathog. 2015, 11, e1004699. [Google Scholar] [CrossRef]
- Day, P.M.; Baker, C.C.; Lowy, D.R.; Schiller, J.T. Establishment of papillomavirus infection is enhanced by promyelocytic leukemia protein (PML) expression. Proc. Natl. Acad. Sci. USA 2004, 101, 14252–14257. [Google Scholar] [CrossRef]
- Guion, L.G.; Sapp, M. The Role of Promyelocytic Leukemia Nuclear Bodies During HPV Infection. Front. Cell. Infect. Microbiol. 2020, 10, 35. [Google Scholar] [CrossRef]
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The biology and life-cycle of human papillomaviruses. Vaccine 2012, 30 (Suppl. S5), F55–F70. [Google Scholar] [CrossRef]
- Sanders, C.M.; Stenlund, A. Recruitment and loading of the E1 initiator protein: An ATP-dependent process catalysed by a transcription factor. EMBO J. 1998, 17, 7044–7055. [Google Scholar] [CrossRef]
- Valle, G.F.; Banks, L. The human papillomavirus (HPV)-6 and HPV-16 E5 proteins co-operate with HPV-16 E7 in the transformation of primary rodent cells. J. Gen. Virol. 1995, 76 Pt 5, 1239–1245. [Google Scholar] [CrossRef]
- Venuti, A.; Paolini, F.; Nasir, L.; Corteggio, A.; Roperto, S.; Campo, M.S.; Borzacchiello, G. Papillomavirus E5: The smallest oncoprotein with many functions. Mol. Cancer 2011, 10, 140. [Google Scholar] [CrossRef]
- Leptak, C.; Ramon y Cajal, S.; Kulke, R.; Horwitz, B.H.; Riese, D.J., 2nd; Dotto, G.P.; DiMaio, D. Tumorigenic transformation of murine keratinocytes by the E5 genes of bovine papillomavirus type 1 and human papillomavirus type 16. J. Virol. 1991, 65, 7078–7083. [Google Scholar] [CrossRef]
- Leechanachai, P.; Banks, L.; Moreau, F.; Matlashewski, G. The E5 gene from human papillomavirus type 16 is an oncogene which enhances growth factor-mediated signal transduction to the nucleus. Oncogene 1992, 7, 19–25. [Google Scholar] [PubMed]
- Pim, D.; Collins, M.; Banks, L. Human papillomavirus type 16 E5 gene stimulates the transforming activity of the epidermal growth factor receptor. Oncogene 1992, 7, 27–32. [Google Scholar] [PubMed]
- Straight, S.W.; Herman, B.; McCance, D.J. The E5 oncoprotein of human papillomavirus type 16 inhibits the acidification of endosomes in human keratinocytes. J. Virol. 1995, 69, 3185–3192. [Google Scholar] [CrossRef] [PubMed]
- Bouvard, V.; Matlashewski, G.; Gu, Z.M.; Storey, A.; Banks, L. The human papillomavirus type 16 E5 gene cooperates with the E7 gene to stimulate proliferation of primary cells and increases viral gene expression. Virology 1994, 203, 73–80. [Google Scholar] [CrossRef]
- Venuti, A.; Salani, D.; Poggiali, F.; Manni, V.; Bagnato, A. The E5 oncoprotein of human papillomavirus type 16 enhances endothelin-1-induced keratinocyte growth. Virology 1998, 248, 1–5. [Google Scholar] [CrossRef][Green Version]
- Thomsen, P.; van Deurs, B.; Norrild, B.; Kayser, L. The HPV16 E5 oncogene inhibits endocytic trafficking. Oncogene 2000, 19, 6023–6032. [Google Scholar] [CrossRef]
- Kabsch, K.; Alonso, A. The human papillomavirus type 16 E5 protein impairs TRAIL- and FasL-mediated apoptosis in HaCaT cells by different mechanisms. J. Virol. 2002, 76, 12162–12172. [Google Scholar] [CrossRef]
- Zhang, B.; Spandau, D.F.; Roman, A. E5 protein of human papillomavirus type 16 protects human foreskin keratinocytes from UV B-irradiation-induced apoptosis. J. Virol. 2002, 76, 220–231. [Google Scholar] [CrossRef]
- Ashrafi, G.H.; Haghshenas, M.R.; Marchetti, B.; O’Brien, P.M.; Campo, M.S. E5 protein of human papillomavirus type 16 selectively downregulates surface HLA class I. Int. J. Cancer 2005, 113, 276–283. [Google Scholar] [CrossRef]
- Genther Williams, S.M.; Disbrow, G.L.; Schlegel, R.; Lee, D.; Threadgill, D.W.; Lambert, P.F. Requirement of epidermal growth factor receptor for hyperplasia induced by E5, a high-risk human papillomavirus oncogene. Cancer Res. 2005, 65, 6534–6542. [Google Scholar] [CrossRef]
- Kim, S.H.; Oh, J.M.; No, J.H.; Bang, Y.J.; Juhnn, Y.S.; Song, Y.S. Involvement of NF-kappaB and AP-1 in COX-2 upregulation by human papillomavirus 16 E5 oncoprotein. Carcinogenesis 2009, 30, 753–757. [Google Scholar] [CrossRef]
- Oh, J.M.; Kim, S.H.; Lee, Y.I.; Seo, M.; Kim, S.Y.; Song, Y.S.; Kim, W.H.; Juhnn, Y.S. Human papillomavirus E5 protein induces expression of the EP4 subtype of prostaglandin E2 receptor in cyclic AMP response element-dependent pathways in cervical cancer cells. Carcinogenesis 2009, 30, 141–149. [Google Scholar] [CrossRef]
- Sudarshan, S.R.; Schlegel, R.; Liu, X. The HPV-16 E5 protein represses expression of stress pathway genes XBP-1 and COX-2 in genital keratinocytes. Biochem. Biophys. Res. Commun. 2010, 399, 617–622. [Google Scholar] [CrossRef]
- Pedroza-Saavedra, A.; Lam, E.W.; Esquivel-Guadarrama, F.; Gutierrez-Xicotencatl, L. The human papillomavirus type 16 E5 oncoprotein synergizes with EGF-receptor signaling to enhance cell cycle progression and the down-regulation of p27(Kip1). Virology 2010, 400, 44–52. [Google Scholar] [CrossRef]
- Oh, J.M.; Kim, S.H.; Cho, E.A.; Song, Y.S.; Kim, W.H.; Juhnn, Y.S. Human papillomavirus type 16 E5 protein inhibits hydrogen-peroxide-induced apoptosis by stimulating ubiquitin-proteasome-mediated degradation of Bax in human cervical cancer cells. Carcinogenesis 2010, 31, 402–410. [Google Scholar] [CrossRef]
- Hu, L.; Potapova, T.A.; Li, S.; Rankin, S.; Gorbsky, G.J.; Angeletti, P.C.; Ceresa, B.P. Expression of HPV16 E5 produces enlarged nuclei and polyploidy through endoreplication. Virology 2010, 405, 342–351. [Google Scholar] [CrossRef]
- Greco, D.; Kivi, N.; Qian, K.; Leivonen, S.K.; Auvinen, P.; Auvinen, E. Human papillomavirus 16 E5 modulates the expression of host microRNAs. PLoS ONE 2011, 6, e21646. [Google Scholar] [CrossRef]
- Ranieri, D.; Belleudi, F.; Magenta, A.; Torrisi, M.R. HPV16 E5 expression induces switching from FGFR2b to FGFR2c and epithelial-mesenchymal transition. Int. J. Cancer 2015, 137, 61–72. [Google Scholar] [CrossRef]
- Wasson, C.W.; Morgan, E.L.; Muller, M.; Ross, R.L.; Hartley, M.; Roberts, S.; Macdonald, A. Human papillomavirus type 18 E5 oncogene supports cell cycle progression and impairs epithelial differentiation by modulating growth factor receptor signalling during the virus life cycle. Oncotarget 2017, 8, 103581–103600. [Google Scholar] [CrossRef]
- Scott, M.L.; Coleman, D.T.; Kelly, K.C.; Carroll, J.L.; Woodby, B.; Songock, W.K.; Cardelli, J.A.; Bodily, J.M. Human papillomavirus type 16 E5-mediated upregulation of Met in human keratinocytes. Virology 2018, 519, 1–11. [Google Scholar] [CrossRef]
- Hochmann, J.; Parietti, F.; Martinez, J.; Lopez, A.C.; Carreno, M.; Quijano, C.; Boccardo, E.; Sichero, L.; Moller, M.N.; Mirazo, S.; et al. Human papillomavirus type 18 E5 oncoprotein cooperates with E6 and E7 in promoting cell viability and invasion and in modulating the cellular redox state. Mem. Inst. Oswaldo Cruz 2020, 115, e190405. [Google Scholar] [CrossRef] [PubMed]
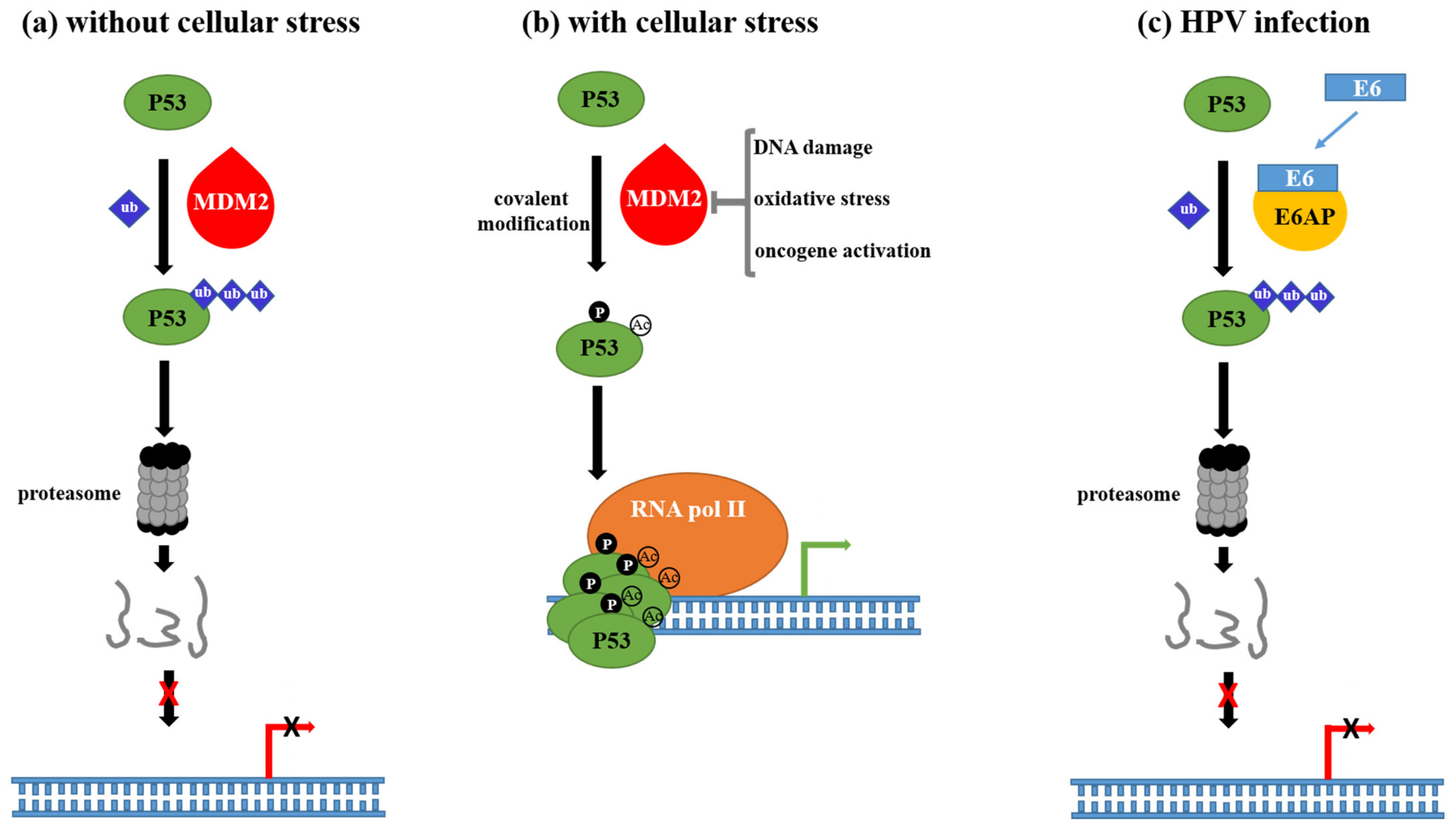
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. p53 mutations in human cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef]
- Scheffner, M.; Takahashi, T.; Huibregtse, J.M.; Minna, J.D.; Howley, P.M. Interaction of the human papillomavirus type 16 E6 oncoprotein with wild-type and mutant human p53 proteins. J. Virol. 1992, 66, 5100–5105. [Google Scholar] [CrossRef]
- Zimmermann, H.; Degenkolbe, R.; Bernard, H.U.; O’Connor, M.J. The human papillomavirus type 16 E6 oncoprotein can down-regulate p53 activity by targeting the transcriptional coactivator CBP/p300. J. Virol. 1999, 73, 6209–6219. [Google Scholar] [CrossRef]
- Thomas, M.C.; Chiang, C.M. E6 oncoprotein represses p53-dependent gene activation via inhibition of protein acetylation independently of inducing p53 degradation. Mol. Cell 2005, 17, 251–264. [Google Scholar] [CrossRef]
- Wallace, N.A.; Robinson, K.; Howie, H.L.; Galloway, D.A. HPV 5 and 8 E6 abrogate ATR activity resulting in increased persistence of UVB induced DNA damage. PLoS Pathog. 2012, 8, e1002807. [Google Scholar] [CrossRef]
- Hasche, D.; Vinzon, S.E.; Rosl, F. Cutaneous Papillomaviruses and Non-melanoma Skin Cancer: Causal Agents or Innocent Bystanders? Front. Microbiol. 2018, 9, 874. [Google Scholar] [CrossRef]
- Kiyono, T.; Foster, S.A.; Koop, J.I.; McDougall, J.K.; Galloway, D.A.; Klingelhutz, A.J. Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature 1998, 396, 84–88. [Google Scholar] [CrossRef]
- Van Doorslaer, K.; Burk, R.D. Association between hTERT activation by HPV E6 proteins and oncogenic risk. Virology 2012, 433, 216–219. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.T.; Kyo, S.; Laimins, L.A. Telomerase activation by human papillomavirus type 16 E6 protein: Induction of human telomerase reverse transcriptase expression through Myc and GC-rich Sp1 binding sites. J. Virol. 2001, 75, 5559–5566. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Watney, E.; McDougall, J.K. Telomerase activity and expression of telomerase RNA component and telomerase catalytic subunit gene in cervical cancer. Am. J. Pathol. 1998, 153, 857–864. [Google Scholar] [CrossRef]
- Weinberg, R.A. The retinoblastoma protein and cell cycle control. Cell 1995, 81, 323–330. [Google Scholar] [CrossRef]
- Boyer, S.N.; Wazer, D.E.; Band, V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 1996, 56, 4620–4624. [Google Scholar]
- Shan, B.; Lee, W.H. Deregulated expression of E2F-1 induces S-phase entry and leads to apoptosis. Mol. Cell. Biol. 1994, 14, 8166–8173. [Google Scholar] [CrossRef]
- Zerfass-Thome, K.; Zwerschke, W.; Mannhardt, B.; Tindle, R.; Botz, J.W.; Jansen-Durr, P. Inactivation of the cdk inhibitor p27KIP1 by the human papillomavirus type 16 E7 oncoprotein. Oncogene 1996, 13, 2323–2330. [Google Scholar]
- McIntyre, M.C.; Ruesch, M.N.; Laimins, L.A. Human papillomavirus E7 oncoproteins bind a single form of cyclin E in a complex with cdk2 and p107. Virology 1996, 215, 73–82. [Google Scholar] [CrossRef]
- Funk, J.O.; Waga, S.; Harry, J.B.; Espling, E.; Stillman, B.; Galloway, D.A. Inhibition of CDK activity and PCNA-dependent DNA replication by p21 is blocked by interaction with the HPV-16 E7 oncoprotein. Genes Dev. 1997, 11, 2090–2100. [Google Scholar] [CrossRef]
- Massimi, P.; Pim, D.; Banks, L. Human papillomavirus type 16 E7 binds to the conserved carboxy-terminal region of the TATA box binding protein and this contributes to E7 transforming activity. J. Gen. Virol. 1997, 78 Pt 10, 2607–2613. [Google Scholar] [CrossRef]
- Berezutskaya, E.; Bagchi, S. The human papillomavirus E7 oncoprotein functionally interacts with the S4 subunit of the 26 S proteasome. J. Biol. Chem. 1997, 272, 30135–30140. [Google Scholar] [CrossRef]
- Lee, J.O.; Russo, A.A.; Pavletich, N.P. Structure of the retinoblastoma tumour-suppressor pocket domain bound to a peptide from HPV E7. Nature 1998, 391, 859–865. [Google Scholar] [CrossRef]
- Luscher-Firzlaff, J.M.; Westendorf, J.M.; Zwicker, J.; Burkhardt, H.; Henriksson, M.; Muller, R.; Pirollet, F.; Luscher, B. Interaction of the fork head domain transcription factor MPP2 with the human papilloma virus 16 E7 protein: Enhancement of transformation and transactivation. Oncogene 1999, 18, 5620–5630. [Google Scholar] [CrossRef]
- Brehm, A.; Ohbo, K.; Zwerschke, W.; Botquin, V.; Jansen-Durr, P.; Scholer, H.R. Synergism with germ line transcription factor Oct-4: Viral oncoproteins share the ability to mimic a stem cell-specific activity. Mol. Cell. Biol. 1999, 19, 2635–2643. [Google Scholar] [CrossRef]
- Park, J.S.; Kim, E.J.; Kwon, H.J.; Hwang, E.S.; Namkoong, S.E.; Um, S.J. Inactivation of interferon regulatory factor-1 tumor suppressor protein by HPV E7 oncoprotein. Implication for the E7-mediated immune evasion mechanism in cervical carcinogenesis. J. Biol. Chem. 2000, 275, 6764–6769. [Google Scholar] [CrossRef]
- Bernat, A.; Avvakumov, N.; Mymryk, J.S.; Banks, L. Interaction between the HPV E7 oncoprotein and the transcriptional coactivator p300. Oncogene 2003, 22, 7871–7881. [Google Scholar] [CrossRef]
- Avvakumov, N.; Torchia, J.; Mymryk, J.S. Interaction of the HPV E7 proteins with the pCAF acetyltransferase. Oncogene 2003, 22, 3833–3841. [Google Scholar] [CrossRef]
- He, W.; Staples, D.; Smith, C.; Fisher, C. Direct activation of cyclin-dependent kinase 2 by human papillomavirus E7. J. Virol. 2003, 77, 10566–10574. [Google Scholar] [CrossRef]
- McLaughlin-Drubin, M.E.; Huh, K.W.; Munger, K. Human papillomavirus type 16 E7 oncoprotein associates with E2F6. J. Virol. 2008, 82, 8695–8705. [Google Scholar] [CrossRef]
- Todorovic, B.; Nichols, A.C.; Chitilian, J.M.; Myers, M.P.; Shepherd, T.G.; Parsons, S.J.; Barrett, J.W.; Banks, L.; Mymryk, J.S. The human papillomavirus E7 proteins associate with p190RhoGAP and alter its function. J. Virol. 2014, 88, 3653–3663. [Google Scholar] [CrossRef]
- Doorbar, J.; Ely, S.; Sterling, J.; McLean, C.; Crawford, L. Specific interaction between HPV-16 E1-E4 and cytokeratins results in collapse of the epithelial cell intermediate filament network. Nature 1991, 352, 824–827. [Google Scholar] [CrossRef]
- Nayar, R.; Wilbur, D.C. The Bethesda System for Reporting Cervical Cytology: A Historical Perspective. Acta Cytol. 2017, 61, 359–372. [Google Scholar] [CrossRef]
- Giuliano, A.R.; Sedjo, R.L.; Roe, D.J.; Harri, R.; Baldwi, S.; Papenfuss, M.R.; Abrahamsen, M.; Inserra, P. Clearance of oncogenic human papillomavirus (HPV) infection: Effect of smoking (United States). Cancer Causes Control 2002, 13, 839–846. [Google Scholar] [CrossRef]
- Oh, H.Y.; Seo, S.S.; Kim, M.K.; Lee, D.O.; Chung, Y.K.; Lim, M.C.; Kim, J.Y.; Lee, C.W.; Park, S.Y. Synergistic effect of viral load and alcohol consumption on the risk of persistent high-risk human papillomavirus infection. PLoS ONE 2014, 9, e104374. [Google Scholar] [CrossRef]
- Boccardo, E.; Lepique, A.P.; Villa, L.L. The role of inflammation in HPV carcinogenesis. Carcinogenesis 2010, 31, 1905–1912. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Gerritsen, M.E. Physiological and pathophysiological roles of eicosanoids in the microcirculation. Cardiovasc. Res. 1996, 32, 720–732. [Google Scholar] [CrossRef]
- Narumiya, S. Physiology and pathophysiology of prostanoid receptors. Proc. Jpn. Academy. Ser. B Phys. Biol. Sci. 2007, 83, 296–319. [Google Scholar] [CrossRef]
- Diao, G.; Huang, J.; Zheng, X.; Sun, X.; Tian, M.; Han, J.; Guo, J. Prostaglandin E2 serves a dual role in regulating the migration of dendritic cells. Int. J. Mol. Med. 2021, 47, 207–218. [Google Scholar] [CrossRef]
- Li, Y.; Wei, Y.; Zheng, F.; Guan, Y.; Zhang, X. Prostaglandin E2 in the Regulation of Water Transport in Renal Collecting Ducts. Int. J. Mol. Sci. 2017, 18, 2539. [Google Scholar] [CrossRef]
- Bryson, T.D.; Harding, P. Prostaglandin E2 EP receptors in cardiovascular disease: An update. Biochem. Pharmacol. 2022, 195, 114858. [Google Scholar] [CrossRef] [PubMed]
- Heeney, A.; Rogers, A.C.; Mohan, H.; Mc Dermott, F.; Baird, A.W.; Winter, D.C. Prostaglandin E2 receptors and their role in gastrointestinal motility—Potential therapeutic targets. Prostaglandins Other Lipid Mediat. 2021, 152, 106499. [Google Scholar] [CrossRef] [PubMed]
- Dey, I.; Lejeune, M.; Chadee, K. Prostaglandin E2 receptor distribution and function in the gastrointestinal tract. Br. J. Pharmacol. 2006, 149, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Tilley, S.L.; Hartney, J.M.; Erikson, C.J.; Jania, C.; Nguyen, M.; Stock, J.; McNeisch, J.; Valancius, C.; Panettieri, R.A., Jr.; Penn, R.B.; et al. Receptors and pathways mediating the effects of prostaglandin E2 on airway tone. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L599–L606. [Google Scholar] [CrossRef]
- Robertson, R.P. Molecular regulation of prostaglandin synthesis Implications for endocrine systems. Trends Endocrinol. Metab. TEM 1995, 6, 293–297. [Google Scholar] [CrossRef]
- Niringiyumukiza, J.D.; Cai, H.; Xiang, W. Prostaglandin E2 involvement in mammalian female fertility: Ovulation, fertilization, embryo development and early implantation. Reprod. Biol. Endocrinol. RBE 2018, 16, 43. [Google Scholar] [CrossRef]
- Li, M.; Thompson, D.D.; Paralkar, V.M. Prostaglandin E(2) receptors in bone formation. Int. Orthop. 2007, 31, 767–772. [Google Scholar] [CrossRef]
- Durand, E.M.; Zon, L.I. Newly emerging roles for prostaglandin E2 regulation of hematopoiesis and hematopoietic stem cell engraftment. Curr. Opin. Hematol. 2010, 17, 308–312. [Google Scholar] [CrossRef]
- Kalinski, P. Regulation of immune responses by prostaglandin E2. J. Immunol. 2012, 188, 21–28. [Google Scholar] [CrossRef]
- Kawahara, K.; Hohjoh, H.; Inazumi, T.; Tsuchiya, S.; Sugimoto, Y. Prostaglandin E2-induced inflammation: Relevance of prostaglandin E receptors. Biochim. Biophys. Acta 2015, 1851, 414–421. [Google Scholar] [CrossRef]
- Kawabata, A. Prostaglandin E2 and pain—An update. Biol. Pharm. Bull. 2011, 34, 1170–1173. [Google Scholar] [CrossRef]
- Coceani, F.; Akarsu, E.S. Prostaglandin E2 in the pathogenesis of fever. An update. Ann. N. Y. Acad. Sci. 1998, 856, 76–82. [Google Scholar] [CrossRef]
- Smith, W.L.; Dewitt, D.L. Prostaglandin endoperoxide H synthases-1 and -2. Adv. Immunol. 1996, 62, 167–215. [Google Scholar] [CrossRef]
- Sreeramkumar, V.; Fresno, M.; Cuesta, N. Prostaglandin E2 and T cells: Friends or foes? Immunol. Cell Biol. 2012, 90, 579–586. [Google Scholar] [CrossRef]
- Samuelsson, B.; Goldyne, M.; Granstrom, E.; Hamberg, M.; Hammarstrom, S.; Malmsten, C. Prostaglandins and thromboxanes. Annu. Rev. Biochem. 1978, 47, 997–1029. [Google Scholar] [CrossRef]
- Gijon, M.A.; Leslie, C.C. Regulation of arachidonic acid release and cytosolic phospholipase A2 activation. J. Leukoc. Biol. 1999, 65, 330–336. [Google Scholar] [CrossRef]
- Hawkey, C.J. COX-1 and COX-2 inhibitors. Best Pract. Res. Clin. Gastroenterol. 2001, 15, 801–820. [Google Scholar] [CrossRef]
- Kirkby, N.S.; Chan, M.V.; Zaiss, A.K.; Garcia-Vaz, E.; Jiao, J.; Berglund, L.M.; Verdu, E.F.; Ahmetaj-Shala, B.; Wallace, J.L.; Herschman, H.R.; et al. Systematic study of constitutive cyclooxygenase-2 expression: Role of NF-kappaB and NFAT transcriptional pathways. Proc. Natl. Acad. Sci. USA 2016, 113, 434–439. [Google Scholar] [CrossRef]
- Smith, W.L.; Marnett, L.J.; DeWitt, D.L. Prostaglandin and thromboxane biosynthesis. Pharmacol. Ther. 1991, 49, 153–179. [Google Scholar] [CrossRef]
- Ritter, C.A.; Jedlitschky, G.; Meyer zu Schwabedissen, H.; Grube, M.; Kock, K.; Kroemer, H.K. Cellular export of drugs and signaling molecules by the ATP-binding cassette transporters MRP4 (ABCC4) and MRP5 (ABCC5). Drug Metab. Rev. 2005, 37, 253–278. [Google Scholar] [CrossRef]
- Kochel, T.J.; Reader, J.C.; Ma, X.; Kundu, N.; Fulton, A.M. Multiple drug resistance-associated protein (MRP4) exports prostaglandin E2 (PGE2) and contributes to metastasis in basal/triple negative breast cancer. Oncotarget 2017, 8, 6540–6554. [Google Scholar] [CrossRef]
- Na, H.K.; Park, J.M.; Lee, H.G.; Lee, H.N.; Myung, S.J.; Surh, Y.J. 15-Hydroxyprostaglandin dehydrogenase as a novel molecular target for cancer chemoprevention and therapy. Biochem. Pharmacol. 2011, 82, 1352–1360. [Google Scholar] [CrossRef]
- Narumiya, S.; Sugimoto, Y.; Ushikubi, F. Prostanoid receptors: Structures, properties, and functions. Physiol. Rev. 1999, 79, 1193–1226. [Google Scholar] [CrossRef]
- Ushikubi, F.; Hirata, M.; Narumiya, S. Molecular biology of prostanoid receptors; an overview. J. Lipid Mediat. Cell Signal. 1995, 12, 343–359. [Google Scholar] [CrossRef]
- Duncan, A.M.; Anderson, L.L.; Funk, C.D.; Abramovitz, M.; Adam, M. Chromosomal localization of the human prostanoid receptor gene family. Genomics 1995, 25, 740–742. [Google Scholar] [CrossRef]
- Smock, S.L.; Pan, L.C.; Castleberry, T.A.; Lu, B.; Mather, R.J.; Owen, T.A. Cloning, structural characterization, and chromosomal localization of the gene encoding the human prostaglandin E(2) receptor EP2 subtype. Gene 1999, 237, 393–402. [Google Scholar] [CrossRef]
- Foord, S.M.; Marks, B.; Stolz, M.; Bufflier, E.; Fraser, N.J.; Lee, M.G. The structure of the prostaglandin EP4 receptor gene and related pseudogenes. Genomics 1996, 35, 182–188. [Google Scholar] [CrossRef]
- Sugimoto, Y.; Narumiya, S. Prostaglandin E receptors. J. Biol. Chem. 2007, 282, 11613–11617. [Google Scholar] [CrossRef]
- Abramovitz, M.; Adam, M.; Boie, Y.; Carriere, M.; Denis, D.; Godbout, C.; Lamontagne, S.; Rochette, C.; Sawyer, N.; Tremblay, N.M.; et al. The utilization of recombinant prostanoid receptors to determine the affinities and selectivities of prostaglandins and related analogs. Biochim. Biophys. Acta 2000, 1483, 285–293. [Google Scholar] [CrossRef]
- Katoh, H.; Watabe, A.; Sugimoto, Y.; Ichikawa, A.; Negishi, M. Characterization of the signal transduction of prostaglandin E receptor EP1 subtype in cDNA-transfected Chinese hamster ovary cells. Biochim. Biophys. Acta 1995, 1244, 41–48. [Google Scholar] [CrossRef]
- O’Callaghan, G.; Houston, A. Prostaglandin E2 and the EP receptors in malignancy: Possible therapeutic targets? Br. J. Pharmacol. 2015, 172, 5239–5250. [Google Scholar] [CrossRef] [PubMed]
- Fujino, H.; West, K.A.; Regan, J.W. Phosphorylation of glycogen synthase kinase-3 and stimulation of T-cell factor signaling following activation of EP2 and EP4 prostanoid receptors by prostaglandin E2. J. Biol. Chem. 2002, 277, 2614–2619. [Google Scholar] [CrossRef] [PubMed]
- Nishigaki, N.; Negishi, M.; Ichikawa, A. Two Gs-coupled prostaglandin E receptor subtypes, EP2 and EP4, differ in desensitization and sensitivity to the metabolic inactivation of the agonist. Mol. Pharmacol. 1996, 50, 1031–1037. [Google Scholar] [PubMed]
- Chun, K.S.; Lao, H.C.; Trempus, C.S.; Okada, M.; Langenbach, R. The prostaglandin receptor EP2 activates multiple signaling pathways and beta-arrestin1 complex formation during mouse skin papilloma development. Carcinogenesis 2009, 30, 1620–1627. [Google Scholar] [CrossRef] [PubMed]
- Fujino, H.; Xu, W.; Regan, J.W. Prostaglandin E2 induced functional expression of early growth response factor-1 by EP4, but not EP2, prostanoid receptors via the phosphatidylinositol 3-kinase and extracellular signal-regulated kinases. J. Biol. Chem. 2003, 278, 12151–12156. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, T. Prostanoid receptor EP2 as a therapeutic target. J. Med. Chem. 2014, 57, 4454–4465. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Kudo, I. Recent advances in molecular biology and physiology of the prostaglandin E2-biosynthetic pathway. Prog. Lipid Res. 2004, 43, 3–35. [Google Scholar] [CrossRef]
- Hester, A.; Ritzer, M.; Kuhn, C.; Schmoeckel, E.; Mayr, D.; Kolben, T.; Dannecker, C.; Mahner, S.; Jeschke, U.; Kolben, T.M. The role of EP3-receptor expression in cervical dysplasia. J. Cancer Res. Clin. Oncol. 2019, 145, 313–319. [Google Scholar] [CrossRef]
- Sales, K.J.; Katz, A.A.; Davis, M.; Hinz, S.; Soeters, R.P.; Hofmeyr, M.D.; Millar, R.P.; Jabbour, H.N. Cyclooxygenase-2 expression and prostaglandin E(2) synthesis are up-regulated in carcinomas of the cervix: A possible autocrine/paracrine regulation of neoplastic cell function via EP2/EP4 receptors. J. Clin. Endocrinol. Metab. 2001, 86, 2243–2249. [Google Scholar] [CrossRef]
- Sales, K.J.; Katz, A.A.; Howard, B.; Soeters, R.P.; Millar, R.P.; Jabbour, H.N. Cyclooxygenase-1 is up-regulated in cervical carcinomas: Autocrine/paracrine regulation of cyclooxygenase-2, prostaglandin e receptors, and angiogenic factors by cyclooxygenase-1. Cancer Res. 2002, 62, 424–432. [Google Scholar]
- Sales, K.J.; Katz, A.A.; Millar, R.P.; Jabbour, H.N. Seminal plasma activates cyclooxygenase-2 and prostaglandin E2 receptor expression and signalling in cervical adenocarcinoma cells. Mol. Hum. Reprod. 2002, 8, 1065–1070. [Google Scholar] [CrossRef][Green Version]
- Sales, K.J.; Sutherland, J.R.; Jabbour, H.N.; Katz, A.A. Seminal plasma induces angiogenic chemokine expression in cervical cancer cells and regulates vascular function. Biochim. Biophys. Acta 2012, 1823, 1789–1795. [Google Scholar] [CrossRef]
- Schmoeckel, E.; Fraungruber, P.; Kuhn, C.; Jeschke, U.; Mahner, S.; Kolben, T.M.; Kolben, T.; Vilsmaier, T.; Hester, A.; Heidegger, H.H. The role of EP-2 receptor expression in cervical intraepithelial neoplasia. Histochem. Cell Biol. 2020, 154, 655–662. [Google Scholar] [CrossRef]
- Dietlmeier, S.; Ye, Y.; Kuhn, C.; Vattai, A.; Vilsmaier, T.; Schroder, L.; Kost, B.P.; Gallwas, J.; Jeschke, U.; Mahner, S.; et al. The prostaglandin receptor EP2 determines prognosis in EP3-negative and galectin-3-high cervical cancer cases. Sci. Rep. 2020, 10, 1154. [Google Scholar] [CrossRef]
- Ye, Y.; Peng, L.; Vattai, A.; Deuster, E.; Kuhn, C.; Dannecker, C.; Mahner, S.; Jeschke, U.; von Schonfeldt, V.; Heidegger, H.H. Prostaglandin E2 receptor 3 (EP3) signaling promotes migration of cervical cancer via urokinase-type plasminogen activator receptor (uPAR). J. Cancer Res. Clin. Oncol. 2020, 146, 2189–2203. [Google Scholar] [CrossRef]
- Sonoshita, M.; Takaku, K.; Sasaki, N.; Sugimoto, Y.; Ushikubi, F.; Narumiya, S.; Oshima, M.; Taketo, M.M. Acceleration of intestinal polyposis through prostaglandin receptor EP2 in Apc(Delta 716) knockout mice. Nat. Med. 2001, 7, 1048–1051. [Google Scholar] [CrossRef]
- Yoshida, S.; Amano, H.; Hayashi, I.; Kitasato, H.; Kamata, M.; Inukai, M.; Yoshimura, H.; Majima, M. COX-2/VEGF-dependent facilitation of tumor-associated angiogenesis and tumor growth in vivo. Lab. Investig. 2003, 83, 1385–1394. [Google Scholar] [CrossRef]
- Ma, X.; Aoki, T.; Tsuruyama, T.; Narumiya, S. Definition of Prostaglandin E2-EP2 Signals in the Colon Tumor Microenvironment That Amplify Inflammation and Tumor Growth. Cancer Res. 2015, 75, 2822–2832. [Google Scholar] [CrossRef]
- Oshima, H.; Popivanova, B.K.; Oguma, K.; Kong, D.; Ishikawa, T.O.; Oshima, M. Activation of epidermal growth factor receptor signaling by the prostaglandin E(2) receptor EP4 pathway during gastric tumorigenesis. Cancer Sci. 2011, 102, 713–719. [Google Scholar] [CrossRef]
- Okuyama, T.; Ishihara, S.; Sato, H.; Rumi, M.A.; Kawashima, K.; Miyaoka, Y.; Suetsugu, H.; Kazumori, H.; Cava, C.F.; Kadowaki, Y.; et al. Activation of prostaglandin E2-receptor EP2 and EP4 pathways induces growth inhibition in human gastric carcinoma cell lines. J. Lab. Clin. Med. 2002, 140, 92–102. [Google Scholar] [CrossRef]
- Grace, V.M.B.; Wilson, D.D.; Anushya, R.; Siddikuzzaman. Regulation of inflammation and COX-2 gene expression in benzo (a) pyrene induced lung carcinogenesis in mice by all trans retinoic acid (ATRA). Life Sci. 2021, 285, 119967. [Google Scholar] [CrossRef]
- Kim, J.I.; Lakshmikanthan, V.; Frilot, N.; Daaka, Y. Prostaglandin E2 promotes lung cancer cell migration via EP4-betaArrestin1-c-Src signalsome. Mol. Cancer Res. 2010, 8, 569–577. [Google Scholar] [CrossRef]
- Krysan, K.; Reckamp, K.L.; Dalwadi, H.; Sharma, S.; Rozengurt, E.; Dohadwala, M.; Dubinett, S.M. Prostaglandin E2 activates mitogen-activated protein kinase/Erk pathway signaling and cell proliferation in non-small cell lung cancer cells in an epidermal growth factor receptor-independent manner. Cancer Res. 2005, 65, 6275–6281. [Google Scholar] [CrossRef]
- Bai, X.; Yang, Q.; Shu, W.; Wang, J.; Zhang, L.; Ma, J.; Xia, S.; Zhang, M.; Cheng, S.; Wang, Y.; et al. Prostaglandin E2 upregulates beta1 integrin expression via the E prostanoid 1 receptor/nuclear factor kappa-light-chain-enhancer of activated B cells pathway in non-small-cell lung cancer cells. Mol. Med. Rep. 2014, 9, 1729–1736. [Google Scholar] [CrossRef]
- Li, L.; Lv, Y.; Yan, D. Inhibition of Ep3 attenuates migration and promotes apoptosis of non-small cell lung cancer cells via suppression of TGF-beta/Smad signaling. Oncol. Lett. 2018, 16, 5645–5654. [Google Scholar] [CrossRef]
- Harati, R.; Mabondzo, A.; Tlili, A.; Khoder, G.; Mahfood, M.; Hamoudi, R. Combinatorial targeting of microRNA-26b and microRNA-101 exerts a synergistic inhibition on cyclooxygenase-2 in brain metastatic triple-negative breast cancer cells. Breast Cancer Res. Treat. 2021, 187, 695–713. [Google Scholar] [CrossRef]
- Tian, J.; Wang, V.; Wang, N.; Khadang, B.; Boudreault, J.; Bakdounes, K.; Ali, S.; Lebrun, J.J. Identification of MFGE8 and KLK5/7 as mediators of breast tumorigenesis and resistance to COX-2 inhibition. Breast Cancer Res. 2021, 23, 23. [Google Scholar] [CrossRef]
- Semmlinger, A.; von Schoenfeldt, V.; Wolf, V.; Meuter, A.; Kolben, T.M.; Kolben, T.; Zeder-Goess, C.; Weis, F.; Gallwas, J.; Wuerstlein, R.; et al. EP3 (prostaglandin E2 receptor 3) expression is a prognostic factor for progression-free and overall survival in sporadic breast cancer. BMC Cancer 2018, 18, 431. [Google Scholar] [CrossRef]
- Cheuk, I.W.; Shin, V.Y.; Siu, M.T.; Tsang, J.Y.; Ho, J.C.; Chen, J.; Tse, G.M.; Wang, X.; Kwong, A. Association of EP2 receptor and SLC19A3 in regulating breast cancer metastasis. Am. J. Cancer Res. 2015, 5, 3389–3399. [Google Scholar]
- Majumder, M.; Xin, X.; Liu, L.; Tutunea-Fatan, E.; Rodriguez-Torres, M.; Vincent, K.; Postovit, L.M.; Hess, D.; Lala, P.K. COX-2 Induces Breast Cancer Stem Cells via EP4/PI3K/AKT/NOTCH/WNT Axis. Stem Cells 2016, 34, 2290–2305. [Google Scholar] [CrossRef]
- Thorat, M.A.; Morimiya, A.; Mehrotra, S.; Konger, R.; Badve, S.S. Prostanoid receptor EP1 expression in breast cancer. Mod. Pathol. 2008, 21, 15–21. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Thanan, R.; Murata, M.; Ma, N.; Hammam, O.; Wishahi, M.; El Leithy, T.; Hiraku, Y.; Oikawa, S.; Kawanishi, S. Nuclear localization of COX-2 in relation to the expression of stemness markers in urinary bladder cancer. Mediat. Inflamm. 2012, 2012, 165879. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, A.; Vattai, A.; Furst, S.; Vilsmaier, T.; Kuhn, C.; Schmoeckel, E.; Mayr, D.; Dannecker, C.; Mahner, S.; Jeschke, U.; et al. EP4 as a Negative Prognostic Factor in Patients with Vulvar Cancer. Cancers 2021, 13, 1410. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Choi, E.M.; Kim, S.R.; Park, J.H.; Kim, H.; Ha, K.S.; Kim, Y.M.; Kim, S.S.; Choe, M.; Kim, J.I.; et al. Cyclooxygenase-2 promotes cell proliferation, migration and invasion in U2OS human osteosarcoma cells. Exp. Mol. Med. 2007, 39, 469–476. [Google Scholar] [CrossRef]
- Urakawa, H.; Nishida, Y.; Naruse, T.; Nakashima, H.; Ishiguro, N. Cyclooxygenase-2 overexpression predicts poor survival in patients with high-grade extremity osteosarcoma: A pilot study. Clin. Orthop. Relat. Res. 2009, 467, 2932–2938. [Google Scholar] [CrossRef]
- Chen, H.; Cai, W.; Chu, E.S.H.; Tang, J.; Wong, C.C.; Wong, S.H.; Sun, W.; Liang, Q.; Fang, J.; Sun, Z.; et al. Hepatic cyclooxygenase-2 overexpression induced spontaneous hepatocellular carcinoma formation in mice. Oncogene 2017, 36, 4415–4426. [Google Scholar] [CrossRef]
- Xu, S.; Zhou, W.; Ge, J.; Zhang, Z. Prostaglandin E2 receptor EP4 is involved in the cell growth and invasion of prostate cancer via the cAMPPKA/PI3KAkt signaling pathway. Mol. Med. Rep. 2018, 17, 4702–4712. [Google Scholar] [CrossRef]
- ElAttar, T.M.; Lin, H.S. Inhibition of human oral squamous carcinoma cell (SCC-25) proliferation by prostaglandin E2 and vitamin E succinate. J. Oral Pathol. Med. 1993, 22, 425–427. [Google Scholar] [CrossRef]
- Kawanishi, S.; Ohnishi, S.; Ma, N.; Hiraku, Y.; Murata, M. Crosstalk between DNA Damage and Inflammation in the Multiple Steps of Carcinogenesis. Int. J. Mol. Sci. 2017, 18, 1808. [Google Scholar] [CrossRef]
- Landskron, G.; De la Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic inflammation and cytokines in the tumor microenvironment. J. Immunol. Res. 2014, 2014, 149185. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Subbaramaiah, K.; Dannenberg, A.J. Cyclooxygenase-2 transcription is regulated by human papillomavirus 16 E6 and E7 oncoproteins: Evidence of a corepressor/coactivator exchange. Cancer Res. 2007, 67, 3976–3985. [Google Scholar] [CrossRef]
- Georgescu, S.R.; Mitran, C.I.; Mitran, M.I.; Caruntu, C.; Sarbu, M.I.; Matei, C.; Nicolae, I.; Tocut, S.M.; Popa, M.I.; Tampa, M. New Insights in the Pathogenesis of HPV Infection and the Associated Carcinogenic Processes: The Role of Chronic Inflammation and Oxidative Stress. J. Immunol. Res. 2018, 2018, 5315816. [Google Scholar] [CrossRef]
- Williams, V.M.; Filippova, M.; Soto, U.; Duerksen-Hughes, P.J. HPV-DNA integration and carcinogenesis: Putative roles for inflammation and oxidative stress. Future Virol. 2011, 6, 45–57. [Google Scholar] [CrossRef]
- Ye, Y.; Wang, X.; Jeschke, U.; von Schonfeldt, V. COX-2-PGE2-EPs in gynecological cancers. Arch. Gynecol. Obstet. 2020, 301, 1365–1375. [Google Scholar] [CrossRef]
- Jabbour, H.N.; Milne, S.A.; Williams, A.R.; Anderson, R.A.; Boddy, S.C. Expression of COX-2 and PGE synthase and synthesis of PGE(2) in endometrial adenocarcinoma: A possible autocrine/paracrine regulation of neoplastic cell function via EP2/EP4 receptors. Br. J. Cancer 2001, 85, 1023–1031. [Google Scholar] [CrossRef]
- Hsu, H.H.; Lin, Y.M.; Shen, C.Y.; Shibu, M.A.; Li, S.Y.; Chang, S.H.; Lin, C.C.; Chen, R.J.; Viswanadha, V.P.; Shih, H.N.; et al. Prostaglandin E2-Induced COX-2 Expressions via EP2 and EP4 Signaling Pathways in Human LoVo Colon Cancer Cells. Int. J. Mol. Sci. 2017, 18, 1132. [Google Scholar] [CrossRef]
- Alcocer-Gonzalez, J.M.; Berumen, J.; Tamez-Guerra, R.; Bermudez-Morales, V.; Peralta-Zaragoza, O.; Hernandez-Pando, R.; Moreno, J.; Gariglio, P.; Madrid-Marina, V. In vivo expression of immunosuppressive cytokines in human papillomavirus-transformed cervical cancer cells. Viral. Immunol. 2006, 19, 481–491. [Google Scholar] [CrossRef]
- Hirsch, I.; Caux, C.; Hasan, U.; Bendriss-Vermare, N.; Olive, D. Impaired Toll-like receptor 7 and 9 signaling: From chronic viral infections to cancer. Trends Immunol. 2010, 31, 391–397. [Google Scholar] [CrossRef]
- Hasan, U.A.; Bates, E.; Takeshita, F.; Biliato, A.; Accardi, R.; Bouvard, V.; Mansour, M.; Vincent, I.; Gissmann, L.; Iftner, T.; et al. TLR9 expression and function is abolished by the cervical cancer-associated human papillomavirus type 16. J. Immunol. 2007, 178, 3186–3197. [Google Scholar] [CrossRef]
- Hasan, U.A.; Zannetti, C.; Parroche, P.; Goutagny, N.; Malfroy, M.; Roblot, G.; Carreira, C.; Hussain, I.; Muller, M.; Taylor-Papadimitriou, J.; et al. The human papillomavirus type 16 E7 oncoprotein induces a transcriptional repressor complex on the Toll-like receptor 9 promoter. J. Exp. Med. 2013, 210, 1369–1387. [Google Scholar] [CrossRef]
- Barnard, P.; Payne, E.; McMillan, N.A. The human papillomavirus E7 protein is able to inhibit the antiviral and anti-growth functions of interferon-alpha. Virology 2000, 277, 411–419. [Google Scholar] [CrossRef]
- Beglin, M.; Melar-New, M.; Laimins, L. Human papillomaviruses and the interferon response. J. Interf. Cytokine Res. 2009, 29, 629–635. [Google Scholar] [CrossRef]
- Huang, J.; Diao, G.; Zhang, Q.; Chen, Y.; Han, J.; Guo, J. E6regulated overproduction of prostaglandin E2 may inhibit migration of dendritic cells in human papillomavirus 16positive cervical lesions. Int. J. Oncol. 2020, 56, 921–931. [Google Scholar] [CrossRef]
- Ferreira, A.R.; Ramalho, A.C.; Marques, M.; Ribeiro, D. The Interplay between Antiviral Signalling and Carcinogenesis in Human Papillomavirus Infections. Cancers 2020, 12, 646. [Google Scholar] [CrossRef]
- Florez-Grau, G.; Cabezon, R.; Borgman, K.J.E.; Espana, C.; Lozano, J.J.; Garcia-Parajo, M.F.; Benitez-Ribas, D. Up-regulation of EP2 and EP3 receptors in human tolerogenic dendritic cells boosts the immunosuppressive activity of PGE2. J. Leukoc. Biol. 2017, 102, 881–895. [Google Scholar] [CrossRef]
- Kundu, N.; Ma, X.; Holt, D.; Goloubeva, O.; Ostrand-Rosenberg, S.; Fulton, A.M. Antagonism of the prostaglandin E receptor EP4 inhibits metastasis and enhances NK function. Breast Cancer Res. Treat. 2009, 117, 235–242. [Google Scholar] [CrossRef]
- Mao, Y.; Sarhan, D.; Steven, A.; Seliger, B.; Kiessling, R.; Lundqvist, A. Inhibition of tumor-derived prostaglandin-e2 blocks the induction of myeloid-derived suppressor cells and recovers natural killer cell activity. Clin. Cancer Res. 2014, 20, 4096–4106. [Google Scholar] [CrossRef]
- Obermajer, N.; Muthuswamy, R.; Lesnock, J.; Edwards, R.P.; Kalinski, P. Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid-derived suppressor cells. Blood 2011, 118, 5498–5505. [Google Scholar] [CrossRef]
- Wu, L.; Liu, H.; Guo, H.; Wu, Q.; Yu, S.; Qin, Y.; Wang, G.; Wu, Q.; Zhang, R.; Wang, L.; et al. Circulating and tumor-infiltrating myeloid-derived suppressor cells in cervical carcinoma patients. Oncol. Lett. 2018, 15, 9507–9515. [Google Scholar] [CrossRef]
- Ching, M.M.; Reader, J.; Fulton, A.M. Eicosanoids in Cancer: Prostaglandin E2 Receptor 4 in Cancer Therapeutics and Immunotherapy. Front. Pharmacol. 2020, 11, 819. [Google Scholar] [CrossRef] [PubMed]
- Mosenden, R.; Tasken, K. Cyclic AMP-mediated immune regulation--overview of mechanisms of action in T cells. Cell Signal. 2011, 23, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Alfonso, L.; Ai, G.; Spitale, R.C.; Bhat, G.J. Molecular targets of aspirin and cancer prevention. Br. J. Cancer 2014, 111, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Friel, G.; Liu, C.S.; Kolomeyevskaya, N.V.; Hampras, S.S.; Kruszka, B.; Schmitt, K.; Cannioto, R.A.; Lele, S.B.; Odunsi, K.O.; Moysich, K.B. Aspirin and Acetaminophen Use and the Risk of Cervical Cancer. J. Low. Genit. Tract Dis. 2015, 19, 189–193. [Google Scholar] [CrossRef]
- Ahmadi, R.; Karimi Ghezeli, Z.; Gravand, F.; Naghshineh, M. The Effect of Aspirin and Ibuprofen on the Proliferation of Cervical Cancer Cells (HeLa) Compared to Non-Cancerous Cells (HEK 293) in Cell Culture Medium. Qom Univ. Med. Sci. J. 2018, 12, 16–24. [Google Scholar] [CrossRef]
- Kim, K.Y.; Seol, J.Y.; Jeon, G.A.; Nam, M.J. The combined treatment of aspirin and radiation induces apoptosis by the regulation of bcl-2 and caspase-3 in human cervical cancer cell. Cancer Lett. 2003, 189, 157–166. [Google Scholar] [CrossRef]
- Xiang, S.; Sun, Z.; He, Q.; Yan, F.; Wang, Y.; Zhang, J. Aspirin inhibits ErbB2 to induce apoptosis in cervical cancer cells. Med. Oncol. 2010, 27, 379–387. [Google Scholar] [CrossRef]
- Sakonlaya, D.; Tapanadechopone, P.; Poomkokruk, A.; Charoenvilaisiri, S. Do NSAIDs inhibit growth of precancerous cervical cells in vitro? J. Med. Assoc. Thai. 2012, 95 (Suppl. S1), S65–S73. [Google Scholar]
- Soriano-Hernandez, A.D.; Madrigal-Perez, D.; Galvan-Salazar, H.R.; Martinez-Fierro, M.L.; Valdez-Velazquez, L.L.; Espinoza-Gomez, F.; Vazquez-Vuelvas, O.F.; Olmedo-Buenrostro, B.A.; Guzman-Esquivel, J.; Rodriguez-Sanchez, I.P.; et al. Anti-inflammatory drugs and uterine cervical cancer cells: Antineoplastic effect of meclofenamic acid. Oncol. Lett. 2015, 10, 2574–2578. [Google Scholar] [CrossRef]
- Kolawole, O.R.; Kashfi, K. NSAIDs and Cancer Resolution: New Paradigms beyond Cyclooxygenase. Int. J. Mol. Sci. 2022, 23, 1432. [Google Scholar] [CrossRef]
- Kim, S.H.; Song, S.H.; Kim, S.G.; Chun, K.S.; Lim, S.Y.; Na, H.K.; Kim, J.W.; Surh, Y.J.; Bang, Y.J.; Song, Y.S. Celecoxib induces apoptosis in cervical cancer cells independent of cyclooxygenase using NF-kappaB as a possible target. J. Cancer Res. Clin. Oncol. 2004, 130, 551–560. [Google Scholar] [CrossRef]
- Ferrandina, G.; Ranelletti, F.O.; Legge, F.; Lauriola, L.; Salutari, V.; Gessi, M.; Testa, A.C.; Werner, U.; Navarra, P.; Tringali, G.; et al. Celecoxib modulates the expression of cyclooxygenase-2, ki67, apoptosis-related marker, and microvessel density in human cervical cancer: A pilot study. Clin. Cancer Res. 2003, 9, 4324–4331. [Google Scholar]
- Farley, J.H.; Truong, V.; Goo, E.; Uyehara, C.; Belnap, C.; Larsen, W.I. A randomized double-blind placebo-controlled phase II trial of the cyclooxygenase-2 inhibitor Celecoxib in the treatment of cervical dysplasia. Gynecol. Oncol. 2006, 103, 425–430. [Google Scholar] [CrossRef]
- Rader, J.S.; Sill, M.W.; Beumer, J.H.; Lankes, H.A.; Benbrook, D.M.; Garcia, F.; Trimble, C.; Tate Thigpen, J.; Lieberman, R.; Zuna, R.E.; et al. A stratified randomized double-blind phase II trial of celecoxib for treating patients with cervical intraepithelial neoplasia: The potential predictive value of VEGF serum levels: An NRG Oncology/Gynecologic Oncology Group study. Gynecol. Oncol. 2017, 145, 291–297. [Google Scholar] [CrossRef]
- Nakata, E.; Mason, K.A.; Hunter, N.; Husain, A.; Raju, U.; Liao, Z.; Ang, K.K.; Milas, L. Potentiation of tumor response to radiation or chemoradiation by selective cyclooxygenase-2 enzyme inhibitors. Int. J. Radiat. Oncol. Biol. Phys. 2004, 58, 369–375. [Google Scholar] [CrossRef]
- Raju, U.; Nakata, E.; Yang, P.; Newman, R.A.; Ang, K.K.; Milas, L. In vitro enhancement of tumor cell radiosensitivity by a selective inhibitor of cyclooxygenase-2 enzyme: Mechanistic considerations. Int. J. Radiat. Oncol. Biol. Phys. 2002, 54, 886–894. [Google Scholar] [CrossRef]
- Herrera, F.G.; Chan, P.; Doll, C.; Milosevic, M.; Oza, A.; Syed, A.; Pintilie, M.; Levin, W.; Manchul, L.; Fyles, A. A prospective phase I-II trial of the cyclooxygenase-2 inhibitor celecoxib in patients with carcinoma of the cervix with biomarker assessment of the tumor microenvironment. Int. J. Radiat. Oncol. Biol. Phys. 2007, 67, 97–103. [Google Scholar] [CrossRef]
- Gaffney, D.K.; Winter, K.; Dicker, A.P.; Miller, B.; Eifel, P.J.; Ryu, J.; Avizonis, V.; Fromm, M.; Greven, K. A Phase II study of acute toxicity for Celebrex (celecoxib) and chemoradiation in patients with locally advanced cervical cancer: Primary endpoint analysis of RTOG 0128. Int. J. Radiat. Oncol. Biol. Phys. 2007, 67, 104–109. [Google Scholar] [CrossRef]
- Barnes, P.J. Anti-inflammatory actions of glucocorticoids: Molecular mechanisms. Clin. Sci. 1998, 94, 557–572. [Google Scholar] [CrossRef]
- Grunberg, S.M. Antiemetic activity of corticosteroids in patients receiving cancer chemotherapy: Dosing, efficacy, and tolerability analysis. Ann. Oncol. 2007, 18, 233–240. [Google Scholar] [CrossRef]
- Chen, Q.M.; Alexander, D.; Sun, H.; Xie, L.; Lin, Y.; Terrand, J.; Morrissy, S.; Purdom, S. Corticosteroids inhibit cell death induced by doxorubicin in cardiomyocytes: Induction of antiapoptosis, antioxidant, and detoxification genes. Mol. Pharmacol. 2005, 67, 1861–1873. [Google Scholar] [CrossRef]
- Zhang, C.; Beckermann, B.; Kallifatidis, G.; Liu, Z.; Rittgen, W.; Edler, L.; Buchler, P.; Debatin, K.M.; Buchler, M.W.; Friess, H.; et al. Corticosteroids induce chemotherapy resistance in the majority of tumour cells from bone, brain, breast, cervix, melanoma and neuroblastoma. Int. J. Oncol. 2006, 29, 1295–1301. [Google Scholar] [CrossRef]
- Kamradt, M.C.; Mohideen, N.; Krueger, E.; Walter, S.; Vaughan, A.T. Inhibition of radiation-induced apoptosis by dexamethasone in cervical carcinoma cell lines depends upon increased HPV E6/E7. Br. J. Cancer 2000, 82, 1709–1716. [Google Scholar] [CrossRef]
- Shi, M.; Du, L.; Liu, D.; Qian, L.; Hu, M.; Yu, M.; Yang, Z.; Zhao, M.; Chen, C.; Guo, L.; et al. Glucocorticoid regulation of a novel HPV-E6-p53-miR-145 pathway modulates invasion and therapy resistance of cervical cancer cells. J. Pathol. 2012, 228, 148–157. [Google Scholar] [CrossRef]
- Bromberg-White, J.L.; Meyers, C. Comparison of the basal and glucocorticoid-inducible activities of the upstream regulatory regions of HPV18 and HPV31 in multiple epithelial cell lines. Virology 2003, 306, 197–202. [Google Scholar] [CrossRef]
- Crusz, S.M.; El-Shakankery, K.; Miller, R.E. Targeting HPV in gynaecological cancers—Current status, ongoing challenges and future directions. Womens Health 2020, 16, 1745506520961709. [Google Scholar] [CrossRef]
- Hellner, K.; Munger, K. Human papillomaviruses as therapeutic targets in human cancer. J. Clin. Oncol. 2011, 29, 1785–1794. [Google Scholar] [CrossRef]
- Liu, Y.; Li, H.; Pi, R.; Yang, Y.; Zhao, X.; Qi, X. Current strategies against persistent human papillomavirus infection (Review). Int. J. Oncol. 2019, 55, 570–584. [Google Scholar] [CrossRef]
- Flickinger, J.C., Jr.; Rodeck, U.; Snook, A.E. Listeria monocytogenes as a Vector for Cancer Immunotherapy: Current Understanding and Progress. Vaccines 2018, 6, 48. [Google Scholar] [CrossRef]
- Gunn, G.R.; Zubair, A.; Peters, C.; Pan, Z.K.; Wu, T.C.; Paterson, Y. Two Listeria monocytogenes vaccine vectors that express different molecular forms of human papilloma virus-16 (HPV-16) E7 induce qualitatively different T cell immunity that correlates with their ability to induce regression of established tumors immortalized by HPV-16. J. Immunol. 2001, 167, 6471–6479. [Google Scholar] [CrossRef] [PubMed]
- Maciag, P.C.; Radulovic, S.; Rothman, J. The first clinical use of a live-attenuated Listeria monocytogenes vaccine: A Phase I safety study of Lm-LLO-E7 in patients with advanced carcinoma of the cervix. Vaccine 2009, 27, 3975–3983. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, P.; Bouillette-Marussig, M.; Hens, A.; De Coster, I.; Depuydt, C.; Goubier, A.; Van Tendeloo, V.; Cools, N.; Goossens, H.; Hercend, T.; et al. GTL001, A Therapeutic Vaccine for Women Infected with Human Papillomavirus 16 or 18 and Normal Cervical Cytology: Results of a Phase I Clinical Trial. Clin. Cancer Res. 2016, 22, 3238–3248. [Google Scholar] [CrossRef] [PubMed]
- Esquerre, M.; Bouillette-Marussig, M.; Goubier, A.; Momot, M.; Gonindard, C.; Keller, H.; Navarro, A.; Bissery, M.C. GTL001, a bivalent therapeutic vaccine against human papillomavirus 16 and 18, induces antigen-specific CD8+ T cell responses leading to tumor regression. PLoS ONE 2017, 12, e0174038. [Google Scholar] [CrossRef]
- Simsova, M.; Sebo, P.; Leclerc, C. The adenylate cyclase toxin from Bordetella pertussis--a novel promising vehicle for antigen delivery to dendritic cells. Int. J. Med. Microbiol. 2004, 293, 571–576. [Google Scholar] [CrossRef]
- Yang, A.; Farmer, E.; Lin, J.; Wu, T.C.; Hung, C.F. The current state of therapeutic and T cell-based vaccines against human papillomaviruses. Virus Res. 2017, 231, 148–165. [Google Scholar] [CrossRef]
- Boursnell, M.E.; Rutherford, E.; Hickling, J.K.; Rollinson, E.A.; Munro, A.J.; Rolley, N.; McLean, C.S.; Borysiewicz, L.K.; Vousden, K.; Inglis, S.C. Construction and characterisation of a recombinant vaccinia virus expressing human papillomavirus proteins for immunotherapy of cervical cancer. Vaccine 1996, 14, 1485–1494. [Google Scholar] [CrossRef]
- Borysiewicz, L.K.; Fiander, A.; Nimako, M.; Man, S.; Wilkinson, G.W.; Westmoreland, D.; Evans, A.S.; Adams, M.; Stacey, S.N.; Boursnell, M.E.; et al. A recombinant vaccinia virus encoding human papillomavirus types 16 and 18, E6 and E7 proteins as immunotherapy for cervical cancer. Lancet 1996, 347, 1523–1527. [Google Scholar] [CrossRef]
- Baldwin, P.J.; van der Burg, S.H.; Boswell, C.M.; Offringa, R.; Hickling, J.K.; Dobson, J.; Roberts, J.S.; Latimer, J.A.; Moseley, R.P.; Coleman, N.; et al. Vaccinia-expressed human papillomavirus 16 and 18 e6 and e7 as a therapeutic vaccination for vulval and vaginal intraepithelial neoplasia. Clin. Cancer Res. 2003, 9, 5205–5213. [Google Scholar]
- Maldonado, L.; Teague, J.E.; Morrow, M.P.; Jotova, I.; Wu, T.C.; Wang, C.; Desmarais, C.; Boyer, J.D.; Tycko, B.; Robins, H.S.; et al. Intramuscular therapeutic vaccination targeting HPV16 induces T cell responses that localize in mucosal lesions. Sci. Transl. Med. 2014, 6, 221ra213. [Google Scholar] [CrossRef]
- Peng, S.; Ferrall, L.; Gaillard, S.; Wang, C.; Chi, W.Y.; Huang, C.H.; Roden, R.B.S.; Wu, T.C.; Chang, Y.N.; Hung, C.F. Development of DNA Vaccine Targeting E6 and E7 Proteins of Human Papillomavirus 16 (HPV16) and HPV18 for Immunotherapy in Combination with Recombinant Vaccinia Boost and PD-1 Antibody. mBio 2021, 12, e03224-20. [Google Scholar] [CrossRef]
- Lamikanra, A.; Pan, Z.K.; Isaacs, S.N.; Wu, T.C.; Paterson, Y. Regression of established human papillomavirus type 16 (HPV-16) immortalized tumors in vivo by vaccinia viruses expressing different forms of HPV-16 E7 correlates with enhanced CD8(+) T-cell responses that home to the tumor site. J. Virol. 2001, 75, 9654–9664. [Google Scholar] [CrossRef]
- Volz, A.; Sutter, G. Modified Vaccinia Virus Ankara: History, Value in Basic Research, and Current Perspectives for Vaccine Development. Adv. Virus Res. 2017, 97, 187–243. [Google Scholar] [CrossRef]
- Rosales, C.; Graham, V.V.; Rosas, G.A.; Merchant, H.; Rosales, R. A recombinant vaccinia virus containing the papilloma E2 protein promotes tumor regression by stimulating macrophage antibody-dependent cytotoxicity. Cancer Immunol. Immunother. 2000, 49, 347–360. [Google Scholar] [CrossRef]
- Valdez Graham, V.; Sutter, G.; Jose, M.V.; Garcia-Carranca, A.; Erfle, V.; Moreno Mendoza, N.; Merchant, H.; Rosales, R. Human tumor growth is inhibited by a vaccinia virus carrying the E2 gene of bovine papillomavirus. Cancer 2000, 88, 1650–1662. [Google Scholar] [CrossRef]
- Corona Gutierrez, C.M.; Tinoco, A.; Navarro, T.; Contreras, M.L.; Cortes, R.R.; Calzado, P.; Reyes, L.; Posternak, R.; Morosoli, G.; Verde, M.L.; et al. Therapeutic vaccination with MVA E2 can eliminate precancerous lesions (CIN 1, CIN 2, and CIN 3) associated with infection by oncogenic human papillomavirus. Hum. Gene Ther. 2004, 15, 421–431. [Google Scholar] [CrossRef]
- Garcia-Hernandez, E.; Gonzalez-Sanchez, J.L.; Andrade-Manzano, A.; Contreras, M.L.; Padilla, S.; Guzman, C.C.; Jimenez, R.; Reyes, L.; Morosoli, G.; Verde, M.L.; et al. Regression of papilloma high-grade lesions (CIN 2 and CIN 3) is stimulated by therapeutic vaccination with MVA E2 recombinant vaccine. Cancer Gene Ther. 2006, 13, 592–597. [Google Scholar] [CrossRef]
- Rosales, R.; Lopez-Contreras, M.; Rosales, C.; Magallanes-Molina, J.R.; Gonzalez-Vergara, R.; Arroyo-Cazarez, J.M.; Ricardez-Arenas, A.; Del Follo-Valencia, A.; Padilla-Arriaga, S.; Guerrero, M.V.; et al. Regression of human papillomavirus intraepithelial lesions is induced by MVA E2 therapeutic vaccine. Hum. Gene Ther. 2014, 25, 1035–1049. [Google Scholar] [CrossRef]
- Brun, J.L.; Dalstein, V.; Leveque, J.; Mathevet, P.; Raulic, P.; Baldauf, J.J.; Scholl, S.; Huynh, B.; Douvier, S.; Riethmuller, D.; et al. Regression of high-grade cervical intraepithelial neoplasia with TG4001 targeted immunotherapy. Am. J. Obstet. Gynecol. 2011, 204, 169.e1–169.e8. [Google Scholar] [CrossRef]
- Gomez-Gutierrez, J.G.; Elpek, K.G.; Montes de Oca-Luna, R.; Shirwan, H.; Sam Zhou, H.; McMasters, K.M. Vaccination with an adenoviral vector expressing calreticulin-human papillomavirus 16 E7 fusion protein eradicates E7 expressing established tumors in mice. Cancer Immunol. Immunother. 2007, 56, 997–1007. [Google Scholar] [CrossRef]
- Ho, W.; Gao, M.; Li, F.; Li, Z.; Zhang, X.Q.; Xu, X. Next-Generation Vaccines: Nanoparticle-Mediated DNA and mRNA Delivery. Adv. Healthc. Mater. 2021, 10, e2001812. [Google Scholar] [CrossRef]
- Bagarazzi, M.L.; Yan, J.; Morrow, M.P.; Shen, X.; Parker, R.L.; Lee, J.C.; Giffear, M.; Pankhong, P.; Khan, A.S.; Broderick, K.E.; et al. Immunotherapy against HPV16/18 generates potent TH1 and cytotoxic cellular immune responses. Sci. Transl. Med. 2012, 4, 155ra138. [Google Scholar] [CrossRef] [PubMed]
- Morrow, M.P.; Kraynyak, K.A.; Sylvester, A.J.; Dallas, M.; Knoblock, D.; Boyer, J.D.; Yan, J.; Vang, R.; Khan, A.S.; Humeau, L.; et al. Clinical and Immunologic Biomarkers for Histologic Regression of High-Grade Cervical Dysplasia and Clearance of HPV16 and HPV18 after Immunotherapy. Clin. Cancer Res. 2018, 24, 276–294. [Google Scholar] [CrossRef] [PubMed]
- Bhuyan, P.K.; Dallas, M.; Kraynyak, K.; Herring, T.; Morrow, M.; Boyer, J.; Duff, S.; Kim, J.; Weiner, D.B. Durability of response to VGX-3100 treatment of HPV16/18 positive cervical HSIL. Hum. Vaccines Immunother. 2021, 17, 1288–1293. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Li, M.; Zhao, C.; Shen, D.; Liu, L.; Zhang, X.; Wei, L. Therapeutic DNA Vaccines against HPV-Related Malignancies: Promising Leads from Clinical Trials. Viruses 2022, 14, 239. [Google Scholar] [CrossRef]
- Kim, T.J.; Jin, H.T.; Hur, S.Y.; Yang, H.G.; Seo, Y.B.; Hong, S.R.; Lee, C.W.; Kim, S.; Woo, J.W.; Park, K.S.; et al. Clearance of persistent HPV infection and cervical lesion by therapeutic DNA vaccine in CIN3 patients. Nat. Commun. 2014, 5, 5317. [Google Scholar] [CrossRef]
- Choi, Y.J.; Hur, S.Y.; Kim, T.J.; Hong, S.R.; Lee, J.K.; Cho, C.H.; Park, K.S.; Woo, J.W.; Sung, Y.C.; Suh, Y.S.; et al. A Phase II, Prospective, Randomized, Multicenter, Open-Label Study of GX-188E, an HPV DNA Vaccine, in Patients with Cervical Intraepithelial Neoplasia 3. Clin. Cancer Res. 2020, 26, 1616–1623. [Google Scholar] [CrossRef]
- Celis, E.; Sette, A.; Grey, H.M. Epitope selection and development of peptide based vaccines to treat cancer. Semin. Cancer Biol. 1995, 6, 329–336. [Google Scholar] [CrossRef]
- Kenter, G.G.; Welters, M.J.; Valentijn, A.R.; Lowik, M.J.; Berends-van der Meer, D.M.; Vloon, A.P.; Essahsah, F.; Fathers, L.M.; Offringa, R.; Drijfhout, J.W.; et al. Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N. Engl. J. Med. 2009, 361, 1838–1847. [Google Scholar] [CrossRef]
- Welters, M.J.; Kenter, G.G.; de Vos van Steenwijk, P.J.; Lowik, M.J.; Berends-van der Meer, D.M.; Essahsah, F.; Stynenbosch, L.F.; Vloon, A.P.; Ramwadhdoebe, T.H.; Piersma, S.J.; et al. Success or failure of vaccination for HPV16-positive vulvar lesions correlates with kinetics and phenotype of induced T-cell responses. Proc. Natl. Acad. Sci. USA 2010, 107, 11895–11899. [Google Scholar] [CrossRef]
- van Poelgeest, M.I.; Welters, M.J.; van Esch, E.M.; Stynenbosch, L.F.; Kerpershoek, G.; van Persijn van Meerten, E.L.; van den Hende, M.; Lowik, M.J.; Berends-van der Meer, D.M.; Fathers, L.M.; et al. HPV16 synthetic long peptide (HPV16-SLP) vaccination therapy of patients with advanced or recurrent HPV16-induced gynecological carcinoma, a phase II trial. J. Transl. Med. 2013, 11, 88. [Google Scholar] [CrossRef]
- de Vos van Steenwijk, P.J.; Ramwadhdoebe, T.H.; Lowik, M.J.; van der Minne, C.E.; Berends-van der Meer, D.M.; Fathers, L.M.; Valentijn, A.R.; Oostendorp, J.; Fleuren, G.J.; Hellebrekers, B.W.; et al. A placebo-controlled randomized HPV16 synthetic long-peptide vaccination study in women with high-grade cervical squamous intraepithelial lesions. Cancer Immunol. Immunother. 2012, 61, 1485–1492. [Google Scholar] [CrossRef]
- de Vos van Steenwijk, P.J.; van Poelgeest, M.I.; Ramwadhdoebe, T.H.; Lowik, M.J.; Berends-van der Meer, D.M.; van der Minne, C.E.; Loof, N.M.; Stynenbosch, L.F.; Fathers, L.M.; Valentijn, A.R.; et al. The long-term immune response after HPV16 peptide vaccination in women with low-grade pre-malignant disorders of the uterine cervix: A placebo-controlled phase II study. Cancer Immunol. Immunother. 2014, 63, 147–160. [Google Scholar] [CrossRef]
- Greenfield, W.W.; Stratton, S.L.; Myrick, R.S.; Vaughn, R.; Donnalley, L.M.; Coleman, H.N.; Mercado, M.; Moerman-Herzog, A.M.; Spencer, H.J.; Andrews-Collins, N.R.; et al. A phase I dose-escalation clinical trial of a peptide-based human papillomavirus therapeutic vaccine with Candida skin test reagent as a novel vaccine adjuvant for treating women with biopsy-proven cervical intraepithelial neoplasia 2/3. Oncoimmunology 2015, 4, e1031439. [Google Scholar] [CrossRef]
- de Jong, A.; O’Neill, T.; Khan, A.Y.; Kwappenberg, K.M.; Chisholm, S.E.; Whittle, N.R.; Dobson, J.A.; Jack, L.C.; St Clair Roberts, J.A.; Offringa, R.; et al. Enhancement of human papillomavirus (HPV) type 16 E6 and E7-specific T-cell immunity in healthy volunteers through vaccination with TA-CIN, an HPV16 L2E7E6 fusion protein vaccine. Vaccine 2002, 20, 3456–3464. [Google Scholar] [CrossRef]
- Gambhira, R.; Gravitt, P.E.; Bossis, I.; Stern, P.L.; Viscidi, R.P.; Roden, R.B. Vaccination of healthy volunteers with human papillomavirus type 16 L2E7E6 fusion protein induces serum antibody that neutralizes across papillomavirus species. Cancer Res. 2006, 66, 11120–11124. [Google Scholar] [CrossRef]
- Karanam, B.; Gambhira, R.; Peng, S.; Jagu, S.; Kim, D.J.; Ketner, G.W.; Stern, P.L.; Adams, R.J.; Roden, R.B. Vaccination with HPV16 L2E6E7 fusion protein in GPI-0100 adjuvant elicits protective humoral and cell-mediated immunity. Vaccine 2009, 27, 1040–1049. [Google Scholar] [CrossRef]
- Yuan, J.; Ni, G.; Wang, T.; Mounsey, K.; Cavezza, S.; Pan, X.; Liu, X. Genital warts treatment: Beyond imiquimod. Hum. Vaccines Immunother. 2018, 14, 1815–1819. [Google Scholar] [CrossRef]
- Bilu, D.; Sauder, D.N. Imiquimod: Modes of action. Br. J. Dermatol. 2003, 149 (Suppl. S66), 5–8. [Google Scholar] [CrossRef]
- Daayana, S.; Elkord, E.; Winters, U.; Pawlita, M.; Roden, R.; Stern, P.L.; Kitchener, H.C. Phase II trial of imiquimod and HPV therapeutic vaccination in patients with vulval intraepithelial neoplasia. Br. J. Cancer 2010, 102, 1129–1136. [Google Scholar] [CrossRef]
- Archambault, J.; Melendy, T. Targeting human papillomavirus genome replication for antiviral drug discovery. Antivir. Ther. 2013, 18, 271–283. [Google Scholar] [CrossRef]
- Cundy, K.C. Clinical pharmacokinetics of the antiviral nucleotide analogues cidofovir and adefovir. Clin. Pharmacokinet. 1999, 36, 127–143. [Google Scholar] [CrossRef]
- Johnson, J.A.; Gangemi, J.D. Selective inhibition of human papillomavirus-induced cell proliferation by (S)-1-[3-hydroxy-2-(phosphonylmethoxy)propyl]cytosine. Antimicrob. Agents Chemother. 1999, 43, 1198–1205. [Google Scholar] [CrossRef]
- Abdulkarim, B.; Sabri, S.; Deutsch, E.; Chagraoui, H.; Maggiorella, L.; Thierry, J.; Eschwege, F.; Vainchenker, W.; Chouaib, S.; Bourhis, J. Antiviral agent Cidofovir restores p53 function and enhances the radiosensitivity in HPV-associated cancers. Oncogene 2002, 21, 2334–2346. [Google Scholar] [CrossRef]
- Yang, J.; Dai, L.X.; Chen, M.; Li, B.; Ding, N.; Li, G.; Liu, Y.Q.; Li, M.Y.; Wang, B.N.; Shi, X.L.; et al. Inhibition of antiviral drug cidofovir on proliferation of human papillomavirus-infected cervical cancer cells. Exp. Ther. Med. 2016, 12, 2965–2973. [Google Scholar] [CrossRef]
- Mertens, B.; Nogueira, T.; Stranska, R.; Naesens, L.; Andrei, G.; Snoeck, R. Cidofovir is active against human papillomavirus positive and negative head and neck and cervical tumor cells by causing DNA damage as one of its working mechanisms. Oncotarget 2016, 7, 47302–47318. [Google Scholar] [CrossRef]
- Andrei, G.; Snoeck, R.; Piette, J.; Delvenne, P.; De Clercq, E. Inhibiting effects of cidofovir (HPMPC) on the growth of the human cervical carcinoma (SiHa) xenografts in athymic nude mice. Oncol. Res. 1998, 10, 533–539. [Google Scholar]
- Van Pachterbeke, C.; Bucella, D.; Rozenberg, S.; Manigart, Y.; Gilles, C.; Larsimont, D.; Vanden Houte, K.; Reynders, M.; Snoeck, R.; Bossens, M. Topical treatment of CIN 2+ by cidofovir: Results of a phase II, double-blind, prospective, placebo-controlled study. Gynecol. Oncol. 2009, 115, 69–74. [Google Scholar] [CrossRef]
- Deutsch, E.; Haie-Meder, C.; Bayar, M.A.; Mondini, M.; Laporte, M.; Mazeron, R.; Adam, J.; Varga, A.; Vassal, G.; Magne, N.; et al. Phase I trial evaluating the antiviral agent Cidofovir in combination with chemoradiation in cervical cancer patients. Oncotarget 2016, 7, 25549–25557. [Google Scholar] [CrossRef]
- Wang, Y.; Coulombe, R.; Cameron, D.R.; Thauvette, L.; Massariol, M.J.; Amon, L.M.; Fink, D.; Titolo, S.; Welchner, E.; Yoakim, C.; et al. Crystal structure of the E2 transactivation domain of human papillomavirus type 11 bound to a protein interaction inhibitor. J. Biol. Chem. 2004, 279, 6976–6985. [Google Scholar] [CrossRef]
- White, P.W.; Faucher, A.M.; Massariol, M.J.; Welchner, E.; Rancourt, J.; Cartier, M.; Archambault, J. Biphenylsulfonacetic acid inhibitors of the human papillomavirus type 6 E1 helicase inhibit ATP hydrolysis by an allosteric mechanism involving tyrosine 486. Antimicrob. Agents Chemother. 2005, 49, 4834–4842. [Google Scholar] [CrossRef]
- Chen, J.; Gu, W.; Yang, L.; Chen, C.; Shao, R.; Xu, K.; Xu, Z.P. Nanotechnology in the management of cervical cancer. Rev. Med. Virol. 2015, 25 (Suppl. S1), 72–83. [Google Scholar] [CrossRef] [PubMed]
- Jain, K.K. Nanomedicine: Application of nanobiotechnology in medical practice. Med. Princ. Pract. 2008, 17, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Menon, I.; Bagwe, P.; Gomes, K.B.; Bajaj, L.; Gala, R.; Uddin, M.N.; D’Souza, M.J.; Zughaier, S.M. Microneedles: A New Generation Vaccine Delivery System. Micromachines 2021, 12, 435. [Google Scholar] [CrossRef] [PubMed]
- Meyer, B.K.; Kendall, M.A.F.; Williams, D.M.; Bett, A.J.; Dubey, S.; Gentzel, R.C.; Casimiro, D.; Forster, A.; Corbett, H.; Crichton, M.; et al. Immune response and reactogenicity of an unadjuvanted intradermally delivered human papillomavirus vaccine using a first generation Nanopatch in rhesus macaques: An exploratory, pre-clinical feasibility assessment. Vaccine X 2019, 2, 100030. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, S.; Guo, F.; Zhang, W.; Wang, Y.; Pan, Y. Induction of apoptosis by chitosan/HPV16 E7 siRNA complexes in cervical cancer cells. Mol. Med. Rep. 2013, 7, 998–1002. [Google Scholar] [CrossRef] [PubMed]
- Saengkrit, N.; Sanitrum, P.; Woramongkolchai, N.; Saesoo, S.; Pimpha, N.; Chaleawlert-Umpon, S.; Tencomnao, T.; Puttipipatkhachorn, S. The PEI-introduced CS shell/PMMA core nanoparticle for silencing the expression of E6/E7 oncogenes in human cervical cells. Carbohydr. Polym. 2012, 90, 1323–1329. [Google Scholar] [CrossRef]
- Xu, C.; Liu, W.; Hu, Y.; Li, W.; Di, W. Bioinspired tumor-homing nanoplatform for co-delivery of paclitaxel and siRNA-E7 to HPV-related cervical malignancies for synergistic therapy. Theranostics 2020, 10, 3325–3339. [Google Scholar] [CrossRef]




| Process | References |
|---|---|
| HPV16 E5 triggers malignant transformation of murine keratinocytes | [20] |
| HPV16 E5 leads to cell growth in low serum and anchorage-independent growth of murine fibroblasts | [21] |
| HPV16 E5 stimulates the transforming activity of the epidermal growth factor receptor and lengthens receptor action by delaying its degradation | [22,23] |
| HPV16 E5 gene cooperates with E7 to stimulate cell proliferation and increases viral gene expression | [24] |
| HPV16 E5 enhances endothelin-1-induced keratinocyte growth | [25] |
| HPV16 E5 inhibits endocytic trafficking | [26] |
| HPV16 E5 impairs apoptosis in the early stages of viral infection in human keratinocytes | [27] |
| HPV16 E5 protects human foreskin keratinocytes from UV radiation-induced apoptosis | [28] |
| HPV16 E5 down-regulates surface HLA class I allowing persistent infection by avoiding host immune clearance | [29] |
| EGFR cooperates with HPV16 E5 to induce hyperplasia in mice | [30] |
| HPV16 E5 up-regulates COX-2 by a mechanism dependent on NF-kB and AP1 | [31] |
| HPV16 E5 increases PTGER4 receptor for PGE2 in cervical cancer cells | [32] |
| HPV16 E5 represses the expression of stress pathway genes -XBP-1 and COX-2 in genital keratinocytes | [33] |
| HPV16 E5 synergizes EGFR signaling to enhance cell cycle progression and down-regulation of p27 | [34] |
| HPV16 E5 inhibits apoptosis by proteasome-dependent degradation of Bax in human cervical cancer cells | [35] |
| Expression of HPV16 E5 produces enlarged nuclei and polyploidy in human keratinocytes | [36] |
| HPV16 E5 modulates the expression of host microRNAs miR-146a, miR-203, and miR-324-5p, and their target genes | [37] |
| HPV16 E5 induces switching from FGFR2b to FGFR2c and epithelial–mesenchymal transition | [38] |
| HPV18 E5 supports cell cycle progression and impairs epithelial differentiation by modulating EGFR signaling | [39] |
| HPV16 E5 increases MET, a growth factor receptor critical for tumor progression in human keratinocytes | [40] |
| HPV18 E5 cooperates with E6 and E7 in promoting cell invasion and in modulating the cellular redox state | [41] |
| Protein Name (Symbol, Common Name) | Consequence of Interaction with E7 | Reference |
|---|---|---|
| Cyclin-dependent kinase inhibitor 1B (CDKN1B, p27) | A cyclin-dependent kinase inhibitor. Inactivation of p27 by E7 promotes cell cycle S phase entry | [57] |
| Cyclin E1 (CCNE1, cyclin E) | A modulator of the cell cycle that functions as a regulatory subunit of CDK2. Enhanced kinase activity mediated by E7 interaction favors cell cycle G1/S transition | [58] |
| Cyclin-dependent kinase inhibitor 1A (CDKN1A, p21) | Another cyclin-dependent kinase inhibitor. E7 interaction with p21 promotes pRB phosphorylation by activated CDK2-cyclin A, enabling cell cycle progression | [59] |
| TATA-box binding protein (TBP, TFIID) | A critical factor in transcription initiation. Interaction between E7 and TBP participates in the transformation of epithelial cells | [60] |
| Proteasome 26S subunit, ATPase 4 (PSMC4, S4 subunit of the 26S proteasome) | An ATPase essential for protein turnover by the 26S proteasome. Upon interaction with E7, this protein might participate in pRB degradation by 26S proteasome favoring in this way the cell cycle progression | [61] |
| Retinoblastoma (pRB) RB transcriptional corepressor like 1 (RBL1, p107) RB transcriptional corepressor like 2 (RBL2, p130) | Hypophosphorylated pRB, p107, and p130 tumor suppressors inhibit E2F-mediated transcription initiation. Interaction of these proteins with E7 alleviates transcriptional inhibition promoting premature entry into the S-phase of the cell cycle | [62] |
| Fork head box M1 (FOXM1, fork head domain transcription factor MPP2) | A transcription factor involved in cell proliferation regulation. E7 enhances the transactivation and transformation properties of matrix metallopeptidase (MMP)-2 | [63] |
| POU class 5 homeobox 1 (POU5F1, OCT4) | OCT4 is a transcription factor essential for stem cell pluripotency and embryonic development. E7 expression in differentiated cells stimulates OCT4 activity | [64] |
| Interferon regulatory factor 1 (IRF1, IRF-1) | A tumor suppressor gene with transcriptional regulation activity involved in immune responses. E7 direct inactivation of IRF1 promotes immune evasion of HPV in cancer | [65] |
| E1A binding protein P300 (EP300, Transcriptional coactivator P300) | A general transcriptional coactivator. By binding to P300, E7 impaired transcriptional regulation | [66] |
| Lysine acetyltransferase 2B (KAT2B, PCAF) | Another general transcriptional coactivator. E7 interaction reduces acetyltransferase activity impairing transcriptional regulation | [67] |
| Cyclin A2 (CCNA2, cyclin A) | A critical cell cycle regulator whose function activates cyclin-dependent kinase 2 (CDK2). E7 promotes cell cycle transition through G1/S and G2/M by activation of CDK2/cyclin A | [68] |
| E2F transcription factor 6 (E2F6, transcription factor E2F6) | E2F6 is a transcription factor that negatively regulates transcription. Interaction between E2F6 and E7 abrogates inhibitory action of E2F6, which extends the S-phase | [69] |
| Rho GTPase activating protein 35 (ARHGAP35, p190RhoGAP) | A GTPase activating protein for RhoA. Binding of E7 alters actin cytoskeleton dynamics and cell migration | [70] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Quiroz, J.; Vázquez-Almazán, B.; García-Becerra, R.; Díaz, L.; Avila, E. The Interaction of Human Papillomavirus Infection and Prostaglandin E2 Signaling in Carcinogenesis: A Focus on Cervical Cancer Therapeutics. Cells 2022, 11, 2528. https://doi.org/10.3390/cells11162528
García-Quiroz J, Vázquez-Almazán B, García-Becerra R, Díaz L, Avila E. The Interaction of Human Papillomavirus Infection and Prostaglandin E2 Signaling in Carcinogenesis: A Focus on Cervical Cancer Therapeutics. Cells. 2022; 11(16):2528. https://doi.org/10.3390/cells11162528
Chicago/Turabian StyleGarcía-Quiroz, Janice, Bismarck Vázquez-Almazán, Rocío García-Becerra, Lorenza Díaz, and Euclides Avila. 2022. "The Interaction of Human Papillomavirus Infection and Prostaglandin E2 Signaling in Carcinogenesis: A Focus on Cervical Cancer Therapeutics" Cells 11, no. 16: 2528. https://doi.org/10.3390/cells11162528
APA StyleGarcía-Quiroz, J., Vázquez-Almazán, B., García-Becerra, R., Díaz, L., & Avila, E. (2022). The Interaction of Human Papillomavirus Infection and Prostaglandin E2 Signaling in Carcinogenesis: A Focus on Cervical Cancer Therapeutics. Cells, 11(16), 2528. https://doi.org/10.3390/cells11162528