
Uridine Diphosphate Glucuronosyl Transferase 2B28 (UGT2B28) Promotes Tumor Progression and Is Elevated in African American Prostate Cancer Patients

 , , , ,
, , , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Lines
2.3. Cell Culture
2.4. Lentiviral Transduction
2.5. Organoid Forming Assay
2.6. Perturbation of Androgen Signaling in the Organoid Assay
2.7. Immunohistochemistry Analysis
2.7.1. University of Michigan Prostate Cancer Tissue Microarrays
2.7.2. Baylor College of Medicine Prostate Cancer Tissue Microarrays
2.8. Immunofluorescence
2.9. Mouse Xenograft Studies
2.10. Quantitative Real-Time Polymerase Chain Reaction
2.11. Sample Preparation for Mass Spectrometry
2.12. Flux Measurement and Analysis of UGT Co-Substrates
2.13. Statistical Analysis
3. Results
3.1. UGT2B28 Expression Is Associated with Advanced Disease and Elevated in AA PCa
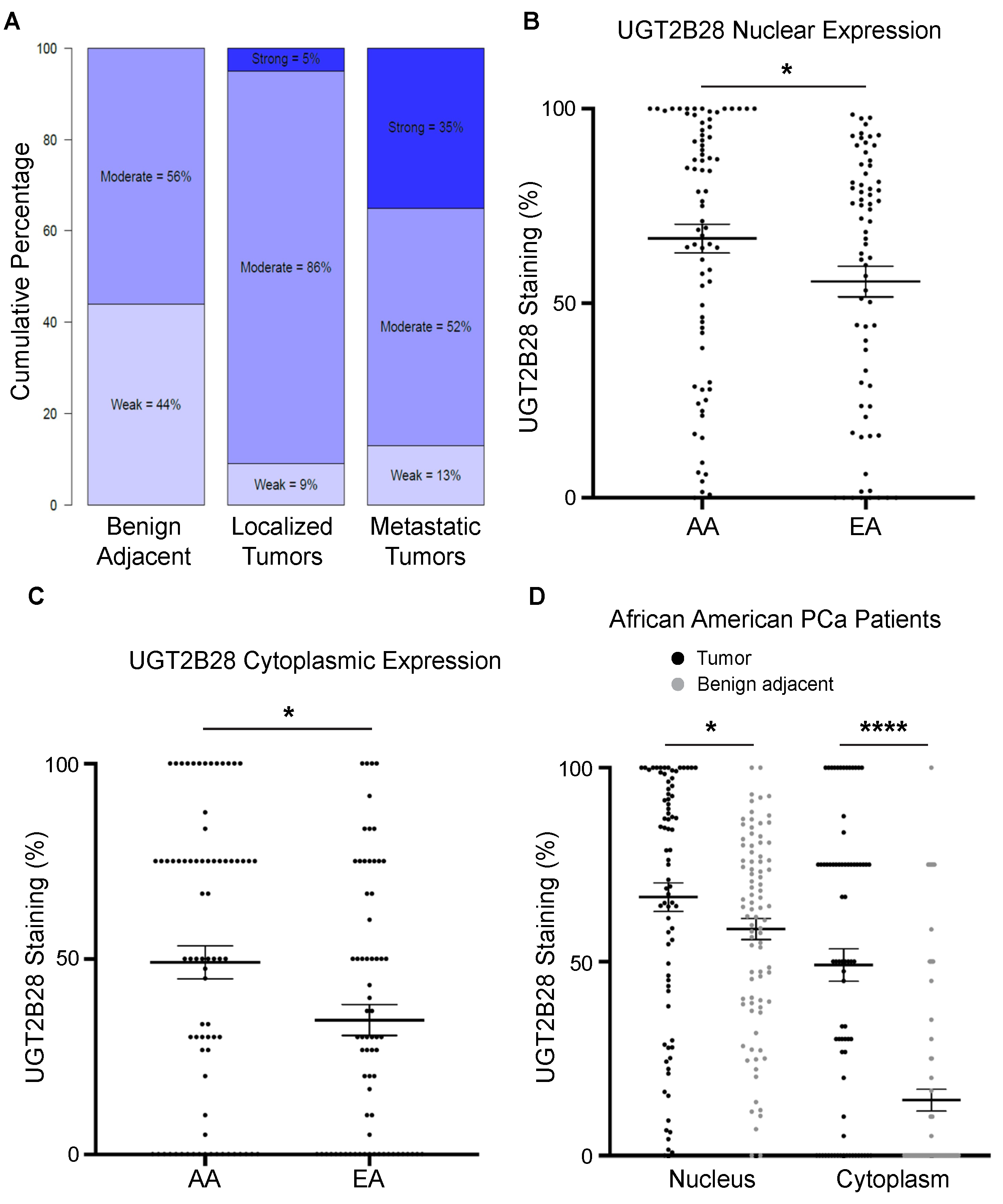
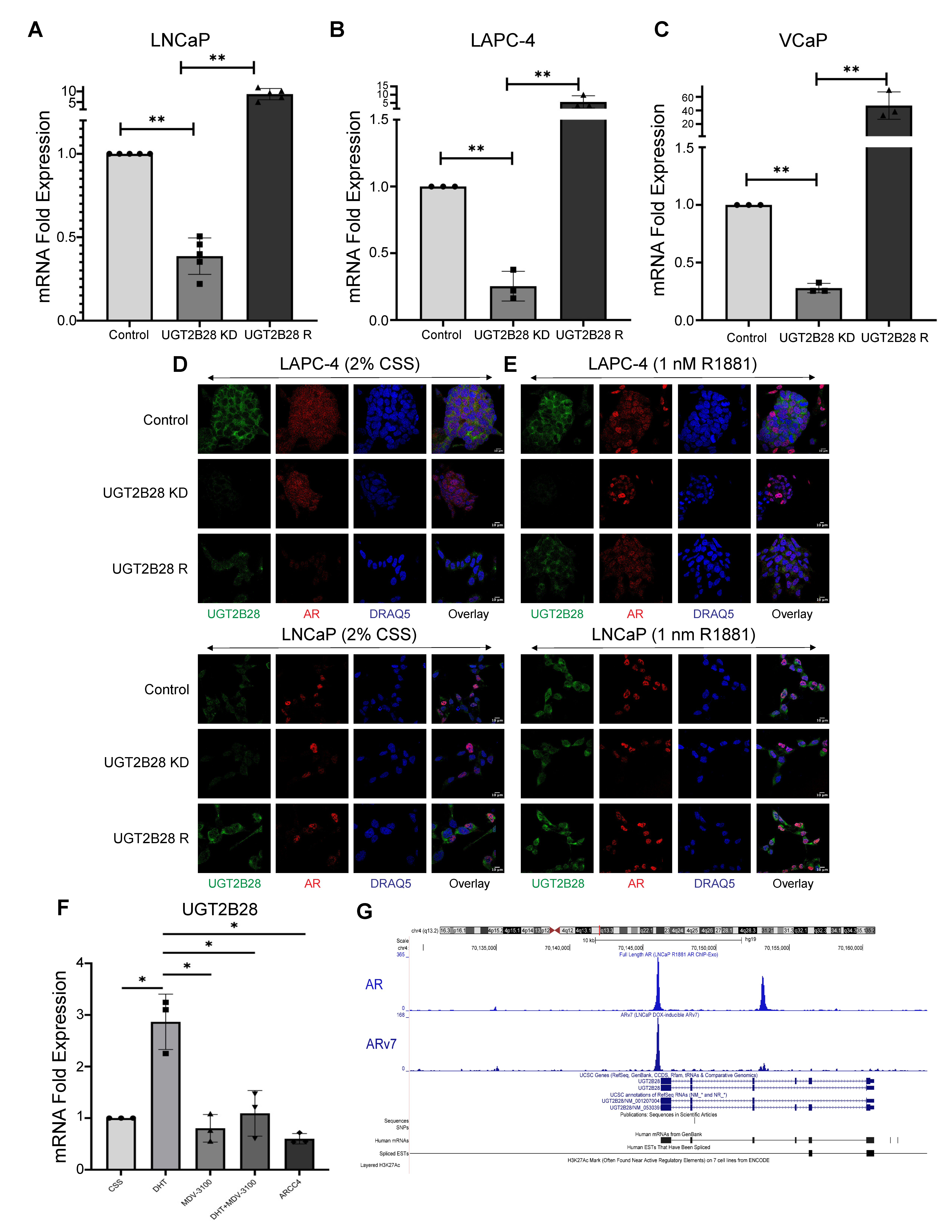
3.2. Androgen Receptor Regulates UGT2B28 Expression in PCa
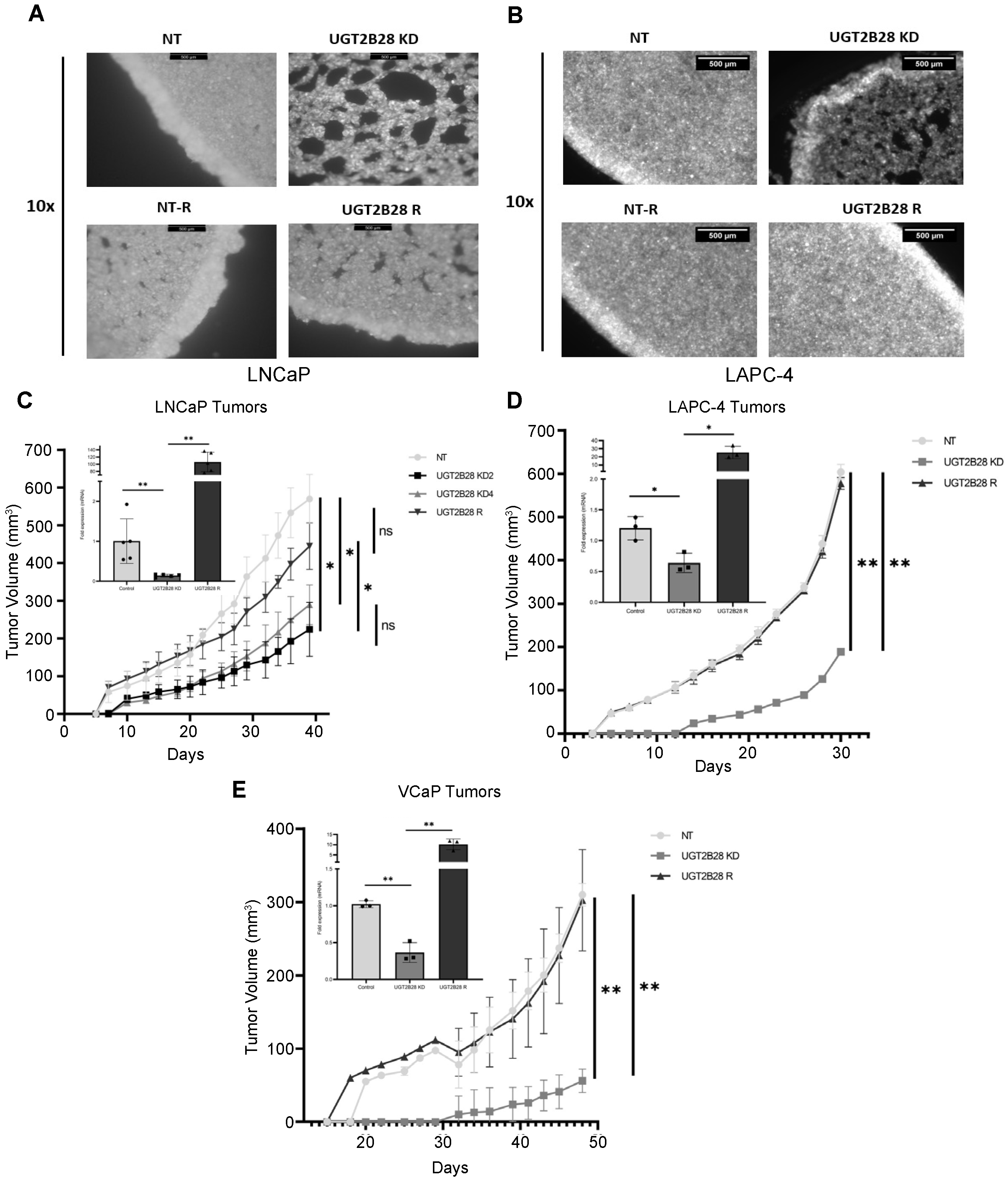
3.3. UGT2B28 Deficiency Impairs Organoid Formation in AD Cells In Vitro
3.4. UGT2B28 Deficiency Prolongs Tumor Take and Delays Tumor Growth In Vivo
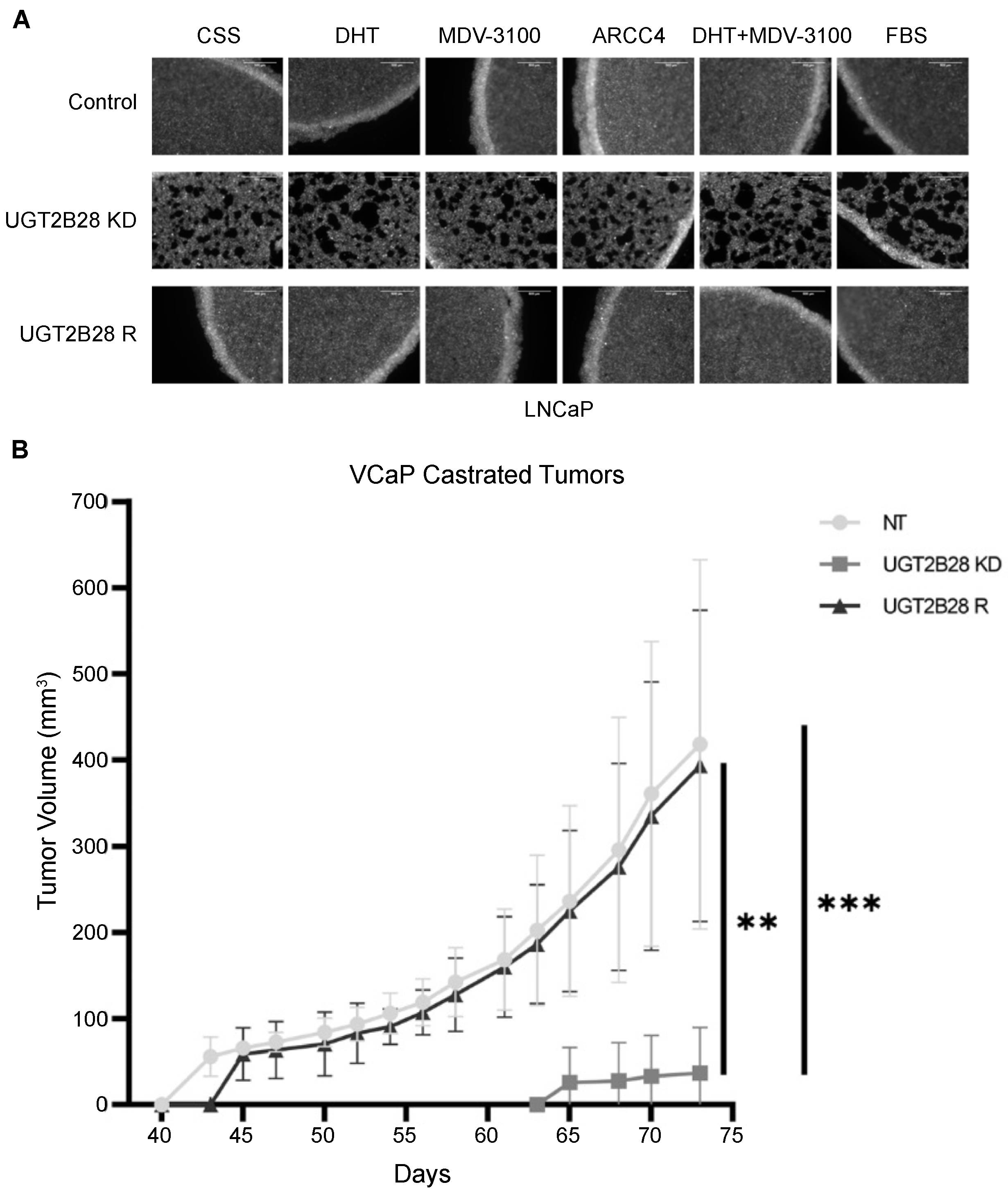
3.5. Effects of UGT2B28 on Organoid Formation and Tumor Growth Is Not Dependent on Ligand-Driven AR Signaling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Sreekumar, A.; Poisson, L.M.; Rajendiran, T.M.; Khan, A.P.; Cao, Q.; Yu, J.; Laxman, B.; Mehra, R.; Lonigro, R.J.; Li, Y.; et al. Metabolomic Profiles Delineate Potential Role for Sarcosine in Prostate Cancer Progression. Nature 2009, 457, 910–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Putluri, N.; Shojaie, A.; Vasu, V.T.; Nalluri, S.; Vareed, S.K.; Putluri, V.; Vivekanandan-Giri, A.; Byun, J.; Pennathur, S.; Sana, T.R.; et al. Metabolomic Profiling Reveals a Role for Androgen in Activating Amino Acid Metabolism and Methylation in Prostate Cancer Cells. PLoS ONE 2011, 6, E21417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaushik, A.K.; Vareed, S.K.; Basu, S.; Putluri, V.; Putluri, N.; Panzitt, K.; Brennan, C.A.; Chinnaiyan, A.M.; Vergara, I.A.; Erho, N.; et al. Metabolomic Profiling Identifies Biochemical Pathways Associated with Castration-Resistant Prostate Cancer. J. Proteome. Res. 2014, 13, 1088–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasgupta, S.; Putluri, N.; Long, W.; Zhang, B.; Wang, J.; Kaushik, A.K.; Arnold, J.M.; Bhowmik, S.K.; Stashi, E.; Brennan, C.A.; et al. Coactivator Src-2-Dependent Metabolic Reprogramming Mediates Prostate Cancer Survival and Metastasis. J. Clin. Invest. 2015, 125, 1174–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafi, A.A.; Putluri, V.; Arnold, J.M.; Tsouko, E.; Maity, S.; Roberts, J.M.; Coarfa, C.; Frigo, D.E.; Putluri, N.; Sreekumar, A.; et al. Differential Regulation of Metabolic Pathways by Androgen Receptor (Ar) and Its Constitutively Active Splice Variant, Ar-V7, in Prostate Cancer Cells. Oncotarget 2015, 6, 31997–32012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalogera, E.; Pistos, C.; Provatopoulou, X.; Athanaselis, S.; Spiliopoulou, C.; Gounaris, A. Androgen Glucuronides Analysis by Liquid Chromatography Tandem-Mass Spectrometry: Could It Raise New Perspectives in the Diagnostic Field of Hormone-Dependent Malignancies? J. Chromatogr. B. Analyt. Technol. Biomed. Life Sci. 2013, 940, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Paquet, S.; Fazli, L.; Grosse, L.; Verreault, M.; Tetu, B.; Rennie, P.S.; Belanger, A.; Barbier, O. Differential Expression of the Androgen-Conjugating Ugt2b15 and Ugt2b17 Enzymes in Prostate Tumor Cells During Cancer Progression. J. Clin. Endocrinol. Metab. 2012, 97, E428–E432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belledant, A.; Hovington, H.; Garcia, L.; Caron, P.; Brisson, H.; Villeneuve, L.; Simonyan, D.; Tetu, B.; Fradet, Y.; Lacombe, L.; et al. The Ugt2b28 Sex-Steroid Inactivation Pathway is a Regulator of Steroidogenesis and Modifies the Risk of Prostate Cancer Progression. Eur. Urol. 2016, 69, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Barron, D.A.; San Martin, R.; Chan, K.S.; Tran, L.L.; Yang, F.; Ressler, S.J.; Rowley, D.R. Runx1 is Essential for Mesenchymal Stem Cell Proliferation and Myofibroblast Differentiation. Proc. Natl. Acad. Sci. USA 2014, 111, 16389–16394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaushik, A.K.; Shojaie, A.; Panzitt, K.; Sonavane, R.; Venghatakrishnan, H.; Manikkam, M.; Zaslavsky, A.; Putluri, V.; Vasu, V.T.; Zhang, Y.; et al. Inhibition of the Hexosamine Biosynthetic Pathway Promotes Castration-Resistant Prostate Cancer. Nat. Commun. 2016, 7, 11612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Schwartzman, J.; Gibbs, A.; Lisac, R.; Kleinschmidt, R.; Wilmot, B.; Bottomly, D.; Coleman, I.; Nelson, P.; Mcweeney, S.; et al. Androgen Receptor Promotes Ligand-Independent Prostate Cancer Progression through C-Myc Upregulation. PLoS ONE 2013, 8, E63563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, J.M.; Gu, F.; Ambati, C.R.; Rasaily, U.; Ramirez-Pena, E.; Joseph, R.; Manikkam, M.; San Martin, R.; Charles, C.; Pan, Y.; et al. Udp-Glucose 6-Dehydrogenase Regulates Hyaluronic Acid Production And Promotes Breast Cancer Progression. Oncogene 2020, 39, 3089–3101. [Google Scholar] [CrossRef] [PubMed]
- Stern, R. Hyaluronidases in Cancer Biology. Semin. Cancer Biol. 2008, 18, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Wang, L.; Liang, X.; Liu, X.; Wu, H.; Liang, Z. High-Molecular-Weight Hyaluronan Produced By Activated Pancreatic Stellate Cells Promotes Pancreatic Cancer Cell Migration Via Paracrine Signaling. Biochem. Biophys Res. Commun. 2019, 515, 493–498. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ravindran, A.; Krieger, K.L.; Kaushik, A.K.; Hovington, H.; Mehdi, S.; Piyarathna, D.W.B.; Putluri, V.; Basil, P.; Rasaily, U.; Gu, F.; et al. Uridine Diphosphate Glucuronosyl Transferase 2B28 (UGT2B28) Promotes Tumor Progression and Is Elevated in African American Prostate Cancer Patients. Cells 2022, 11, 2329. https://doi.org/10.3390/cells11152329
Ravindran A, Krieger KL, Kaushik AK, Hovington H, Mehdi S, Piyarathna DWB, Putluri V, Basil P, Rasaily U, Gu F, et al. Uridine Diphosphate Glucuronosyl Transferase 2B28 (UGT2B28) Promotes Tumor Progression and Is Elevated in African American Prostate Cancer Patients. Cells. 2022; 11(15):2329. https://doi.org/10.3390/cells11152329
Chicago/Turabian StyleRavindran, Anindita, Kimiko L. Krieger, Akash K. Kaushik, Hélène Hovington, Sadia Mehdi, Danthasinghe Waduge Badrajee Piyarathna, Vasanta Putluri, Paul Basil, Uttam Rasaily, Franklin Gu, and et al. 2022. "Uridine Diphosphate Glucuronosyl Transferase 2B28 (UGT2B28) Promotes Tumor Progression and Is Elevated in African American Prostate Cancer Patients" Cells 11, no. 15: 2329. https://doi.org/10.3390/cells11152329
APA StyleRavindran, A., Krieger, K. L., Kaushik, A. K., Hovington, H., Mehdi, S., Piyarathna, D. W. B., Putluri, V., Basil, P., Rasaily, U., Gu, F., Dang, T., Choi, J. M., Sonavane, R., Jung, S. Y., Wang, L., Mehra, R., Weigel, N. L., Putluri, N., Rowley, D. R., ... Sreekumar, A. (2022). Uridine Diphosphate Glucuronosyl Transferase 2B28 (UGT2B28) Promotes Tumor Progression and Is Elevated in African American Prostate Cancer Patients. Cells, 11(15), 2329. https://doi.org/10.3390/cells11152329