
Hsp90: From Cellular to Organismal Proteostasis


Abstract
1. Cellular Proteostasis
1.1. The Cytoplasmic Heat Shock Response (HSR)
1.2. The Unfolded Protein Response of the Endoplasmic Reticulum (UPRER)
1.3. Mitochondrial UPR (UPRMT)
2. The Hsp90 Chaperone
2.1. Hsp90 Isoforms and Structure
2.2. Hsp90 Function
2.3. The Hsp90 Chaperone Cycle and Its Regulation
3. Hsp90 in Intracellular Proteostasis
4. Hsp90 in the Extracellular Space and Proteostasis
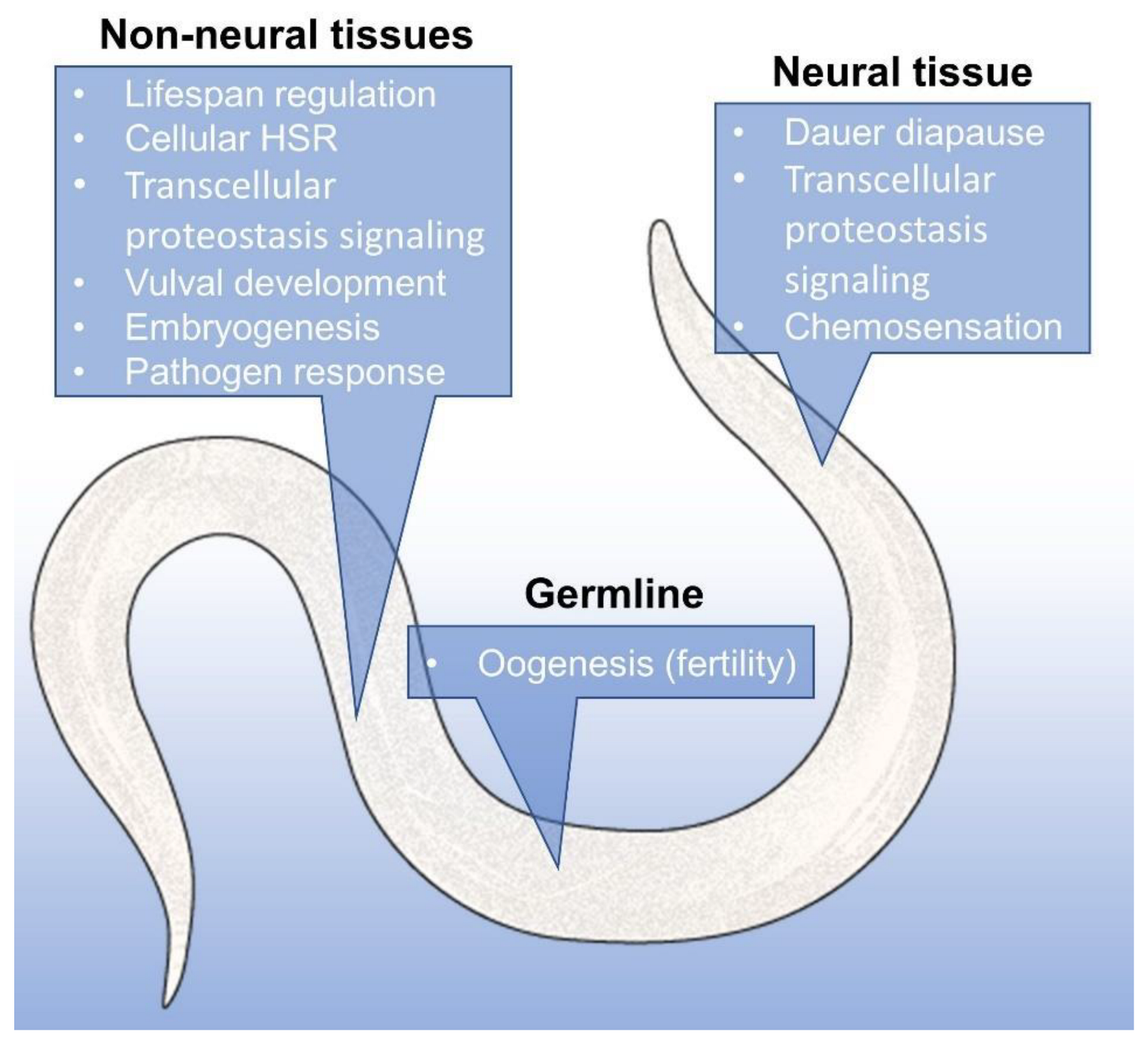
5. Hsp90 Is a Signal and Executor in Organismal Proteostasis
6. Hsp90 Is a Capacitor of the Evolution of the Proteome
7. Hsp90 as a Target in Diseases of Proteostasis
8. Outlook and Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hansen, M.; Taubert, S.; Crawford, D.; Libina, N.; Lee, S.-J.; Kenyon, C. Lifespan extension by conditions that inhibit translation in Caenorhabditis elegans. Aging Cell 2007, 6, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Kmiecik, S.W.; Mayer, M.P. Molecular mechanisms of heat shock factor 1 regulation. Trends Biochem. Sci. 2021, 47, 218–234. [Google Scholar] [CrossRef] [PubMed]
- Westerheide, S.D.; Anckar, J.; Stevens, S.M.; Sistonen, L.; Morimoto, R.I. Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science 2009, 323, 1063–1066. [Google Scholar] [CrossRef]
- Marcu, M.G.; Doyle, M.; Bertolotti, A.; Ron, D.; Hendershot, L.; Neckers, L. Heat Shock Protein 90 Modulates the Unfolded Protein Response by Stabilizing IRE1α. Mol. Cell. Biol. 2002, 22, 8506–8513. [Google Scholar] [CrossRef] [PubMed]
- Whitesell, L.; Lindquist, S.L. HSP90 and the chaperoning of cancer. Nat. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Csermely, P.; Schnaider, T.; So, C.; Prohászka, Z.; Nardai, G. The 90-kDa Molecular Chaperone Family: Structure, Function, and Clinical Applications. A Comprehensive Review. Pharmacol. Ther. 1998, 79, 129–168. [Google Scholar] [CrossRef]
- Hoter, A.; El-Sabban, M.E.; Naim, H.Y. The HSP90 Family: Structure, regulation, function, and implications in health and disease. Int. J. Mol. Sci. 2018, 19, 2560. [Google Scholar] [CrossRef]
- Buchner, J.; Li, J. Structure, Function and Regulation of the Hsp90 Machinery. Biomed. J. 2013, 36, 106–117. [Google Scholar] [CrossRef]
- Johnson, J.L. Evolution and function of diverse Hsp90 homologs and cochaperone proteins. Biochim. Biophys. Acta 2012, 1823, 607–613. [Google Scholar] [CrossRef]
- Morano, K.A.; Santoro, N.; Koch, K.A.; Thiele, D.J. A trans-Activation Domain in Yeast Heat Shock Transcription Factor Is Essential for Cell Cycle Progression during Stress. Mol. Cell. Biol. 1999, 19, 402–411. [Google Scholar] [CrossRef]
- Borkovich, K.A.; Farrelly, F.W.; Finkelstein, D.B.; Taulien, J.; Lindquist, S. Hsp82 Is an Essential Protein that is Required in Higher Concentrations for Growth of Cells at Higher Temperatures. Mol. Cell. Biol. 1989, 9, 3919–3930. [Google Scholar] [CrossRef] [PubMed]
- Biebl, M.M.; Buchner, J. Structure, function, and regulation of the hsp90 machinery. Cold Spring Harb. Perspect. Biol. 2019, 11, a034017. [Google Scholar] [CrossRef] [PubMed]
- Millson, S.H.; Truman, A.W.; Rácz, A.; Hu, B.; Panaretou, B.; Nuttall, J.; Mollapour, M.; Söti, C.; Piper, P.W. Expressed as the sole Hsp90 of yeast, the α and β isoforms of human Hsp90 differ with regard to their capacities for activation of certain client proteins, whereas only Hsp90β generates sensitivity to the Hsp90 inhibitor radicicol. FEBS J. 2007, 274, 4453–4463. [Google Scholar] [CrossRef] [PubMed]
- Sreedhar, A.S.; Kalmár, É.; Csermely, P.; Shen, Y.-F. Hsp90 isoforms: Functions, expression and clinical importance. FEBS Lett. 2004, 562, 11–15. [Google Scholar] [CrossRef]
- Prodromou, C. Mechanisms of Hsp90 regulation. Biochem. J. 2016, 473, 2439–2452. [Google Scholar] [CrossRef]
- Koyasu, S.; Nishida, E.; Kadowaki, T.; Matsuzaki, F.; Iida, K.; Harada, F.; Kasuga, M.; Sakai, H.; Yahara, I. Two mammalian heat shock proteins, HSP90 and HSP100, are actin-binding proteins. Proc. Natl. Acad. Sci. USA 1986, 83, 8054–8058. [Google Scholar] [CrossRef]
- Nardai, G.; Schnaider, T.; Söti, C.; Ryan, M.; Hoj, P.B.; Somogyi, J.; Csermely, P. Characterization of the 90 kDa heat shock protein (HSP90)-associated ATP/GTPase. J. Biosci. 1996, 21, 179–190. [Google Scholar] [CrossRef]
- Yamamoto, M.; Takahashi, Y.; Inano, K.; Horigome, T.; Sugano, H. Characterization of the Hydrophobic Region of Heat Shock Protein 90. J. Biochem. 1991, 110, 141–145. [Google Scholar] [CrossRef]
- Jackson, S.E. Hsp90: Structure and Function. In Molecular Chaperones; Springer: Berlin/Heidelberg, Germany, 2013; Volume 328, pp. 155–240. [Google Scholar] [CrossRef]
- Tsutsumi, S.; Mollapour, M.; Graf, C.; Lee, C.T.; Scroggins, B.T.; Xu, W.; Haslerova, L.; Hessling, M.; Konstantinova, A.A.; Trepel, J.B.; et al. Hsp90 charged-linker truncation reverses the functional consequences of weakened hydrophobic contacts in the N domain. Nat. Struct. Mol. Biol. 2009, 16, 1141–1147. [Google Scholar] [CrossRef]
- Shiau, A.K.; Harris, S.F.; Southworth, D.R.; Agard, D.A. Structural Analysis of E. coli hsp90 Reveals Dramatic Nucleotide-Dependent Conformational Rearrangements. Cell 2006, 127, 329–340. [Google Scholar] [CrossRef]
- Dutta, R.; Inouye, M. GHKL, an emergent ATPase/kinase superfamily. Trends Biochem. Sci. 2000, 25, 24–28. [Google Scholar] [CrossRef]
- Minami, Y.; Kawasaki, H.; Suzuki, K.; Yahara, I. The calmodulin-binding domain of the mouse 90-kDa heat shock protein. J. Biol. Chem. 1993, 268, 9604–9610. [Google Scholar] [CrossRef]
- Bouhouche-Chatelier, I.; Chadli, A.; Catelli, M.-G. The N-terminal adenosine triphosphate binding domain of Hsp90 is necessary and sufficient for interaction with estrogen receptor. Cell Stress Chaperon. 2001, 6, 297–305. [Google Scholar] [CrossRef]
- Marcu, M.G.; Schulte, T.W.; Neckers, L. Novobiocin and Related Coumarins and Depletion of Heat Shock Protein 90-Dependent Signaling Proteins. JNCI J. Natl. Cancer Inst. 2000, 92, 242–248. [Google Scholar] [CrossRef]
- Sőti, C.; Vermes, A.; Haystead, T.A.J.; Csermely, P. Comparative analysis of the ATP-binding sites of Hsp90 by nucleotide affinity cleavage: A distinct nucleotide specificity of the C-terminal ATP-binding site. JBIC J. Biol. Inorg. Chem. 2003, 270, 2421–2428. [Google Scholar] [CrossRef]
- Söti, C.; Rácz, A.; Csermely, P. A nucleotide-dependent molecular switch controls ATP binding at the C-terminal domain of Hsp90. N-terminal nucleotide binding unmasks a C-terminal binding pocket. J. Biol. Chem. 2002, 277, 7066–7075. [Google Scholar] [CrossRef]
- Garg, G.; Khandelwal, A.; Blagg, B.S. Anticancer Inhibitors of Hsp90 Function: Beyond the Usual Suspects, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2016; Volume 129. [Google Scholar]
- Crunden, J.L.; Diezmann, S. Hsp90 interaction networks in fungi—Tools and techniques. FEMS Yeast Res. 2021, 21, foab054. [Google Scholar] [CrossRef]
- Taipale, M.; Krykbaeva, I.; Koeva, M.; Kayatekin, C.; Westover, K.D.; Karras, G.I.; Lindquist, S. Quantitative Analysis of Hsp90-Client Interactions Reveals Principles of Substrate Recognition. Cell 2012, 150, 987–1001. [Google Scholar] [CrossRef]
- Echeverria, P.; Bernthaler, A.; Dupuis, P.M.J.; Mayer, B.; Picard, D. An Interaction Network Predicted from Public Data as a Discovery Tool: Application to the Hsp90 Molecular Chaperone Machine. PLoS ONE 2011, 6, e26044. [Google Scholar] [CrossRef]
- Taipale, M.; Jarosz, D.F.; Lindquist, S. HSP90 at the hub of protein homeostasis: Emerging mechanistic insights. Nat. Rev. Mol. Cell Biol. 2010, 11, 515–528. [Google Scholar] [CrossRef]
- Breitkreutz, B.-J.; Stark, C.; Reguly, T.; Boucher, L.; Breitkreutz, A.; Livstone, M.; Oughtred, R.; Lackner, D.H.; Bähler, J.; Wood, V.; et al. The BioGRID Interaction Database: 2008 update. Nucleic Acids Res. 2008, 36, 637–640. [Google Scholar] [CrossRef] [PubMed]
- Sreedhar, A.S.; Mihály, K.; Pató, B.; Schnaider, T.; Steták, A.; Kis-Petik, K.; Fidy, J.; Simonics, T.; Maráz, A.; Csermely, P. Hsp90 inhibition accelerates cell lysis: Anti-Hsp90 ribozyme reveals a complex mechanism of Hsp90 inhibitors involving both superoxide- and Hsp90-dependent events. J. Biol. Chem. 2003, 278, 35231–35240. [Google Scholar] [CrossRef] [PubMed]
- Clerico, E.M.; Tilitsky, J.M.; Meng, W.; Gierasch, L.M. How Hsp70 Molecular Machines Interact with Their Substrates to Mediate Diverse Physiological Functions. J. Mol. Biol. 2015, 427, 1575–1588. [Google Scholar] [CrossRef] [PubMed]
- Grammatikakis, N.; Lin, J.-H.; Grammatikakis, A.; Tsichlis, P.N.; Cochran, B.H. p50cdc37 Acting in Concert with Hsp90 Is Required for Raf-1 Function. Mol. Cell. Biol. 1999, 19, 1661–1672. [Google Scholar] [CrossRef]
- Polier, S.; Samant, R.; Clarke, P.; Workman, P.; Prodromou, C.; Pearl, L.H. ATP-competitive inhibitors block protein kinase recruitment to the Hsp90-Cdc37 system. Nat. Chem. Biol. 2013, 9, 307–312. [Google Scholar] [CrossRef]
- Boczek, E.E.; Reefschläger, L.G.; Dehling, M.; Struller, T.J.; Häusler, E.; Seidl, A.; Kaila, V.R.I.; Buchner, J. Conformational processing of oncogenic v-Src kinase by the molecular chaperone Hsp90. Proc. Natl. Acad. Sci. USA 2015, 112, E3189–E3198. [Google Scholar] [CrossRef]
- Eckl, J.M.; Daake, M.; Schwartz, S.; Richter, K. Nucleotide-Free sB-Raf is Preferentially Bound by Hsp90 and Cdc37 In Vitro. J. Mol. Biol. 2016, 428, 4185–4196. [Google Scholar] [CrossRef]
- Pratt, W.B.; Dittmar, K.D. Studies with Purified Chaperones Advance the Understanding of the Mechanism of Glucocorticoid Receptor–hsp90 Heterocomplex Assembly. Trends Endocrinol. Metab. 1998, 9, 244–252. [Google Scholar] [CrossRef]
- Kirschke, E.; Goswami, D.; Southworth, D.; Griffin, P.R.; Agard, D.A. Glucocorticoid Receptor Function Regulated by Coordinated Action of the Hsp90 and Hsp70 Chaperone Cycles. Cell 2014, 157, 1685–1697. [Google Scholar] [CrossRef]
- Lorenz, O.R.; Freiburger, L.; Rutz, D.A.; Krause, M.; Zierer, B.K.; Alvira, S.; Cuéllar, J.; Valpuesta, J.M.; Madl, T.; Sattler, M.; et al. Modulation of the Hsp90 Chaperone Cycle by a Stringent Client Protein. Mol. Cell 2014, 53, 941–953. [Google Scholar] [CrossRef]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Walter, S.; Buchner, J. Molecular chaperones—Cellular machines for protein folding. Angew. Chem.-Int. Ed. 2002, 41, 1098–1113. [Google Scholar] [CrossRef]
- Chaudhury, S.; Welch, T.R.; Blagg, B.S.J. Hsp90 as a Target for Drug Development. ChemMedChem 2006, 1, 1331–1340. [Google Scholar] [CrossRef] [PubMed]
- Kosano, H.; Stensgard, B.; Charlesworth, M.C.; McMahon, N.; Toft, D. The Assembly of Progesterone Receptor-hsp90 Complexes Using Purified Proteins. J. Biol. Chem. 1998, 273, 32973–32979. [Google Scholar] [CrossRef] [PubMed]
- Prodromou, C.; Panaretou, B.; Chohan, S.; Siligardi, G.; O’Brien, R.; Ladbury, J.E.; Roe, M.; Piper, P.W.; Pearl, L.H. The ATPase cycle of Hsp90 drives a molecular clamp’ via transient dimerization of the N-terminal domains. EMBO J. 2000, 19, 4383–4392. [Google Scholar] [CrossRef]
- Ali, M.M.U.; Roe, S.M.; Vaughan, C.K.; Meyer, P.; Panaretou, B.; Piper, P.W.; Prodromou, C.; Pearl, L.H. Crystal structure of an Hsp90–nucleotide–p23/Sba1 closed chaperone complex. Nature 2006, 440, 1013–1017. [Google Scholar] [CrossRef]
- Li, J.; Soroka, J.; Buchner, J. The Hsp90 chaperone machinery: Conformational dynamics and regulation by co-chaperones. Biochim. Biophys. Acta 2012, 1823, 624–635. [Google Scholar] [CrossRef]
- Müller, L.; Schaupp, A.; Walerych, D.; Wegele, H.; Buchner, J. Hsp90 Regulates the Activity of Wild Type p53 under Physiological and Elevated Temperatures. J. Biol. Chem. 2004, 279, 48846–48854. [Google Scholar] [CrossRef]
- Karagöz, G.E.; Duarte, A.M.; Akoury, E.; Ippel, H.; Biernat, J.; Luengo, T.M.; Radli, M.; Didenko, T.; Nordhues, B.A.; Veprintsev, D.B.; et al. Hsp90-Tau Complex Reveals Molecular Basis for Specificity in Chaperone Action. Cell 2014, 156, 963–974. [Google Scholar] [CrossRef]
- Hagn, F.; Lagleder, S.; Retzlaff, M.; Rohrberg, J.; Demmer, O.; Richter, K.; Buchner, J.; Kessler, H. Structural analysis of the interaction between Hsp90 and the tumor suppressor protein p53. Nat. Struct. Mol. Biol. 2010, 18, 1086–1093. [Google Scholar] [CrossRef]
- Park, S.J.; Kostic, M.; Dyson, H.J. Dynamic Interaction of Hsp90 with Its Client Protein p53. J. Mol. Biol. 2011, 411, 158–173. [Google Scholar] [CrossRef] [PubMed]
- Roe, S.M.; Prodromou, C.; O’Brien, R.; Ladbury, J.E.; Piper, P.W.; Pearl, L.H. Structural Basis for Inhibition of the Hsp90 Molecular Chaperone by the Antitumor Antibiotics Radicicol and Geldanamycin. J. Med. Chem. 1999, 42, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Sharp, S.; Workman, P. Inhibitors of the HSP90 Molecular Chaperone: Current Status. Adv. Cancer Res. 2006, 95, 323–348. [Google Scholar] [CrossRef]
- Mayer, M.P.; Le Breton, L. Hsp90: Breaking the Symmetry. Mol. Cell 2015, 58, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Röhl, A.; Rohrberg, J.; Buchner, J. The chaperone Hsp90: Changing partners for demanding clients. Trends Biochem. Sci. 2013, 38, 253–262. [Google Scholar] [CrossRef]
- Scroggins, B.T.; Neckers, L. Post-translational modification of heat-shock protein 90: Impact on chaperone function. Expert Opin. Drug Discov. 2007, 2, 1403–1414. [Google Scholar] [CrossRef]
- Soroka, J.; Wandinger, S.K.; Mäusbacher, N.; Schreiber, T.; Richter, K.; Daub, H.; Buchner, J. Conformational Switching of the Molecular Chaperone Hsp90 via Regulated Phosphorylation. Mol. Cell 2012, 45, 517–528. [Google Scholar] [CrossRef]
- Wandinger, S.K.; Suhre, M.; Wegele, H.; Buchner, J. The phosphatase Ppt1 is a dedicated regulator of the molecular chaperone Hsp90. EMBO J. 2006, 25, 367–376. [Google Scholar] [CrossRef]
- Kovacs, J.J.; Murphy, P.J.M.; Gaillard, S.; Zhao, X.; Wu, J.-T.; Nicchitta, C.V.; Yoshida, M.; Toft, D.O.; Pratt, W.B.; Yao, T.-P. HDAC6 Regulates Hsp90 Acetylation and Chaperone-Dependent Activation of Glucocorticoid Receptor. Mol. Cell 2005, 18, 601–607. [Google Scholar] [CrossRef]
- Bali, P.; Pranpat, M.; Bradner, J.; Balasis, M.; Fiskus, W.; Guo, F.; Rocha, K.; Kumaraswamy, S.; Boyapalle, S.; Atadja, P.; et al. Inhibition of Histone Deacetylase 6 Acetylates and Disrupts the Chaperone Function of Heat Shock Protein 90: A novel basis for antileukemia activity of histone deacetylase inhibitors. J. Biol. Chem. 2005, 280, 26729–26734. [Google Scholar] [CrossRef]
- García-Cardeña, G.; Fan, R.; Shah, V.; Sorrentino, R.; Cirino, G.; Papapetropoulos, A.; Sessa, W.C. Dynamic activation of endothelial nitric oxide synthase by Hsp90. Nature 1998, 392, 821–824. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Ruiz, A.; Villanueva, L.; González de Orduña, C.; López-Ferrer, D.; Higueras, M.Á.; Tarín, C.; Rodríguez-Crespo, I.; Vázquez, J.; Lamas, S. S-nitrosylation of Hsp90 promotes the inhibition of its ATPase and endothelial nitric oxide synthase regulatory activities. Proc. Natl. Acad. Sci. USA 2005, 24, 8525–8530. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Beebe, K.; Chavez, J.D.; Boysen, M.; Lu, Y.; Zuehlke, A.D.; Keramisanou, D.; Trepel, J.B.; Prodromou, C.; Mayer, M.P.; et al. Hsp90 middle domain phosphorylation initiates a complex conformational program to recruit the ATPase-stimulating cochaperone Aha1. Nat. Commun. 2019, 10, 2574. [Google Scholar] [CrossRef] [PubMed]
- Somogyvari, M.; Gecse, E.; Sőti, C. DAF-21/Hsp90 is required for C. elegans longevity by ensuring DAF-16/FOXO isoform A function. Sci. Rep. 2018, 8, 12048. [Google Scholar] [CrossRef]
- Hajdú, G.; Gecse, E.; Taisz, I.; Móra, I.; Sőti, C. Toxic stress-specific cytoprotective responses regulate learned behavioral decisions in C. elegans. BMC Biol. 2021, 19, 26. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Somogyvári, M.; Sőti, C. Hsp90 Stabilizes SIRT1 Orthologs in Mammalian Cells and C. elegans. Int. J. Mol. Sci. 2018, 19, 3661. [Google Scholar] [CrossRef]
- Donzé, O.; Picard, D. Hsp90 Binds and Regulates Gcn2, the Ligand-Inducible Kinase of the α Subunit of Eukaryotic Translation Initiation Factor 2. Mol. Cell. Biol. 2000, 20, 1897. [Google Scholar] [CrossRef]
- Donzé, O.; Abbas-Terki, T.; Picard, D. The Hsp90 chaperone complex is both a facilitator and a repressor of the dsRNA-dependent kinase PKR. EMBO J. 2001, 20, 3771–3780. [Google Scholar] [CrossRef]
- Berwal, S.K.; Bhatia, V.; Bendre, A.; Suresh, C.; Chatterjee, S.; Pal, J.K. Activation of HRI is mediated by Hsp90 during stress through modulation of the HRI-Hsp90 complex. Int. J. Biol. Macromol. 2018, 118, 1604–1613. [Google Scholar] [CrossRef]
- McClellan, A.J.; Xia, Y.; Deutschbauer, A.M.; Davis, R.W.; Gerstein, M.; Frydman, J. Diverse Cellular Functions of the Hsp90 Molecular Chaperone Uncovered Using Systems Approaches. Cell 2007, 131, 121–135. [Google Scholar] [CrossRef]
- Wang, X.; Venable, J.; LaPointe, P.; Hutt, D.M.; Koulov, A.V.; Coppinger, J.; Gurkan, C.; Kellner, W.; Matteson, J.; Plutner, H.; et al. Hsp90 Cochaperone Aha1 Downregulation Rescues Misfolding of CFTR in Cystic Fibrosis. Cell 2006, 127, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Wang, T.; Araujo, T.L.S.; Sharma, S.; Brodsky, J.L.; Chiosis, G. Adapting to stress—Chaperome networks in cancer. Nat. Cancer 2018, 18, 562–575. [Google Scholar] [CrossRef] [PubMed]
- Taldone, T.; Wang, T.; Rodina, A.; Pillarsetty, N.V.K.; Digwal, C.S.; Sharma, S.; Yan, P.; Joshi, S.; Pagare, P.P.; Bolaender, A.; et al. A Chemical Biology Approach to the Chaperome in Cancer—HSP90 and Beyond. Cold Spring Harb. Perspect. Biol. 2019, 12, a034116. [Google Scholar] [CrossRef] [PubMed]
- Solís, E.J.; Pandey, J.P.; Zheng, X.; Jin, D.X.; Gupta, P.B.; Airoldi, E.M.; Pincus, D.; Denic, V. Defining the Essential Function of Yeast Hsf1 Reveals a Compact Transcriptional Program for Maintaining Eukaryotic Proteostasis. Mol. Cell 2016, 63, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Frumkin, A.; Dror, S.; Pokrzywa, W.; Bar-Lavan, Y.; Karady, I.; Hoppe, T.; Ben-Zvi, A. Challenging muscle homeostasis uncovers novel chaperone interactions in Caenorhabditis elegans. Front. Mol. Biosci. 2014, 1, 21. [Google Scholar] [CrossRef]
- Biebl, M.M.; Lopez, A.; Rehn, A.; Freiburger, L.; Lawatscheck, J.; Blank, B.; Sattler, M.; Buchner, J. Structural elements in the flexible tail of the co-chaperone p23 coordinate client binding and progression of the Hsp90 chaperone cycle. Nat. Commun. 2021, 12, 828. [Google Scholar] [CrossRef]
- Li, T.; Jiang, H.-L.; Tong, Y.-G.; Lu, J.-J. Targeting the Hsp90-Cdc37-client protein interaction to disrupt Hsp90 chaperone machinery. J. Hematol. Oncol. 2018, 11, 59. [Google Scholar] [CrossRef]
- Thirumalaikumar, V.P.; Gorka, M.; Schulz, K.; Masclaux-Daubresse, C.; Sampathkumar, A.; Skirycz, A.; Vierstra, R.D.; Balazadeh, S. Selective autophagy regulates heat stress memory in Arabidopsis by NBR1-mediated targeting of HSP90.1 and ROF1. Autophagy 2021, 17, 2184–2199. [Google Scholar] [CrossRef]
- McKeen, H.D.; Byrne, C.; Jithesh, P.V.; Donley, C.; Valentine, A.; Yakkundi, A.; O’Rourke, M.; Swanton, C.; McCarthy, H.O.; Hirst, D.G.; et al. FKBPL Regulates Estrogen Receptor Signaling and Determines Response to Endocrine Therapy. Cancer Res. 2010, 70, 1090–1100. [Google Scholar] [CrossRef]
- Crevel, G.; Bennett, D.; Cotterill, S. The Human TPR Protein TTC4 Is a Putative Hsp90 Co-Chaperone Which Interacts with CDC6 and Shows Alterations in Transformed Cells. PLoS ONE 2008, 3, e0001737. [Google Scholar] [CrossRef]
- Zhao, Q.; Yang, J.; Chen, H.; Li, J.; Que, L.; Zhu, G.; Liu, L.; Ha, T.; Chen, Q.; Li, C.; et al. Peli1 induction impairs cardiac microvascular endothelium through Hsp90 dissociation from IRE1α. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2019, 1865, 2606–2617. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Binder, R.J.; Ramalingam, T.; Srivastava, P.K. CD91 Is a Common Receptor for Heat Shock Proteins gp96, hsp90, hsp70, and Calreticulin. Immunity 2001, 14, 303–313. [Google Scholar] [CrossRef]
- Calderwood, S.K.; Gong, J.; Murshid, A. Extracellular HSPs: The Complicated Roles of Extracellular HSPs in Immunity. Front. Immunol. 2016, 7, 159. [Google Scholar] [CrossRef] [PubMed]
- El Hamidieh, A.; Grammatikakis, N.; Patsavoudi, E. Cell Surface Cdc37 Participates in Extracellular HSP90 Mediated Cancer Cell Invasion. PLoS ONE 2012, 7, e42722. [Google Scholar] [CrossRef] [PubMed]
- Baker-Williams, A.J.; Hashmi, F.; Budzyński, M.A.; Woodford, M.R.; Gleicher, S.; Himanen, S.V.; Makedon, A.M.; Friedman, D.; Cortes, S.; Namek, S.; et al. Co-chaperones TIMP2 and AHA1 Competitively Regulate Extracellular HSP90:Client MMP2 Activity and Matrix Proteolysis. Cell Rep. 2019, 28, 1894–1906.e6. [Google Scholar] [CrossRef]
- Sims, J.D.; McCready, J.; Jay, D.G. Extracellular Heat Shock Protein (Hsp)70 and Hsp90α Assist in Matrix Metalloproteinase-2 Activation and Breast Cancer Cell Migration and Invasion. PLoS ONE 2011, 6, e18848. [Google Scholar] [CrossRef]
- Lee, K.-J.; Kim, Y.M.; Kim, D.Y.; Jeoung, O.; Han, K.; Lee, S.-T.; Lee, Y.-S.; Park, K.H.; Park, J.H.; Kim, D.J.; et al. Release of heat shock protein 70 (Hsp70) and the effects of extracellular Hsp70 on matric metalloproteinase-9 expression in human monocytic U937 cells. Exp. Mol. Med. 2006, 38, 364–374. [Google Scholar] [CrossRef]
- Chakraborty, A.; Boel, N.M.-E.; Edkins, A.L. HSP90 Interacts with the Fibronectin N-terminal Domains and Increases Matrix Formation. Cells 2020, 9, 272. [Google Scholar] [CrossRef]
- Luparello, C.; Sirchia, R.; Pupello, D. PTHrP [67–86] regulates the expression of stress proteins in breast cancer cells inducing modifications in urokinase-plasminogen activator and MMP-1 expression. J. Cell Sci. 2003, 116, 2421–2430. [Google Scholar] [CrossRef]
- Song, X.; Wang, X.; Zhuo, W.; Shi, H.; Feng, D.; Sun, Y.; Liang, Y.; Fu, Y.; Zhou, D.; Luo, Y. The Regulatory Mechanism of Extracellular Hsp90α on Matrix Metalloproteinase-2 Processing and Tumor Angiogenesis. J. Biol. Chem. 2010, 285, 40039–40049. [Google Scholar] [CrossRef]
- Correia, A.L.; Mori, H.; Chen, E.I.; Schmitt, F.C.; Bissell, M.J. The hemopexin domain of MMP3 is responsible for mammary epithelial invasion and morphogenesis through extracellular interaction with HSP90β. Genes Dev. 2013, 27, 805–817. [Google Scholar] [CrossRef] [PubMed]
- Stellas, D.; El Hamidieh, A.; Patsavoudi, E. Monoclonal antibody 4C5 prevents activation of MMP2 and MMP9 by disrupting their interaction with extracellular HSP90 and inhibits formation of metastatic breast cancer cell deposits. BMC Cell Biol. 2010, 11, 51. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Chang, C.; Li, W. The role of secreted heat shock protein-90 (Hsp90) in wound healing—How could it shape future therapeutics? Expert Rev. Proteom. 2017, 14, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Liao, D.-F.; Jin, Z.-G.; Baas, A.S.; Daum, G.; Gygi, S.P.; Aebersold, R.; Berk, B.C. Purification and Identification of Secreted Oxidative Stress-induced Factors from Vascular Smooth Muscle Cells. J. Biol. Chem. 2000, 275, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Hightower, L.E.; Guidon, P.T., Jr. Selective release from cultured mammalian cells of heat-shock (stress) proteins that resemble glia-axon transfer proteins. J. Cell. Physiol. 1989, 138, 257–266. [Google Scholar] [CrossRef]
- Clayton, A.; Turkes, A.; Navabi, H.; Mason, M.D.; Tabi, Z. Induction of heat shock proteins in B-cell exosomes. J. Cell Sci. 2005, 118, 3631–3638. [Google Scholar] [CrossRef]
- Yu, X.; Harris, S.L.; Levine, A.J. The Regulation of Exosome Secretion: A Novel Function of the p53 Protein. Cancer Res. 2006, 66, 4795–4801. [Google Scholar] [CrossRef]
- Li, W.; Li, Y.; Guan, S.; Fan, J.; Cheng, C.-F.; Bright, A.M.; Chinn, C.; Chen, M.; Woodley, D.T. Extracellular heat shock protein-90α: Linking hypoxia to skin cell motility and wound healing. EMBO J. 2007, 26, 1221–1233. [Google Scholar] [CrossRef]
- Woodley, D.T.; Fan, J.; Cheng, C.-F.; Li, Y.; Chen, M.; Bu, G.; Li, W. Participation of the lipoprotein receptor LRP1 in hypoxia-HSP90α autocrine signaling to promote keratinocyte migration. J. Cell Sci. 2009, 122, 1495–1498. [Google Scholar] [CrossRef]
- Cheng, C.-F.; Fan, J.; Fedesco, M.; Guan, S.; Li, Y.; Bandyopadhyay, B.; Bright, A.M.; Yerushalmi, D.; Liang, M.; Chen, M.; et al. Transforming Growth Factor α (TGFα)-Stimulated Secretion of HSP90α: Using the Receptor LRP-1/CD91 To Promote Human Skin Cell Migration against a TGFβ-Rich Environment during Wound Healing. Mol. Cell. Biol. 2008, 28, 3344–3358. [Google Scholar] [CrossRef]
- Chen, J.-S.; Hsu, Y.-M.; Chen, C.-C.; Chen, L.-L.; Lee, C.-C.; Huang, T.-S. Secreted Heat Shock Protein 90α Induces Colorectal Cancer Cell Invasion through CD91/LRP-1 and NF-κB-mediated Integrin αV Expression. J. Biol. Chem. 2010, 285, 25458–25466. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.-Y.; Tsai, M.-C.; Wu, Y.-P.; Wang, R.Y.L. Identification of heat-shock protein 90 beta in Japanese encephalitis virus-induced secretion proteins. J. Gen. Virol. 2011, 92, 2803–2809. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Kulkarni, A.B. Extracellular heat shock protein HSP90β secreted by MG63 osteosarcoma cells inhibits activation of latent TGF-β1. Biochem. Biophys. Res. Commun. 2010, 398, 525–531. [Google Scholar] [CrossRef] [PubMed]
- McCready, J.; Sims, J.D.; Chan, D.; Jay, D.G. Secretion of extracellular hsp90α via exosomes increases cancer cell motility: A role for plasminogen activation. BMC Cancer 2010, 10, 294. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, T.; Suzuki, M.; Fujikake, N.; Popiel, H.A.; Kikuchi, H.; Futaki, S.; Wada, K.; Nagai, Y. Intercellular chaperone transmission via exosomes contributes to maintenance of protein homeostasis at the organismal level. Proc. Natl. Acad. Sci. USA 2015, 112, E2497–E2506. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Gee, H.Y.; Lee, M.G. Unconventional protein secretion—New insights into the pathogenesis and therapeutic targets of human diseases. J. Cell Sci. 2018, 131, jcs213686. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Song, X.; Zhuo, W.; Fu, Y.; Shi, H.; Liang, Y.; Tong, M.; Chang, G.; Luo, Y. The regulatory mechanism of Hsp90α secretion and its function in tumor malignancy. Proc. Natl. Acad. Sci. USA 2009, 106, 21288–21293. [Google Scholar] [CrossRef]
- Mambula, S.S.; Calderwood, S.K. Heat Shock Protein 70 Is Secreted from Tumor Cells by a Nonclassical Pathway Involving Lysosomal Endosomes. J. Immunol. 2006, 177, 7849–7857. [Google Scholar] [CrossRef]
- Eguchi, T.; Sogawa, C.; Ono, K.; Matsumoto, M.; Tran, M.T.; Okusha, Y.; Lang, B.J.; Okamoto, K.; Calderwood, S.K. Cell Stress Induced Stressome Release Including Damaged Membrane Vesicles and Extracellular HSP90 by Prostate Cancer Cells. Cells 2020, 9, 755. [Google Scholar] [CrossRef]
- Facciponte, J.G.; Wang, X.-Y.; MacDonald, I.J.; Park, J.-E.; Arnouk, H.; Grimm, M.J.; Li, Y.; Kim, H.; Manjili, M.H.; Easton, D.P.; et al. Heat shock proteins HSP70 and GP96: Structural insights. Cancer Immunol. Immunother. 2006, 55, 339–346. [Google Scholar] [CrossRef]
- Calderwood, S.K.; Murshid, A.; Gong, J. Heat Shock Proteins: Conditional Mediators of Inflammation in Tumor Immunity. Front. Immunol. 2012, 3, 75. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, S.K.; Borges, T.J.; Eguchi, T.; Lang, B.J.; Murshid, A.; Okusha, Y.; Prince, T.L. Extracellular Hsp90 and protection of neuronal cells through Nrf2. Biochem. Soc. Trans. 2021, 49, 2299–2306. [Google Scholar] [CrossRef] [PubMed]
- Jayaprakash, P.; Dong, H.; Zou, M.; Bhatia, A.; O’Brien, K.; Chen, M.; Woodley, D.T.; Li, W. Hsp90α and Hsp90β Co-Operate a Stress-Response Mechanism to Cope with Hypoxia and Nutrient Paucity during Wound Healing. J. Cell Sci. 2015, 128, 1475–1480. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, D.; Jones, L.M.; Good, S.; Miles, J.; Vijayabaskar, M.; Aston, R.; Smith, C.E.; Westhead, D.; van Oosten-Hawle, P. A PQM-1-Mediated Response Triggers Transcellular Chaperone Signaling and Regulates Organismal Proteostasis. Cell Rep. 2018, 23, 3905–3919. [Google Scholar] [CrossRef] [PubMed]
- Berendzen, K.M.; Durieux, J.; Shao, L.-W.; Tian, Y.; Kim, H.-E.; Wolff, S.; Liu, Y.; Dillin, A. Neuroendocrine Coordination of Mitochondrial Stress Signaling and Proteostasis. Cell 2016, 166, 1553–1563.e10. [Google Scholar] [CrossRef] [PubMed]
- Poggio, P.; Sorge, M.; Seclì, L.; Brancaccio, M. Extracellular HSP90 Machineries Build Tumor Microenvironment and Boost Cancer Progression. Front. Cell Dev. Biol. 2021, 9, 735529. [Google Scholar] [CrossRef]
- Lee, S.-J.; Kenyon, C. Regulation of the Longevity Response to Temperature by Thermosensory Neurons in Caenorhabditis elegans. Curr. Biol. 2009, 19, 715–722. [Google Scholar] [CrossRef]
- Prahlad, V.; Cornelius, T.; Morimoto, R.I. Regulation of the Cellular Heat Shock Response in Caenorhabditis elegans by Thermosensory Neurons. Science 2008, 320, 811–814. [Google Scholar] [CrossRef] [PubMed]
- Sugi, T.; Nishida, Y.; Mori, I. Regulation of behavioral plasticity by systemic temperature signaling in Caenorhabditis elegans. Nat. Neurosci. 2011, 14, 984–992. [Google Scholar] [CrossRef]
- Demontis, F.; Perrimon, N. FOXO/4E-BP Signaling in Drosophila Muscles Regulates Organism-wide Proteostasis during Aging. Cell 2010, 143, 813–825. [Google Scholar] [CrossRef]
- Shaw, W.M.; Luo, S.; Landis, J.; Ashraf, J.; Murphy, C.T. The C. elegans TGF-β Dauer Pathway Regulates Longevity via Insulin Signaling. Curr. Biol. 2007, 17, 1635–1645. [Google Scholar] [CrossRef]
- Tewari, M.; Hu, P.; Ahn, J.S.; Ayivi-Guedehoussou, N.; Vidalain, P.-O.; Li, S.; Milstein, S.; Armstrong, C.M.; Boxem, M.; Butler, M.D.; et al. Systematic Interactome Mapping and Genetic Perturbation Analysis of a C. elegans TGF-β Signaling Network. Mol. Cell 2004, 13, 469–482. [Google Scholar] [CrossRef]
- Durieux, J.; Wolff, S.; Dillin, A. The Cell-Non-Autonomous Nature of Electron Transport Chain-Mediated Longevity. Cell 2011, 144, 79–91. [Google Scholar] [CrossRef]
- Taylor, R.C.; Dillin, A. XBP-1 Is a Cell-Nonautonomous Regulator of Stress Resistance and Longevity. Cell 2013, 153, 1435–1447. [Google Scholar] [CrossRef]
- Shemesh, N.; Shai, N.; Ben-Zvi, A. Germline stem cell arrest inhibits the collapse of somatic proteostasis early in Caenorhabditis elegans adulthood. Aging Cell 2013, 12, 814–822. [Google Scholar] [CrossRef]
- van Oosten-Hawle, P.; Porter, R.S.; Morimoto, R.I. Regulation of Organismal Proteostasis by Transcellular Chaperone Signaling. Cell 2013, 153, 1366–1378. [Google Scholar] [CrossRef]
- Fawcett, T.W.; Sylvester, S.L.; Sarge, K.D.; Morimoto, R.I.; Holbrook, N.J. Effects of neurohormonal stress and aging on the activation of mammalian heat shock factor 1. J. Biol. Chem. 1994, 269, 32272–32278. [Google Scholar] [CrossRef]
- Tawe, W.N.; Eschbach, M.-L.; Walter, R.D.; Henkle-Dührsen, K. Identification of stress-responsive genes in Caenorhabditis elegans using RT-PCR differential display. Nucleic Acids Res. 1998, 26, 1621–1627. [Google Scholar] [CrossRef]
- Shpigel, N.; Shemesh, N.; Kishner, M.; Ben-Zvi, A. Dietary restriction and gonadal signaling differentially regulate post-development quality control functions in Caenorhabditis elegans. Aging Cell 2019, 18, e12891. [Google Scholar] [CrossRef]
- Rutherford, S.L.; Lindquist, S. Hsp90 as a capacitor for morphological evolution. Nature 1998, 396, 336–342. [Google Scholar] [CrossRef]
- Yahara, I. The role of HSP90 in evolution. Genes Cells 1999, 4, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Lenski, R.E.; Barrick, J.E.; Ofria, C.; Levin, S. Balancing Robustness and Evolvability. PLoS Biol. 2006, 4, e428. [Google Scholar] [CrossRef] [PubMed]
- Burga, A.; Casanueva, M.O.; Lehner, B. Predicting mutation outcome from early stochastic variation in genetic interaction partners. Nature 2011, 480, 250–253. [Google Scholar] [CrossRef] [PubMed]
- Casanueva, M.O.; Burga, A.; Lehner, B. Fitness Trade-Offs and Environmentally Induced Mutation Buffering in Isogenic C. elegans. Science 2012, 335, 82–85. [Google Scholar] [CrossRef]
- Waddington, C.H. Genetic Assimilation of an Acquired Character. Evolution 2011, 7, 118–126. Available online: http://www.jstor.org/stable/2405747 (accessed on 12 June 2022). [CrossRef]
- Zabinsky, R.A.; Mason, G.A.; Queitsch, C.; Jarosz, D.F. It’s not magic–Hsp90 and its effects on genetic and epigenetic variation. Semin. Cell Dev. Biol. 2019, 88, 21–35. [Google Scholar] [CrossRef]
- Mittelman, D.; Sykoudis, K.; Hersh, M.; Lin, Y.; Wilson, J.H. Hsp90 modulates CAG repeat instability in human cells. Cell Stress Chaperon. 2010, 15, 753–759. [Google Scholar] [CrossRef]
- Karam, J.A.; Parikh, R.Y.; Nayak, D.; Rosenkranz, D.; Gangaraju, V.K. Co-chaperone Hsp70/Hsp90-organizing protein (Hop) is required for transposon silencing and Piwi-interacting RNA (piRNA) biogenesis. J. Biol. Chem. 2017, 292, 6039–6046. [Google Scholar] [CrossRef]
- Sawarkar, R.; Paro, R. Hsp90@chromatin.nucleus: An emerging hub of a networker. Trends Cell Biol. 2013, 23, 193–201. [Google Scholar] [CrossRef]
- Sollars, V.; Lu, X.; Xiao, L.; Wang, X.; Garfinkel, M.D.; Ruden, D.M. Evidence for an epigenetic mechanism by which Hsp90 acts as a capacitor for morphological evolution. Nat. Genet. 2003, 33, 70–74. [Google Scholar] [CrossRef]
- Zohn, I.E. Hsp90 and complex birth defects: A plausible mechanism for the interaction of genes and environment. Neurosci. Lett. 2020, 716, 134680. [Google Scholar] [CrossRef] [PubMed]
- Lacey, T.; Lacey, H. Linking hsp90′s role as an evolutionary capacitator to the development of cancer. Cancer Treat. Res. Commun. 2021, 28, 100400. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.T.; Csermely, P.; Sőti, C. Hsp90 chaperones PPARγ and regulates differentiation and survival of 3T3-L1 adipocytes. Cell Death Differ. 2013, 20, 1654–1663. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, M.C.; Gekakis, N. Hsp90 modulates PPARγ activity in a mouse model of nonalcoholic fatty liver disease. J. Lipid Res. 2014, 55, 1702–1710. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.-C.; Leu, S.-Y.; Chen, S.-Y.; Kung, C.-W.; Lee, Y.-M.; Liu, Y.-P.; Yen, M.-H.; Cheng, P.-Y. 17-DMAG, an Hsp90 inhibitor, ameliorates ovariectomy-induced obesity in rats. Life Sci. 2019, 232, 116672. [Google Scholar] [CrossRef] [PubMed]
- Lallier, M.; Marchandet, L.; Moukengue, B.; Charrier, C.; Baud’Huin, M.; Verrecchia, F.; Ory, B.; Lamoureux, F. Molecular Chaperones in Osteosarcoma: Diagnosis and Therapeutic Issues. Cells 2021, 10, 754. [Google Scholar] [CrossRef]
- Calderwood, S.K.; Khaleque, A.; Sawyer, D.B.; Ciocca, D.R. Heat shock proteins in cancer: Chaperones of tumorigenesis. Trends Biochem. Sci. 2006, 31, 164–172. [Google Scholar] [CrossRef]
- Ory, B.; Baud’Huin, M.; Verrecchia, F.; Royer, B.B.-L.; Quillard, T.; Amiaud, J.; Battaglia, S.; Heymann, D.; Redini, F.; Lamoureux, F. Blocking HSP90 Addiction Inhibits Tumor Cell Proliferation, Metastasis Development, and Synergistically Acts with Zoledronic Acid to Delay Osteosarcoma Progression. Clin. Cancer Res. 2016, 22, 2520–2533. [Google Scholar] [CrossRef]
- Zheng, D.; Liu, W.; Xie, W.; Huang, G.; Jiang, Q.; Yang, Y.; Huang, J.; Xing, Z.; Yuan, M.; Wei, M.; et al. AHA1 upregulates IDH1 and metabolic activity to promote growth and metastasis and predicts prognosis in osteosarcoma. Signal Transduct. Target. Ther. 2021, 6, 25. [Google Scholar] [CrossRef]
- Shu, X.; Liu, H.; Zhen, R.; Jie, Y.; Chen, L.; Qi, H.; Wang, C.; Wang, R.; Chen, D.; Ran, Y. Hsp90 inhibitor 17-AAG inhibits stem cell-like properties and chemoresistance in osteosarcoma cells via the Hedgehog signaling pathway. Oncol. Rep. 2020, 44, 313–324. [Google Scholar] [CrossRef]
- Kim, A.; Lu, Y.; Okuno, S.H.; Reinke, D.; Maertens, O.; Perentesis, J.; Basu, M.; Wolters, P.L.; De Raedt, T.; Chawla, S.; et al. Targeting Refractory Sarcomas and Malignant Peripheral Nerve Sheath Tumors in a Phase I/II Study of Sirolimus in Combination with Ganetespib (SARC023). Sarcoma 2020, 2020, 5784876. [Google Scholar] [CrossRef] [PubMed]
- Chinn, D.C.; Holland, W.S.; Yoon, J.M.; Zwerdling, T.; Mack, P.C. Anti-Tumor Activity of the HSP90 Inhibitor SNX-2112 in Pediatric Cancer Cell Lines Danielle. Pediatr. Blood Cancer 2012, 58, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Koay, Y.C.; McAlpine, S.R. How Selective are Hsp90 Inhibitors for Cancer Cells over Normal Cells? ChemMedChem 2017, 12, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Koay, Y.C.; McAlpine, S.R. Redefining the phenotype of Heat shock protein 90 (Hsp90) inhibitors. Chem.–Eur. J. 2017, 23, 2010–2013. [Google Scholar] [CrossRef]
- Lee, C.; Park, H.-K.; Jeong, H.; Lim, J.; Lee, A.-J.; Cheon, K.Y.; Kim, C.-S.; Thomas, A.P.; Bae, B.; Kim, N.D.; et al. Development of a Mitochondria-Targeted Hsp90 Inhibitor Based on the Crystal Structures of Human TRAP1. J. Am. Chem. Soc. 2015, 137, 4358–4367. [Google Scholar] [CrossRef]
- Sattin, S.; Tao, J.; Vettoretti, G.; Moroni, E.; Pennati, M.; Lopergolo, A.; Morelli, L.; Bugatti, A.; Zuehlke, A.; Moses, M.; et al. Activation of Hsp90 Enzymatic Activity and Conformational Dynamics through Rationally Designed Allosteric Ligands. Chem.–Eur. J. 2015, 21, 13598–13608. [Google Scholar] [CrossRef]
- D’Annessa, I.; Sattin, S.; Tao, J.; Pennati, M.; Martin, C.S.; Moroni, E.; Rasola, A.; Zaffaroni, N.; Agard, D.A.; Bernardi, A.; et al. Design of Allosteric Stimulators of the Hsp90 ATPase as New Anticancer Leads. Chem.-Eur. J. 2017, 23, 5188–5192. [Google Scholar] [CrossRef]
- Roe, M.; Wahab, B.; Török, Z.; Horváth, I.; Vigh, L.; Prodromou, C. Dihydropyridines Allosterically Modulate Hsp90 Providing a Novel Mechanism for Heat Shock Protein Co-induction and Neuroprotection. Front. Mol. Biosci. 2018, 5, 51. [Google Scholar] [CrossRef]
- Wang, Z. Aging and Aging-Related Diseases; Springer: Berlin/Heidelberg, Germany, 2018. [Google Scholar]
- Ho, S.W.; Tsui, Y.T.C.; Wong, T.T.; Cheung, S.K.-K.; Goggins, W.B.; Yi, L.M.; Cheng, K.K.; Baum, L. Effects of 17-allylamino-17-demethoxygeldanamycin (17-AAG) in transgenic mouse models of frontotemporal lobar degeneration and Alzheimer’s disease. Transl. Neurodegener. 2013, 2, 24. [Google Scholar] [CrossRef]
- Gallo, K.A. Targeting HSP90 to Halt Neurodegeneration. Chem. Biol. 2006, 13, 115–116. [Google Scholar] [CrossRef]
- Nachman, E.; Wentink, A.S.; Madiona, K.; Bousset, L.; Katsinelos, T.; Allinson, K.; Kampinga, H.; McEwan, W.A.; Jahn, T.R.; Melki, R.; et al. Disassembly of Tau fibrils by the human Hsp70 disaggregation machinery generates small seeding-competent species. J. Biol. Chem. 2020, 295, 9676–9690. [Google Scholar] [CrossRef] [PubMed]
- Tittelmeier, J.; Sandhof, C.A.; Ries, H.M.; Druffel-Augustin, S.; Mogk, A.; Bukau, B.; Nussbaum-Krammer, C. The HSP110/HSP70 disaggregation system generates spreading-competent toxic α-synuclein species. EMBO J. 2020, 39, e103954. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Bergkvist, L.; Kumar, R.; Winblad, B.; Pavlov, P.F. Targeting Chaperone/Co-Chaperone Interactions with Small Molecules: A Novel Approach to Tackle Neurodegenerative Diseases. Cells 2021, 10, 2596. [Google Scholar] [CrossRef]
- Kurop, M.K.; Huyen, C.M.; Kelly, J.H.; Blagg, B.S. The heat shock response and small molecule regulators. Eur. J. Med. Chem. 2021, 226, 113846. [Google Scholar] [CrossRef] [PubMed]
- Koopman, M.B.; Rüdiger, S.G.D. Alzheimer Cells on Their Way to Derailment Show Selective Changes in Protein Quality Control Network. Front. Mol. Biosci. 2020, 7, 214. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. Available online: http://www.sciencedirect.com/science/article/pii/S0092867413006454 (accessed on 11 September 2014). [CrossRef]
- Janssens, G.E.; Lin, X.-X.; Millan-Ariño, L.; Kavšek, A.; Sen, I.; Seinstra, R.I.; Stroustrup, N.; Nollen, E.A.; Riedel, C.G. Transcriptomics-Based Screening Identifies Pharmacological Inhibition of Hsp90 as a Means to Defer Aging. Cell Rep. 2019, 27, 467–480.e6. [Google Scholar] [CrossRef]
- Fuentealba, M.; Dönertaş, H.M.; Williams, R.; Labbadia, J.; Thornton, J.M.; Partridge, L. Using the drug-protein interactome to identify anti-ageing compounds for humans. PLOS Comput. Biol. 2019, 15, e1006639. [Google Scholar] [CrossRef]
- Hsu, A.-L.; Murphy, C.T.; Kenyon, C. Regulation of Aging and Age-Related Disease by DAF-16 and Heat-Shock Factor. Science 2003, 300, 1142–1145. [Google Scholar] [CrossRef]
- Garigan, D.; Hsu, A.-L.; Fraser, A.G.; Kamath, R.S.; Ahringer, J.; Kenyon, C. Genetic Analysis of Tissue Aging in Caenorhabditis elegans: A Role for Heat-Shock Factor and Bacterial Proliferation. Genetics 2002, 161, 1101–1112. [Google Scholar] [CrossRef]
- Zhang, M.; Poplawski, M.; Yen, K.; Cheng, H.; Bloss, E.; Zhu, X.; Patel, H.; Mobbs, C.V. Role of CBP and SATB-1 in Aging, Dietary Restriction, and Insulin-Like Signaling. PLoS Biol. 2009, 7, e1000245. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hsp90 Interactors in Proteostasis | ||||
|---|---|---|---|---|
| Location | Process Affected | Name | Function | Reference |
| Intracellular | HSR | HSF1 | Transcription factor | [76] |
| Hsp70 | Chaperone | [76] | ||
| Hsp40 | Chaperone | [76] | ||
| p23 | Co-chaperone | [77,78] | ||
| Aha1 | Co-chaperone | [77] | ||
| Cdc37 | Co-chaperone | [79] | ||
| FKBP1 | Co-chaperone | [80] | ||
| FKBPL | Co-chaperone | [81] | ||
| TTC4 | Co-chaperone | [82] | ||
| Hop | Co-chaperone | [77] | ||
| UPRER | PERK | Kinase, client | [4] | |
| IRE1 | Kinase, client | [4,83] | ||
| UPRMT | GCN2 | Kinase, client | [69] | |
| Transcription regulation | SIRT1 | Deacethylase, client | [68] | |
| Insulin/IGF1 signaling | DAF-16 | Transcription factor | [66] | |
| Extracellular | Wound healing/inflammation | CD91/LRP1 | Receptor | [84] |
| APC | LOX-1 | Receptor | [85] | |
| SREC-1 | Receptor | [85] | ||
| Cell migration | Cdc37 | Co-chaperone | [86] | |
| Activating Hsp90–MMP2 complex | Aha1 | Co-chaperone | [87] | |
| MMP2 activation | p23 | Co-chaperone | [88] | |
| Hop | Co-chaperone | [87,88] | ||
| Hsp70 | Chaperone | [88,89] | ||
| ECM formation | Fibronectin | ECM glycoprotein, client | [90] | |
| Cell migration/metastasis Formation | MMP1 | Endopeptidase, client | [91] | |
| MMP2 | Endopeptidase, client | [92] | ||
| MMP3 | Endopeptidase, client | [93] | ||
| MMP9 | Endopeptidase, client | [94] | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Somogyvári, M.; Khatatneh, S.; Sőti, C. Hsp90: From Cellular to Organismal Proteostasis. Cells 2022, 11, 2479. https://doi.org/10.3390/cells11162479
Somogyvári M, Khatatneh S, Sőti C. Hsp90: From Cellular to Organismal Proteostasis. Cells. 2022; 11(16):2479. https://doi.org/10.3390/cells11162479
Chicago/Turabian StyleSomogyvári, Milán, Saba Khatatneh, and Csaba Sőti. 2022. "Hsp90: From Cellular to Organismal Proteostasis" Cells 11, no. 16: 2479. https://doi.org/10.3390/cells11162479
APA StyleSomogyvári, M., Khatatneh, S., & Sőti, C. (2022). Hsp90: From Cellular to Organismal Proteostasis. Cells, 11(16), 2479. https://doi.org/10.3390/cells11162479