
Interplay of Developmental Hippo–Notch Signaling Pathways with the DNA Damage Response in Prostate Cancer

 , , ,
, , ,  and
and
Abstract
1. Introduction
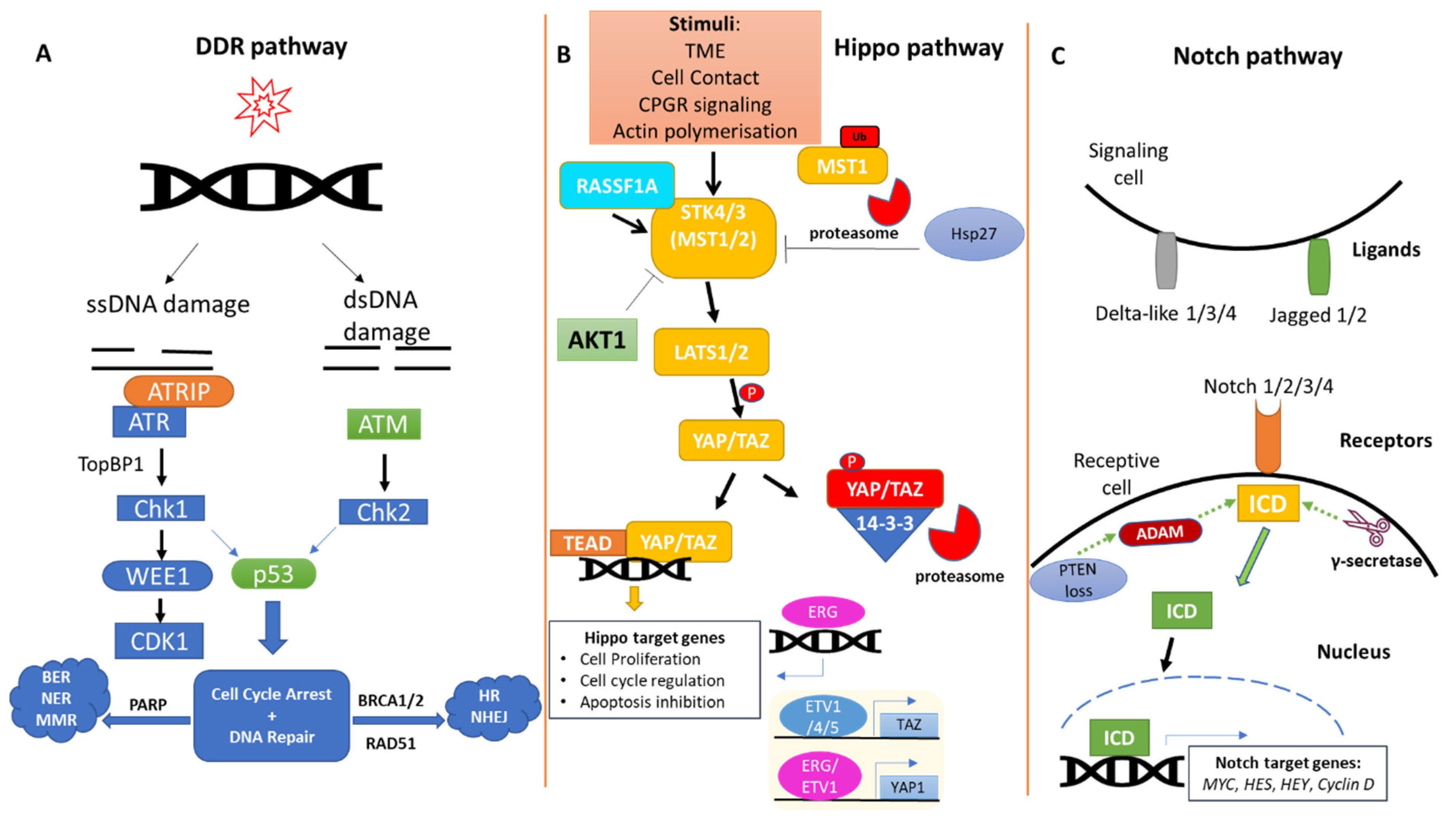
2. The DDR Pathway in Prostate Cancer
3. The Hippo Pathway in Prostate Cancer
4. The Notch Pathway in Prostate Cancer
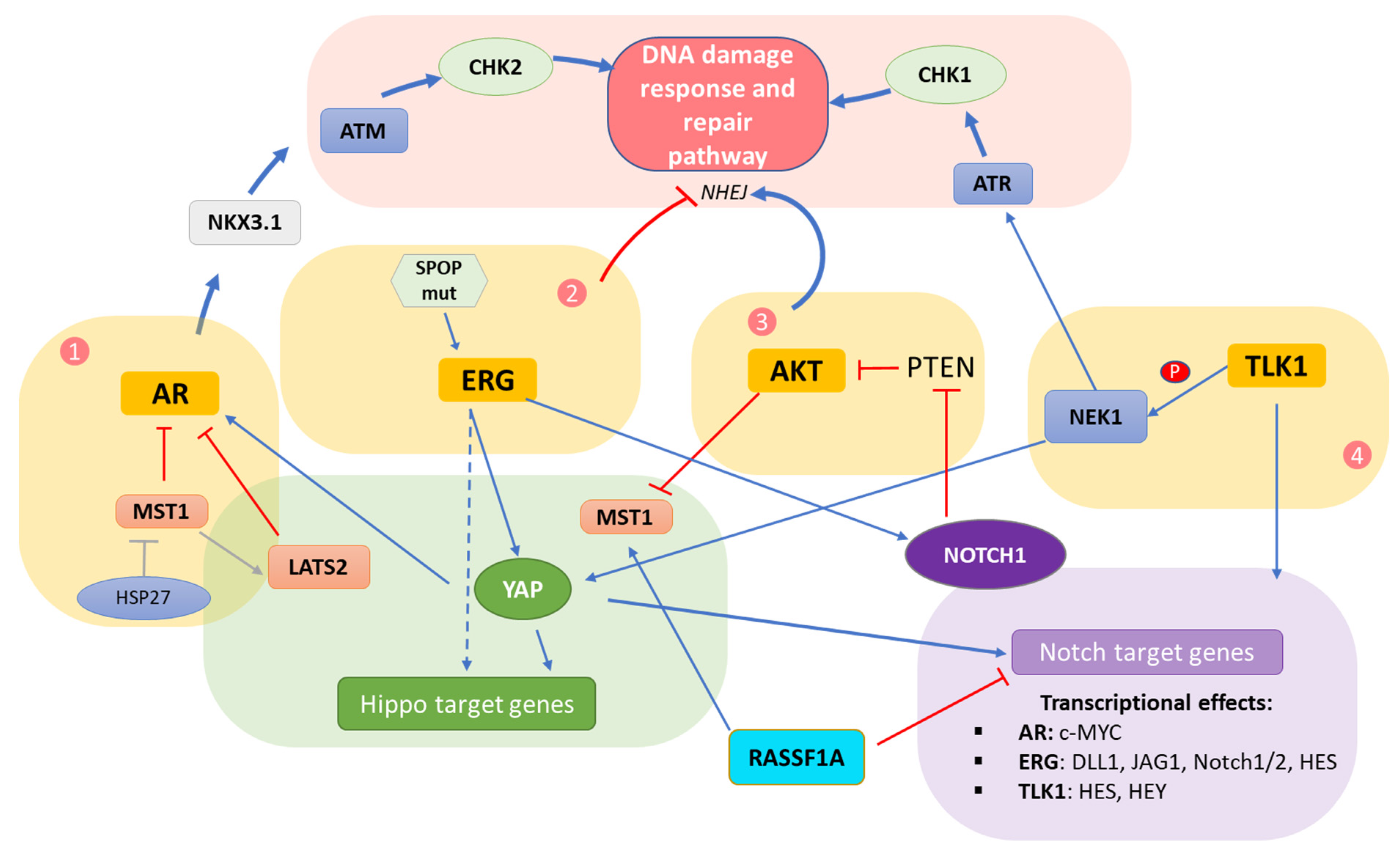
5. Interplay of DDR/Hippo/Notch Pathways in Prostate Cancer
5.1. Crosstalk Involving the AR
5.2. Crosstalk Involving the ERG Oncoprotein
5.3. Crosstalk within the AKT Hub
5.4. Crosstalk Involving TLK/NEK
5.5. Direct Hippo–Notch Pathway Crosstalk
5.6. Potential Therapeutic Implications
6. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics. CA A Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- E MacLean, H.; Chu, S.; Warne, G.L.; Zajac, J.D. Related individuals with different androgen receptor gene deletions. J. Clin. Investig. 1993, 91, 1123–1128. [Google Scholar] [CrossRef] [PubMed]
- E Massie, C.; Lynch, A.; Ramos-Montoya, A.; Boren, J.; Stark, R.; Fazli, L.; Warren, A.; Scott, H.; Madhu, B.; Sharma, N.; et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011, 30, 2719–2733. [Google Scholar] [CrossRef]
- Hååg, P.; Bektic, J.; Bartsch, G.; Klocker, H.; Eder, I.E. Androgen receptor down regulation by small interference RNA induces cell growth inhibition in androgen sensitive as well as in androgen independent prostate cancer cells. J. Steroid Biochem. Mol. Biol. 2005, 96, 251–258. [Google Scholar] [CrossRef]
- Tan, Y.G.; Poon, R.J.; Pang, L.J.; Villanueva, A.; Huang, H.H.; Chen, K.; Ng, T.K.; Tay, K.J.; Ho, H.S.; Yuen, J.S. Comparative study of surgical orchidectomy and medical castration in treatment efficacy, adverse effects and cost based on a large prospective metastatic prostate cancer registry. Urol. Oncol. Semin. Orig. Investig. 2020, 38, 682.e1–682.e9. [Google Scholar] [CrossRef]
- Clinton, T.N.; Woldu, S.L.; Raj, G.V. Degarelix versus luteinizing hormone-releasing hormone agonists for the treatment of prostate cancer. Expert Opin. Pharmacother. 2017, 18, 825–832. [Google Scholar] [CrossRef]
- Sciarra, A.; Busetto, G.M.; Salciccia, S.; Del Giudice, F.; Maggi, M.; Crocetto, F.; Ferro, M.; De Berardinis, E.; Scarpa, R.M.; Porpiglia, F.; et al. Does Exist a Differential Impact of Degarelix Versus LHRH Agonists on Cardiovascular Safety? Evidences From Randomized and Real-World Studies. Front. Endocrinol. 2021, 12, 695170. [Google Scholar] [CrossRef] [PubMed]
- Albertsen, P.C.; Hanley, J.A.; Gleason, D.F.; Barry, M.J. Competing Risk Analysis of Men Aged 55 to 74 Years at Diagnosis Managed Conservatively for Clinically Localized Prostate Cancer. JAMA 1998, 280, 975–980. [Google Scholar] [CrossRef]
- Saraon, P.; Drabovich, A.P.; Jarvi, K.A.; Diamandis, E.P. Mechanisms of Androgen-Independent Prostate Cancer. EJIFCC 2014, 25, 42–54. [Google Scholar]
- Kirby, M.; Hirst, C.; Crawford, E.D. Characterising the castration-resistant prostate cancer population: A systematic review. Int. J. Clin. Pract. 2011, 65, 1180–1192. [Google Scholar] [CrossRef]
- Papaspyropoulos, A.; Lagopati, N.; Mourkioti, I.; Angelopoulou, A.; Kyriazis, S.; Liontos, M.; Gorgoulis, V.; Kotsinas, A. Regulatory and Functional Involvement of Long Non-Coding RNAs in DNA Double-Strand Break Repair Mechanisms. Cells 2021, 10, 1506. [Google Scholar] [CrossRef] [PubMed]
- Komseli, E.-S.; Pateras, I.S.; Krejsgaard, T.; Stawiski, K.; Rizou, S.V.; Polyzos, A.; Roumelioti, F.-M.; Chiourea, M.; Mourkioti, I.; Paparouna, E.; et al. A prototypical non-malignant epithelial model to study genome dynamics and concurrently monitor micro-RNAs and proteins in situ during oncogene-induced senescence. BMC Genom. 2018, 19, 37. [Google Scholar] [CrossRef]
- Pateras, I.S.; Havaki, S.; Nikitopoulou, X.; Vougas, K.; Townsend, P.A.; Panayiotidis, M.I.; Georgakilas, A.G.; Gorgoulis, V.G. The DNA damage response and immune signaling alliance: Is it good or bad? Nature decides when and where. Pharmacol. Ther. 2015, 154, 36–56. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Van Gent, D.C.; Incrocci, L.; Van Weerden, W.M.; Nonnekens, J. Role of the DNA damage response in prostate cancer formation, progression and treatment. Prostate Cancer Prostatic Dis. 2019, 23, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Feldmann, G.; Huang, J.; Wu, S.; Zhang, N.; Comerford, S.A.; Gayyed, M.F.; Anders, R.A.; Maitra, A.; Pan, D. Elucidation of a Universal Size-Control Mechanism in Drosophila and Mammals. Cell 2007, 130, 1120–1133. [Google Scholar] [CrossRef] [PubMed]
- Coffey, K. Targeting the Hippo Pathway in Prostate Cancer: What’s New? Cancers 2021, 13, 611. [Google Scholar] [CrossRef]
- Leong, K.G.; Gao, W.-Q. The Notch pathway in prostate development and cancer. Differentiation 2008, 76, 699–716. [Google Scholar] [CrossRef]
- Wang, L.; Zi, H.; Luo, Y.; Liu, T.; Zheng, H.; Xie, C.; Wang, X.; Huang, X. Inhibition of Notch pathway enhances the anti-tumor effect of docetaxel in prostate cancer stem-like cells. Stem Cell Res. Ther. 2020, 11, 1–8. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef]
- Hakem, R. DNA-damage repair; the good, the bad, and the ugly. EMBO J. 2008, 27, 589–605. [Google Scholar] [CrossRef]
- Gorgoulis, V.G.; Pefani, D.-E.; Pateras, I.S.; Trougakos, I.P. Integrating the DNA damage and protein stress responses during cancer development and treatment. J. Pathol. 2018, 246, 12–40. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed]
- Pećina-Šlaus, N.; Kafka, A.; Salamon, I.; Bukovac, A. Mismatch Repair Pathway, Genome Stability and Cancer. Front. Mol. Biosci. 2020, 7, 122. [Google Scholar] [CrossRef] [PubMed]
- Li, G.-M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2007, 18, 85–98. [Google Scholar] [CrossRef]
- Steinbeck, R.G. Dysplasia in view of the cell cycle. Eur. J. Histochem. 2004, 48, 203–211. [Google Scholar]
- Vitkin, N.; Nersesian, S.; Siemens, D.R.; Koti, M. The Tumor Immune Contexture of Prostate Cancer. Front. Immunol. 2019, 10, 603. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 162, 454. [Google Scholar] [CrossRef]
- Pritchard, C.C.; Mateo, J.; Walsh, M.F.; De Sarkar, N.; Abida, W.; Beltran, H.; Garofalo, A.; Gulati, R.; Carreira, S.; Eeles, R.; et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N. Engl. J. Med. 2016, 375, 443–453. [Google Scholar] [CrossRef]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative Genomic Profiling of Human Prostate Cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef]
- Muller, P.A.J.; Vousden, K.H. p53 mutations in cancer. Nature 2013, 15, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Rachmat, R.; Enyioma, S.; Ghose, A.; Revythis, A.; Boussios, S. BRCA Mutations in Prostate Cancer: Assessment, Implications and Treatment Considerations. Int. J. Mol. Sci. 2021, 22, 12628. [Google Scholar] [CrossRef] [PubMed]
- Schiewer, M.J.; Knudsen, K.E. DNA Damage Response in Prostate Cancer. Cold Spring Harb. Perspect. Med. 2018, 9, a030486. [Google Scholar] [CrossRef] [PubMed]
- Schlomm, T.; Iwers, L.; Kirstein, P.; Jessen, B.; Köllermann, J.; Minner, S.; Passow-Drolet, A.; Mirlacher, M.; Milde-Langosch, K.; Graefen, M.; et al. Clinical significance of p53 alterations in surgically treated prostate cancers. Mod. Pathol. 2008, 21, 1371–1378. [Google Scholar] [CrossRef] [PubMed]
- A Eastham, J.; Stapleton, A.M.; E Gousse, A.; Timme, T.L.; Yang, G.; Slawin, K.M.; Wheeler, T.M.; Scardino, P.T.; Thompson, T.C. Association of p53 mutations with metastatic prostate cancer. Clin. Cancer Res. 1995, 1, 1111–1118. [Google Scholar]
- Armenia, J.; Wankowicz, S.A.M.; Liu, D.; Gao, J.; Kundra, R.; Reznik, E.; Chatila, W.K.; Chakravarty, D.; Han, G.C.; Coleman, I.; et al. The long tail of oncogenic drivers in prostate cancer. Nat. Genet. 2018, 50, 645–651. [Google Scholar] [CrossRef]
- Grasso, C.S.; Wu, Y.-M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef]
- Mateo, J.; Boysen, G.; Barbieri, C.E.; Bryant, H.E.; Castro, E.; Nelson, P.S.; Olmos, D.; Pritchard, C.C.; Rubin, M.A.; de Bono, J.S. DNA Repair in Prostate Cancer: Biology and Clinical Implications. Eur. Urol. 2017, 71, 417–425. [Google Scholar] [CrossRef]
- Dey, A.; Varelas, X.; Guan, K.-L. Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat. Rev. Drug Discov. 2020, 19, 480–494. [Google Scholar] [CrossRef]
- Cunningham, R.; Hansen, C.G. The Hippo pathway in cancer: YAP/TAZ and TEAD as therapeutic targets in cancer. Clin. Sci. 2022, 136, 197–222. [Google Scholar] [CrossRef]
- Papaspyropoulos, A.; Bradley, L.; Thapa, A.; Leung, C.Y.; Toskas, K.; Koennig, D.; Pefani, D.-E.; Raso, C.; Grou, C.; Hamilton, G.; et al. RASSF1A uncouples Wnt from Hippo signalling and promotes YAP mediated differentiation via p73. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Lagopati, N.; Belogiannis, K.; Angelopoulou, A.; Papaspyropoulos, A.; Gorgoulis, V. Non-Canonical Functions of the ARF Tumor Suppressor in Development and Tumorigenesis. Biomolecules 2021, 11, 86. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Moroishi, T.; Guan, K.-L. Mechanisms of Hippo pathway regulation. Genes Dev. 2016, 30, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Salem, O.; Hansen, C.G. The Hippo Pathway in Prostate Cancer. Cells 2019, 8, 370. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Mei, L.; Fan, X.; Tang, C.; Ji, X.; Hu, X.; Shi, W.; Qian, Y.; Hussain, M.; Wu, J.; et al. Phosphodiesterase 5/protein kinase G signal governs stemness of prostate cancer stem cells through Hippo pathway. Cancer Lett. 2016, 378, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Collak, F.K.; Demir, U.; Sagir, F. YAP1 Is Involved in Tumorigenic Properties of Prostate Cancer Cells. Pathol. Oncol. Res. 2019, 26, 867–876. [Google Scholar] [CrossRef]
- Lin, M.; Bu, C.; He, Q.; Gu, J.; Wang, H.; Feng, N.; Jiang, S.W. TAZ is overexpressed in prostate cancers and regulates the proliferation, migration and apoptosis of prostate cancer PC3 cells. Oncol. Rep. 2020, 44, 747–756. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Tretiakova, M.S.; Silvis, M.R.; Lucas, J.; Klezovitch, O.; Coleman, I.; Bolouri, H.; Kutyavin, V.I.; Morrissey, C.; True, L.D.; et al. ERG Activates the YAP1 Transcriptional Program and Induces the Development of Age-Related Prostate Tumors. Cancer Cell 2015, 27, 797–808. [Google Scholar] [CrossRef]
- Kim, T.-D.; Jin, F.; Shin, S.; Oh, S.; Lightfoot, S.A.; Grande, J.P.; Johnson, A.J.; van Deursen, J.M.; Wren, J.; Janknecht, R. Histone demethylase JMJD2A drives prostate tumorigenesis through transcription factor ETV1. J. Clin. Investig. 2016, 126, 706–720. [Google Scholar] [CrossRef]
- Liu, C.-Y.; Yu, T.; Huang, Y.; Cui, L.; Hong, W. ETS (E26 transformation-specific) up-regulation of the transcriptional co-activator TAZ promotes cell migration and metastasis in prostate cancer. J. Biol. Chem. 2017, 292, 9420–9430. [Google Scholar] [CrossRef]
- Vahid, S.; Thaper, D.; Gibson, K.F.; Bishop, J.L.; Zoubeidi, A. Molecular chaperone Hsp27 regulates the Hippo tumor suppressor pathway in cancer. Sci. Rep. 2016, 6, 31842. [Google Scholar] [CrossRef] [PubMed]
- Ren, A.; Yan, G.; You, B.; Sun, J. Down-regulation of mammalian sterile 20-like kinase 1 by heat shock protein 70 mediates cisplatin resistance in prostate cancer cells. Cancer Res. 2008, 68, 2266–2274. [Google Scholar] [CrossRef] [PubMed]
- Hsu, T.-I.; Hsu, C.-H.; Lee, K.-H.; Lin, J.-T.; Chen, C.-S.; Chang, K.-C.; Su, C.-Y.; Hsiao, M.; Lu, P.-J. MicroRNA-18a is elevated in prostate cancer and promotes tumorigenesis through suppressing STK4 in vitro and in vivo. Oncogenesis 2014, 3, e99. [Google Scholar] [CrossRef] [PubMed]
- Cinar, B.; Fang, P.-K.; Lutchman, M.; Di Vizio, D.; Adam, R.M.; Pavlova, N.; Rubin, M.A.; Yelick, P.C.; Freeman, M.R. The pro-apoptotic kinase Mst1 and its caspase cleavage products are direct inhibitors of Akt1. EMBO J. 2007, 26, 4523–4534. [Google Scholar] [CrossRef] [PubMed]
- Mohseni, M.; Sun, J.; Lau, A.; Curtis, S.; Goldsmith, J.; Fox, V.L.; Wei, C.; Frazier, M.; Samson, O.; Wong, K.K.; et al. A genetic screen identifies an LKB1–MARK signalling axis controlling the Hippo–YAP pathway. Nat. Cell Biol. 2013, 16, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Hermanova, I.; Zúñiga-García, P.; Caro-Maldonado, A.; Fernandez-Ruiz, S.; Salvador, F.; Martín-Martín, N.; Zabala-Letona, A.; Nuñez-Olle, M.; Torrano, V.; Camacho, L.; et al. Genetic manipulation of LKB1 elicits lethal metastatic prostate cancer. J. Exp. Med. 2020, 217, e20191787. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, L.; Wan, D.; Zhou, L.; Zheng, S.; Lin, S.; Qiao, Y. Extracellular matrix and its therapeutic potential for cancer treatment. Signal Transduct. Target. Ther. 2021, 6, 153. [Google Scholar] [CrossRef]
- Dupont, S. Role of YAP/TAZ in cell-matrix adhesion-mediated signalling and mechanotransduction. Exp. Cell Res. 2016, 343, 42–53. [Google Scholar] [CrossRef]
- Gaspar, P.; Tapon, N. Sensing the local environment: Actin architecture and Hippo signalling. Curr. Opin. Cell Biol. 2014, 31, 74–83. [Google Scholar] [CrossRef]
- Luthold, C.; Hallal, T.; Labbé, D.P.; Bordeleau, F. The Extracellular Matrix Stiffening: A Trigger of Prostate Cancer Progression and Castration Resistance? Cancers 2022, 14, 2887. [Google Scholar] [CrossRef]
- Zhao, B.; Li, L.; Wang, L.; Wang, C.-Y.; Yu, J.; Guan, K.-L. Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev. 2012, 26, 54–68. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-C.; Lin, S.-C.; Yu, G.; Zhu, M.; Song, J.H.; Rivera, K.; Pappin, D.J.; Logothetis, C.J.; Panaretakis, T.; Wang, G.; et al. Prostate tumor-induced stromal reprogramming generates Tenascin C that promotes prostate cancer metastasis through YAP/TAZ inhibition. Oncogene 2022, 41, 757–769. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-Y.; Huang, S.-P.; Lin, V.C.; Yu, C.-C.; Chang, T.-Y.; Juang, S.-H.; Bao, B.-Y. Genetic variants in the Hippo pathway predict biochemical recurrence after radical prostatectomy for localized prostate cancer. Sci. Rep. 2015, 5, 8556. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-D.; Leow, C.C.; Zha, J.; Tang, Z.; Modrusan, Z.; Radtke, F.; Aguet, M.; de Sauvage, F.J.; Gao, W.-Q. Notch signaling is required for normal prostatic epithelial cell proliferation and differentiation. Dev. Biol. 2006, 290, 66–80. [Google Scholar] [CrossRef] [PubMed]
- Villaronga, M.A.; Bevan, C.L.; Belandia, B. Notch Signaling: A Potential Therapeutic Target in Prostate Cancer. Curr. Cancer Drug Targets 2008, 8, 566–580. [Google Scholar] [CrossRef][Green Version]
- Hori, K.; Sen, A.; Artavanis-Tsakonas, S. Notch signaling at a glance. J. Cell Sci. 2013, 126, 2135–2140. [Google Scholar] [CrossRef]
- Shou, J.; Ross, S.; Koeppen, H.; De Sauvage, F.J.; Gao, W.Q. Dynamics of notch expression during murine prostate development and tumorigenesis. Cancer Res. 2001, 61, 7291–7297. [Google Scholar]
- Zhang, L.; Sha, J.; Yang, G.; Huang, X.; Bo, J.; Huang, Y. Activation of Notch pathway is linked with epithelial-mesenchymal transition in prostate cancer cells. Cell Cycle 2017, 16, 999–1007. [Google Scholar] [CrossRef]
- Rice, M.A.; Hsu, E.-C.; Aslan, M.; Ghoochani, A.; Su, A.; Stoyanova, T. Loss of Notch1 Activity Inhibits Prostate Cancer Growth and Metastasis and Sensitizes Prostate Cancer Cells to Antiandrogen Therapies. Mol. Cancer Ther. 2019, 18, 1230–1242. [Google Scholar] [CrossRef]
- Farah, E.; Li, C.; Cheng, L.; Kong, Y.; Lanman, N.A.; Pascuzzi, P.; Lorenz, G.R.; Zhang, Y.; Ahmad, N.; Li, L.; et al. NOTCH signaling is activated in and contributes to resistance in enzalutamide-resistant prostate cancer cells. J. Biol. Chem. 2019, 294, 8543–8554. [Google Scholar] [CrossRef]
- Cui, D.; Dai, J.; Keller, J.M.; Mizokami, A.; Xia, S.; Keller, E.T. Notch Pathway Inhibition Using PF-03084014, a γ-Secretase Inhibitor (GSI), Enhances the Antitumor Effect of Docetaxel in Prostate Cancer. Clin. Cancer Res. 2015, 21, 4619–4629. [Google Scholar] [CrossRef] [PubMed]
- Scorey, N.; Fraser, S.P.; Patel, P.; Pridgeon, C.; Dallman, M.J.; A Djamgoz, M.B. Notch signalling and voltage-gated Na+ channel activity in human prostate cancer cells: Independent modulation of in vitro motility. Prostate Cancer Prostatic Dis. 2006, 9, 399–406. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Marignol, L.; Rivera-Figueroa, K.; Lynch, T.; Hollywood, D. Hypoxia, notch signalling, and prostate cancer. Nat. Rev. Urol. 2013, 10, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Santagata, S.; Demichelis, F.; Riva, A.; Varambally, S.; Hofer, M.D.; Kutok, J.L.; Kim, R.; Tang, J.; Montie, J.E.; Chinnaiyan, A.M.; et al. JAGGED1 Expression Is Associated with Prostate Cancer Metastasis and Recurrence. Cancer Res. 2004, 64, 6854–6857. [Google Scholar] [CrossRef]
- Puca, L.; Gavyert, K.; Sailer, V.; Conteduca, V.; Dardenne, E.; Sigouros, M.; Isse, K.; Kearney, M.; Vosoughi, A.; Fernandez, L.; et al. Delta-like protein 3 expression and therapeutic targeting in neuroendocrine prostate cancer. Sci. Transl. Med. 2019, 11, eaav0891. [Google Scholar] [CrossRef]
- Kwon, O.-J.; Zhang, L.; Wang, J.; Su, Q.; Feng, Q.; Zhang, X.H.; Mani, S.A.; Paulter, R.; Creighton, C.J.; Ittmann, M.M.; et al. Notch promotes tumor metastasis in a prostate-specific Pten-null mouse model. J. Clin. Investig. 2016, 126, 2626–2641. [Google Scholar] [CrossRef]
- Su, Q.; Zhang, B.; Zhang, L.; Dang, T.; Rowley, D.; Ittmann, M.; Xin, L. Jagged1 upregulation in prostate epithelial cells promotes formation of reactive stroma in the Pten null mouse model for prostate cancer. Oncogene 2016, 36, 618–627. [Google Scholar] [CrossRef]
- Revandkar, A.; Perciato, M.L.; Toso, A.; Alajati, A.; Chen, J.; Gerber, H.; Dimitrov, M.; Rinaldi, A.; Delaleu, N.; Pasquini, E.; et al. Inhibition of Notch pathway arrests PTEN-deficient advanced prostate cancer by triggering p27-driven cellular senescence. Nat. Commun. 2016, 7, 13719. [Google Scholar] [CrossRef]
- Li, J.; Al-Azzawi, F. Mechanism of androgen receptor action. Maturitas 2009, 63, 142–148. [Google Scholar] [CrossRef]
- Dagar, M.; Singh, J.P.; Dagar, G.; Tyagi, R.K.; Bagchi, G. Phosphorylation of HSP90 by protein kinase A is essential for the nuclear translocation of androgen receptor. J. Biol. Chem. 2019, 294, 8699–8710. [Google Scholar] [CrossRef]
- van de Wijngaart, D.J.; Dubbink, H.J.; van Royen, M.E.; Trapman, J.; Jenster, G. Androgen receptor coregulators: Recruitment via the coactivator binding groove. Mol. Cell. Endocrinol. 2012, 352, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Davey, R.A.; Grossmann, M. Androgen Receptor Structure, Function and Biology: From Bench to Bedside. Clin. Biochem. Rev. 2016, 37, 3–15. [Google Scholar] [PubMed]
- Kuser-Abali, G.; Alptekin, A.; Lewis, M.J.; Garraway, I.P.; Cinar, B. YAP1 and AR interactions contribute to the switch from androgen-dependent to castration-resistant growth in prostate cancer. Nat. Commun. 2015, 6, 8126. [Google Scholar] [CrossRef] [PubMed]
- Powzaniuk, M.; McElwee-Witmer, S.; Vogel, R.L.; Hayami, T.; Rutledge, S.J.; Chen, F.; Harada, S.-I.; Schmidt, A.; Rodan, G.A.; Freedman, L.P.; et al. The LATS2/KPM Tumor Suppressor Is a Negative Regulator of the Androgen Receptor. Mol. Endocrinol. 2004, 18, 2011–2023. [Google Scholar] [CrossRef]
- Bai, S.; Cao, S.; Jin, L.; Kobelski, M.; Schouest, B.; Wang, X.; Ungerleider, N.; Baddoo, M.; Zhang, W.; Corey, E.; et al. A positive role of c-Myc in regulating androgen receptor and its splice variants in prostate cancer. Oncogene 2019, 38, 4977–4989. [Google Scholar] [CrossRef]
- Cinar, B.; Collak, F.K.; Lopez, D.; Akgul, S.; Mukhopadhyay, N.K.; Kilicarslan, M.; Gioeli, D.G.; Freeman, M.R. MST1 Is a Multifunctional Caspase-Independent Inhibitor of Androgenic Signaling. Cancer Res. 2011, 71, 4303–4313. [Google Scholar] [CrossRef]
- Bowen, C.; Ju, J.-H.; Lee, J.-H.; Paull, T.T.; Gelmann, E.P. Functional Activation of ATM by the Prostate Cancer Suppressor NKX3.1. Cell Rep. 2013, 4, 516–529. [Google Scholar] [CrossRef]
- Hamilton, G.; Yee, K.S.; Scrace, S.; O’Neill, E. ATM Regulates a RASSF1A-Dependent DNA Damage Response. Curr. Biol. 2009, 19, 2020–2025. [Google Scholar] [CrossRef]
- Pefani, D.E.; Tognoli, M.L.; Ercan, D.P.; Gorgoulis, V.; O’Neill, E. MST2 kinase suppresses rDNA transcription in response to DNA damage by phosphorylating nucleolar histone H2B. EMBO J. 2018, 37, e98760. [Google Scholar] [CrossRef]
- van der Weyden, L.; Papaspyropoulos, A.; Poulogiannis, G.; Rust, A.G.; Rashid, M.; Adams, D.J.; Arends, M.J.; O’Neill, E. Loss of Rassf1a Synergizes with Deregulated Runx2 Signaling in Tumorigenesis. Cancer Res. 2012, 72, 3817–3827. [Google Scholar] [CrossRef]
- Goodwin, J.F.; Schiewer, M.J.; Dean, J.L.; Schrecengost, R.S.; de Leeuw, R.; Han, S.; Ma, T.; Den, R.B.; Dicker, A.P.; Feng, F.Y.; et al. A Hormone–DNA Repair Circuit Governs the Response to Genotoxic Insult. Cancer Discov. 2013, 3, 1254–1271. [Google Scholar] [CrossRef] [PubMed]
- Mani, R.-S.; Tomlins, S.A.; Callahan, K.; Ghosh, A.; Nyati, M.K.; Varambally, S.; Palanisamy, N.; Chinnaiyan, A.M. Induced Chromosomal Proximity and Gene Fusions in Prostate Cancer. Science 2009, 326, 1230. [Google Scholar] [CrossRef] [PubMed]
- Taniue, K.; Akimitsu, N. Fusion Genes and RNAs in Cancer Development. Non-Coding RNA 2021, 7, 10. [Google Scholar] [CrossRef]
- Gan, W.; Dai, X.; Lunardi, A.; Li, Z.; Inuzuka, H.; Liu, P.; Varmeh, S.; Zhang, J.; Cheng, L.; Sun, Y.; et al. SPOP Promotes Ubiquitination and Degradation of the ERG Oncoprotein to Suppress Prostate Cancer Progression. Mol. Cell 2015, 59, 917–930. [Google Scholar] [CrossRef] [PubMed]
- Klezovitch, O.; Risk, M.; Coleman, I.; Lucas, J.M.; Null, M.; True, L.D.; Nelson, P.S.; Vasioukhin, V. A causal role for ERG in neoplastic transformation of prostate epithelium. Proc. Natl. Acad. Sci. USA 2008, 105, 2105–2110. [Google Scholar] [CrossRef]
- Chatterjee, P.; Choudhary, G.S.; Alswillah, T.; Xiong, X.; Heston, W.D.; Magi-Galluzzi, C.; Zhang, J.; Klein, E.A.; Almasan, A. The TMPRSS2–ERG Gene Fusion Blocks XRCC4-Mediated Nonhomologous End-Joining Repair and Radiosensitizes Prostate Cancer Cells to PARP Inhibition. Mol. Cancer Ther. 2015, 14, 1896–1906. [Google Scholar] [CrossRef]
- Imada, E.L.; Sanchez, D.F.; Dinalankara, W.; Vidotto, T.; Ebot, E.M.; Tyekucheva, S.; Franco, G.R.; Mucci, L.A.; Loda, L.; Schaeffer, E.M.; et al. Transcriptional landscape of PTEN loss in primary prostate cancer. BMC Cancer 2021, 21, 856. [Google Scholar] [CrossRef] [PubMed]
- Hong, Z.; Zhang, W.; Ding, D.; Huang, Z.; Yan, Y.; Cao, W.; Pan, Y.; Hou, X.; Weroha, S.J.; Karnes, R.J.; et al. DNA Damage Promotes TMPRSS2-ERG Oncoprotein Destruction and Prostate Cancer Suppression via Signaling Converged by GSK3β and WEE1. Mol. Cell 2020, 79, 1008–1023.e4. [Google Scholar] [CrossRef]
- Shorning, B.Y.; Dass, M.S.; Smalley, M.J.; Pearson, H.B. The PI3K-AKT-mTOR Pathway and Prostate Cancer: At the Crossroads of AR, MAPK, and WNT Signaling. Int. J. Mol. Sci. 2020, 21, 4507. [Google Scholar] [CrossRef]
- Mohamed, A.A.; Tan, S.-H.; Xavier, C.P.; Katta, S.; Huang, W.; Ravindranath, L.; Jamal, M.; Li, H.; Srivastava, M.; Srivatsan, E.S.; et al. Synergistic Activity with NOTCH Inhibition and Androgen Ablation in ERG-Positive Prostate Cancer Cells. Mol. Cancer Res. 2017, 15, 1308–1317. [Google Scholar] [CrossRef]
- Kron, K.J.; Murison, A.; Zhou, S.; Huang, V.; Yamaguchi, T.N.; Shiah, Y.-J.; Fraser, M.; Van der Kwast, T.; Boutros, P.C.; Bristow, R.; et al. TMPRSS2–ERG fusion co-opts master transcription factors and activates NOTCH signaling in primary prostate cancer. Nat. Genet. 2017, 49, 1336–1345. [Google Scholar] [CrossRef] [PubMed]
- Boysen, G.; E Barbieri, C.; Prandi, D.; Blattner, M.; Chae, S.-S.; Dahija, A.; Nataraj, S.; Huang, D.; Marotz, C.; Xu, L.; et al. SPOP mutation leads to genomic instability in prostate cancer. Elife 2015, 4, e09207. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Ren, S.; Murphy, S.J.; Dalangood, S.; Chang, C.; Pang, X.; Cui, Y.; Wang, L.; Pan, Y.; Zhang, X.; et al. Truncated ERG Oncoproteins from TMPRSS2-ERG Fusions Are Resistant to SPOP-Mediated Proteasome Degradation. Mol. Cell 2015, 59, 904–916. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, M.; Higashiyama, S. The Roles of SPOP in DNA Damage Response and DNA Replication. Int. J. Mol. Sci. 2020, 21, 7293. [Google Scholar] [CrossRef] [PubMed]
- Hjorth-Jensen, K.; Maya-Mendoza, A.; Dalgaard, N.; O Sigurðsson, J.; Bartek, J.; Iglesias-Gato, D.; Olsen, J.V.; Flores-Morales, A. SPOP promotes transcriptional expression of DNA repair and replication factors to prevent replication stress and genomic instability. Nucleic Acids Res. 2018, 46, 9891. [Google Scholar] [CrossRef]
- Bernasocchi, T.; El Tekle, G.; Bolis, M.; Mutti, A.; Vallerga, A.; Brandt, L.P.; Spriano, F.; Svinkina, T.; Zoma, M.; Ceserani, V.; et al. Dual functions of SPOP and ERG dictate androgen therapy responses in prostate cancer. Nat. Commun. 2021, 12, 734. [Google Scholar] [CrossRef]
- Szymonowicz, K.; Oeck, S.; Malewicz, N.; Jendrossek, V. New Insights into Protein Kinase B/Akt Signaling: Role of Localized Akt Activation and Compartment-Specific Target Proteins for the Cellular Radiation Response. Cancers 2018, 10, 78. [Google Scholar] [CrossRef]
- Xu, N.; Lao, Y.; Zhang, Y.; Gillespie, D.A. Akt: A Double-Edged Sword in Cell Proliferation and Genome Stability. J. Oncol. 2012, 2012, 951724. [Google Scholar] [CrossRef]
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 425. [Google Scholar] [CrossRef]
- Cham, J.; Venkateswaran, A.R.; Bhangoo, M. Targeting the PI3K-AKT-mTOR Pathway in Castration Resistant Prostate Cancer: A Review Article. Clin. Genitourin. Cancer 2021, 19, 563.e561–563.e567. [Google Scholar] [CrossRef]
- Romano, D.; Matallanas, D.; Weitsman, G.; Preisinger, C.; Ng, T.; Kolch, W. Proapoptotic Kinase MST2 Coordinates Signaling Crosstalk between RASSF1A, Raf-1, and Akt. Cancer Res. 2010, 70, 1195–1203. [Google Scholar] [CrossRef] [PubMed]
- Conley-LaComb, M.K.; Saliganan, A.; Kandagatla, P.; Chen, Y.Q.; Cher, M.L.; Chinni, S.R. PTEN loss mediated Akt activation promotes prostate tumor growth and metastasis via CXCL12/CXCR4 signaling. Mol. Cancer 2013, 12, 85. [Google Scholar] [CrossRef] [PubMed]
- Silljé, H.; Takahashi, K.; Tanaka, K.; Van Houwe, G.; Nigg, E. Mammalian homologues of the plant Tousled gene code for cell-cycle-regulated kinases with maximal activities linked to ongoing DNA replication. EMBO J. 1999, 18, 5691–5702. [Google Scholar] [CrossRef] [PubMed]
- De Benedetti, A. The Tousled-Like Kinases as Guardians of Genome Integrity. ISRN Mol. Biol. 2012, 2012, 627596. [Google Scholar] [CrossRef]
- Segura-Bayona, S.; Stracker, T.H. The Tousled-like kinases regulate genome and epigenome stability: Implications in development and disease. Experientia 2019, 76, 3827–3841. [Google Scholar] [CrossRef]
- Sunavala-Dossabhoy, G. Preserving salivary gland physiology against genotoxic damage-the Tousled way. Oral Dis. 2018, 24, 1390–1398. [Google Scholar] [CrossRef]
- Singh, V.; Connelly, Z.M.; Shen, X.; De Benedetti, A. Identification of the proteome complement of humanTLK1 reveals it binds and phosphorylates NEK1 regulating its activity. Cell Cycle 2017, 16, 915–926. [Google Scholar] [CrossRef]
- Polci, R.; Peng, A.; Chen, P.-L.; Riley, D.J.; Chen, Y. NIMA-Related Protein Kinase 1 Is Involved Early in the Ionizing Radiation-Induced DNA Damage Response. Cancer Res. 2004, 64, 8800–8803. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, P.-L.; Chen, C.-F.; Jiang, X.; Riley, D.J. Never-in-mitosis related Kinase 1 functions in DNA damage response and checkpoint control. Cell Cycle 2008, 7, 3194–3201. [Google Scholar] [CrossRef]
- Liu, S.; Ho, C.K.; Ouyang, J.; Zou, L. Nek1 kinase associates with ATR–ATRIP and primes ATR for efficient DNA damage signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 2175–2180. [Google Scholar] [CrossRef]
- Khalil, I.; De Benedetti, A. Tousled-like kinase 1: A novel factor with multifaceted role in mCRPC progression and development of therapy resistance. Cancer Drug Resist 2022, 5, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Yim, H.; Sung, C.K.; You, J.; Tian, Y.; Benjamin, T. Nek1 and TAZ interact to maintain normal levels of polycystin 2. J. Am. Soc. Nephrol. 2011, 22, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Khalil, M.I.; Ghosh, I.; Singh, V.; Chen, J.; Zhu, H.; De Benedetti, A. NEK1 Phosphorylation of YAP Promotes Its Stabilization and Transcriptional Output. Cancers 2020, 12, 3666. [Google Scholar] [CrossRef] [PubMed]
- Zagurovskaya, M.; Shareef, M.M.; Das, A.; Reeves, A.; Gupta, S.; Sudol, M.; Bedford, M.T.; Prichard, J.; Mohiuddin, M.; Ahmed, M.M. EGR-1 forms a complex with YAP-1 and upregulates Bax expression in irradiated prostate carcinoma cells. Oncogene 2009, 28, 1121–1131. [Google Scholar] [CrossRef]
- Bertrand, F.E.; McCubrey, J.A.; Angus, C.W.; Nutter, J.M.; Sigounas, G. NOTCH and PTEN in prostate cancer. Adv. Biol. Regul. 2014, 56, 51–65. [Google Scholar] [CrossRef]
- Papaspyropoulos, A.; Angelopoulou, A.; Mourkioti, I.; Polyzou, A.; Pankova, D.; Toskas, K.; Lanfredini, S.; A Pantazaki, A.; Lagopati, N.; Kotsinas, A.; et al. RASSF1A disrupts the NOTCH signaling axis via SNURF/RNF4-mediated ubiquitination of HES1. EMBO Rep. 2021, 23, e51287. [Google Scholar] [CrossRef]
- Grawenda, A.M.; O’Neill, E. Clinical utility of RASSF1A methylation in human malignancies. Br. J. Cancer 2015, 113, 372–381. [Google Scholar] [CrossRef]
- Amin, K.S.; Banerjee, P.P. The cellular functions of RASSF1A and its inactivation in prostate cancer. J. Carcinog. 2012, 11, 3. [Google Scholar] [CrossRef]
- Gibault, F.; Sturbaut, M.; Bailly, F.; Melnyk, P.; Cotelle, P. Targeting Transcriptional Enhanced Associate Domains (TEADs). J. Med. Chem. 2017, 61, 5057–5072. [Google Scholar] [CrossRef]
- Nouri, K.; Azad, T.; Lightbody, E.; Khanal, P.; Nicol, C.J.; Yang, X. A kinome-wide screen using a NanoLuc LATS luminescent biosensor identifies ALK as a novel regulator of the Hippo pathway in tumorigenesis and immune evasion. FASEB J. 2019, 33, 12487–12499. [Google Scholar] [CrossRef]
- Carneiro, B.A.; Pamarthy, S.; Shah, A.N.; Sagar, V.; Unno, K.; Han, H.; Yang, X.J.; Costa, R.B.; Nagy, R.J.; Lanman, R.B.; et al. Anaplastic Lymphoma Kinase Mutation (ALK F1174C) in Small Cell Carcinoma of the Prostate and Molecular Response to Alectinib. Clin. Cancer Res. 2018, 24, 2732–2739. [Google Scholar] [CrossRef]
- Ji, J.; Chen, W.; Lian, W.; Chen, R.; Yang, J.; Zhang, Q.; Weng, Q.; Khan, Z.; Hu, J.; Chen, X.; et al. (S)-crizotinib reduces gastric cancer growth through oxidative DNA damage and triggers pro-survival akt signal. Cell Death Dis. 2018, 9, 660. [Google Scholar] [CrossRef] [PubMed]
- Schultze, A.; Fiedler, W. Therapeutic potential and limitations of new FAK inhibitors in the treatment of cancer. Expert Opin. Investig. Drugs 2010, 19, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.-J.; Constanzo, J.D.; Venkateswaran, N.; Melegari, M.; Ilcheva, M.; Morales, J.C.; Skoulidis, F.; Heymach, J.V.; Boothman, D.A.; Scaglioni, P.P. Focal Adhesion Kinase Regulates the DNA Damage Response and Its Inhibition Radiosensitizes Mutant KRAS Lung Cancer. Clin. Cancer Res. 2016, 22, 5851–5863. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Amin, K.S.; Jagadeesh, S.; Baishay, G.; Rao, P.G.; Barua, N.C.; Bhattacharya, S.; Banerjee, P.P. Mahanine restores RASSF1A expression by down-regulating DNMT1 and DNMT3B in prostate cancer cells. Mol. Cancer 2013, 12, 99. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Y.; Zhou, D.; Wang, K.; Wang, X.; Wang, X.; Jiang, Y.; Zhao, M.; Yu, R.; Zhou, X. Radiation-induced YAP activation confers glioma radioresistance via promoting FGF2 transcription and DNA damage repair. Oncogene 2021, 40, 4580–4591. [Google Scholar] [CrossRef]
- Peng, S.; Sen, B.; Mazumdar, T.; Byers, L.A.; Diao, L.; Wang, J.; Tong, P.; Giri, U.; Heymach, J.V.; Kadara, H.N.; et al. Dasatinib induces DNA damage and activates DNA repair pathways leading to senescence in non-small cell lung cancer cell lines with kinase-inactivating BRAF mutations. Oncotarget 2015, 7, 565–579. [Google Scholar] [CrossRef]
- Efimova, E.V.; Ricco, N.; Labay, E.; Mauceri, H.J.; Flor, A.C.; Ramamurthy, A.; Sutton, H.G.; Weichselbaum, R.R.; Kron, S.J. HMG-CoA Reductase Inhibition Delays DNA Repair and Promotes Senescence After Tumor Irradiation. Mol. Cancer Ther. 2018, 17, 407–418. [Google Scholar] [CrossRef]
- Jayaprakash, K.T.; Hussein, M.; Shaffer, R.; Michael, A.; Nisbet, A.; Ajaz, M. In Vitro Evaluation of Notch Inhibition to Enhance Efficacy of Radiation Therapy in Melanoma. Adv. Radiat. Oncol. 2020, 6, 100622. [Google Scholar] [CrossRef]
- Wei, C.; Li, X. Verteporfin inhibits cell proliferation and induces apoptosis in different subtypes of breast cancer cell lines without light activation. BMC Cancer 2020, 20, 1042. [Google Scholar] [CrossRef]
- Yan, J.; Tang, D. Prostate cancer stem-like cells proliferate slowly and resist etoposide-induced cytotoxicity via enhancing DNA damage response. Exp. Cell Res. 2014, 328, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Tsao, T.; Beretov, J.; Ni, J.; Bai, X.; Bucci, J.; Graham, P.; Li, Y. Cancer stem cells in prostate cancer radioresistance. Cancer Lett. 2019, 465, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Ewald, J.A.; Desotelle, J.A.; Wilding, G.; Jarrard, D.F. Therapy-Induced Senescence in Cancer. J. Natl. Cancer Inst. 2010, 102, 1536–1546. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Feng, W.; Li, H.; Gardner, D.; Luo, Y.; Wang, Y.; Liu, L.; Meng, A.; Sharpless, N.E.; Zhou, D. Total body irradiation causes long-term mouse BM injury via induction of HSC premature senescence in an Ink4a- and Arf-independent manner. Blood 2014, 123, 3105–3115. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; di Fagagna, F.D. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2020, 22, 75–95. [Google Scholar] [CrossRef]
- Mavrogonatou, E.; Pratsinis, H.; Kletsas, D. The role of senescence in cancer development. Semin. Cancer Biol. 2019, 62, 182–191. [Google Scholar] [CrossRef]
- Malaquin, N.; Vancayseele, A.; Gilbert, S.; Antenor-Habazac, L.; Olivier, M.-A.; Brahem, Z.A.A.; Saad, F.; Delouya, G.; Rodier, F. DNA Damage- But Not Enzalutamide-Induced Senescence in Prostate Cancer Promotes Senolytic Bcl-xL Inhibitor Sensitivity. Cells 2020, 9, 1593. [Google Scholar] [CrossRef]
- Angelopoulou, A.; Papaspyropoulos, A.; Papantonis, A.; Gorgoulis, V.G. CRISPR-Cas9-mediated induction of large chromosomal inversions in human bronchial epithelial cells. STAR Protoc. 2022, 3, 101257. [Google Scholar] [CrossRef]
- Zampetidis, C.P.; Galanos, P.; Angelopoulou, A.; Zhu, Y.; Polyzou, A.; Karamitros, T.; Kotsinas, A.; Lagopati, N.; Mourkioti, I.; Mirzazadeh, R.; et al. A recurrent chromosomal inversion suffices for driving escape from oncogene-induced senescence via subTAD reorganization. Mol. Cell 2021, 81, 4907–4923.e8. [Google Scholar] [CrossRef]
- Parisotto, M.; Grelet, E.; El Bizri, R.; Metzger, D. Senescence controls prostatic neoplasia driven by Pten loss. Mol. Cell. Oncol. 2018, 6, 1511205. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.L.; Tchkonia, T. Senolytic drugs: From discovery to translation. J. Intern. Med. 2020, 288, 518–536. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lankhorst, L.; Bernards, R. Exploiting senescence for the treatment of cancer. Nat. Cancer 2022, 22, 340–355. [Google Scholar] [CrossRef] [PubMed]
- Tzekaki, E.E.; Geromichalos, G.; Lavrentiadou, S.N.; Tsantarliotou, M.P.; Pantazaki, A.A.; Papaspyropoulos, A. Oleuropein is a natural inhibitor of PAI-1-mediated proliferation in human ER-/PR- breast cancer cells. Breast Cancer Res. Treat. 2021, 186, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Tzekaki, E.E.; Papaspyropoulos, A.; Tsolaki, M.; Lazarou, E.; Kozori, M.; Pantazaki, A. Restoration of BMI1 levels after the administration of early harvest extra virgin olive oil as a therapeutic strategy against Alzheimer’s disease. Exp. Gerontol. 2020, 144, 111178. [Google Scholar] [CrossRef]
- Evangelou, K.; Bartkova, J.; Kotsinas, A.; Pateras, I.S.; Liontos, M.; Velimezi, G.; Kosar, M.; Liloglou, T.; Trougakos, I.P.; Dyrskjøt, L.; et al. The DNA damage checkpoint precedes activation of ARF in response to escalating oncogenic stress during tumorigenesis. Cell Death Differ. 2013, 20, 1485–1497. [Google Scholar] [CrossRef]
- Tsaridou, S.; Velimezi, G.; Willenbrock, F.; Chatzifrangkeskou, M.; Elsayed, W.; Panagopoulos, A.; Karamitros, D.; Gorgoulis, V.; Lygerou, Z.; Roukos, V.; et al. 53BP1-mediated recruitment of RASSF1A to ribosomal DNA breaks promotes local ATM signaling. EMBO Rep. 2022, 23, e54483. [Google Scholar] [CrossRef]
- Bissell, M.J.; Hines, W.C. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat. Med. 2011, 17, 320–329. [Google Scholar] [CrossRef]
- Gunaydin, G. CAFs Interacting With TAMs in Tumor Microenvironment to Enhance Tumorigenesis and Immune Evasion. Front. Oncol. 2021, 11, 668349. [Google Scholar] [CrossRef]
- Zanconato, F.; Battilana, G.; Cordenonsi, M.; Piccolo, S. YAP/TAZ as therapeutic targets in cancer. Curr. Opin. Pharmacol. 2016, 29, 26–33. [Google Scholar] [CrossRef]
- Nowell, C.S.; Odermatt, P.D.; Azzolin, L.; Hoehnel, S.; Wagner, E.F.; Fantner, G.E.; Lutolf, M.P.; Barrandon, Y.; Piccolo, S.; Radtke, F. Chronic inflammation imposes aberrant cell fate in regenerating epithelia through mechanotransduction. Nat. Cell Biol. 2015, 18, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Panciera, T.; Azzolin, L.; Cordenonsi, M.; Piccolo, S. Mechanobiology of YAP and TAZ in physiology and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Elbadawy, M.; Abugomaa, A.; Yamawaki, H.; Usui, T.; Sasaki, K. Development of Prostate Cancer Organoid Culture Models in Basic Medicine and Translational Research. Cancers 2020, 12, 777. [Google Scholar] [CrossRef] [PubMed]
- Papaspyropoulos, A.; Tsolaki, M.; Foroglou, N.; Pantazaki, A.A. Modeling and Targeting Alzheimer’s Disease With Organoids. Front. Pharmacol. 2020, 11, 396. [Google Scholar] [CrossRef] [PubMed]
- Sachs, N.; Papaspyropoulos, A.; Zomer-van Ommen, D.D.; Heo, I.; Böttinger, L.; Klay, D.; Weeber, F.; Huelsz-Prince, G.; Iakobachvili, N.; Amatngalim, G.D.; et al. Long-term expanding human airway organoids for disease modeling. EMBO J. 2019, 38, e100300. [Google Scholar] [CrossRef]
- Wiener, D.J.; Basak, O.; Asra, P.; Boonekamp, K.E.; Kretzschmar, K.; Papaspyropoulos, A.; Clevers, H. Establishment and characterization of a canine keratinocyte organoid culture system. Vet. Dermatol. 2018, 29, 375-e126. [Google Scholar] [CrossRef] [PubMed]
- Puca, L.; Bareja, R.; Prandi, D.; Shaw, R.; Benelli, M.; Karthaus, W.R.; Hess, J.; Sigouros, M.; Donoghue, A.; Kossai, M.; et al. Patient derived organoids to model rare prostate cancer phenotypes. Nat. Commun. 2018, 9, 2404. [Google Scholar] [CrossRef]
- Gao, D.; Vela, I.; Sboner, A.; Iaquinta, P.J.; Karthaus, W.R.; Gopalan, A.; Dowling, C.; Wanjala, J.N.; Undvall, E.A.; Arora, V.K.; et al. Organoid Cultures Derived from Patients with Advanced Prostate Cancer. Cell 2014, 159, 176–187. [Google Scholar] [CrossRef]
- Vela, I.; Chen, Y. Prostate cancer organoids: A potential new tool for testing drug sensitivity. Expert Rev. Anticancer Ther. 2015, 15, 261–263. [Google Scholar] [CrossRef]
- Karkampouna, S.; La Manna, F.; Benjak, A.; Kiener, M.; De Menna, M.; Zoni, E.; Grosjean, J.; Klima, I.; Garofoli, A.; Bolis, M.; et al. Patient-derived xenografts and organoids model therapy response in prostate cancer. Nat. Commun. 2021, 12, 1117. [Google Scholar] [CrossRef]
- Drost, J.; Karthaus, W.R.; Gao, D.; Driehuis, E.; Sawyers, C.L.; Chen, Y.; Clevers, H. Organoid culture systems for prostate epithelial and cancer tissue. Nat. Protoc. 2016, 11, 347–358. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Drug | Target | Effect | Implication with DDR Pathway |
|---|---|---|---|
| Crizotinib | Hippo Pathway | Inhibition of ALK | ATM phosphorylation and activation in response to oxidative DNA damage [132] |
| FAK inhibitors | Hippo Pathway | Abrogation of YAP activity | Induction of senescence and activation of DNA damage pathways [134] |
| DNMT inhibitors | Hippo Pathway | Demethylation of RASSF1A promoter | Mahanine: Inactivation of AKT, proteasomal degradation of DNMT1 and DNMT3B, demethylation and restoration of RASSF1A normal expression [135] |
| Verteporfin | Hippo Pathway | Induction of YAP sequestration in cytoplasm and inhibition of YAP–TEAD interaction | Inhibition of DNA repair and apoptosis following IR [136] |
| Dasatinib | Hippo Pathway | Induction of YAP/TAZ proteasomal degradation | Induction of DNA damage and senescence [137] |
| Statins | Hippo Pathway | Induction of YAP sequestration in cytoplasm | Pitavastatin: Inhibition of DNA repair and induction of senescence in vitro and in vivo [138] |
| GSIs | Notch Pathway | Inhibition of NICD production | RO4929097 + Radiation: Inhibition of the DDR pathway [139] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mourkioti, I.; Angelopoulou, A.; Belogiannis, K.; Lagopati, N.; Potamianos, S.; Kyrodimos, E.; Gorgoulis, V.; Papaspyropoulos, A. Interplay of Developmental Hippo–Notch Signaling Pathways with the DNA Damage Response in Prostate Cancer. Cells 2022, 11, 2449. https://doi.org/10.3390/cells11152449
Mourkioti I, Angelopoulou A, Belogiannis K, Lagopati N, Potamianos S, Kyrodimos E, Gorgoulis V, Papaspyropoulos A. Interplay of Developmental Hippo–Notch Signaling Pathways with the DNA Damage Response in Prostate Cancer. Cells. 2022; 11(15):2449. https://doi.org/10.3390/cells11152449
Chicago/Turabian StyleMourkioti, Ioanna, Andriani Angelopoulou, Konstantinos Belogiannis, Nefeli Lagopati, Spyridon Potamianos, Efthymios Kyrodimos, Vassilis Gorgoulis, and Angelos Papaspyropoulos. 2022. "Interplay of Developmental Hippo–Notch Signaling Pathways with the DNA Damage Response in Prostate Cancer" Cells 11, no. 15: 2449. https://doi.org/10.3390/cells11152449
APA StyleMourkioti, I., Angelopoulou, A., Belogiannis, K., Lagopati, N., Potamianos, S., Kyrodimos, E., Gorgoulis, V., & Papaspyropoulos, A. (2022). Interplay of Developmental Hippo–Notch Signaling Pathways with the DNA Damage Response in Prostate Cancer. Cells, 11(15), 2449. https://doi.org/10.3390/cells11152449