
An Epigenetic Role of Mitochondria in Cancer

 ,
, 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Mitochondria Function in Epigenetic Regulation
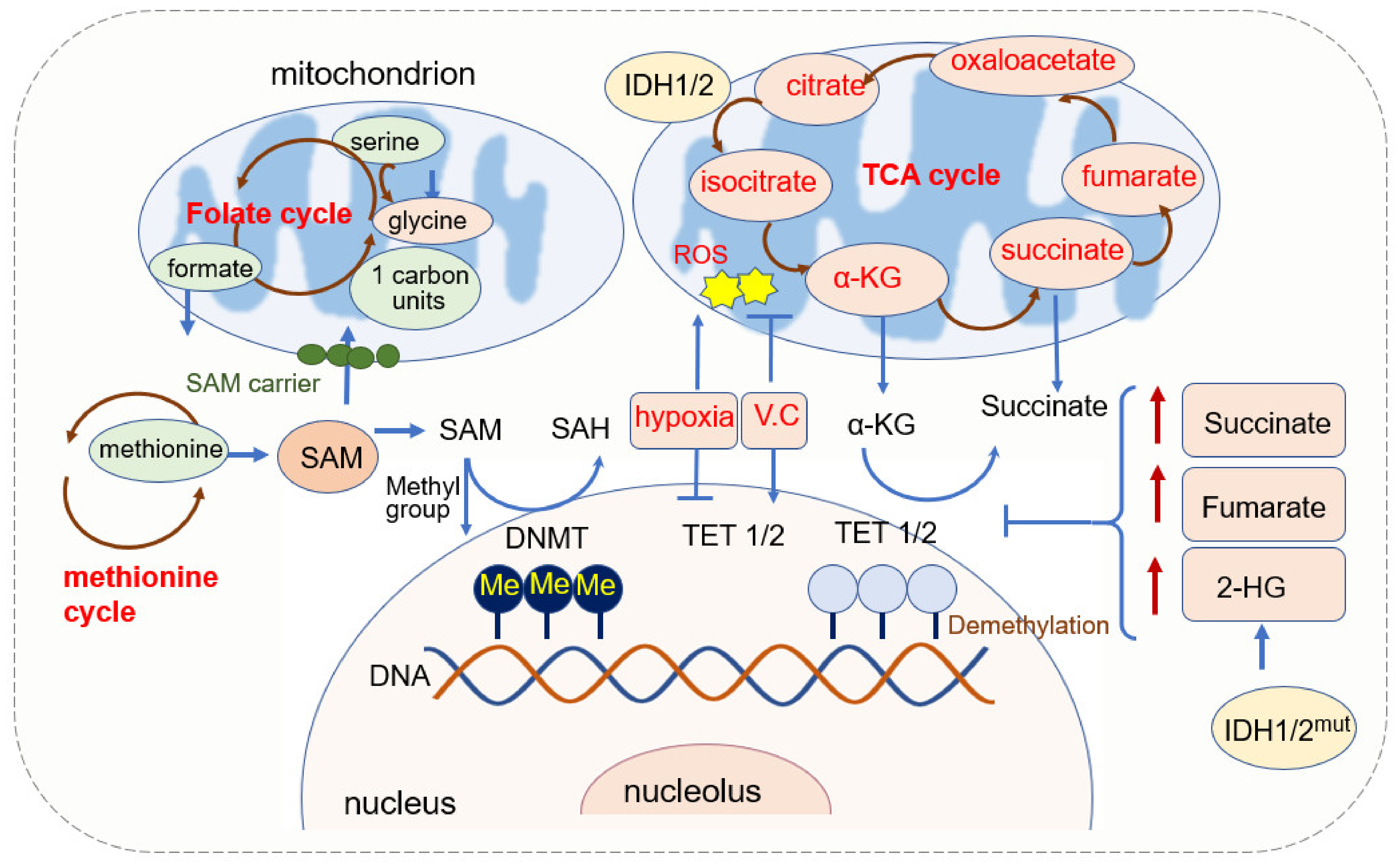
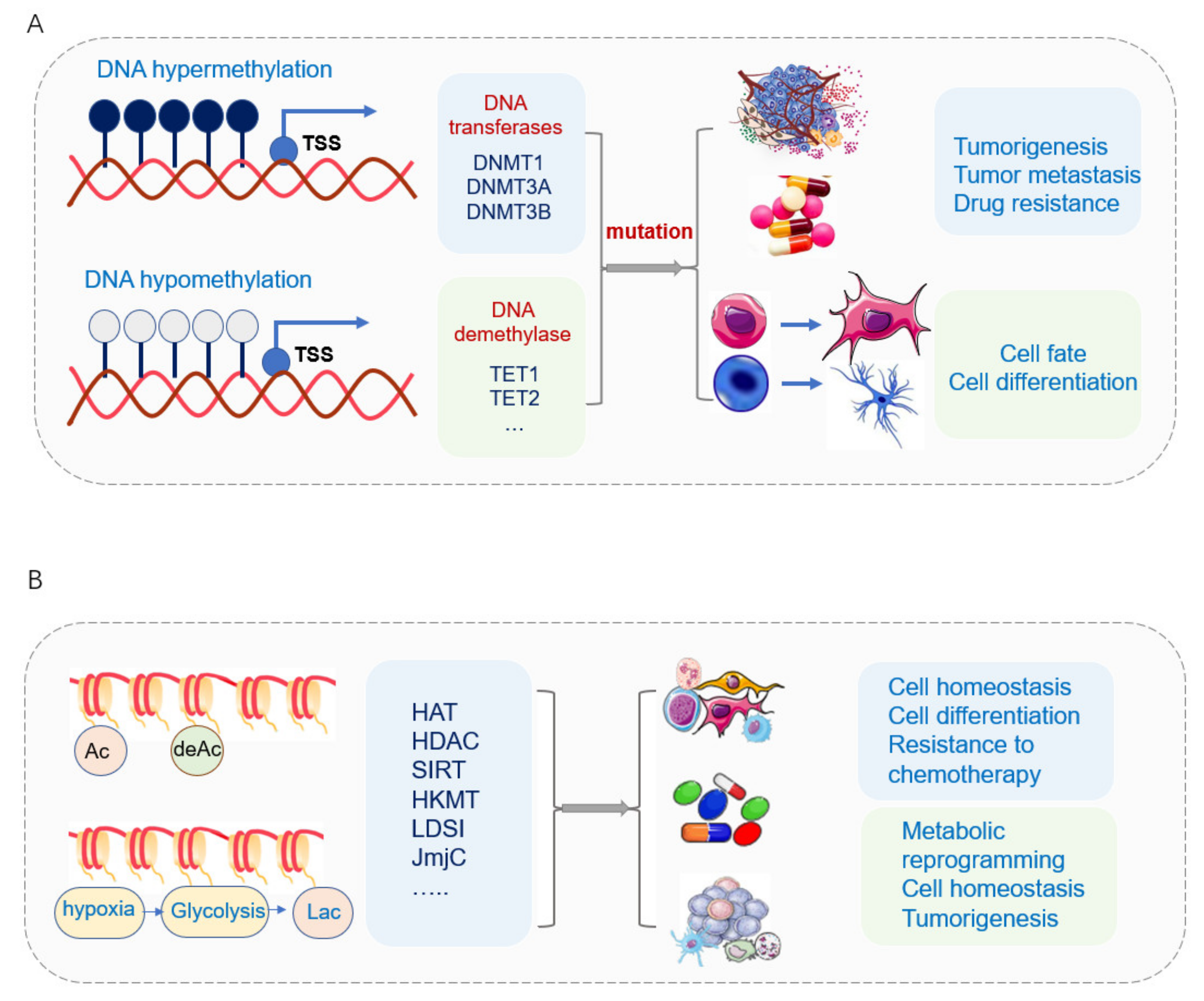
2.1. Mitochondria and DNA Methylation
2.1.1. SAM Source and Mitochondrial Regulation of SAM
2.1.2. α-KG Source and Its Function in DNA Methylation
2.1.3. Mitochondrial ROS and DNA Methylation
2.1.4. Mitochondria Dysfunction and DNA Methylation
2.2. Mitochondria and RNA Methylation
2.2.1. The Effect of Mitochondria-Associated Metabolites on RNA Methylation
2.2.2. The Roles of RNA Methylation in Mitochondria
2.3. Mitochondria and Histone Modifications
2.3.1. Mitochondria and Histone Acetylation
- a.
- Concept and enzymes of histone acetylation
- b.
- Function of histone acetylation
- c.
- Mitochondria regulate histone acetylation
- d.
- NAD+ function in histone acetylation
- e.
- Acetyl-CoA function in histone acetylation
2.3.2. Mitochondria and Histone Methylation
- a.
- Histone methylation
- b.
- Enzymes of histone methylation
- c.
- Mitochondrial metabolites and histone methylation
2.3.3. Mitochondria and Histone Succinylation
- a.
- Concept of histone succinylation
- b.
- Enzymes of histone succinylation
- c.
- Mitochondrial metabolites and histone succinylation
2.3.4. Mitochondria and Histone O-GlcNAcylation
- a.
- Concept of O-GlcNAcylation
- b.
- Enzymes of O-GlcNAcylation
- c.
- Mitochondria and O-GlcNAcylation
- d.
- O-GlcNAcylation and other modifications
2.3.5. Mitochondria and Histone Lactylation
- a.
- Concept and function of histone lactylation
- b.
- Regulation of histone lactylation
2.4. Epigenetic Modification Regulates Mitochondrial Function
3. Epigenetics in Normal Development and Tumorigenesis
3.1. DNA Methylation in Cell Development and Tumorigenesis
3.1.1. DNA Methylation in Normal Development
3.1.2. DNA Methylation in Tumorigenesis
3.1.3. DNA Methylation in Cancer Stem Cell
3.2. Histone Acetylation and Methylation in Normal Development and Tumorigenesis
3.2.1. Histone Acetylation and Methylation in Normal Development
3.2.2. Histone Acetylation and Methylation in Tumorigenesis
3.2.3. Mitochondrial Metabolites Contribute to Normal Development and Tumorigenesis via Regulation of Epigenetics
4. Reagents and Drugs Targeting Mitochondria and Epigenetics for Cancer Treatment
4.1. Targeting Mitochondrial Metabolites and Enzymes in TCA in Cancer
4.2. Targeting Epigenetics in Cancer
4.2.1. Targeting DNA Methylation
- a.
- Targeting DNMTs
- b.
- Targeting demethylases
4.2.2. Targeting Histone Methylation
4.2.3. Targeting Histone Acetylation
4.2.4. Targeting Histone Lactylation
5. Conclusions and Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lane, N.; Martin, W. The energetics of genome complexity. Nature 2010, 467, 929–934. [Google Scholar] [CrossRef] [PubMed]
- Archibald, J.M. Endosymbiosis and Eukaryotic Cell Evolution. Curr. Biol. 2015, 25, R911–R921. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.P.; Price, N.L.; Ling, A.J.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 2013, 155, 1624–1638. [Google Scholar] [CrossRef]
- Janssen, J.J.E.; Grefte, S.; Keijer, J.; de Boer, V.C.J. Mito-Nuclear Communication by Mitochondrial Metabolites and Its Regulation by B-Vitamins. Front. Physiol. 2019, 10, 78. [Google Scholar] [CrossRef]
- Rigon, M.; Townley, A.R.; Campanella, M. Mitochondria ensure immune surveillance by retro-communication with the nucleus. Cell Metab. 2021, 33, 853–855. [Google Scholar] [CrossRef]
- Scatena, R. Mitochondria and drugs. Adv. Exp. Med. Biol. 2012, 942, 329–346. [Google Scholar] [CrossRef]
- Liu, Y.E.; Shi, Y.F. Mitochondria as a target in cancer treatment. Medcomm 2020, 1, 129–139. [Google Scholar] [CrossRef]
- Chen, K.; Zhang, J.; Beeraka, N.M.; Tang, C.; Babayeva, Y.V.; Sinelnikov, M.Y.; Zhang, X.; Zhang, J.; Liu, J.; Reshetov, I.V.; et al. Advances in the Prevention and Treatment of Obesity-Driven Effects in Breast Cancers. Front. Oncol. 2022, 12, 820968. [Google Scholar] [CrossRef]
- Chen, K.; Lu, P.; Beeraka, N.M.; Sukocheva, O.A.; Madhunapantula, S.V.; Liu, J.; Sinelnikov, M.Y.; Nikolenko, V.N.; Bulygin, K.V.; Mikhaleva, L.M.; et al. Mitochondrial mutations and mitoepigenetics: Focus on regulation of oxidative stress-induced responses in breast cancers. Semin. Cancer Biol. 2022, 83, 556–569. [Google Scholar] [CrossRef] [PubMed]
- Matilainen, O.; Quiros, P.M.; Auwerx, J. Mitochondria and Epigenetics—Crosstalk in Homeostasis and Stress. Trends Cell Biol. 2017, 27, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Waddington, C.H. The Epigenotype. Int. J. Epidemiol. 2011, 41, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Holliday, R.; Pugh, J.E. DNA modification mechanisms and gene activity during development. Science 1975, 187, 226–232. [Google Scholar] [CrossRef]
- Riggs, A.D. X-Inactivation, Differentiation, and DNA Methylation. Cytogenet. Genome Res. 1975, 14, 9–25. [Google Scholar] [CrossRef]
- Sun, L.; Zhang, H.; Gao, P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell 2021, 13, 877–919. [Google Scholar] [CrossRef]
- Daniel, M.; Tollefsbol, T.O. Epigenetic linkage of aging, cancer and nutrition. J. Exp. Biol. 2015, 218, 59–70. [Google Scholar] [CrossRef]
- Martin, S.L.; Hardy, T.M.; Tollefsbol, T.O. Medicinal chemistry of the epigenetic diet and caloric restriction. Curr. Med. Chem. 2013, 20, 4050–4059. [Google Scholar] [CrossRef]
- Ross, S.A.; Dwyer, J.; Umar, A.; Kagan, J.; Verma, M.; Van Bemmel, D.M.; Dunn, B.K. Introduction: Diet, epigenetic events and cancer prevention. Nutr. Rev. 2008, 66 (Suppl. S1), S1–S6. [Google Scholar] [CrossRef]
- Wallace, D.C.; Fan, W. Energetics, epigenetics, mitochondrial genetics. Mitochondrion 2010, 10, 12–31. [Google Scholar] [CrossRef]
- Zhang, L.; Lu, Q.; Chang, C. Epigenetics in Health and Disease. Adv. Exp. Med. Biol. 2020, 1253, 3–55. [Google Scholar] [CrossRef] [PubMed]
- Bowden, G.T.; Schneider, B.; Domann, R.; Kulesz-Martin, M. Oncogene activation and tumor suppressor gene inactivation during multistage mouse skin carcinogenesis. Cancer Res. 1994, 54, 1882s–1885s. [Google Scholar] [PubMed]
- Solomon, H.; Brosh, R.; Buganim, Y.; Rotter, V. Inactivation of the p53 tumor suppressor gene and activation of the Ras oncogene: Cooperative events in tumorigenesis. Discov. Med. 2010, 9, 448–454. [Google Scholar] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.K. DNA Damage, Mutagenesis and Cancer. Int. J. Mol. Sci. 2018, 19, 970. [Google Scholar] [CrossRef]
- Blackadar, C.B. Historical review of the causes of cancer. World J. Clin. Oncol. 2016, 7, 54–86. [Google Scholar] [CrossRef]
- Wu, F.; Fan, J.; He, Y.; Xiong, A.; Yu, J.; Li, Y.; Zhang, Y.; Zhao, W.; Zhou, F.; Li, W.; et al. Single-cell profiling of tumor heterogeneity and the microenvironment in advanced non-small cell lung cancer. Nat. Commun. 2021, 12, 2540. [Google Scholar] [CrossRef]
- Walcher, L.; Kistenmacher, A.K.; Suo, H.; Kitte, R.; Dluczek, S.; Strauss, A.; Blaudszun, A.R.; Yevsa, T.; Fricke, S.; Kossatz-Boehlert, U. Cancer Stem Cells-Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front. Immunol. 2020, 11, 1280. [Google Scholar] [CrossRef]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; Ridgway, R.A.; van Es, J.H.; van de Wetering, M.; Begthel, H.; van den Born, M.; Danenberg, E.; Clarke, A.R.; Sansom, O.J.; Clevers, H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009, 457, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.Q.; Gibson, P.; Currle, D.S.; Tong, Y.; Richardson, R.J.; Bayazitov, I.T.; Poppleton, H.; Zakharenko, S.; Ellison, D.W.; Gilbertson, R.J. Prominin 1 marks intestinal stem cells that are susceptible to neoplastic transformation. Nature 2009, 457, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Roskams, T. Liver stem cells and their implication in hepatocellular and cholangiocarcinoma. Oncogene 2006, 25, 3818–3822. [Google Scholar] [CrossRef]
- Toh, T.B.; Lim, J.J.; Chow, E.K. Epigenetics in cancer stem cells. Mol. Cancer 2017, 16, 29. [Google Scholar] [CrossRef]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer’s Achilles’ heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef]
- Miranda Furtado, C.L.; Dos Santos Luciano, M.C.; Silva Santos, R.D.; Furtado, G.P.; Moraes, M.O.; Pessoa, C. Epidrugs: Targeting epigenetic marks in cancer treatment. Epigenetics 2019, 14, 1164–1176. [Google Scholar] [CrossRef]
- Reik, W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 2007, 447, 425–432. [Google Scholar] [CrossRef]
- Cai, S.F.; Levine, R.L. Genetic and epigenetic determinants of AML pathogenesis. Semin. Hematol. 2019, 56, 84–89. [Google Scholar] [CrossRef]
- Ilango, S.; Paital, B.; Jayachandran, P.; Padma, P.R.; Nirmaladevi, R. Epigenetic alterations in cancer. Front. Biosci. 2020, 25, 1058–1109. [Google Scholar] [CrossRef]
- Mazor, T.; Pankov, A.; Song, J.S.; Costello, J.F. Intratumoral Heterogeneity of the Epigenome. Cancer Cell 2016, 29, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, E.N.; Scaffidi, P. Epigenetics and Cancer Stem Cells: Unleashing, Hijacking, and Restricting Cellular Plasticity. Trends Cancer 2017, 3, 372–386. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.K.; Jirtle, R.L. Imprinting evolution and the price of silence. Bioessays 2003, 25, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Hampton, D.D.; Jirtle, R.L. Imprinting evolution and human health. Mamm. Genome 2009, 20, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Epigenetic gene silencing in cancer: The DNA hypermethylome. Hum. Mol. Genet. 2007, 16, R50–R59. [Google Scholar] [CrossRef] [PubMed]
- Marusyk, A.; Almendro, V.; Polyak, K. Intra-tumour heterogeneity: A looking glass for cancer? Nat. Rev. Cancer 2012, 12, 323–334. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef]
- Shackleton, M.; Quintana, E.; Fearon, E.R.; Morrison, S.J. Heterogeneity in cancer: Cancer stem cells versus clonal evolution. Cell 2009, 138, 822–829. [Google Scholar] [CrossRef]
- Peng, Y.; Liu, H.; Liu, J.; Long, J. Post-translational modifications on mitochondrial metabolic enzymes in cancer. Free Radic. Biol. Med. 2022, 179, 11–23. [Google Scholar] [CrossRef]
- Dai, Z.; Ramesh, V.; Locasale, J.W. The evolving metabolic landscape of chromatin biology and epigenetics. Nat. Rev. Genet. 2020, 21, 737–753. [Google Scholar] [CrossRef]
- Zhu, J.; Thompson, C.B. Metabolic regulation of cell growth and proliferation. Nat. Rev. Mol. Cell Biol. 2019, 20, 436–450. [Google Scholar] [CrossRef] [PubMed]
- Zhou, V.W.; Goren, A.; Bernstein, B.E. Charting histone modifications and the functional organization of mammalian genomes. Nat. Rev. Genet. 2011, 12, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Margueron, R.; Reinberg, D. Chromatin structure and the inheritance of epigenetic information. Nat. Rev. Genet. 2010, 11, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Zhang, X.; Liu, C.; Liu, Q.; Chai, H.; Luo, Y.; Li, S. Role of Mitochondria in Neurodegenerative Diseases: From an Epigenetic Perspective. Front. Cell Dev. Biol. 2021, 9, 688789. [Google Scholar] [CrossRef] [PubMed]
- Paik, W.K.; Paik, D.C.; Kim, S. Historical review: The field of protein methylation. Trends Biochem. Sci. 2007, 32, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Bird, A.P. DNA methylation and the frequency of CpG in animal DNA. Nucleic Acids Res. 1980, 8, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Chen, T. DNA Methylation Reprogramming during Mammalian Development. Genes 2019, 10, 257. [Google Scholar] [CrossRef]
- Li, C.; Fan, Y.; Li, G.; Xu, X.; Duan, J.; Li, R.; Kang, X.; Ma, X.; Chen, X.; Ke, Y.; et al. DNA methylation reprogramming of functional elements during mammalian embryonic development. Cell Discov. 2018, 4, 41. [Google Scholar] [CrossRef]
- Kulis, M.; Esteller, M. DNA methylation and cancer. Adv. Genet. 2010, 70, 27–56. [Google Scholar] [CrossRef]
- Bestor, T.H. The DNA methyltransferases of mammals. Hum. Mol. Genet. 2000, 9, 2395–2402. [Google Scholar] [CrossRef]
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018, 19, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, K.X.; Lu, G.Z.; Zhao, X.H. Research progress on 5hmC and TET dioxygenases in neurodevelopment and neurological diseases. Yi Chuan 2017, 39, 1138–1149. [Google Scholar] [CrossRef]
- Matuleviciute, R.; Cunha, P.P.; Johnson, R.S.; Foskolou, I.P. Oxygen regulation of TET enzymes. FEBS J. 2021, 288, 7143–7161. [Google Scholar] [CrossRef] [PubMed]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-Hydroxyglutarate Is a Competitive Inhibitor of alpha-Ketoglutarate-Dependent Dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Tang, J.; Lai, M.; Zhang, H. 5-Hydroxymethylcytosine and disease. Mutat. Res. Rev. Mutat. Res. 2014, 762, 167–175. [Google Scholar] [CrossRef]
- Valinluck, V.; Sowers, L.C. Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Res. 2007, 67, 946–950. [Google Scholar] [CrossRef]
- Bhutani, N.; Burns, D.M.; Blau, H.M. DNA demethylation dynamics. Cell 2011, 146, 866–872. [Google Scholar] [CrossRef]
- Jin, S.G.; Kadam, S.; Pfeifer, G.P. Examination of the specificity of DNA methylation profiling techniques towards 5-methylcytosine and 5-hydroxymethylcytosine. Nucleic Acids Res. 2010, 38, e125. [Google Scholar] [CrossRef]
- Ouyang, Y.; Wu, Q.; Li, J.; Sun, S.; Sun, S. S-adenosylmethionine: A metabolite critical to the regulation of autophagy. Cell Prolif. 2020, 53, e12891. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. S-Adenosylmethionine. Int. J. Biochem. Cell Biol. 2000, 32, 391–395. [Google Scholar] [CrossRef]
- Agrimi, G.; Di Noia, M.A.; Marobbio, C.M.; Fiermonte, G.; Lasorsa, F.M.; Palmieri, F. Identification of the human mitochondrial S-adenosylmethionine transporter: Bacterial expression, reconstitution, functional characterization and tissue distribution. Biochem. J. 2004, 379, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Mato, J.M.; Alvarez, L.; Ortiz, P.; Pajares, M.A. S-adenosylmethionine synthesis: Molecular mechanisms and clinical implications. Pharmacol. Ther. 1997, 73, 265–280. [Google Scholar] [CrossRef]
- Finkelstein, J.D. Methionine metabolism in mammals. J. Nutr. Biochem. 1990, 1, 228–237. [Google Scholar] [CrossRef]
- Mentch, S.J.; Mehrmohamadi, M.; Huang, L.; Liu, X.J.; Gupta, D.; Mattocks, D.; Padilla, P.G.; Ables, G.; Bamman, M.M.; Thalacker-Mercer, A.E.; et al. Histone Methylation Dynamics and Gene Regulation Occur through the Sensing of One-Carbon Metabolism. Cell Metab. 2015, 22, 861–873. [Google Scholar] [CrossRef] [PubMed]
- Ducker, G.S.; Chen, L.; Morscher, R.J.; Ghergurovich, J.M.; Esposito, M.; Teng, X.; Kang, Y.; Rabinowitz, J.D. Reversal of Cytosolic One-Carbon Flux Compensates for Loss of the Mitochondrial Folate Pathway. Cell Metab. 2016, 24, 640–641. [Google Scholar] [CrossRef] [PubMed]
- Martelli, M.P.; Martino, G.; Cardinali, V.; Falini, B.; Martinelli, G.; Cerchione, C. Enasidenib and ivosidenib in AML. Minerva Med. 2020, 111, 411–426. [Google Scholar] [CrossRef]
- Liu, S.; He, L.; Yao, K. The Antioxidative Function of Alpha-Ketoglutarate and Its Applications. Biomed. Res. Int. 2018, 2018, 3408467. [Google Scholar] [CrossRef]
- Baracco, E.E.; Castoldi, F.; Durand, S.; Enot, D.P.; Tadic, J.; Kainz, K.; Madeo, F.; Chery, A.; Izzo, V.; Maiuri, M.C.; et al. alpha-Ketoglutarate inhibits autophagy. Aging 2019, 11, 3418–3431. [Google Scholar] [CrossRef]
- Asadi Shahmirzadi, A.; Edgar, D.; Liao, C.Y.; Hsu, Y.M.; Lucanic, M.; Asadi Shahmirzadi, A.; Wiley, C.D.; Gan, G.; Kim, D.E.; Kasler, H.G.; et al. Alpha-Ketoglutarate, an Endogenous Metabolite, Extends Lifespan and Compresses Morbidity in Aging Mice. Cell Metab. 2020, 32, 447–456.e6. [Google Scholar] [CrossRef] [PubMed]
- Long, L.H.; Halliwell, B. Artefacts in cell culture: Alpha-Ketoglutarate can scavenge hydrogen peroxide generated by ascorbate and epigallocatechin gallate in cell culture media. Biochem. Biophys. Res. Commun. 2011, 406, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Tran, K.A.; Dillingham, C.M.; Sridharan, R. The role of alpha-ketoglutarate-dependent proteins in pluripotency acquisition and maintenance. J. Biol. Chem. 2019, 294, 5408–5419. [Google Scholar] [CrossRef] [PubMed]
- Baksh, S.C.; Finley, L.W.S. Metabolic Coordination of Cell Fate by alpha-Ketoglutarate-Dependent Dioxygenases. Trends Cell Biol. 2021, 31, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr.; McKnight, S.L. Influence of metabolism on epigenetics and disease. Cell 2013, 153, 56–69. [Google Scholar] [CrossRef]
- Yang, Q.; Liang, X.; Sun, X.; Zhang, L.; Fu, X.; Rogers, C.J.; Berim, A.; Zhang, S.; Wang, S.; Wang, B.; et al. AMPK/alpha-Ketoglutarate Axis Dynamically Mediates DNA Demethylation in the Prdm16 Promoter and Brown Adipogenesis. Cell Metab. 2016, 24, 542–554. [Google Scholar] [CrossRef]
- Raffel, S.; Falcone, M.; Kneisel, N.; Hansson, J.; Wang, W.; Lutz, C.; Bullinger, L.; Poschet, G.; Nonnenmacher, Y.; Barnert, A.; et al. BCAT1 restricts alphaKG levels in AML stem cells leading to IDHmut-like DNA hypermethylation. Nature 2017, 551, 384–388. [Google Scholar] [CrossRef]
- Sciacovelli, M.; Goncalves, E.; Johnson, T.I.; Zecchini, V.R.; da Costa, A.S.H.; Gaude, E.; Drubbel, A.V.; Theobald, S.J.; Abbo, S.R.; Tran, M.G.B.; et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 2016, 537, 544–547. [Google Scholar] [CrossRef]
- Xiao, M.T.; Yang, H.; Xu, W.; Ma, S.H.; Lin, H.P.; Zhu, H.G.; Liu, L.X.; Liu, Y.; Yang, C.; Xu, Y.H.; et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Gene Dev. 2012, 26, 1326–1338. [Google Scholar] [CrossRef]
- Laukka, T.; Mariani, C.J.; Ihantola, T.; Cao, J.Z.; Hokkanen, J.; Kaelin, W.G., Jr.; Godley, L.A.; Koivunen, P. Fumarate and Succinate Regulate Expression of Hypoxia-inducible Genes via TET Enzymes. J. Biol. Chem. 2016, 291, 4256–4265. [Google Scholar] [CrossRef]
- Pollard, P.J.; Briere, J.J.; Alam, N.A.; Barwell, J.; Barclay, E.; Wortham, N.C.; Hunt, T.; Mitchell, M.; Olpin, S.; Moat, S.J.; et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum. Mol. Genet. 2005, 14, 2231–2239. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, A.S.; de Graaff, M.A.; Briaire-de Bruijn, I.H.; Ras, C.; Seifar, R.M.; van Minderhout, I.; Cornelisse, C.J.; Hogendoorn, P.C.; Breuning, M.H.; Suijker, J.; et al. Inactivation of SDH and FH cause loss of 5hmC and increased H3K9me3 in paraganglioma/pheochromocytoma and smooth muscle tumors. Oncotarget 2015, 6, 38777–38788. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Thienpont, B.; Galle, E.; Lambrechts, D. TET enzymes as oxygen-dependent tumor suppressors: Exciting new avenues for cancer management. Epigenomics 2016, 8, 1445–1448. [Google Scholar] [CrossRef]
- Giorgio, M.; Trinei, M.; Migliaccio, E.; Pelicci, P.G. Hydrogen peroxide: A metabolic by-product or a common mediator of ageing signals? Nat. Rev. Mol. Cell Biol. 2007, 8, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Dan Dunn, J.; Alvarez, L.A.; Zhang, X.; Soldati, T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. 2015, 6, 472–485. [Google Scholar] [CrossRef]
- Li, R.; Jia, Z.; Trush, M.A. Defining ROS in Biology and Medicine. React. Oxyg. Species 2016, 1, 9–21. [Google Scholar] [CrossRef]
- Ziech, D.; Franco, R.; Pappa, A.; Panayiotidis, M.I. Reactive oxygen species (ROS)—Induced genetic and epigenetic alterations in human carcinogenesis. Mutat. Res. 2011, 711, 167–173. [Google Scholar] [CrossRef]
- Quan, X.; Lim, S.O.; Jung, G. Reactive oxygen species downregulate catalase expression via methylation of a CpG island in the Oct-1 promoter. FEBS Lett. 2011, 585, 3436–3441. [Google Scholar] [CrossRef]
- Lim, S.O.; Gu, J.M.; Kim, M.S.; Kim, H.S.; Park, Y.N.; Park, C.K.; Cho, J.W.; Park, Y.M.; Jung, G. Epigenetic changes induced by reactive oxygen species in hepatocellular carcinoma: Methylation of the E-cadherin promoter. Gastroenterology 2008, 135, 2128–2140.e8. [Google Scholar] [CrossRef]
- Wu, Q.; Ni, X. ROS-mediated DNA methylation pattern alterations in carcinogenesis. Curr. Drug Targets 2015, 16, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Afanas’ev, I. New nucleophilic mechanisms of ros-dependent epigenetic modifications: Comparison of aging and cancer. Aging Dis. 2014, 5, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Luo, J.Y. SIRT1 Regulates UV-Induced DNA Repair through Deacetylating XPA. Mol. Cell 2010, 39, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.A.; Zhang, R.; Kim, G.Y.; Bae, S.C.; Hyun, J.W. Epigenetic changes induced by oxidative stress in colorectal cancer cells: Methylation of tumor suppressor RUNX3. Tumour Biol. 2012, 33, 403–412. [Google Scholar] [CrossRef]
- Fuks, F.; Burgers, W.A.; Brehm, A.; Hughes-Davies, L.; Kouzarides, T. DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat. Genet. 2000, 24, 88–91. [Google Scholar] [CrossRef]
- Fuks, F. DNA methylation and histone modifications: Teaming up to silence genes. Curr. Opin. Genet. Dev. 2005, 15, 490–495. [Google Scholar] [CrossRef]
- Delatte, B.; Jeschke, J.; Defrance, M.; Bachman, M.; Creppe, C.; Calonne, E.; Bizet, M.; Deplus, R.; Marroqui, L.; Libin, M.; et al. Genome-wide hydroxymethylcytosine pattern changes in response to oxidative stress. Sci. Rep. 2015, 5, 12714. [Google Scholar] [CrossRef]
- Kriaucionis, S.; Heintz, N. The Nuclear DNA Base 5-Hydroxymethylcytosine Is Present in Purkinje Neurons and the Brain. Science 2009, 324, 929–930. [Google Scholar] [CrossRef]
- Chia, N.; Wang, L.; Lu, X.; Senut, M.C.; Brenner, C.; Ruden, D.M. Hypothesis: Environmental regulation of 5-hydroxymethylcytosine by oxidative stress. Epigenetics 2011, 6, 853–856. [Google Scholar] [CrossRef]
- Cimmino, L.; Dolgalev, I.; Wang, Y.; Yoshimi, A.; Martin, G.H.; Wang, J.; Ng, V.; Xia, B.; Witkowski, M.T.; Mitchell-Flack, M.; et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell 2017, 170, 1079–1095.e20. [Google Scholar] [CrossRef]
- Minor, E.A.; Court, B.L.; Young, J.I.; Wang, G. Ascorbate induces ten-eleven translocation (Tet) methylcytosine dioxygenase-mediated generation of 5-hydroxymethylcytosine. J. Biol. Chem. 2013, 288, 13669–13674. [Google Scholar] [CrossRef] [PubMed]
- Yin, R.; Mao, S.Q.; Zhao, B.; Chong, Z.; Yang, Y.; Zhao, C.; Zhang, D.; Huang, H.; Gao, J.; Li, Z.; et al. Ascorbic acid enhances Tet-mediated 5-methylcytosine oxidation and promotes DNA demethylation in mammals. J. Am. Chem. Soc. 2013, 135, 10396–10403. [Google Scholar] [CrossRef] [PubMed]
- Thienpont, B.; Steinbacher, J.; Zhao, H.; D’Anna, F.; Kuchnio, A.; Ploumakis, A.; Ghesquiere, B.; Van Dyck, L.; Boeckx, B.; Schoonjans, L.; et al. Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature 2016, 537, 63–68. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, A.R.; Rouleau, J.; Szyf, M. Regulation of DNA methylation by the Ras signaling pathway. J. Biol. Chem. 1995, 270, 11327–11337. [Google Scholar] [CrossRef]
- Ploumakis, A.; Coleman, M.L. OH, the Places You’ll Go! Hydroxylation, Gene Expression, and Cancer. Mol. Cell 2015, 58, 729–741. [Google Scholar] [CrossRef]
- Singh, K.K. Mitochondria damage checkpoint in apoptosis and genome stability. FEMS Yeast Res. 2004, 5, 127–132. [Google Scholar] [CrossRef]
- Minocherhomji, S.; Tollefsbol, T.O.; Singh, K.K. Mitochondrial regulation of epigenetics and its role in human diseases. Epigenetics 2012, 7, 326–334. [Google Scholar] [CrossRef]
- Smiraglia, D.J.; Kulawiec, M.; Bistulfi, G.L.; Gupta, S.G.; Singh, K.K. A novel role for mitochondria in regulating epigenetic modification in the nucleus. Cancer Biol. Ther. 2008, 7, 1182–1190. [Google Scholar] [CrossRef]
- Gardiner-Garden, M.; Frommer, M. CpG islands in vertebrate genomes. J. Mol. Biol. 1987, 196, 261–282. [Google Scholar] [CrossRef]
- Matsuda, S.; Yasukawa, T.; Sakaguchi, Y.; Ichiyanagi, K.; Unoki, M.; Gotoh, K.; Fukuda, K.; Sasaki, H.; Suzuki, T.; Kang, D. Accurate estimation of 5-methylcytosine in mammalian mitochondrial DNA. Sci. Rep. 2018, 8, 5801. [Google Scholar] [CrossRef]
- Mechta, M.; Ingerslev, L.R.; Fabre, O.; Picard, M.; Barres, R. Evidence Suggesting Absence of Mitochondrial DNA Methylation. Front. Genet. 2017, 8, 166. [Google Scholar] [CrossRef] [PubMed]
- Hong, E.E.; Okitsu, C.Y.; Smith, A.D.; Hsieh, C.L. Regionally specific and genome-wide analyses conclusively demonstrate the absence of CpG methylation in human mitochondrial DNA. Mol. Cell Biol. 2013, 33, 2683–2690. [Google Scholar] [CrossRef] [PubMed]
- Shock, L.S.; Thakkar, P.V.; Peterson, E.J.; Moran, R.G.; Taylor, S.M. DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc. Natl. Acad. Sci. USA 2011, 108, 3630–3635. [Google Scholar] [CrossRef] [PubMed]
- Sharma, J.; Kumari, R.; Bhargava, A.; Tiwari, R.; Mishra, P.K. Mitochondrial-induced Epigenetic Modifications: From Biology to Clinical Translation. Curr. Pharm. Des. 2021, 27, 159–176. [Google Scholar] [CrossRef] [PubMed]
- Bicci, I.; Calabrese, C.; Golder, Z.J.; Gomez-Duran, A.; Chinnery, P.F. Single-molecule mitochondrial DNA sequencing shows no evidence of CpG methylation in human cells and tissues. Nucleic Acids Res. 2021, 49, 12757–12768. [Google Scholar] [CrossRef]
- Yang, B.; Wang, J.Q.; Tan, Y.; Yuan, R.; Chen, Z.S.; Zou, C. RNA methylation and cancer treatment. Pharm. Res. 2021, 174, 105937. [Google Scholar] [CrossRef]
- Jonkhout, N.; Tran, J.; Smith, M.A.; Schonrock, N.; Mattick, J.S.; Novoa, E.M. The RNA modification landscape in human disease. RNA 2017, 23, 1754–1769. [Google Scholar] [CrossRef]
- Desrosiers, R.; Friderici, K.; Rottman, F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc. Natl. Acad. Sci. USA 1974, 71, 3971–3975. [Google Scholar] [CrossRef]
- Perry, R.P.; Kelley, D.E. Existence of methylated messenger RNA in mouse L cells. Cell 1974, 1, 37–42. [Google Scholar] [CrossRef]
- Zaccara, S.; Ries, R.J.; Jaffrey, S.R. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 608–624. [Google Scholar] [CrossRef]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Su, R.; Weng, H.; Huang, H.; Li, Z.; Chen, J. RNA N6-methyladenosine modification in cancers: Current status and perspectives. Cell Res. 2018, 28, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Huo, F.C.; Zhu, Z.M.; Pei, D.S. N6-methyladenosine (m6 A) RNA modification in human cancer. Cell Prolif. 2020, 53, e12921. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wei, L.H.; Wang, Y.X.; Xiao, Y.; Liu, J.; Zhang, W.; Yan, N.; Amu, G.; Tang, X.J.; Zhang, L.; et al. Structural insights into FTO’s catalytic mechanism for the demethylation of multiple RNA substrates. Proc. Natl. Acad. Sci. USA 2019, 116, 2919–2924. [Google Scholar] [CrossRef]
- Liu, Y.; You, Y.; Lu, Z.; Yang, J.; Li, P.; Liu, L.; Xu, H.; Niu, Y.; Cao, X. N6-methyladenosine RNA modification-mediated cellular metabolism rewiring inhibits viral replication. Science 2019, 365, 1171–1176. [Google Scholar] [CrossRef]
- Li, Z.J.; Weng, H.Y.; Su, R.; Weng, X.C.; Zuo, Z.X.; Li, C.Y.; Huang, H.L.; Nachtergaele, S.; Dong, L.; Hu, C.; et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N-6-Methyladenosine RNA Demethylase. Cancer Cell 2017, 31, 127–141. [Google Scholar] [CrossRef]
- Su, R.; Li, Z.J.; Weng, H.Y.; Weng, X.C.; Zuo, Z.X.; Li, C.Y.; Huang, H.L.; Hu, C.; Qin, X.; Tang, L.C.; et al. Fto Plays an Oncogenic Role in Acute Myeloid Leukemia As a N-6-Methyladenosine RNA Demethylase. Blood 2016, 128, 2706. [Google Scholar] [CrossRef]
- Scholler, E.; Marks, J.; Marchand, V.; Bruckmann, A.; Powell, C.A.; Reichold, M.; Mutti, C.D.; Dettmer, K.; Feederle, R.; Huttelmaier, S.; et al. Balancing of mitochondrial translation through METTL8-mediated m3C modification of mitochondrial tRNAs. Mol. Cell 2021, 81, 4810–4825. [Google Scholar] [CrossRef]
- Morscher, R.J.; Ducker, G.S.; Li, S.H.; Mayer, J.A.; Gitai, Z.; Sperl, W.; Rabinowitz, J.D. Mitochondrial translation requires folate-dependent tRNA methylation. Nature 2018, 554, 128–132. [Google Scholar] [CrossRef]
- Delaunay, S.; Pascual, G.; Feng, B.; Klann, K.; Behm, M.; Hotz-Wagenblatt, A.; Richter, K.; Zaoui, K.; Herpel, E.; Munch, C.; et al. Mitochondrial RNA modifications shape metabolic plasticity in metastasis. Nature 2022, 607, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Vilardo, E.; Amman, F.; Toth, U.; Kotter, A.; Helm, M.; Rossmanith, W. Functional characterization of the human tRNA methyltransferases TRMT10A and TRMT10B. Nucleic Acids Res. 2020, 48, 6157–6169. [Google Scholar] [CrossRef] [PubMed]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Gong, F.; Miller, K.M. Histone methylation and the DNA damage response. Mutat. Res. Rev. Mutat. Res. 2019, 780, 37–47. [Google Scholar] [CrossRef]
- Mazzio, E.A.; Soliman, K.F. Basic concepts of epigenetics: Impact of environmental signals on gene expression. Epigenetics 2012, 7, 119–130. [Google Scholar] [CrossRef]
- Brownell, J.E.; Allis, C.D. Special HATs for special occasions: Linking histone acetylation to chromatin assembly and gene activation. Curr. Opin. Genet. Dev. 1996, 6, 176–184. [Google Scholar] [CrossRef]
- Tessarz, P.; Kouzarides, T. Histone core modifications regulating nucleosome structure and dynamics. Nat. Rev. Mol. Cell Biol. 2014, 15, 703–708. [Google Scholar] [CrossRef]
- Diallo, I.; Seve, M.; Cunin, V.; Minassian, F.; Poisson, J.F.; Michelland, S.; Bourgoin-Voillard, S. Current trends in protein acetylation analysis. Expert Rev. Proteom. 2019, 16, 139–159. [Google Scholar] [CrossRef]
- Shahbazian, M.D.; Grunstein, M. Functions of site-specific histone acetylation and deacetylation. Annu. Rev. Biochem. 2007, 76, 75–100. [Google Scholar] [CrossRef]
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81–120. [Google Scholar] [CrossRef]
- Swigut, T.; Wysocka, J. H3K27 demethylases, at long last. Cell 2007, 131, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef] [PubMed]
- Akimova, T.; Beier, U.H.; Liu, Y.; Wang, L.; Hancock, W.W. Histone/protein deacetylases and T-cell immune responses. Blood 2012, 119, 2443–2451. [Google Scholar] [CrossRef] [PubMed]
- Kida, Y.; Goligorsky, M.S. Sirtuins, Cell Senescence, and Vascular Aging. Can. J. Cardiol. 2016, 32, 634–641. [Google Scholar] [CrossRef]
- Gertler, A.A.; Cohen, H.Y. SIRT6, a protein with many faces. Biogerontology 2013, 14, 629–639. [Google Scholar] [CrossRef]
- Imai, S.; Yoshino, J. The importance of NAMPT/NAD/SIRT1 in the systemic regulation of metabolism and ageing. Diabetes Obes. Metab. 2013, 15 (Suppl. S3), 26–33. [Google Scholar] [CrossRef]
- Berger, S.L. The complex language of chromatin regulation during transcription. Nature 2007, 447, 407–412. [Google Scholar] [CrossRef]
- Demetriadou, C.; Kirmizis, A. Histone Acetyltransferases in Cancer: Guardians or Hazards? Crit. Rev. Oncog. 2017, 22, 195–218. [Google Scholar] [CrossRef]
- Eberharter, A.; Becker, P.B. Histone acetylation: A switch between repressive and permissive chromatin—Second in review series on chromatin dynamics. EMBO Rep. 2002, 3, 224–229. [Google Scholar] [CrossRef]
- Kurdistani, S.K. Histone modifications in cancer biology and prognosis. Prog. Drug Res. 2011, 67, 91–106. [Google Scholar] [CrossRef]
- Shen, Y.; Wei, W.; Zhou, D.X. Histone Acetylation Enzymes Coordinate Metabolism and Gene Expression. Trends Plant Sci. 2015, 20, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, K.; Oi, R.; Nojima, S.; Morii, E. Mitochondria govern histone acetylation in colorectal cancer. J. Pathol. 2022, 256, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.B.; Karpova, A.; Gritsenko, M.A.; Kyle, J.E.; Cao, S.; Li, Y.; Rykunov, D.; Colaprico, A.; Rothstein, J.H.; Hong, R.; et al. Proteogenomic and metabolomic characterization of human glioblastoma. Cancer Cell 2021, 39, 509–528.e20. [Google Scholar] [CrossRef] [PubMed]
- McBrian, M.A.; Behbahan, I.S.; Ferrari, R.; Su, T.; Huang, T.W.; Li, K.; Hong, C.S.; Christofk, H.R.; Vogelauer, M.; Seligson, D.B.; et al. Histone acetylation regulates intracellular pH. Mol. Cell 2013, 49, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2009, 9, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Krainer, A.R.; Mayeda, A.; Kozak, D.; Binns, G. Functional expression of cloned human splicing factor SF2: Homology to RNA-binding proteins, U1 70K, and Drosophila splicing regulators. Cell 1991, 66, 383–394. [Google Scholar] [CrossRef]
- Jiang, J.; Zhang, Y.; Krainer, A.R.; Xu, R.M. Crystal structure of human p32, a doughnut-shaped acidic mitochondrial matrix protein. Proc. Natl. Acad. Sci. USA 1999, 96, 3572–3577. [Google Scholar] [CrossRef] [PubMed]
- Ghebrehiwet, B.; Geisbrecht, B.V.; Xu, X.; Savitt, A.G.; Peerschke, E.I.B. The C1q Receptors: Focus on gC1qR/p33 (C1qBP, p32, HABP-1)(1). Semin. Immunol. 2019, 45, 101338. [Google Scholar] [CrossRef] [PubMed]
- Fogal, V.; Richardson, A.D.; Karmali, P.P.; Scheffler, I.E.; Smith, J.W.; Ruoslahti, E. Mitochondrial p32 Protein Is a Critical Regulator of Tumor Metabolism via Maintenance of Oxidative Phosphorylation. Mol. Cell. Biol. 2010, 30, 1303–1318. [Google Scholar] [CrossRef] [PubMed]
- Yagi, M.; Uchiumi, T.; Takazaki, S.; Okuno, B.; Nomura, M.; Yoshida, S.; Kanki, T.; Kang, D. p32/gC1qR is indispensable for fetal development and mitochondrial translation: Importance of its RNA-binding ability. Nucleic Acids Res. 2012, 40, 9717–9737. [Google Scholar] [CrossRef] [PubMed]
- Zapata-Perez, R.; Wanders, R.J.A.; van Karnebeek, C.D.M.; Houtkooper, R.H. NAD(+) homeostasis in human health and disease. Embo Mol. Med. 2021, 13, e13943. [Google Scholar] [CrossRef] [PubMed]
- Ansari, H.R.; Raghava, G.P. Identification of NAD interacting residues in proteins. BMC Bioinform. 2010, 11, 160. [Google Scholar] [CrossRef]
- Chambon, P.; Weill, J.D.; Mandel, P. Nicotinamide mononucleotide activation of new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochem. Biophys. Res. Commun. 1963, 11, 39–43. [Google Scholar] [CrossRef]
- Bai, P. Biology of Poly(ADP-Ribose) Polymerases: The Factotums of Cell Maintenance. Mol. Cell 2015, 58, 947–958. [Google Scholar] [CrossRef]
- Gupte, R.; Liu, Z.; Kraus, W.L. PARPs and ADP-ribosylation: Recent advances linking molecular functions to biological outcomes. Genes Dev. 2017, 31, 101–126. [Google Scholar] [CrossRef]
- Imai, S.; Armstrong, C.M.; Kaeberlein, M.; Guarente, L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 2000, 403, 795–800. [Google Scholar] [CrossRef]
- Imai, S.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 2012, 13, 225–238. [Google Scholar] [CrossRef]
- Thakur, C.; Chen, F. Connections between metabolism and epigenetics in cancers. Semin. Cancer Biol. 2019, 57, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Sinclair, D.A.; Ellis, J.L.; Steegborn, C. Sirtuin activators and inhibitors: Promises, achievements, and challenges. Pharm. Ther. 2018, 188, 140–154. [Google Scholar] [CrossRef] [PubMed]
- Murayama, A.; Ohmori, K.; Fujimura, A.; Minami, H.; Yasuzawa-Tanaka, K.; Kuroda, T.; Oie, S.; Daitoku, H.; Okuwaki, M.; Nagata, K.; et al. Epigenetic control of rDNA loci in response to intracellular energy status. Cell 2008, 133, 627–639. [Google Scholar] [CrossRef]
- Menzies, K.J.; Zhang, H.; Katsyuba, E.; Auwerx, J. Protein acetylation in metabolism—Metabolites and cofactors. Nat. Rev. Endocrinol. 2016, 12, 43–60. [Google Scholar] [CrossRef]
- Pietrocola, F.; Galluzzi, L.; Bravo-San Pedro, J.M.; Madeo, F.; Kroemer, G. Acetyl coenzyme A: A central metabolite and second messenger. Cell Metab. 2015, 21, 805–821. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Tu, B.P. Acetyl-CoA and the regulation of metabolism: Mechanisms and consequences. Curr. Opin. Cell Biol. 2015, 33, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Srere, P.A. The citrate cleavage enzyme. I. Distribution and purification. J. Biol. Chem. 1959, 234, 2544–2547. [Google Scholar] [CrossRef]
- Horner, D.S.; Hirt, R.P.; Embley, T.M. A single eubacterial origin of eukaryotic pyruvate: Ferredoxin oxidoreductase genes: Implications for the evolution of anaerobic eukaryotes. Mol. Biol. Evol. 1999, 16, 1280–1291. [Google Scholar] [CrossRef]
- Sutendra, G.; Kinnaird, A.; Dromparis, P.; Paulin, R.; Stenson, T.H.; Haromy, A.; Hashimoto, K.; Zhang, N.; Flaim, E.; Michelakis, E.D. A Nuclear Pyruvate Dehydrogenase Complex Is Important for the Generation of Acetyl-CoA and Histone Acetylation. Cell 2014, 158, 84–97. [Google Scholar] [CrossRef]
- Choudhary, C.; Weinert, B.T.; Nishida, Y.; Verdin, E.; Mann, M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 536–550. [Google Scholar] [CrossRef]
- Cai, L.; Sutter, B.M.; Li, B.; Tu, B.P. Acetyl-CoA Induces Cell Growth and Proliferation by Promoting the Acetylation of Histones at Growth Genes. Mol. Cell 2011, 42, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Tu, B.P. Protein acetylation as a means to regulate protein function in tune with metabolic state. Biochem. Soc. T 2014, 42, 1037–1042. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Tu, B.P. On Acetyl-CoA as a Gauge of Cellular Metabolic State. In Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2011; Volume 76, pp. 195–202. [Google Scholar] [CrossRef]
- Mews, P.; Egervari, G.; Nativio, R.; Sidoli, S.; Donahue, G.; Lombroso, S.I.; Alexander, D.C.; Riesche, S.L.; Heller, E.A.; Nestler, E.J.; et al. Alcohol metabolism contributes to brain histone acetylation. Nature 2019, 574, 717–721. [Google Scholar] [CrossRef] [PubMed]
- Mews, P.; Donahue, G.; Drake, A.M.; Luczak, V.; Abel, T.; Berger, S.L. Acetyl-CoA synthetase regulates histone acetylation and hippocampal memory. Nature 2017, 546, 381–386. [Google Scholar] [CrossRef]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, Z.; Jia, J.; Du, T.; Zhang, N.; Tang, Y.; Fang, Y.; Fang, D. Overview of Histone Modification. Adv. Exp. Med. Biol. 2021, 1283, 1–16. [Google Scholar] [CrossRef]
- Black, J.C.; Van Rechem, C.; Whetstine, J.R. Histone lysine methylation dynamics: Establishment, regulation, and biological impact. Mol. Cell 2012, 48, 491–507. [Google Scholar] [CrossRef]
- Yang, Y.; Bedford, M.T. Protein arginine methyltransferases and cancer. Nat. Rev. Cancer 2013, 13, 37–50. [Google Scholar] [CrossRef]
- Bedford, M.T.; Clarke, S.G. Protein Arginine Methylation in Mammals: Who, What, and Why. Mol. Cell 2009, 33, 1–13. [Google Scholar] [CrossRef]
- Blanc, R.S.; Richard, S. Arginine Methylation: The Coming of Age. Mol. Cell 2017, 65, 8–24. [Google Scholar] [CrossRef]
- Dimitrova, E.; Turberfield, A.H.; Klose, R.J. Histone demethylases in chromatin biology and beyond. EMBO Rep. 2015, 16, 1620–1639. [Google Scholar] [CrossRef] [PubMed]
- Kooistra, S.M.; Helin, K. Molecular mechanisms and potential functions of histone demethylases. Nat. Rev. Mol. Cell Biol. 2012, 13, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.F.; Yu, H.T. Structural insights into histone lysine demethylation. Curr. Opin. Struc. Biol. 2010, 20, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, Y.; Fang, J.; Erdjument-Bromage, H.; Warren, M.E.; Borchers, C.H.; Tempst, P.; Zhang, Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature 2006, 439, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Whetstine, J.R.; Nottke, A.; Lan, F.; Huarte, M.; Smolikov, S.; Chen, Z.; Spooner, E.; Li, E.; Zhang, G.; Colaiacovo, M.; et al. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell 2006, 125, 467–481. [Google Scholar] [CrossRef]
- Teperino, R.; Schoonjans, K.; Auwerx, J. Histone Methyl Transferases and Demethylases; Can They Link Metabolism and Transcription? Cell Metab. 2010, 12, 321–327. [Google Scholar] [CrossRef]
- Yun, J.; Song, S.H.; Park, J.; Kim, H.P.; Yoon, Y.K.; Lee, K.H.; Han, S.W.; Oh, D.Y.; Im, S.A.; Bang, Y.J.; et al. Gene silencing of EREG mediated by DNA methylation and histone modification in human gastric cancers. Lab. Investig. 2012, 92, 1033–1044. [Google Scholar] [CrossRef]
- Kang, H.S.; Lee, J.H.; Oh, K.J.; Lee, E.W.; Han, B.S.; Park, K.Y.; Suh, J.M.; Min, J.K.; Chi, S.W.; Lee, S.C.; et al. IDH1-dependent alpha-KG regulates brown fat differentiation and function by modulating histone methylation. Metabolism 2020, 105, 154173. [Google Scholar] [CrossRef]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef]
- Ulanovskaya, O.A.; Zuhl, A.M.; Cravatt, B.F. NNMT promotes epigenetic remodeling in cancer by creating a metabolic methylation sink. Nat. Chem. Biol. 2013, 9, 300–306. [Google Scholar] [CrossRef]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Dai, J.; Dai, L.; Tan, M.; Cheng, Z.; Wu, Y.; Boeke, J.D.; Zhao, Y. Lysine succinylation and lysine malonylation in histones. Mol. Cell Proteom. 2012, 11, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Weinert, B.T.; Scholz, C.; Wagner, S.A.; Iesmantavicius, V.; Su, D.; Daniel, J.A.; Choudhary, C. Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation. Cell Rep. 2013, 4, 842–851. [Google Scholar] [CrossRef]
- Du, J.; Zhou, Y.; Su, X.; Yu, J.J.; Khan, S.; Jiang, H.; Kim, J.; Woo, J.; Kim, J.H.; Choi, B.H.; et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 2011, 334, 806–809. [Google Scholar] [CrossRef] [PubMed]
- Rosen, R.; Becher, D.; Buttner, K.; Biran, D.; Hecker, M.; Ron, E.Z. Probing the active site of homoserine trans-succinylase. FEBS Lett. 2004, 577, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.H.; Tan, M.J.; Xie, Z.Y.; Dai, L.Z.; Chen, Y.; Zhao, Y.M. Identification of lysine succinylation as a new post-translational modification. Nat. Chem. Biol. 2011, 7, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Sabari, B.R.; Zhang, D.; Allis, C.D.; Zhao, Y.M. Metabolic regulation of gene expression through histone acylations. Nat.Rev. Mol. Cell Biol. 2017, 18, 90–101. [Google Scholar] [CrossRef]
- Wang, Y.G.; Guo, Y.R.; Liu, K.; Yin, Z.; Liu, R.; Xia, Y.; Tan, L.; Yang, P.Y.; Lee, J.H.; Li, X.J.; et al. KAT2A coupled with the alpha-KGDH complex acts as a histone H3 succinyltransferase. Nature 2017, 552, 273–277. [Google Scholar] [CrossRef]
- Zhao, S.; Zhang, X.; Li, H. Beyond histone acetylation-writing and erasing histone acylations. Curr. Opin. Struct. Biol. 2018, 53, 169–177. [Google Scholar] [CrossRef]
- Yang, G.; Yuan, Y.; Yuan, H.; Wang, J.; Yun, H.; Geng, Y.; Zhao, M.; Li, L.; Weng, Y.; Liu, Z.; et al. Histone acetyltransferase 1 is a succinyltransferase for histones and non-histones and promotes tumorigenesis. EMBO Rep. 2021, 22, e50967. [Google Scholar] [CrossRef]
- Peng, C.; Lu, Z.; Xie, Z.; Cheng, Z.; Chen, Y.; Tan, M.; Luo, H.; Zhang, Y.; He, W.; Yang, K.; et al. The first identification of lysine malonylation substrates and its regulatory enzyme. Mol. Cell Proteom. 2011, 10, M111012658. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Chen, Y.; Tishkoff, D.X.; Peng, C.; Tan, M.; Dai, L.; Xie, Z.; Zhang, Y.; Zwaans, B.M.; Skinner, M.E.; et al. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol. Cell 2013, 50, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Rardin, M.J.; He, W.; Nishida, Y.; Newman, J.C.; Carrico, C.; Danielson, S.R.; Guo, A.; Gut, P.; Sahu, A.K.; Li, B.; et al. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab. 2013, 18, 920–933. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Shi, L.; Yang, S.D.; Yan, R.R.; Zhang, D.; Yang, J.G.; He, L.; Li, W.J.; Yi, X.; Sun, L.Y.; et al. SIRT7 is a histone desuccinylase that functionally links to chromatin compaction and genome stability. Nat. Commun. 2016, 7, 12235. [Google Scholar] [CrossRef] [PubMed]
- Trefely, S.; Lovell, C.D.; Snyder, N.W.; Wellen, K.E. Compartmentalised acyl-CoA metabolism and roles in chromatin regulation. Mol. Metab. 2020, 38, 100941. [Google Scholar] [CrossRef]
- Paine, P.L.; Moore, L.C.; Horowitz, S.B. Nuclear envelope permeability. Nature 1975, 254, 109–114. [Google Scholar] [CrossRef]
- Palmieri, F. The mitochondrial transporter family (SLC25): Physiological and pathological implications. Pflug. Arch. 2004, 447, 689–709. [Google Scholar] [CrossRef]
- Monne, M.; Miniero, D.V.; Iacobazzi, V.; Bisaccia, F.; Fiermonte, G. The mitochondrial oxoglutarate carrier: From identification to mechanism. J. Bioenerg. Biomembr. 2013, 45, 1–13. [Google Scholar] [CrossRef]
- Smestad, J.; Erber, L.; Chen, Y.; Maher, L.J. Chromatin Succinylation Correlates with Active Gene Expression and Is Perturbed by Defective TCA Cycle Metabolism. Iscience 2018, 2, 63–75. [Google Scholar] [CrossRef]
- Holt, G.D.; Snow, C.M.; Senior, A.; Haltiwanger, R.S.; Gerace, L.; Hart, G.W. Nuclear pore complex glycoproteins contain cytoplasmically disposed O-linked N-acetylglucosamine. J. Cell Biol. 1987, 104, 1157–1164. [Google Scholar] [CrossRef]
- Zhu, Y.; Hart, G.W. Targeting O-GlcNAcylation to develop novel therapeutics. Mol. Asp. Med. 2021, 79, 100885. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.B.; Pyo, K.H.; Kim, H.R. Role and Function of O-GlcNAcylation in Cancer. Cancers 2021, 13, 5365. [Google Scholar] [CrossRef] [PubMed]
- Finlay, D.R.; Newmeyer, D.D.; Price, T.M.; Forbes, D.J. Inhibition of in vitro nuclear transport by a lectin that binds to nuclear pores. J. Cell Biol. 1987, 104, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Hanover, J.A.; Cohen, C.K.; Willingham, M.C.; Park, M.K. O-Linked N-Acetylglucosamine Is Attached to Proteins of the Nuclear-Pore—Evidence for Cytoplasmic and Nucleoplasmic Glycoproteins. J. Biol. Chem. 1987, 262, 9887–9894. [Google Scholar] [CrossRef]
- Park, M.K.; D’Onofrio, M.; Willingham, M.C.; Hanover, J.A. A monoclonal antibody against a family of nuclear pore proteins (nucleoporins): O-linked N-acetylglucosamine is part of the immunodeterminant. Proc. Natl. Acad. Sci. USA 1987, 84, 6462–6466. [Google Scholar] [CrossRef]
- Kelly, W.G.; Hart, G.W. Glycosylation of chromosomal proteins: Localization of O-linked N-acetylglucosamine in Drosophila chromatin. Cell 1989, 57, 243–251. [Google Scholar] [CrossRef]
- Love, D.C.; Kochan, J.; Cathey, R.L.; Shin, S.H.; Hanover, J.A. Mitochondrial and nucleocytoplasmic targeting of O-linked GlcNAc transferase. J. Cell Sci. 2003, 116, 647–654. [Google Scholar] [CrossRef]
- Shin, S.H.; Love, D.C.; Hanover, J.A. Elevated O-GlcNAc-dependent signaling through inducible mOGT expression selectively triggers apoptosis. Amino Acids 2011, 40, 885–893. [Google Scholar] [CrossRef]
- Hanover, J.A. Epigenetics Gets Sweeter: O-GlcNAc Joins the “Histone Code”. Chem. Biol. 2010, 17, 1272–1274. [Google Scholar] [CrossRef]
- Sakabe, K.; Wang, Z.H.; Hart, G.W. beta-N-acetylglucosamine (O-GlcNAc) is part of the histone code. Proc. Natl. Acad. Sci. USA 2010, 107, 19915–19920. [Google Scholar] [CrossRef]
- Fujiki, R.; Hashiba, W.; Sekine, H.; Yokoyama, A.; Chikanishi, T.; Ito, S.; Imai, Y.; Kim, J.; He, H.H.; Igarashi, K.; et al. GlcNAcylation of histone H2B facilitates its monoubiquitination. Nature 2011, 480, 557–560. [Google Scholar] [CrossRef]
- Xu, Q.; Yang, C.; Du, Y.; Chen, Y.; Liu, H.; Deng, M.; Zhang, H.; Zhang, L.; Liu, T.; Liu, Q.; et al. AMPK regulates histone H2B O-GlcNAcylation. Nucleic Acids Res. 2014, 42, 5594–5604. [Google Scholar] [CrossRef]
- Dehennaut, V.; Leprince, D.; Lefebvre, T. O-GlcNAcylation, an epigenetic mark. Focus on the histone code, TET family proteins, and polycomb group proteins. Front. Endocrinol. 2014, 5, 155. [Google Scholar] [CrossRef]
- Hanover, J.A.; Krause, M.W.; Love, D.C. The hexosamine signaling pathway: O-GlcNAc cycling in feast or famine. Biochim. Biophys. Acta 2010, 1800, 80–95. [Google Scholar] [CrossRef]
- Fong, J.J.; Nguyen, B.L.; Bridger, R.; Medrano, E.E.; Wells, L.; Pan, S.J.; Sifers, R.N. beta-N-Acetylglucosamine (O-GlcNAc) Is a Novel Regulator of Mitosis-specific Phosphorylations on Histone H3. J. Biol. Chem. 2012, 287, 12195–12203. [Google Scholar] [CrossRef]
- Zhang, S.; Roche, K.; Nasheuer, H.P.; Lowndes, N.F. Modification of histones by sugar beta-N-acetylglucosamine (GlcNAc) occurs on multiple residues, including histone H3 serine 10, and is cell cycle-regulated. J. Biol. Chem. 2011, 286, 37483–37495. [Google Scholar] [CrossRef]
- Sekine, O.; Love, D.C.; Rubenstein, D.S.; Hanover, J.A. Blocking O-linked GlcNAc cycling in Drosophila insulin-producing cells perturbs glucose-insulin homeostasis. J. Biol. Chem. 2010, 285, 38684–38691. [Google Scholar] [CrossRef]
- Sinclair, D.A.R.; Syrzycka, M.; Macauley, M.S.; Rastgardani, T.; Komljenovic, I.; Vocadlo, D.J.; Brock, H.W.; Honda, B.M. Drosophila O-GlcNAc transferase (OGT) is encoded by the Polycomb group (PcG) gene, super sex combs (sxc). Proc. Natl. Acad. Sci. USA 2009, 106, 13427–13432. [Google Scholar] [CrossRef]
- Kassis, J.A.; Kennison, J.A. Recruitment of polycomb complexes: A role for SCM. Mol. Cell Biol. 2010, 30, 2581–2583. [Google Scholar] [CrossRef]
- Haltiwanger, R.S.; Holt, G.D.; Hart, G.W. Enzymatic Addition of O-Glcnac to Nuclear and Cytoplasmic Proteins—Identification of a Uridine Diphospho-N-Acetylglucosamine-Peptide Beta-N-Acetylglucosaminyltransferase. J. Biol. Chem. 1990, 265, 2563–2568. [Google Scholar] [CrossRef]
- Kornfeld, S.; Kornfeld, R.; Neufeld, E.F.; O’Brien, P.J. The Feedback Control of Sugar Nucleotide Biosynthesis in Liver. Proc. Natl. Acad. Sci. USA 1964, 52, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Wellen, K.E.; Thompson, C.B. A two-way street: Reciprocal regulation of metabolism and signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Gut, P.; Verdin, E. The nexus of chromatin regulation and intermediary metabolism. Nature 2013, 502, 489–498. [Google Scholar] [CrossRef]
- Chen, Q.; Chen, Y.; Bian, C.; Fujiki, R.; Yu, X. TET2 promotes histone O-GlcNAcylation during gene transcription. Nature 2013, 493, 561–564. [Google Scholar] [CrossRef]
- Deplus, R.; Delatte, B.; Schwinn, M.K.; Defrance, M.; Mendez, J.; Murphy, N.; Dawson, M.A.; Volkmar, M.; Putmans, P.; Calonne, E.; et al. TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. EMBO J. 2013, 32, 645–655. [Google Scholar] [CrossRef]
- Ito, R.; Katsura, S.; Shimada, H.; Tsuchiya, H.; Hada, M.; Okumura, T.; Sugawara, A.; Yokoyama, A. TET3-OGT interaction increases the stability and the presence of OGT in chromatin. Genes Cells 2014, 19, 52–65. [Google Scholar] [CrossRef]
- Kronlage, M.; Dewenter, M.; Grosso, J.; Fleming, T.; Oehl, U.; Lehmann, L.H.; Falcao-Pires, I.; Leite-Moreira, A.F.; Volk, N.; Grone, H.J.; et al. O-GlcNAcylation of Histone Deacetylase 4 Protects the Diabetic Heart From Failure. Circulation 2019, 140, 580–594. [Google Scholar] [CrossRef]
- Zhu, G.; Tao, T.; Zhang, D.; Liu, X.; Qiu, H.; Han, L.; Xu, Z.; Xiao, Y.; Cheng, C.; Shen, A. O-GlcNAcylation of histone deacetylases 1 in hepatocellular carcinoma promotes cancer progression. Glycobiology 2016, 26, 820–833. [Google Scholar] [CrossRef]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. Histone Lactylation: A New Role for Glucose Metabolism. Trends Biochem. Sci. 2020, 45, 179–182. [Google Scholar] [CrossRef]
- Hui, S.; Ghergurovich, J.M.; Morscher, R.J.; Jang, C.; Teng, X.; Lu, W.; Esparza, L.A.; Reya, T.; Le, Z.; Yanxiang Guo, J.; et al. Glucose feeds the TCA cycle via circulating lactate. Nature 2017, 551, 115–118. [Google Scholar] [CrossRef]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Yu, J.; Chai, P.; Xie, M.; Ge, S.; Ruan, J.; Fan, X.; Jia, R. Histone lactylation drives oncogenesis by facilitating m6A reader protein YTHDF2 expression in ocular melanoma. Genome Biol. 2021, 22, 85. [Google Scholar] [CrossRef]
- Irizarry-Caro, R.A.; McDaniel, M.M.; Overcast, G.R.; Jain, V.G.; Troutman, T.D.; Pasare, C. TLR signaling adapter BCAP regulates inflammatory to reparatory macrophage transition by promoting histone lactylation. Proc. Natl. Acad. Sci. USA 2020, 117, 30628–30638. [Google Scholar] [CrossRef]
- Dai, X.; Lv, X.; Thompson, E.W.; Ostrikov, K.K. Histone lactylation: Epigenetic mark of glycolytic switch. Trends Genet. 2022, 38, 124–127. [Google Scholar] [CrossRef]
- Brooks, G.A. Lactate as a fulcrum of metabolism. Redox Biol. 2020, 35, 101454. [Google Scholar] [CrossRef]
- Passarella, S.; de Bari, L.; Valenti, D.; Pizzuto, R.; Paventi, G.; Atlante, A. Mitochondria and L-lactate metabolism. FEBS Lett. 2008, 582, 3569–3576. [Google Scholar] [CrossRef]
- Glancy, B.; Kane, D.A.; Kavazis, A.N.; Goodwin, M.L.; Willis, W.T.; Gladden, L.B. Mitochondrial lactate metabolism: History and implications for exercise and disease. J. Physiol. 2021, 599, 863–888. [Google Scholar] [CrossRef]
- Chen, Y.J.; Mahieu, N.G.; Huang, X.J.; Singh, M.; Crawford, P.A.; Johnson, S.L.; Gross, R.W.; Schaefer, J.; Patti, G.J. Lactate metabolism is associated with mammalian mitochondria. Nat. Chem. Biol. 2016, 12, 937–943. [Google Scholar] [CrossRef]
- Chen, A.N.; Luo, Y.; Yang, Y.H.; Fu, J.T.; Geng, X.M.; Shi, J.P.; Yang, J. Lactylation, a Novel Metabolic Reprogramming Code: Current Status and Prospects. Front. Immunol. 2021, 12, 688910. [Google Scholar] [CrossRef]
- Kraus, D.; Yang, Q.; Kong, D.; Banks, A.S.; Zhang, L.; Rodgers, J.T.; Pirinen, E.; Pulinilkunnil, T.C.; Gong, F.; Wang, Y.C.; et al. Nicotinamide N-methyltransferase knockdown protects against diet-induced obesity. Nature 2014, 508, 258–262. [Google Scholar] [CrossRef]
- Gao, W.; Liu, J.L.; Lu, X.; Yang, Q. Epigenetic regulation of energy metabolism in obesity. J. Mol. Cell Biol. 2021, 13, 480–499. [Google Scholar] [CrossRef]
- Duteil, D.; Metzger, E.; Willmann, D.; Karagianni, P.; Friedrichs, N.; Greschik, H.; Gunther, T.; Buettner, R.; Talianidis, I.; Metzger, D.; et al. LSD1 promotes oxidative metabolism of white adipose tissue. Nat. Commun. 2014, 5, 4093. [Google Scholar] [CrossRef]
- Barres, R.; Osler, M.E.; Yan, J.; Rune, A.; Fritz, T.; Caidahl, K.; Krook, A.; Zierath, J.R. Non-CpG Methylation of the PGC-1 alpha Promoter through DNMT3B Controls Mitochondrial Density. Cell Metab. 2009, 10, 189–198. [Google Scholar] [CrossRef]
- Barres, R.; Yan, J.; Egan, B.; Treebak, J.T.; Rasmussen, M.; Fritz, T.; Caidahl, K.; Krook, A.; O’Gorman, D.J.; Zierath, J.R. Acute exercise remodels promoter methylation in human skeletal muscle. Cell Metab. 2012, 15, 405–411. [Google Scholar] [CrossRef]
- Vizioli, M.G.; Liu, T.; Miller, K.N.; Robertson, N.A.; Gilroy, K.; Lagnado, A.B.; Perez-Garcia, A.; Kiourtis, C.; Dasgupta, N.; Lei, X.; et al. Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. Genes Dev. 2020, 34, 428–445. [Google Scholar] [CrossRef]
- Shao, L.W.; Peng, Q.; Dong, M.; Gao, K.; Li, Y.; Li, Y.; Li, C.Y.; Liu, Y. Histone deacetylase HDA-1 modulates mitochondrial stress response and longevity. Nat. Commun. 2020, 11, 4639. [Google Scholar] [CrossRef]
- Vaupel, P.; Multhoff, G. Revisiting the Warburg effect: Historical dogma versus current understanding. J. Physiol. 2021, 599, 1745–1757. [Google Scholar] [CrossRef]
- Lin, R.; Tao, R.; Gao, X.; Li, T.; Zhou, X.; Guan, K.L.; Xiong, Y.; Lei, Q.Y. Acetylation stabilizes ATP-citrate lyase to promote lipid biosynthesis and tumor growth. Mol. Cell 2013, 51, 506–518. [Google Scholar] [CrossRef]
- Uyeda, K.; Repa, J.J. Carbohydrate response element binding protein, ChREBP, a transcription factor coupling hepatic glucose utilization and lipid synthesis. Cell Metab. 2006, 4, 107–110. [Google Scholar] [CrossRef]
- Yang, Y.; Gibson, G.E. Succinylation Links Metabolism to Protein Functions. Neurochem. Res. 2019, 44, 2346–2359. [Google Scholar] [CrossRef]
- Zhang, Y.; Bharathi, S.S.; Rardin, M.J.; Lu, J.; Maringer, K.V.; Sims-Lucas, S.; Prochownik, E.V.; Gibson, B.W.; Goetzman, E.S. Lysine desuccinylase SIRT5 binds to cardiolipin and regulates the electron transport chain. J. Biol. Chem. 2017, 292, 10239–10249. [Google Scholar] [CrossRef]
- Lu, J.; Tan, M.; Cai, Q. The Warburg effect in tumor progression: Mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett. 2015, 356, 156–164. [Google Scholar] [CrossRef]
- Zhu, X.X.; Xuan, Z.F.; Chen, J.; Li, Z.Q.; Zheng, S.S.; Song, P.H. How DNA methylation affects the Warburg effect. Int. J. Biol. Sci. 2020, 16, 2029–2041. [Google Scholar] [CrossRef]
- Singh, S.; Narayanan, S.P.; Biswas, K.; Gupta, A.; Ahuja, N.; Yadav, S.; Panday, R.K.; Samaiya, A.; Sharan, S.K.; Shukla, S. Intragenic DNA methylation and BORIS-mediated cancer-specific splicing contribute to the Warburg effect. Proc. Natl. Acad. Sci. USA 2017, 114, 11440–11445. [Google Scholar] [CrossRef]
- Subramaniam, A.V.; Yehya, A.H.S.; Cheng, W.K.; Wang, X.; Oon, C.E. Epigenetics: The master control of endothelial cell fate in cancer. Life Sci. 2019, 232, 116652. [Google Scholar] [CrossRef]
- Jang, S.; Hwang, J.; Jeong, H.S. The Role of Histone Acetylation in Mesenchymal Stem Cell Differentiation. Chonnam. Med. J. 2022, 58, 6–12. [Google Scholar] [CrossRef]
- Prasetyanti, P.R.; Medema, J.P. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol. Cancer 2017, 16, 41. [Google Scholar] [CrossRef]
- Anderson, A.R.; Weaver, A.M.; Cummings, P.T.; Quaranta, V. Tumor morphology and phenotypic evolution driven by selective pressure from the microenvironment. Cell 2006, 127, 905–915. [Google Scholar] [CrossRef]
- Sottoriva, A.; Verhoeff, J.J.; Borovski, T.; McWeeney, S.K.; Naumov, L.; Medema, J.P.; Sloot, P.M.; Vermeulen, L. Cancer stem cell tumor model reveals invasive morphology and increased phenotypical heterogeneity. Cancer Res. 2010, 70, 46–56. [Google Scholar] [CrossRef]
- Waclaw, B.; Bozic, I.; Pittman, M.E.; Hruban, R.H.; Vogelstein, B.; Nowak, M.A. A spatial model predicts that dispersal and cell turnover limit intratumour heterogeneity. Nature 2015, 525, 261–264. [Google Scholar] [CrossRef]
- Clevers, H. The cancer stem cell: Premises, promises and challenges. Nat. Med. 2011, 17, 313–319. [Google Scholar] [CrossRef]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Eun, K.; Ham, S.W.; Kim, H. Cancer stem cell heterogeneity: Origin and new perspectives on CSC targeting. BMB Rep. 2017, 50, 117–125. [Google Scholar] [CrossRef]
- Medema, J.P. Cancer stem cells: The challenges ahead. Nat. Cell Biol. 2013, 15, 338–344. [Google Scholar] [CrossRef]
- Meacham, C.E.; Morrison, S.J. Tumour heterogeneity and cancer cell plasticity. Nature 2013, 501, 328–337. [Google Scholar] [CrossRef]
- Ferrer, A.I.; Trinidad, J.R.; Sandiford, O.; Etchegaray, J.P.; Rameshwar, P. Epigenetic dynamics in cancer stem cell dormancy. Cancer Metastasis Rev. 2020, 39, 721–738. [Google Scholar] [CrossRef]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef]
- Munoz, P.; Iliou, M.S.; Esteller, M. Epigenetic alterations involved in cancer stem Cell Rep.rogramming. Mol. Oncol. 2012, 6, 620–636. [Google Scholar] [CrossRef]
- Pace, L.; Amigorena, S. Epigenetics of T cell fate decision. Curr. Opin. Immunol. 2020, 63, 43–50. [Google Scholar] [CrossRef]
- Saeed, S.; Quintin, J.; Kerstens, H.H.; Rao, N.A.; Aghajanirefah, A.; Matarese, F.; Cheng, S.C.; Ratter, J.; Berentsen, K.; van der Ent, M.A.; et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 2014, 345, 1251086. [Google Scholar] [CrossRef]
- Alvarez-Errico, D.; Vento-Tormo, R.; Sieweke, M.; Ballestar, E. Epigenetic control of myeloid cell differentiation, identity and function. Nat. Rev. Immunol. 2015, 15, 7–17. [Google Scholar] [CrossRef]
- Ziller, M.J.; Gu, H.; Muller, F.; Donaghey, J.; Tsai, L.T.; Kohlbacher, O.; De Jager, P.L.; Rosen, E.D.; Bennett, D.A.; Bernstein, B.E.; et al. Charting a dynamic DNA methylation landscape of the human genome. Nature 2013, 500, 477–481. [Google Scholar] [CrossRef]
- Berdasco, M.; Esteller, M. DNA methylation in stem cell renewal and multipotency. Stem Cell Res. Ther. 2011, 2, 42. [Google Scholar] [CrossRef]
- Neri, F.; Rapelli, S.; Krepelova, A.; Incarnato, D.; Parlato, C.; Basile, G.; Maldotti, M.; Anselmi, F.; Oliviero, S. Intragenic DNA methylation prevents spurious transcription initiation. Nature 2017, 543, 72–77. [Google Scholar] [CrossRef]
- Skvortsova, K.; Stirzaker, C.; Taberlay, P. The DNA methylation landscape in cancer. Essays Biochem. 2019, 63, 797–811. [Google Scholar] [CrossRef]
- Vincent, A.; Van Seuningen, I. Epigenetics, stem cells and epithelial cell fate. Differentiation 2009, 78, 99–107. [Google Scholar] [CrossRef]
- Berdasco, M.; Esteller, M. Aberrant Epigenetic Landscape in Cancer: How Cellular Identity Goes Awry. Dev. Cell 2010, 19, 698–711. [Google Scholar] [CrossRef]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef]
- Monk, M.; Boubelik, M.; Lehnert, S. Temporal and regional changes in DNA methylation in the embryonic, extraembryonic and germ cell lineages during mouse embryo development. Development 1987, 99, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Hodges, E.; Molaro, A.; Dos Santos, C.O.; Thekkat, P.; Song, Q.; Uren, P.J.; Park, J.; Butler, J.; Rafii, S.; McCombie, W.R.; et al. Directional DNA methylation changes and complex intermediate states accompany lineage specificity in the adult hematopoietic compartment. Mol. Cell 2011, 44, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Lagarkova, M.A.; Volchkov, P.Y.; Lyakisheva, A.V.; Philonenko, E.S.; Kiselev, S.L. Diverse epigenetic profile of novel human embryonic stem cell lines. Cell Cycle 2006, 5, 416–420. [Google Scholar] [CrossRef]
- Yeo, S.; Jeong, S.; Kim, J.; Han, J.S.; Han, Y.M.; Kang, Y.K. Characterization of DNA methylation change in stem cell marker genes during differentiation of human embryonic stem cells. Biochem. Biophys. Res. Commun. 2007, 359, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Fouse, S.D.; Shen, Y.; Pellegrini, M.; Cole, S.; Meissner, A.; Van Neste, L.; Jaenisch, R.; Fan, G. Promoter CpG methylation contributes to ES cell gene regulation in parallel with Oct4/Nanog, PcG complex, and histone H3 K4/K27 trimethylation. Cell Stem Cell 2008, 2, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Loh, Y.H.; Wu, Q.; Chew, J.L.; Vega, V.B.; Zhang, W.; Chen, X.; Bourque, G.; George, J.; Leong, B.; Liu, J.; et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat. Genet. 2006, 38, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Boyer, L.A.; Lee, T.I.; Cole, M.F.; Johnstone, S.E.; Levine, S.S.; Zucker, J.P.; Guenther, M.G.; Kumar, R.M.; Murray, H.L.; Jenner, R.G.; et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 2005, 122, 947–956. [Google Scholar] [CrossRef]
- Hattori, N.; Imao, Y.; Nishino, K.; Hattori, N.; Ohgane, J.; Yagi, S.; Tanaka, S.; Shiota, K. Epigenetic regulation of Nanog gene in embryonic stem and trophoblast stem cells. Genes Cells 2007, 12, 387–396. [Google Scholar] [CrossRef]
- Hawkins, R.D.; Hon, G.C.; Lee, L.K.; Ngo, Q.; Lister, R.; Pelizzola, M.; Edsall, L.E.; Kuan, S.; Luu, Y.; Klugman, S.; et al. Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell Stem Cell 2010, 6, 479–491. [Google Scholar] [CrossRef]
- Bibikova, M.; Chudin, E.; Wu, B.; Zhou, L.X.; Garcia, E.W.; Liu, Y.; Shin, S.; Plaia, T.W.; Auerbach, J.M.; Arking, D.E.; et al. Human embryonic stem cells have a unique epigenetic signature. Genome Res. 2006, 16, 1075–1083. [Google Scholar] [CrossRef]
- Mohn, F.; Weber, M.; Rebhan, M.; Roloff, T.C.; Richter, J.; Stadler, M.B.; Bibel, M.; Schubeler, D. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol. Cell 2008, 30, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Shen, Q.; Goderie, S.K.; He, W.; Capela, A.; Davis, A.A.; Temple, S. Timing of CNS cell generation: A programmed sequence of neuron and glial cell production from isolated murine cortical stem cells. Neuron 2000, 28, 69–80. [Google Scholar] [CrossRef]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Tzika, E.; Dreker, T.; Imhof, A. Epigenetics and Metabolism in Health and Disease. Front. Genet. 2018, 9, 361. [Google Scholar] [CrossRef]
- Easwaran, H.; Tsai, H.C.; Baylin, S.B. Cancer epigenetics: Tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol. Cell 2014, 54, 716–727. [Google Scholar] [CrossRef]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef]
- Bergman, Y.; Cedar, H. DNA methylation dynamics in health and disease. Nat. Struct. Mol. Biol. 2013, 20, 274–281. [Google Scholar] [CrossRef]
- Varley, K.E.; Gertz, J.; Bowling, K.M.; Parker, S.L.; Reddy, T.E.; Pauli-Behn, F.; Cross, M.K.; Williams, B.A.; Stamatoyannopoulos, J.A.; Crawford, G.E.; et al. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 2013, 23, 555–567. [Google Scholar] [CrossRef]
- Irizarry, R.A.; Ladd-Acosta, C.; Wen, B.; Wu, Z.J.; Montano, C.; Onyango, P.; Cui, H.M.; Gabo, K.; Rongione, M.; Webster, M.; et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009, 41, 178–186. [Google Scholar] [CrossRef]
- Nazor, K.L.; Altun, G.; Lynch, C.; Tran, H.; Harness, J.V.; Slavin, I.; Garitaonandia, I.; Muller, F.J.; Wang, Y.C.; Boscolo, F.S.; et al. Recurrent Variations in DNA Methylation in Human Pluripotent Stem Cells and Their Differentiated Derivatives. Cell Stem Cell 2012, 10, 620–634. [Google Scholar] [CrossRef]
- Weber, M.; Davies, J.J.; Wittig, D.; Oakeley, E.J.; Haase, M.; Lam, W.L.; Schubeler, D. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat. Genet. 2005, 37, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Meissner, A.; Mikkelsen, T.S.; Gu, H.; Wernig, M.; Hanna, J.; Sivachenko, A.; Zhang, X.; Bernstein, B.E.; Nusbaum, C.; Jaffe, D.B.; et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 2008, 454, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Laurent, L.; Wong, E.; Li, G.; Huynh, T.; Tsirigos, A.; Ong, C.T.; Low, H.M.; Kin Sung, K.W.; Rigoutsos, I.; Loring, J.; et al. Dynamic changes in the human methylome during differentiation. Genome Res. 2010, 20, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Berman, B.P.; Weisenberger, D.J.; Aman, J.F.; Hinoue, T.; Ramjan, Z.; Liu, Y.; Noushmehr, H.; Lange, C.P.; van Dijk, C.M.; Tollenaar, R.A.; et al. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat. Genet. 2011, 44, 40–46. [Google Scholar] [CrossRef]
- Cohen, N.M.; Kenigsberg, E.; Tanay, A. Primate CpG islands are maintained by heterogeneous evolutionary regimes involving minimal selection. Cell 2011, 145, 773–786. [Google Scholar] [CrossRef][Green Version]
- Davuluri, R.V.; Suzuki, Y.; Sugano, S.; Plass, C.; Huang, T.H.M. The functional consequences of alternative promoter use in mammalian genomes. Trends Genet. 2008, 24, 167–177. [Google Scholar] [CrossRef]
- Demircioglu, D.; Cukuroglu, E.; Kindermans, M.; Nandi, T.; Calabrese, C.; Fonseca, N.A.; Kahles, A.; Lehmann, K.V.; Stegle, O.; Brazma, A.; et al. A Pan-cancer Transcriptome Analysis Reveals Pervasive Regulation through Alternative Promoters. Cell 2019, 178, 1465–1477.e17. [Google Scholar] [CrossRef]
- Qamra, A.; Xing, M.; Padmanabhan, N.; Kwok, J.J.T.; Zhang, S.; Xu, C.; Leong, Y.S.; Lee Lim, A.P.; Tang, Q.; Ooi, W.F.; et al. Epigenomic Promoter Alterations Amplify Gene Isoform and Immunogenic Diversity in Gastric Adenocarcinoma. Cancer Discov. 2017, 7, 630–651. [Google Scholar] [CrossRef]
- Sotillo, E.; Barrett, D.M.; Black, K.L.; Bagashev, A.; Oldridge, D.; Wu, G.; Sussman, R.; Lanauze, C.; Ruella, M.; Gazzara, M.R.; et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015, 5, 1282–1295. [Google Scholar] [CrossRef]
- Ma, X.; Liu, Y.; Liu, Y.; Alexandrov, L.B.; Edmonson, M.N.; Gawad, C.; Zhou, X.; Li, Y.; Rusch, M.C.; Easton, J.; et al. Pan-cancer genome and transcriptome analyses of 1699 paediatric leukaemias and solid tumours. Nature 2018, 555, 371–376. [Google Scholar] [CrossRef]
- Grobner, S.N.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, M.; Brabetz, S.; et al. The landscape of genomic alterations across childhood cancers. Nature 2018, 555, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Huether, R.; Dong, L.; Chen, X.; Wu, G.; Parker, M.; Wei, L.; Ma, J.; Edmonson, M.N.; Hedlund, E.K.; Rusch, M.C.; et al. The landscape of somatic mutations in epigenetic regulators across 1000 paediatric cancer genomes. Nat. Commun. 2014, 5, 3630. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Muller, T.; Gessi, M.; Waha, A.; Isselstein, L.J.; Luxen, D.; Freihoff, D.; Freihoff, J.; Becker, A.; Simon, M.; Hammes, J.; et al. Nuclear Exclusion of TET1 Is Associated with Loss of 5-Hydroxymethylcytosine in IDH1 Wild-Type Gliomas. Am. J. Pathol. 2012, 181, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.; Christensen, J.; Pedersen, M.T.; Johansen, J.V.; Cloos, P.A.C.; Rappsilber, J.; Helin, K. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature 2011, 473, 343–348. [Google Scholar] [CrossRef]
- Ficz, G.; Branco, M.R.; Seisenberger, S.; Santos, F.; Krueger, F.; Hore, T.A.; Marques, C.J.; Andrews, S.; Reik, W. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature 2011, 473, 398–402. [Google Scholar] [CrossRef]
- Kuehner, J.N.; Chen, J.; Bruggeman, E.C.; Wang, F.; Li, Y.; Xu, C.; McEachin, Z.T.; Li, Z.; Chen, L.; Hales, C.M.; et al. 5-hydroxymethylcytosine is dynamically regulated during forebrain organoid development and aberrantly altered in Alzheimer’s disease. Cell Rep. 2021, 35, 109042. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Z.; Li, L.; Xu, K.; Ma, Z.; Chow, H.M.; Herrup, K.; Li, J. Selective loss of 5hmC promotes neurodegeneration in the mouse model of Alzheimer’s disease. FASEB J. 2020, 34, 16364–16382. [Google Scholar] [CrossRef]
- Hu, L.L.; Lu, J.Y.; Cheng, J.D.; Rao, Q.H.; Li, Z.; Hou, H.F.; Lou, Z.Y.; Zhang, L.; Li, W.; Gong, W.; et al. Structural insight into substrate preference for TET- mediated oxidation. Nature 2015, 527, 118–122. [Google Scholar] [CrossRef]
- Hu, L.L.; Li, Z.; Cheng, J.D.; Rao, Q.H.; Gong, W.; Liu, M.J.; Shi, Y.J.G.; Zhu, J.Y.; Wang, P.; Xu, Y.H. Crystal Structure of TET2-DNA Complex: Insight into TET-Mediated 5mC Oxidation. Cell 2013, 155, 1545–1555. [Google Scholar] [CrossRef]
- Si, Y.; Liu, J.; Shen, H.; Zhang, C.; Wu, Y.; Huang, Y.; Gong, Z.; Xue, J.; Liu, T. Fisetin decreases TET1 activity and CCNY/CDK16 promoter 5hmC levels to inhibit the proliferation and invasion of renal cancer stem cell. J. Cell. Mol. Med. 2019, 23, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.J.; Jung, S.W.; Huh, S.; Chung, Y.S.; Cho, H.; Kang, H. Alteration of DNA Methylation in Gastric Cancer with Chemotherapy. J. Microbiol. Biotechnol. 2017, 27, 1367–1378. [Google Scholar] [CrossRef] [PubMed]
- Chesnelong, C.; Chaumeil, M.M.; Blough, M.D.; Al-Najjar, M.; Stechishin, O.D.; Chan, J.A.; Pieper, R.O.; Ronen, S.M.; Weiss, S.; Luchman, H.A.; et al. Lactate dehydrogenase A silencing in IDH mutant gliomas. Neuro-Oncol. 2014, 16, 686–695. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.C.; Xu, J.; Bian, X.; Wu, J.L.; Kang, W.; Qian, Y.; Li, W.; Chen, H.; Gou, H.; Liu, D.; et al. In Colorectal Cancer Cells with Mutant KRAS, SLC25A22-Mediated Glutaminolysis Reduces DNA Demethylation to Increase WNT Signaling, Stemness, and Drug Resistance. Gastroenterology 2020, 159, 2163–2180.e6. [Google Scholar] [CrossRef]
- El Helou, R.; Wicinski, J.; Guille, A.; Adelaide, J.; Finetti, P.; Bertucci, F.; Chaffanet, M.; Birnbaum, D.; Charafe-Jauffret, E.; Ginestier, C. Brief reports: A distinct DNA methylation signature defines breast cancer stem cells and predicts cancer outcome. Stem Cells 2014, 32, 3031–3036. [Google Scholar] [CrossRef]
- Lu, R.; Wang, P.; Parton, T.; Zhou, Y.; Chrysovergis, K.; Rockowitz, S.; Chen, W.Y.; Abdel-Wahab, O.; Wade, P.A.; Zheng, D.; et al. Epigenetic Perturbations by Arg882-Mutated DNMT3A Potentiate Aberrant Stem Cell Gene-Expression Program and Acute Leukemia Development. Cancer Cell 2016, 30, 92–107. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, J.; Liu, Y.G.; Sidik, H.; Young, K.H.; Lodish, H.F.; Fleming, M.D. Oncogenic Kras-induced leukemogeneis: Hematopoietic stem cells as the initial target and lineage-specific progenitors as the potential targets for final leukemic transformation. Blood 2009, 113, 1304–1314. [Google Scholar] [CrossRef]
- Pathania, R.; Ramachandran, S.; Elangovan, S.; Padia, R.; Yang, P.Y.; Cinghu, S.; Veeranan-Karmegam, R.; Arjunan, P.; Gnana-Prakasam, J.P.; Sadanand, F.; et al. DNMT1 is essential for mammary and cancer stem cell maintenance and tumorigenesis. Nat. Commun. 2015, 6, 6910. [Google Scholar] [CrossRef]
- Wang, Q.; Liang, N.; Yang, T.; Li, Y.; Li, J.; Huang, Q.; Wu, C.; Sun, L.; Zhou, X.; Cheng, X.; et al. DNMT1-mediated methylation of BEX1 regulates stemness and tumorigenicity in liver cancer. J. Hepatol. 2021, 75, 1142–1153. [Google Scholar] [CrossRef]
- Bostick, M.; Kim, J.K.; Esteve, P.O.; Clark, A.; Pradhan, S.; Jacobsen, S.E. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science 2007, 317, 1760–1764. [Google Scholar] [CrossRef]
- Stathopoulou, A.; Chhetri, J.B.; Ambrose, J.C.; Esteve, P.O.; Ji, L.; Erdjument-Bromage, H.; Zhang, G.; Neubert, T.A.; Pradhan, S.; Herrero, J.; et al. A novel requirement for DROSHA in maintenance of mammalian CG methylation. Nucleic Acids Res. 2017, 45, 9398–9412. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Mao, S.Q.; Ray, C.; Zhang, Y.; Bell, F.T.; Ng, S.F.; Xu, G.L.; Li, X. Differential regulation of genomic imprinting by TET proteins in embryonic stem cells. Stem Cell Res. 2015, 15, 435–443. [Google Scholar] [CrossRef][Green Version]
- Wang, B.C.; Lin, G.H.; Wang, B.; Yan, M.; He, B.; Zhang, W.; Yang, A.K.; Long, Z.J.; Liu, Q. UHRF1 suppression promotes cell differentiation and reduces inflammatory reaction in anaplastic thyroid cancer. Oncotarget 2018, 9, 31945–31957. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Zhang, J.; Guo, Z.Q.; Ma, Q.; Xu, Z.Z.; Zhou, Y.Y.; Xu, Z.Y.; Li, Z.W.; Liu, Y.Q.; Ye, X.J.; et al. Loss of 5-hydroxymethylcytosine is linked to gene body hypermethylation in kidney cancer. Cell Res. 2016, 26, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.H.; Li, H.Q.; Liu, F. DNA Methyltransferase Inhibitors and their Therapeutic Potential. Curr. Top. Med. Chem. 2018, 18, 2448–2457. [Google Scholar] [CrossRef]
- Cramer-Morales, K.L.; Heer, C.D.; Mapuskar, K.A.; Domann, F.E. Succinate Accumulation Links Mitochondrial MnSOD Depletion to Aberrant Nuclear DNA Methylation and Altered Cell Fate. J. Exp. Pathol. 2020, 1, 60–70. [Google Scholar]
- Morris, J.P.t.; Yashinskie, J.J.; Koche, R.; Chandwani, R.; Tian, S.; Chen, C.C.; Baslan, T.; Marinkovic, Z.S.; Sanchez-Rivera, F.J.; Leach, S.D.; et al. alpha-Ketoglutarate links p53 to cell fate during tumour suppression. Nature 2019, 573, 595–599. [Google Scholar] [CrossRef]
- Letouze, E.; Martinelli, C.; Loriot, C.; Burnichon, N.; Abermil, N.; Ottolenghi, C.; Janin, M.; Menara, M.; Nguyen, A.T.; Benit, P.; et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell 2013, 23, 739–752. [Google Scholar] [CrossRef]
- Cordes, T.; Wallace, M.; Michelucci, A.; Divakaruni, A.S.; Sapcariu, S.C.; Sousa, C.; Koseki, H.; Cabrales, P.; Murphy, A.N.; Hiller, K.; et al. Immunoresponsive Gene 1 and Itaconate Inhibit Succinate Dehydrogenase to Modulate Intracellular Succinate Levels. J. Biol. Chem. 2016, 291, 14274–14284. [Google Scholar] [CrossRef]
- Heintzman, N.D.; Hon, G.C.; Hawkins, R.D.; Kheradpour, P.; Stark, A.; Harp, L.F.; Ye, Z.; Lee, L.K.; Stuart, R.K.; Ching, C.W.; et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009, 459, 108–112. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Huebert, D.J.; Bernstein, B.E. Genomic views of chromatin. Curr. Opin. Genet. Dev. 2005, 15, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Moussaieff, A.; Rouleau, M.; Kitsberg, D.; Cohen, M.; Levy, G.; Barasch, D.; Nemirovski, A.; Shen-Orr, S.; Laevsky, I.; Amit, M.; et al. Glycolysis-mediated changes in acetyl-CoA and histone acetylation control the early differentiation of embryonic stem cells. Cell Metab. 2015, 21, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Cliff, T.S.; Wu, T.; Boward, B.R.; Yin, A.; Yin, H.; Glushka, J.N.; Prestegaard, J.H.; Dalton, S. MYC Controls Human Pluripotent Stem Cell Fate Decisions through Regulation of Metabolic Flux. Cell Stem Cell 2017, 21, 502–516. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Gaeta, X.; Sahakyan, A.; Chan, A.B.; Hong, C.S.; Kim, R.; Braas, D.; Plath, K.; Lowry, W.E.; Christofk, H.R. Glycolytic Metabolism Plays a Functional Role in Regulating Human Pluripotent Stem Cell State. Cell Stem Cell 2016, 19, 476–490. [Google Scholar] [CrossRef]
- Lee, J.H.; Hart, S.R.L.; Skalnik, D.G. Histone deacetylase activity is required for embryonic stem cell differentiation. Genesis 2004, 38, 32–38. [Google Scholar] [CrossRef]
- Keller, G. Embryonic stem cell differentiation: Emergence of a new era in biology and medicine. Genes Dev. 2005, 19, 1129–1155. [Google Scholar] [CrossRef]
- Meshorer, E.; Misteli, T. Chromatin in pluripotent embryonic stem cells and differentiation. Nat. Rev. Mol. Cell Biol. 2006, 7, 540–546. [Google Scholar] [CrossRef]
- Golob, J.L.; Paige, S.L.; Muskheli, V.; Pabon, L.; Murry, C.E. Chromatin remodeling during mouse and human embryonic stem cell differentiation. Dev. Dyn. 2008, 237, 1389–1398. [Google Scholar] [CrossRef]
- Inoue, A.; Jiang, L.; Lu, F.; Suzuki, T.; Zhang, Y. Maternal H3K27me3 controls DNA methylation-independent imprinting. Nature 2017, 547, 419–424. [Google Scholar] [CrossRef]
- Meng, T.G.; Zhou, Q.; Ma, X.S.; Liu, X.Y.; Meng, Q.R.; Huang, X.J.; Liu, H.L.; Lei, W.L.; Zhao, Z.H.; Ouyang, Y.C.; et al. PRC2 and EHMT1 regulate H3K27me2 and H3K27me3 establishment across the zygote genome. Nat. Commun. 2020, 11, 6354. [Google Scholar] [CrossRef] [PubMed]
- Daitoku, H.; Hatta, M.; Matsuzaki, H.; Aratani, S.; Ohshima, T.; Miyagishi, M.; Nakajima, T.; Fukamizu, A. Silent information regulator 2 potentiates Foxo1-mediated transcription through its deacetylase activity. Proc. Natl. Acad. Sci. USA 2004, 101, 10042–10047. [Google Scholar] [CrossRef] [PubMed]
- Rokudai, S.; Laptenko, O.; Arnal, S.M.; Taya, Y.; Kitabayashi, I.; Prives, C. MOZ increases p53 acetylation and premature senescence through its complex formation with PML. Proc. Natl. Acad. Sci. USA 2013, 110, 3895–3900. [Google Scholar] [CrossRef] [PubMed]
- Sykes, S.M.; Mellert, H.S.; Holbert, M.A.; Li, K.; Marmorstein, R.; Lane, W.S.; McMahon, S.B. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol. Cell 2006, 24, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Tou, L.; Liu, Q.; Shivdasani, R.A. Regulation of mammalian epithelial differentiation and intestine development by class I histone deacetylases. Mol. Cell Biol. 2004, 24, 3132–3139. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.J.; Byun, D.S.; Popova, N.; Murray, L.B.; L’Italien, K.; Sowa, Y.; Arango, D.; Velcich, A.; Augenlicht, L.H.; Mariadason, J.M. Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J. Biol. Chem. 2006, 281, 13548–13558. [Google Scholar] [CrossRef]
- De Marzo, A.M.; Marchi, V.L.; Yang, E.S.; Veeraswamy, R.; Lin, X.; Nelson, W.G. Abnormal regulation of DNA methyltransferase expression during colorectal carcinogenesis. Cancer Res. 1999, 59, 3855–3860. [Google Scholar]
- Kang, Y.; Kim, Y.W.; Kang, J.; Kim, A. Histone H3K4me1 and H3K27ac play roles in nucleosome eviction and eRNA transcription, respectively, at enhancers. FASEB J. 2021, 35, e21781. [Google Scholar] [CrossRef]
- Azuara, V.; Perry, P.; Sauer, S.; Spivakov, M.; Jorgensen, H.F.; John, R.M.; Gouti, M.; Casanova, M.; Warnes, G.; Merkenschlager, M.; et al. Chromatin signatures of pluripotent cell lines. Nat. Cell Biol. 2006, 8, 532–538. [Google Scholar] [CrossRef]
- Sheikh, B.N.; Akhtar, A. The many lives of KATs—Detectors, integrators and modulators of the cellular environment. Nat. Rev. Genet. 2019, 20, 7–23. [Google Scholar] [CrossRef]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Lu, J.; Guo, Y.; Li, D.; Zhang, Z.M.; Tsai, Y.H.; Pi, W.C.; Ahn, J.H.; Gong, W.; Xiang, Y.; et al. BAHCC1 binds H3K27me3 via a conserved BAH module to mediate gene silencing and oncogenesis. Nat. Genet. 2020, 52, 1384–1396. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Li, Y.; Xi, Y.; Jiang, S.; Stratton, S.; Peng, D.; Tanaka, K.; Ren, Y.; Xia, Z.; Wu, J.; et al. ZMYND11 links histone H3.3K36me3 to transcription elongation and tumour suppression. Nature 2014, 508, 263–268. [Google Scholar] [CrossRef]
- Rabaneda-Bueno, R.; Mena-Montes, B.; Torres-Castro, S.; Torres-Carrillo, N.; Torres-Carrillo, N.M. Advances in Genetics and Epigenetic Alterations in Alzheimer’s Disease: A Notion for Therapeutic Treatment. Genes 2021, 12, 1959. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.; Rei, D.; Guan, J.S.; Wang, W.Y.; Seo, J.; Hennig, K.M.; Nieland, T.J.; Fass, D.M.; Kao, P.F.; Kahn, M.; et al. An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature 2012, 483, 222–226. [Google Scholar] [CrossRef]
- Zhang, K.; Schrag, M.; Crofton, A.; Trivedi, R.; Vinters, H.; Kirsch, W. Targeted proteomics for quantification of histone acetylation in Alzheimer’s disease. Proteomics 2012, 12, 1261–1268. [Google Scholar] [CrossRef]
- Vlaming, H.; McLean, C.M.; Korthout, T.; Alemdehy, M.F.; Hendriks, S.; Lancini, C.; Palit, S.; Klarenbeek, S.; Kwesi-Maliepaard, E.M.; Molenaar, T.M.; et al. Conserved crosstalk between histone deacetylation and H3K79 methylation generates DOT1L-dose dependency in HDAC1-deficient thymic lymphoma. EMBO J. 2019, 38, e101564. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Ye, X.; Tam, W.L.; Shibue, T.; Kaygusuz, Y.; Reinhardt, F.; Ng Eaton, E.; Weinberg, R.A. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature 2015, 525, 256–260. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Marjanovic, N.D.; Lee, T.; Bell, G.; Kleer, C.G.; Reinhardt, F.; D’Alessio, A.C.; Young, R.A.; Weinberg, R.A. Poised Chromatin at the ZEB1 Promoter Enables Breast Cancer Cell Plasticity and Enhances Tumorigenicity. Cell 2013, 154, 61–74. [Google Scholar] [CrossRef]
- Van den Beucken, T.; Koch, E.; Chu, K.; Rupaimoole, R.; Prickaerts, P.; Adriaens, M.; Voncken, J.W.; Harris, A.L.; Buffa, F.M.; Haider, S.; et al. Hypoxia promotes stem cell phenotypes and poor prognosis through epigenetic regulation of DICER. Nat. Commun. 2014, 5, 5203. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, A.; Xudong, Z.; Zhenyu, J.; Saretzki, G. Mitochondrial sirtuins in stem cells and cancer. FEBS J. 2022, 289, 3393–3415. [Google Scholar] [CrossRef] [PubMed]
- Szade, K.; Gulati, G.S.; Chan, C.K.F.; Kao, K.S.; Miyanishi, M.; Marjon, K.D.; Sinha, R.; George, B.M.; Chen, J.Y.; Weissman, I.L. Where Hematopoietic Stem Cells Live: The Bone Marrow Niche. Antioxid. Redox Signal. 2018, 29, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010, 12, 662–667. [Google Scholar] [CrossRef]
- Tseng, A.H.; Shieh, S.S.; Wang, D.L. SIRT3 deacetylates FOXO3 to protect mitochondria against oxidative damage. Free Radic. Biol. Med. 2013, 63, 222–234. [Google Scholar] [CrossRef]
- Brown, K.; Xie, S.; Qiu, X.; Mohrin, M.; Shin, J.; Liu, Y.; Zhang, D.; Scadden, D.T.; Chen, D. SIRT3 reverses aging-associated degeneration. Cell Rep. 2013, 3, 319–327. [Google Scholar] [CrossRef]
- Liu, C.Y.; Huang, Z.H.; Jiang, H.; Shi, F.J. The Sirtuin 3 Expression Profile Is Associated with Pathological and Clinical Outcomes in Colon Cancer Patients. Biomed Res. Int. 2014, 2014, 871263. [Google Scholar] [CrossRef]
- Dong, X.C.; Jing, L.M.; Wang, W.X.; Gao, Y.X. Down-regulation of SIRT3 promotes ovarian carcinoma metastasis. Biochem. Biophys. Res. Commun. 2016, 475, 245–250. [Google Scholar] [CrossRef]
- Min, Z.Y.; Gao, J.M.; Yu, Y. The Roles of Mitochondrial SIRT4 in Cellular Metabolism. Front. Endocrinol. 2019, 9, 783. [Google Scholar] [CrossRef]
- Du, L.; Liu, X.; Ren, Y.; Li, J.; Li, P.; Jiao, Q.; Meng, P.; Wang, F.; Wang, Y.; Wang, Y.S.; et al. Loss of SIRT4 promotes the self-renewal of Breast Cancer Stem Cells. Theranostics 2020, 10, 9458–9476. [Google Scholar] [CrossRef]
- Guan, J.; Jiang, X.; Gai, J.; Sun, X.; Zhao, J.; Li, J.; Li, Y.; Cheng, M.; Du, T.; Fu, L.; et al. Sirtuin 5 regulates the proliferation, invasion and migration of prostate cancer cells through acetyl-CoA acetyltransferase 1. J. Cell. Mol. Med. 2020, 24, 14039–14049. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.Z.; Qi, Y.J.; Wang, L.; Zheng, Z.X.; Zhang, Y.; Zheng, J.F. SIRT5-mediated SDHA desuccinylation promotes clear cell renal cell carcinoma tumorigenesis. Free Radic. Biol. Med. 2019, 134, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Yan, H.; An, S.; Shen, M.; Jia, W.; Zhang, R.; Zhao, L.; Huang, G.; Liu, J. SIRT5-mediated deacetylation of LDHB promotes autophagy and tumorigenesis in colorectal cancer. Mol. Oncol. 2019, 13, 358–375. [Google Scholar] [CrossRef]
- Yang, X.; Wang, Z.; Li, X.; Liu, B.; Liu, M.; Liu, L.; Chen, S.; Ren, M.; Wang, Y.; Yu, M.; et al. SHMT2 Desuccinylation by SIRT5 Drives Cancer Cell Proliferation. Cancer Res. 2018, 78, 372–386. [Google Scholar] [CrossRef] [PubMed]
- Ippolito, L.; Marini, A.; Cavallini, L.; Morandi, A.; Pietrovito, L.; Pintus, G.; Giannoni, E.; Schrader, T.; Puhr, M.; Chiarugi, P.; et al. Metabolic shift toward oxidative phosphorylation in docetaxel resistant prostate cancer cells. Oncotarget 2016, 7, 61890–61904. [Google Scholar] [CrossRef] [PubMed]
- Schopf, B.; Weissensteiner, H.; Schafer, G.; Fazzini, F.; Charoentong, P.; Naschberger, A.; Rupp, B.; Fendt, L.; Bukur, V.; Giese, I.; et al. OXPHOS remodeling in high-grade prostate cancer involves mtDNA mutations and increased succinate oxidation. Nat. Commun. 2020, 11, 1487. [Google Scholar] [CrossRef]
- Ahn, B.H.; Kim, H.S.; Song, S.; Lee, I.H.; Liu, J.; Vassilopoulos, A.; Deng, C.X.; Finkel, T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 14447–14452. [Google Scholar] [CrossRef]
- Zhang, M.; Wu, J.; Sun, R.; Tao, X.; Wang, X.; Kang, Q.; Wang, H.; Zhang, L.; Liu, P.; Zhang, J.; et al. SIRT5 deficiency suppresses mitochondrial ATP production and promotes AMPK activation in response to energy stress. PLoS ONE 2019, 14, e0211796. [Google Scholar] [CrossRef]
- Min, L.H.; Ji, Y.; Bakiri, L.; Qiu, Z.X.; Cen, J.; Chen, X.T.; Chen, L.L.; Scheuch, H.; Zheng, H.; Qin, L.X.; et al. Liver cancer initiation is controlled by AP-1 through SIRT6-dependent inhibition of survivin. Nat. Cell Biol. 2012, 14, 1203–1211. [Google Scholar] [CrossRef]
- Sebastian, C.; Zwaans, B.M.M.; Silberman, D.M.; Gymrek, M.; Goren, A.; Zhong, L.; Ram, O.; Truelove, J.; Guimaraes, A.R.; Toiber, D.; et al. The Histone Deacetylase SIRT6 Is a Tumor Suppressor that Controls Cancer Metabolism. Cell 2012, 151, 1185–1199. [Google Scholar] [CrossRef]
- Kugel, S.; Sebastian, C.; Fitamant, J.; Ross, K.N.; Saha, S.K.; Jain, E.; Gladden, A.; Arora, K.S.; Kato, Y.; Rivera, M.N.; et al. SIRT6 Suppresses Pancreatic Cancer through Control of Lin28b. Cell 2016, 165, 1401–1415. [Google Scholar] [CrossRef] [PubMed]
- Van Meter, M.; Mao, Z.; Gorbunova, V.; Seluanov, A. SIRT6 overexpression induces massive apoptosis in cancer cells but not in normal cells. Cell Cycle 2011, 10, 3153–3158. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wu, M.; Du, H.; Shi, X.; Zhang, T.; Li, J. SIRT6 inhibits colorectal cancer stem cell proliferation by targeting CDC25A. Oncol. Lett. 2018, 15, 5368–5374. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.S.; Wang, H.P.; Li, X.Y.; Chao, T.; Christen, T.T.S.; Christen, S.; Di Conza, G.; Cheng, W.C.; Chou, C.H.; Vavakova, M.; et al. alpha-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol. 2017, 18, 985–994. [Google Scholar] [CrossRef]
- Ishii, M.; Wen, H.; Corsa, C.A.; Liu, T.; Coelho, A.L.; Allen, R.M.; Carson, W.F.t.; Cavassani, K.A.; Li, X.; Lukacs, N.W.; et al. Epigenetic regulation of the alternatively activated macrophage phenotype. Blood 2009, 114, 3244–3254. [Google Scholar] [CrossRef]
- Wierda, R.J.; Goedhart, M.; van Eggermond, M.C.; Muggen, A.F.; Miggelbrink, X.M.; Geutskens, S.B.; van Zwet, E.; Haasnoot, G.W.; van den Elsen, P.J. A role for KMT1c in monocyte to dendritic cell differentiation: Epigenetic regulation of monocyte differentiation. Hum. Immunol. 2015, 76, 431–437. [Google Scholar] [CrossRef]
- Kurotaki, D.; Tamura, T. Transcriptional and Epigenetic Regulation of Innate Immune Cell Development by the Transcription Factor, Interferon Regulatory Factor-8. J. Interferon Cytokine Res. 2016, 36, 433–441. [Google Scholar] [CrossRef]
- Chakrabarty, R.P.; Chandel, N.S. Mitochondria as Signaling Organelles Control Mammalian Stem Cell Fate. Cell Stem Cell 2021, 28, 394–408. [Google Scholar] [CrossRef]
- Das, S.; Morvan, F.; Morozzi, G.; Jourde, B.; Minetti, G.C.; Kahle, P.; Rivet, H.; Brebbia, P.; Toussaint, G.; Glass, D.J.; et al. ATP Citrate Lyase Regulates Myofiber Differentiation and Increases Regeneration by Altering Histone Acetylation. Cell Rep. 2017, 21, 3003–3011. [Google Scholar] [CrossRef]
- Mani, S.; Swargiary, G.; Tyagi, S.; Singh, M.; Jha, N.K.; Singh, K.K. Nanotherapeutic approaches to target mitochondria in cancer. Life Sci. 2021, 281, 119773. [Google Scholar] [CrossRef]
- Yang, L.D.; Youngblood, H.; Wu, C.Y.; Zhang, Q.G. Mitochondria as a target for neuroprotection: Role of methylene blue and photobiomodulation. Transl. Neurodegener. 2020, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.F.; Myers, C.R.; Wang, Y.A.; You, M. Mitochondria as a Novel Target for Cancer Chemoprevention: Emergence of Mitochondrial-targeting Agents. Cancer Prev. Res. 2021, 14, 285–306. [Google Scholar] [CrossRef] [PubMed]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef] [PubMed]
- Willmes, C. Mitochondria—A Powerful Therapeutic Target. Trends Mol. Med. 2020, 26, 1–2. [Google Scholar] [CrossRef]
- Dong, L.F.; Neuzil, J. Mitochondria in cancer: Why mitochondria are a good target for cancer therapy. Prog. Mol. Biol. Transl. Sci. 2014, 127, 211–227. [Google Scholar] [CrossRef]
- Zinovkin, R.A.; Zamyatnin, A.A. Mitochondria-Targeted Drugs. Curr. Mol. Pharm. 2019, 12, 202–214. [Google Scholar] [CrossRef]
- Heller, A.; Brockhoff, G.; Goepferich, A. Targeting drugs to mitochondria. Eur. J. Pharm. Biopharm. 2012, 82, 1–18. [Google Scholar] [CrossRef]
- Ferrin, G.; Linares, C.I.; Muntane, J. Mitochondrial drug targets in cell death and cancer. Curr. Pharm. Des. 2011, 17, 2002–2016. [Google Scholar] [CrossRef]
- Huang, S.; Liu, Y.; Zhang, Y.; Zhang, R.; Zhu, C.; Fan, L.; Pei, G.; Zhang, B.; Shi, Y. Baicalein inhibits SARS-CoV-2/VSV replication with interfering mitochondrial oxidative phosphorylation in a mPTP dependent manner. Signal. Transduct. Target. Ther. 2020, 5, 266. [Google Scholar] [CrossRef]
- Martinez-Reyes, I.; Chandel, N.S. Acetyl-CoA-directed gene transcription in cancer cells. Genes Dev. 2018, 32, 463–465. [Google Scholar] [CrossRef]
- Lee, J.V.; Berry, C.T.; Kim, K.; Sen, P.; Kim, T.; Carrer, A.; Trefely, S.; Zhao, S.; Fernandez, S.; Barney, L.E.; et al. Acetyl-CoA promotes glioblastoma cell adhesion and migration through Ca2+-NFAT signaling. Genes Dev. 2018, 32, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Lei, I.; Tian, S.; Gao, W.; Liu, L.; Guo, Y.; Tang, P.; Chen, E.; Wang, Z. Acetyl-CoA production by specific metabolites promotes cardiac repair after myocardial infarction via histone acetylation. Elife 2021, 10, e60311. [Google Scholar] [CrossRef] [PubMed]
- Carrer, A.; Trefely, S.; Zhao, S.; Campbell, S.L.; Norgard, R.J.; Schultz, K.C.; Sidoli, S.; Parris, J.L.D.; Affronti, H.C.; Sivanand, S.; et al. Acetyl-CoA Metabolism Supports Multistep Pancreatic Tumorigenesis. Cancer Discov. 2019, 9, 416–435. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Rajput, S.; Watabe, K.; Liao, D.F.; Cao, D. Acetyl-CoA carboxylase-a as a novel target for cancer therapy. Front. Biosci. 2010, 2, 515–526. [Google Scholar] [CrossRef]
- Chen, L.; Liu, T.; Zhou, J.; Wang, Y.; Wang, X.; Di, W.; Zhang, S. Citrate synthase expression affects tumor phenotype and drug resistance in human ovarian carcinoma. PLoS ONE 2014, 9, e115708. [Google Scholar] [CrossRef]
- Schlichtholz, B.; Turyn, J.; Goyke, E.; Biernacki, M.; Jaskiewicz, K.; Sledzinski, Z.; Swierczynski, J. Enhanced citrate synthase activity in human pancreatic cancer. Pancreas 2005, 30, 99–104. [Google Scholar] [CrossRef]
- Zaidi, N.; Swinnen, J.V.; Smans, K. ATP-citrate lyase: A key player in cancer metabolism. Cancer Res. 2012, 72, 3709–3714. [Google Scholar] [CrossRef]
- Migita, T.; Narita, T.; Nomura, K.; Miyagi, E.; Inazuka, F.; Matsuura, M.; Ushijima, M.; Mashima, T.; Seimiya, H.; Satoh, Y.; et al. ATP citrate lyase: Activation and therapeutic implications in non-small cell lung cancer. Cancer Res. 2008, 68, 8547–8554. [Google Scholar] [CrossRef]
- Huang, L.; Wang, C.D.; Xu, H.X.; Peng, G.Y. Targeting citrate as a novel therapeutic strategy in cancer treatment. BBA-Rev. Cancer 2020, 1873, 188332. [Google Scholar] [CrossRef]
- Popovici-Muller, J.; Lemieux, R.M.; Artin, E.; Saunders, J.O.; Salituro, F.G.; Travins, J.; Cianchetta, G.; Cai, Z.; Zhou, D.; Cui, D.; et al. Discovery of AG-120 (Ivosidenib): A First-in-Class Mutant IDH1 Inhibitor for the Treatment of IDH1 Mutant Cancers. ACS Med. Chem. Lett. 2018, 9, 300–305. [Google Scholar] [CrossRef]
- Han, S.; Liu, Y.; Cai, S.J.; Qian, M.; Ding, J.; Larion, M.; Gilbert, M.R.; Yang, C. IDH mutation in glioma: Molecular mechanisms and potential therapeutic targets. Br. J. Cancer 2020, 122, 1580–1589. [Google Scholar] [CrossRef] [PubMed]
- McMurry, H.; Fletcher, L.; Traer, E. IDH Inhibitors in AML-Promise and Pitfalls. Curr. Hematol. Malig. Rep. 2021, 16, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Paschka, P.; Schlenk, R.F.; Gaidzik, V.I.; Habdank, M.; Kronke, J.; Bullinger, L.; Spath, D.; Kayser, S.; Zucknick, M.; Gotze, K.; et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J. Clin. Oncol. 2010, 28, 3636–3643. [Google Scholar] [CrossRef] [PubMed]
- Waitkus, M.S.; Diplas, B.H.; Yan, H. Biological Role and Therapeutic Potential of IDH Mutations in Cancer. Cancer Cell 2018, 34, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Borger, D.R.; Tanabe, K.K.; Fan, K.C.; Lopez, H.U.; Fantin, V.R.; Straley, K.S.; Schenkein, D.P.; Hezel, A.F.; Ancukiewicz, M.; Liebman, H.M.; et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist 2012, 17, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; O’Neill, L.A.J. Krebs Cycle Reimagined: The Emerging Roles of Succinate and Itaconate as Signal Transducers. Cell 2018, 174, 780–784. [Google Scholar] [CrossRef]
- Kula-Alwar, D.; Prag, H.A.; Krieg, T. Targeting Succinate Metabolism in Ischemia/Reperfusion Injury. Circulation 2019, 140, 1968–1970. [Google Scholar] [CrossRef]
- Cui, H.T.; Chen, Y.H.; Li, K.; Zhan, R.; Zhao, M.M.; Xu, Y.K.; Lin, Z.Y.; Fu, Y.; He, Q.H.; Tang, P.C.; et al. Untargeted metabolomics identifies succinate as a biomarker and therapeutic target in aortic aneurysm and dissection. Eur. Heart J. 2021, 42, 4373–4385. [Google Scholar] [CrossRef]
- Kornberg, M.D.; Bhargava, P.; Kim, P.M.; Putluri, V.; Snowman, A.M.; Putluri, N.; Calabresi, P.A.; Snyder, S.H. Dimethyl fumarate targets GAPDH and aerobic glycolysis to modulate immunity. Science 2018, 360, 449–453. [Google Scholar] [CrossRef]
- Angiari, S.; O’Neill, L.A. Dimethyl fumarate: Targeting glycolysis to treat MS. Cell Res. 2018, 28, 613–615. [Google Scholar] [CrossRef]
- Waack, I.N.; Himmen, S.; Mueller, F.; Rohrig, R.; Roehrborn, F.; Teloh, J.K.; de Groot, H. L-Malate’s Plasma and Excretion Profile in the Treatment of Moderate and Severe Hemorrhagic Shock in Rats. Biomed Res. Int. 2016, 2016, 5237148. [Google Scholar] [CrossRef] [PubMed]
- Pons, F.; Bellmunt, J. Sunitinib malate in the treatment of urothelial cancer. Expert Opin. Investig. Drugs 2014, 23, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Selinski, J.; Scheibe, R. Malate valves: Old shuttles with new perspectives. Plant Biol. 2019, 21 (Suppl. S1), 21–30. [Google Scholar] [CrossRef] [PubMed]
- Kayamba, F.; Faya, M.; Pooe, O.J.; Kushwaha, B.; Kushwaha, N.D.; Obakachi, V.A.; Nyamori, V.O.; Karpoormath, R. Lactate dehydrogenase and malate dehydrogenase: Potential antiparasitic targets for drug development studies. Bioorg. Med. Chem. 2021, 50, 116458. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.C.; Tian, P.F.; Chen, Z.P.; Yue, D.S.; Liu, C.C.; Li, C.G.; Chen, C.; Zhang, H.; Liu, H.L.; Zhang, Z.F.; et al. Urinary malate dehydrogenase 2 is a new biomarker for early detection of non-small-cell lung cancer. Cancer Sci. 2021, 112, 2349–2360. [Google Scholar] [CrossRef]
- Wiese, E.K.; Hitosugi, S.; Loa, S.T.; Sreedhar, A.; Andres-Beck, L.G.; Kurmi, K.; Pang, Y.P.; Karnitz, L.M.; Gonsalves, W.I.; Hitosugi, T. Enzymatic activation of pyruvate kinase increases cytosolic oxaloacetate to inhibit the Warburg effect. Nat. Metab. 2021, 3, 954–968. [Google Scholar] [CrossRef]
- Tungtur, S.K.; Wilkins, H.M.; Rogers, R.S.; Badawi, Y.; Sage, J.M.; Agbas, A.; Jawdat, O.; Barohn, R.J.; Swerdlow, R.H.; Nishimune, H. Oxaloacetate treatment preserves motor function in SOD1(G93A) mice and normalizes select neuroinflammation-related parameters in the spinal cord. Sci. Rep. 2021, 11, 11051. [Google Scholar] [CrossRef]
- Gou, L.; Lee, J.; Hao, H.; Park, Y.D.; Zhan, Y.; Lu, Z.R. The effect of oxaloacetic acid on tyrosinase activity and structure: Integration of inhibition kinetics with docking simulation. Int. J. Biol. Macromol. 2017, 101, 59–66. [Google Scholar] [CrossRef]
- Regenbrecht, C.R.; Lehrach, H.; Adjaye, J. Stemming cancer: Functional genomics of cancer stem cells in solid tumors. Stem Cell Rev. 2008, 4, 319–328. [Google Scholar] [CrossRef]
- Ganesh, S.; Venkatakrishnan, K.; Tan, B. Quantum scale organic semiconductors for SERS detection of DNA methylation and gene expression. Nat. Commun. 2020, 11, 1135. [Google Scholar] [CrossRef]
- Morera, L.; Lubbert, M.; Jung, M. Targeting histone methyltransferases and demethylases in clinical trials for cancer therapy. Clin. Epigenet. 2016, 8, 57. [Google Scholar] [CrossRef] [PubMed]
- Plass, C.; Pfister, S.M.; Lindroth, A.M.; Bogatyrova, O.; Claus, R.; Lichter, P. Mutations in regulators of the epigenome and their connections to global chromatin patterns in cancer. Nat. Rev. Genet. 2013, 14, 765–780. [Google Scholar] [CrossRef] [PubMed]
- Heerboth, S.; Lapinska, K.; Snyder, N.; Leary, M.; Rollinson, S.; Sarkar, S. Use of epigenetic drugs in disease: An overview. Genet. Epigenet. 2014, 6, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Khan, H.; Vale, C.; Bhagat, T.; Verma, A. Role of DNA methylation in the pathogenesis and treatment of myelodysplastic syndromes. Semin. Hematol. 2013, 50, 16–37. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, E.; Issa, J.P.; Garcia-Manero, G.; Kantarjian, H. Evolution of decitabine development: Accomplishments, ongoing investigations, and future strategies. Cancer 2008, 112, 2341–2351. [Google Scholar] [CrossRef]
- Cheng, J.C.; Matsen, C.B.; Gonzales, F.A.; Ye, W.; Greer, S.; Marquez, V.E.; Jones, P.A.; Selker, E.U. Inhibition of DNA methylation and reactivation of silenced genes by zebularine. J. Natl. Cancer Inst. 2003, 95, 399–409. [Google Scholar] [CrossRef]
- Graca, I.; Sousa, E.J.; Baptista, T.; Almeida, M.; Ramalho-Carvalho, J.; Palmeira, C.; Henrique, R.; Jeronimo, C. Anti-tumoral effect of the non-nucleoside DNMT inhibitor RG108 in human prostate cancer cells. Curr. Pharm. Des. 2014, 20, 1803–1811. [Google Scholar] [CrossRef]
- Borges, S.; Doppler, H.R.; Storz, P. A combination treatment with DNA methyltransferase inhibitors and suramin decreases invasiveness of breast cancer cells. Breast Cancer Res. Treat. 2014, 144, 79–91. [Google Scholar] [CrossRef]
- Suzuki, T.; Tanaka, R.; Hamada, S.; Nakagawa, H.; Miyata, N. Design, synthesis, inhibitory activity, and binding mode study of novel DNA methyltransferase 1 inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 1124–1127. [Google Scholar] [CrossRef]
- Stresemann, C.; Brueckner, B.; Musch, T.; Stopper, H.; Lyko, F. Functional diversity of DNA methyltransferase inhibitors in human cancer cell lines. Cancer Res. 2006, 66, 2794–2800. [Google Scholar] [CrossRef]
- Song, Y.; Zhang, C. Hydralazine inhibits human cervical cancer cell growth in vitro in association with APC demethylation and re-expression. Cancer Chemother. Pharm. 2009, 63, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Duenas-Gonzalez, A.; Lyko, F.; Medina-Franco, J.L. Molecular modeling and molecular dynamics studies of hydralazine with human DNA methyltransferase 1. ChemMedChem 2009, 4, 792–799. [Google Scholar] [CrossRef] [PubMed]
- Datta, J.; Ghoshal, K.; Denny, W.A.; Gamage, S.A.; Brooke, D.G.; Phiasivongsa, P.; Redkar, S.; Jacob, S.T. A new class of quinoline-based DNA hypomethylating agents reactivates tumor suppressor genes by blocking DNA methyltransferase 1 activity and inducing its degradation. Cancer Res. 2009, 69, 4277–4285. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Dominguez, P.; Dell’aversana, C.; Alvarez, R.; Altucci, L.; de Lera, A.R. Synthetic approaches to DNMT inhibitor SGI-1027 and effects on the U937 leukemia cell line. Bioorg. Med. Chem. Lett. 2013, 23, 1631–1635. [Google Scholar] [CrossRef]
- Manara, M.C.; Valente, S.; Cristalli, C.; Nicoletti, G.; Landuzzi, L.; Zwergel, C.; Mazzone, R.; Stazi, G.; Arimondo, P.B.; Pasello, M.; et al. A Quinoline-Based DNA Methyltransferase Inhibitor as a Possible Adjuvant in Osteosarcoma Therapy. Mol. Cancer Ther. 2018, 17, 1881–1892. [Google Scholar] [CrossRef]
- She, S.; Zhao, Y.; Kang, B.; Chen, C.; Chen, X.; Zhang, X.; Chen, W.; Dan, S.; Wang, H.; Wang, Y.J.; et al. Combined inhibition of JAK1/2 and DNMT1 by newly identified small-molecule compounds synergistically suppresses the survival and proliferation of cervical cancer cells. Cell Death Dis. 2020, 11, 724. [Google Scholar] [CrossRef]
- Pappalardi, M.B.; Keenan, K.; Cockerill, M.; Kellner, W.A.; Stowell, A.; Sherk, C.; Wong, K.; Pathuri, S.; Briand, J.; Steidel, M.; et al. Discovery of a first-in-class reversible DNMT1-selective inhibitor with improved tolerability and efficacy in acute myeloid leukemia. Nat. Cancer 2021, 2, 1002–1017. [Google Scholar] [CrossRef]
- Colyn, L.; Barcena-Varela, M.; Alvarez-Sola, G.; Latasa, M.U.; Uriarte, I.; Santamaria, E.; Herranz, J.M.; Santos-Laso, A.; Arechederra, M.; Ruiz de Gauna, M.; et al. Dual Targeting of G9a and DNA Methyltransferase-1 for the Treatment of Experimental Cholangiocarcinoma. Hepatology 2021, 73, 2380–2396. [Google Scholar] [CrossRef]
- Du, S.; Xu, G.; Zou, W.; Xiang, T.; Luo, Z. Effect of dihydroartemisinin on UHRF1 gene expression in human prostate cancer PC-3 cells. Anticancer Drugs 2017, 28, 384–391. [Google Scholar] [CrossRef]
- Unoki, M.; Brunet, J.; Mousli, M. Drug discovery targeting epigenetic codes: The great potential of UHRF1, which links DNA methylation and histone modifications, as a drug target in cancers and toxoplasmosis. Biochem. Pharm. 2009, 78, 1279–1288. [Google Scholar] [CrossRef]
- Li, X.L.; Xu, J.H.; Nie, J.H.; Fan, S.J. Exogenous expression of UHRF1 promotes proliferation and metastasis of breast cancer cells. Oncol. Rep. 2012, 28, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Ferry, L.; Fournier, A.; Tsusaka, T.; Adelmant, G.; Shimazu, T.; Matano, S.; Kirsh, O.; Amouroux, R.; Dohmae, N.; Suzuki, T.; et al. Methylation of DNA Ligase 1 by G9a/GLP Recruits UHRF1 to Replicating DNA and Regulates DNA Methylation. Mol. Cell 2017, 67, 550–565.e5. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.J.; Byun, H.O.; Jee, B.A.; Min, S.; Jeoun, U.W.; Lee, Y.K.; Seo, Y.; Woo, H.G.; Yoon, G. The Ubiquitin-like with PHD and Ring Finger Domains 1 (UHRF1)/DNA Methyltransferase 1 (DNMT1) Axis Is a Primary Regulator of Cell Senescence. J. Biol. Chem. 2017, 292, 3729–3739. [Google Scholar] [CrossRef] [PubMed]
- Amato, R.J.; Stephenson, J.; Hotte, S.; Nemunaitis, J.; Belanger, K.; Reid, G.; Martell, R.E. MG98, a second-generation DNMT1 inhibitor, in the treatment of advanced renal cell carcinoma. Cancer Investig. 2012, 30, 415–421. [Google Scholar] [CrossRef]
- Nandakumar, V.; Vaid, M.; Katiyar, S.K. (-)-Epigallocatechin-3-gallate reactivates silenced tumor suppressor genes, Cip1/p21 and p16INK4a, by reducing DNA methylation and increasing histones acetylation in human skin cancer cells. Carcinogenesis 2011, 32, 537–544. [Google Scholar] [CrossRef]
- Li, Y.C.; Wang, Y.; Li, D.D.; Zhang, Y.; Zhao, T.C.; Li, C.F. Procaine is a specific DNA methylation inhibitor with anti-tumor effect for human gastric cancer. J. Cell. Biochem. 2018, 119, 2440–2449. [Google Scholar] [CrossRef]
- Medina-Franco, J.L.; Caulfield, T. Advances in the computational development of DNA methyltransferase inhibitors. Drug Discov. Today 2011, 16, 418–425. [Google Scholar] [CrossRef]
- Pereira, M.A.; Tao, L.; Liu, Y.; Li, L.; Steele, V.E.; Lubet, R.A. Modulation by budesonide of DNA methylation and mRNA expression in mouse lung tumors. Int. J. Cancer 2007, 120, 1150–1153. [Google Scholar] [CrossRef]
- Alyaqoub, F.S.; Tao, L.; Kramer, P.M.; Steele, V.E.; Lubet, R.A.; Gunning, W.T.; Pereira, M.A. Prevention of mouse lung tumors and modulation of DNA methylation by combined treatment with budesonide and R115777 (ZarnestraMT). Carcinogenesis 2006, 27, 2442–2447. [Google Scholar] [CrossRef][Green Version]
- Neja, S.A. Site-Specific DNA Demethylation as a Potential Target for Cancer Epigenetic Therapy. Epigenet. Insights 2020, 13. [Google Scholar] [CrossRef]
- Cong, B.; Zhang, Q.; Cao, X. The function and regulation of TET2 in innate immunity and inflammation. Protein Cell 2021, 12, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Luo, M.; Pan, K.; Ahmad, T.; Zhou, T.; Miao, Z.; Zhou, H.; Sun, H.; Xu, X.; Namaka, M.; et al. DNA hydroxymethylation changes in response to spinal cord damage in a multiple sclerosis mouse model. Epigenomics 2019, 11, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Nair, N.; Plant, D.; Verstappen, S.M.; Isaacs, J.D.; Morgan, A.W.; Hyrich, K.L.; Barton, A.; Wilson, A.G. MATURA investigators Differential DNA methylation correlates with response to methotrexate in rheumatoid arthritis. Rheumatology 2020, 59, 1364–1371. [Google Scholar] [CrossRef] [PubMed]
- Rohle, D.; Popovici-Muller, J.; Palaskas, N.; Turcan, S.; Grommes, C.; Campos, C.; Tsoi, J.; Clark, O.; Oldrini, B.; Komisopoulou, E.; et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013, 340, 626–630. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, A.; Cruz, M.M.A.; Jyotsana, N.; Sharma, A.; Yun, H.Y.; Gorlich, K.; Wichmann, M.; Schwarzer, A.; Preller, M.; Thol, F.; et al. Mutant IDH1 promotes leukemogenesis in vivo and can be specifically targeted in human AML. Blood 2013, 122, 2877–2887. [Google Scholar] [CrossRef]
- McCabe, M.T.; Mohammad, H.P.; Barbash, O.; Kruger, R.G. Targeting Histone Methylation in Cancer. Cancer J. 2017, 23, 292–301. [Google Scholar] [CrossRef]
- Min, J.R.; Feng, Q.; Li, Z.Z.; Zhang, Y.; Xu, R.M. Structure of the catalytic domain of human DOT1L, a Non-SET domain nucleosomal histone methyltransferase. Cell 2003, 112, 711–723. [Google Scholar] [CrossRef]
- Mao, Y.; Sun, Y.; Wu, Z.; Zheng, J.; Zhang, J.; Zeng, J.; Lee, C.; Kim, J.K. Targeting of histone methyltransferase DOT1L plays a dual role in chemosensitization of retinoblastoma cells and enhances the efficacy of chemotherapy. Cell Death Dis. 2021, 12, 1141. [Google Scholar] [CrossRef]
- Stein, E.M.; Garcia-Manero, G.; Rizzieri, D.A.; Tibes, R.; Berdeja, J.G.; Savona, M.R.; Jongen-Lavrenic, M.; Altman, J.K.; Thomson, B.; Blakemore, S.J.; et al. The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood 2018, 131, 2661–2669. [Google Scholar] [CrossRef]
- Campbell, C.T.; Haladyna, J.N.; Drubin, D.A.; Thomson, T.M.; Maria, M.J.; Yamauchi, T.; Waters, N.J.; Olhava, E.J.; Pollock, R.M.; Smith, J.J.; et al. Mechanisms of Pinometostat (EPZ-5676) Treatment-Emergent Resistance in MLL-Rearranged Leukemia. Mol. Cancer Ther. 2017, 16, 1669–1679. [Google Scholar] [CrossRef]
- Yi, Y.; Ge, S. Targeting the histone H3 lysine 79 methyltransferase DOT1L in MLL-rearranged leukemias. J. Hematol. Oncol. 2022, 15, 35. [Google Scholar] [CrossRef] [PubMed]
- Kanouni, T.; Severin, C.; Cho, R.W.; Yuen, N.Y.; Xu, J.; Shi, L.; Lai, C.; Del Rosario, J.R.; Stansfield, R.K.; Lawton, L.N.; et al. Discovery of CC-90011: A Potent and Selective Reversible Inhibitor of Lysine Specific Demethylase 1 (LSD1). J. Med. Chem. 2020, 63, 14522–14529. [Google Scholar] [CrossRef] [PubMed]
- Minucci, S.; Pelicci, P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Copeland, R.A.; Solomon, M.E.; Richon, V.M. Protein methyltransferases as a target class for drug discovery. Nat. Rev. Drug Discov. 2009, 8, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Janknecht, R. The versatile functions of the transcriptional coactivators p300 and CBP and their roles in disease. Histol. Histopathol. 2002, 17, 657–668. [Google Scholar]
- He, Z.X.; Wei, B.F.; Zhang, X.; Gong, Y.P.; Ma, L.Y.; Zhao, W. Current development of CBP/p300 inhibitors in the last decade. Eur. J. Med. Chem. 2021, 209, 112861. [Google Scholar] [CrossRef]
- Bedford, D.C.; Kasper, L.H.; Fukuyama, T.; Brindle, P.K. Target gene context influences the transcriptional requirement for the KAT3 family of CBP and p300 histone acetyltransferases. Epigenetics 2010, 5, 9–15. [Google Scholar] [CrossRef]
- Cai, L.Y.; Chen, S.J.; Xiao, S.H.; Sun, Q.J.; Ding, C.H.; Zheng, B.N.; Zhu, X.Y.; Liu, S.Q.; Yang, F.; Yang, Y.X.; et al. Targeting p300/CBP Attenuates Hepatocellular Carcinoma Progression through Epigenetic Regulation of Metabolism. Cancer Res. 2021, 81, 860–872. [Google Scholar] [CrossRef]
- Crabb, S.; Plummer, R.; Greystoke, A.; Carter, L.; Pacey, S.; Walter, H.; Coyle, V.M.; Knurowski, T.; Clegg, K.; Ashby, F.; et al. A phase I/IIa study to evaluate the safety and efficacy of CCS1477, a first in clinic inhibitor of p300/CBP, as monotherapy in patients with selected molecular alterations. Ann. Oncol. 2021, 32, S617. [Google Scholar] [CrossRef]
- Knurowski, T.; Clegg, K.; Brooks, N.; Ashby, F.; Pegg, N.A.; West, W.; Walter, H.S.; Somervaille, T.C.P.; Knapper, S.; Davies, A. An Open-Label Phase I/IIa Study to Evaluate the Safety and Efficacy of CCS1477, a First in Clinic Inhibitor of the p300/CPB Bromodomains, As Monotherapy in Patients with Advanced Haematological Malignancies. Blood 2019, 134, 1266. [Google Scholar] [CrossRef]
- Josling, G.A.; Selvarajah, S.A.; Petter, M.; Duffy, M.F. The role of bromodomain proteins in regulating gene expression. Genes 2012, 3, 320–343. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, Y. The Bromodomain and Extra-Terminal Domain (BET) Family: Functional Anatomy of BET Paralogous Proteins. Int. J. Mol. Sci. 2016, 17, 1849. [Google Scholar] [CrossRef] [PubMed]
- Spriano, F.; Stathis, A.; Bertoni, F. Targeting BET bromodomain proteins in cancer: The example of lymphomas. Pharm. Ther. 2020, 215, 107631. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Bose, P.; Dai, Y.; Grant, S. Histone deacetylase inhibitor (HDACI) mechanisms of action: Emerging insights. Pharm. Ther. 2014, 143, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination Therapy With Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front. Oncol. 2018, 8, 92. [Google Scholar] [CrossRef]
- Song, Y.; Wu, F.; Wu, J. Targeting histone methylation for cancer therapy: Enzymes, inhibitors, biological activity and perspectives. J. Hematol. Oncol. 2016, 9, 49. [Google Scholar] [CrossRef] [PubMed]
- Meidhof, S.; Brabletz, S.; Lehmann, W.; Preca, B.T.; Mock, K.; Ruh, M.; Schuler, J.; Berthold, M.; Weber, A.; Burk, U.; et al. ZEB1-associated drug resistance in cancer cells is reversed by the class I HDAC inhibitor mocetinostat. EMBO Mol. Med. 2015, 7, 831–847. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.H.; Xu, X.G.; Yan, S.L.; Sun, Z.; Ying, Y.; Wang, B.K.; Tu, Y.X. Depletion of HDAC1, 7 and 8 by Histone Deacetylase Inhibition Confers Elimination of Pancreatic Cancer Stem Cells in Combination with Gemcitabine. Sci. Rep. 2018, 8, 1621. [Google Scholar] [CrossRef]
- Rivera-Del Valle, N.; Gao, S.; Miller, C.P.; Fulbright, J.; Gonzales, C.; Sirisawad, M.; Steggerda, S.; Wheler, J.; Balasubramanian, S.; Chandra, J. PCI-24781, a Novel Hydroxamic Acid HDAC Inhibitor, Exerts Cytotoxicity and Histone Alterations via Caspase-8 and FADD in Leukemia Cells. Int. J. Cell Biol. 2010, 2010, 207420. [Google Scholar] [CrossRef]
- Morschhauser, F.; Terriou, L.; Coiffier, B.; Bachy, E.; Varga, A.; Kloos, I.; Lelievre, H.; Sarry, A.L.; Depil, S.; Ribrag, V. Phase 1 study of the oral histone deacetylase inhibitor abexinostat in patients with Hodgkin lymphoma, non-Hodgkin lymphoma, or chronic lymphocytic leukaemia. Investig. New Drugs 2015, 33, 423–431. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Chen, C.; Wang, X.; Sun, Y.; Zhang, J.; Chen, J.; Shi, Y. An Epigenetic Role of Mitochondria in Cancer. Cells 2022, 11, 2518. https://doi.org/10.3390/cells11162518
Liu Y, Chen C, Wang X, Sun Y, Zhang J, Chen J, Shi Y. An Epigenetic Role of Mitochondria in Cancer. Cells. 2022; 11(16):2518. https://doi.org/10.3390/cells11162518
Chicago/Turabian StyleLiu, Yu’e, Chao Chen, Xinye Wang, Yihong Sun, Jin Zhang, Juxiang Chen, and Yufeng Shi. 2022. "An Epigenetic Role of Mitochondria in Cancer" Cells 11, no. 16: 2518. https://doi.org/10.3390/cells11162518
APA StyleLiu, Y., Chen, C., Wang, X., Sun, Y., Zhang, J., Chen, J., & Shi, Y. (2022). An Epigenetic Role of Mitochondria in Cancer. Cells, 11(16), 2518. https://doi.org/10.3390/cells11162518