
Anti-Ischemic Effects of PIK3IP1 Are Mediated through Its Interactions with the ETA-PI3Kγ-AKT Axis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Antibodies
2.2. Cell Culture and Treatment
2.3. MTT Assay
2.4. Western Blot Analysis
2.5. TUNEL Assay
2.6. Co-Immunoprecipitation
2.7. Quantitative Real-Time PCR (qRT-PCR)
2.8. Statistics
3. Results
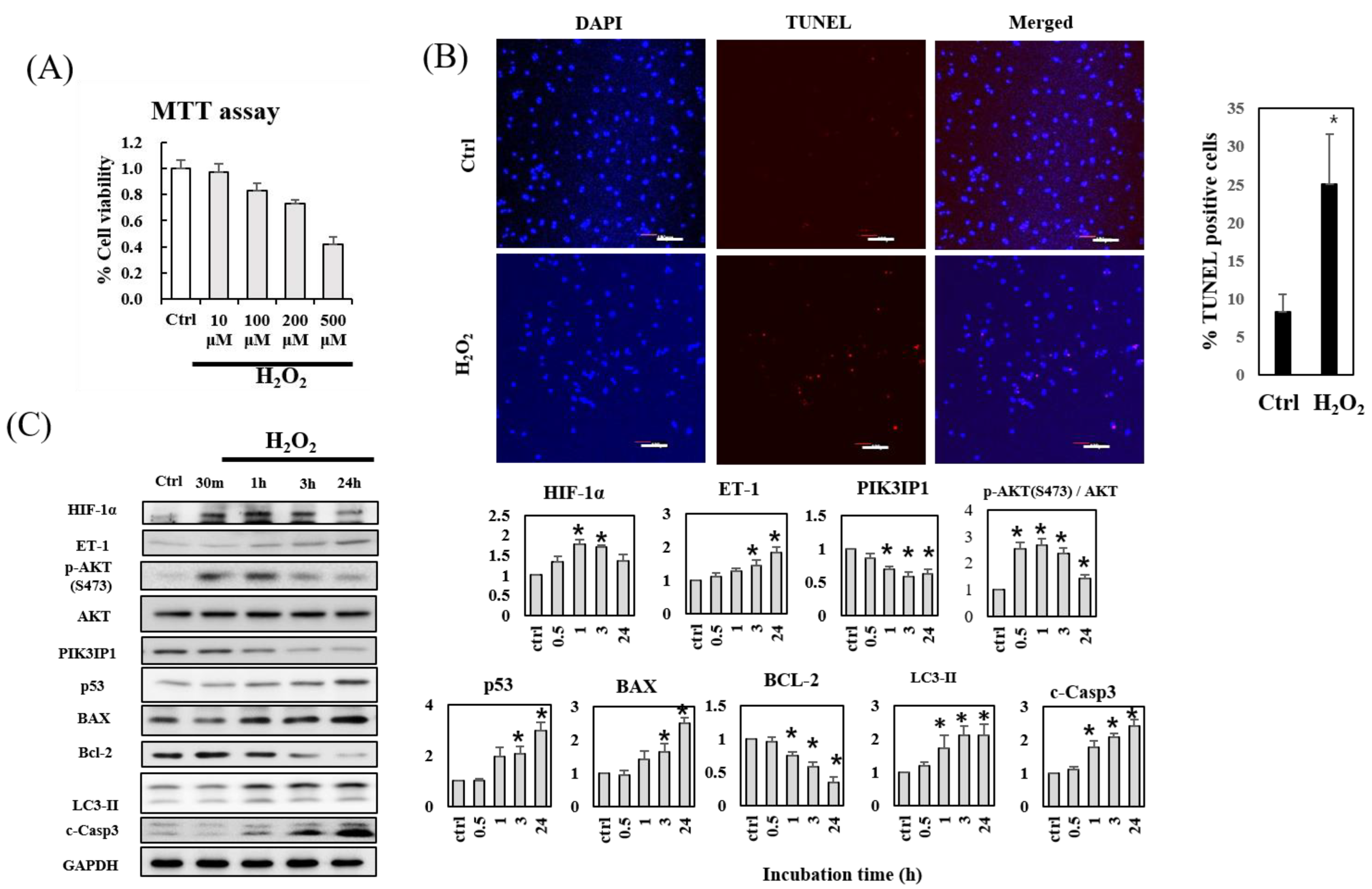
3.1. H2O2-Induced Apoptosis of H9c2 Cardiomyocytes Is Accompanied by Decreased PIK3IP1 and Bcl-2 Expression, but Increased HIF-1α, ET-1, p-AKT, BAX, and Cleaved Caspase-3 Expression
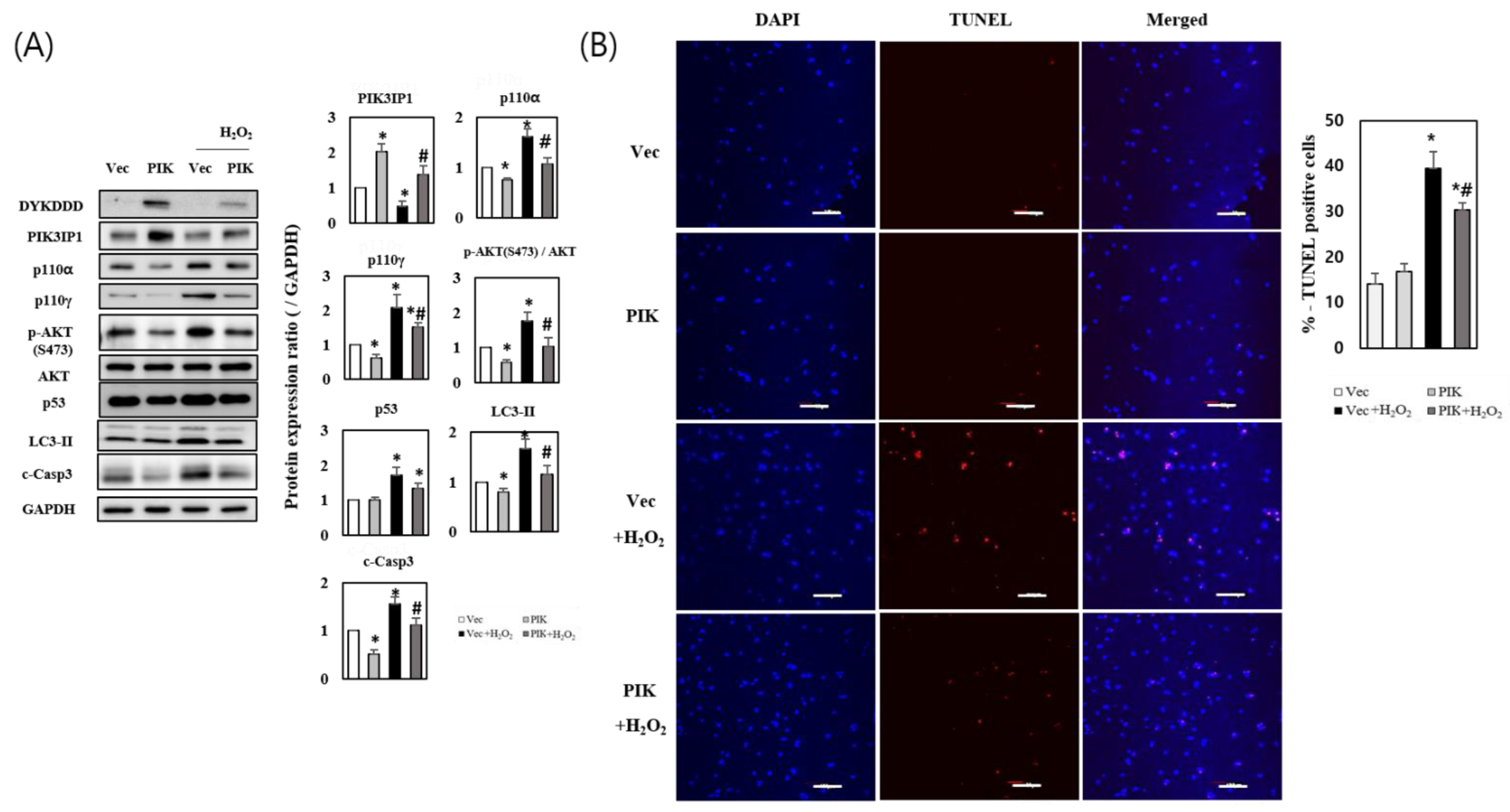
3.2. PIK3IP1 Overexpression Inhibits H2O2-Induced Cell Death via Downregulation of p110α, p110γ, p-AKT, and Cleaved Caspase-3 Expression
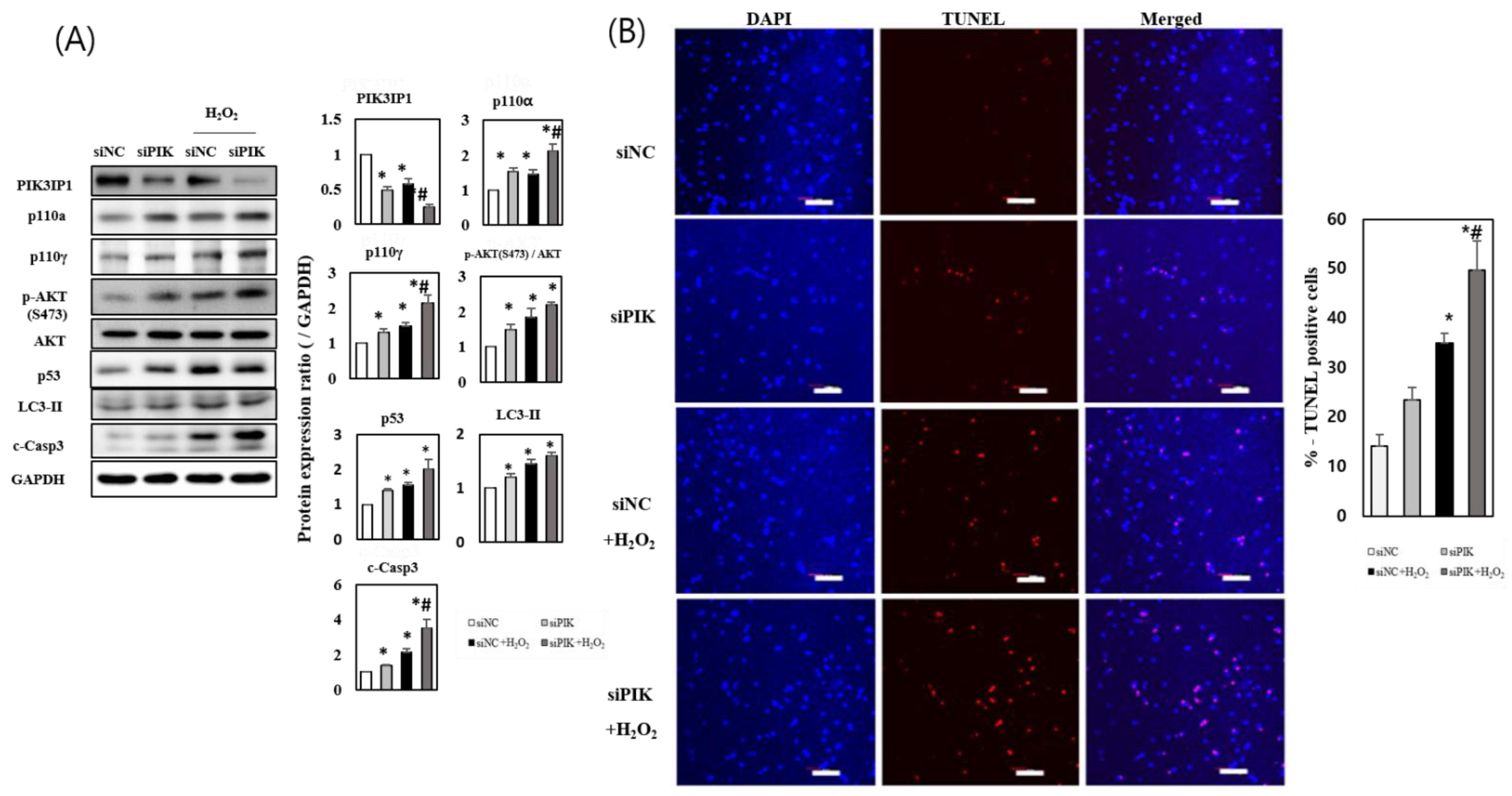
3.3. PIK3IP1 Knockdown Increased H2O2-Induced Cell Death via Upregulation of p110α, p110γ, p-AKT, and Cleaved Caspase-3 Expression
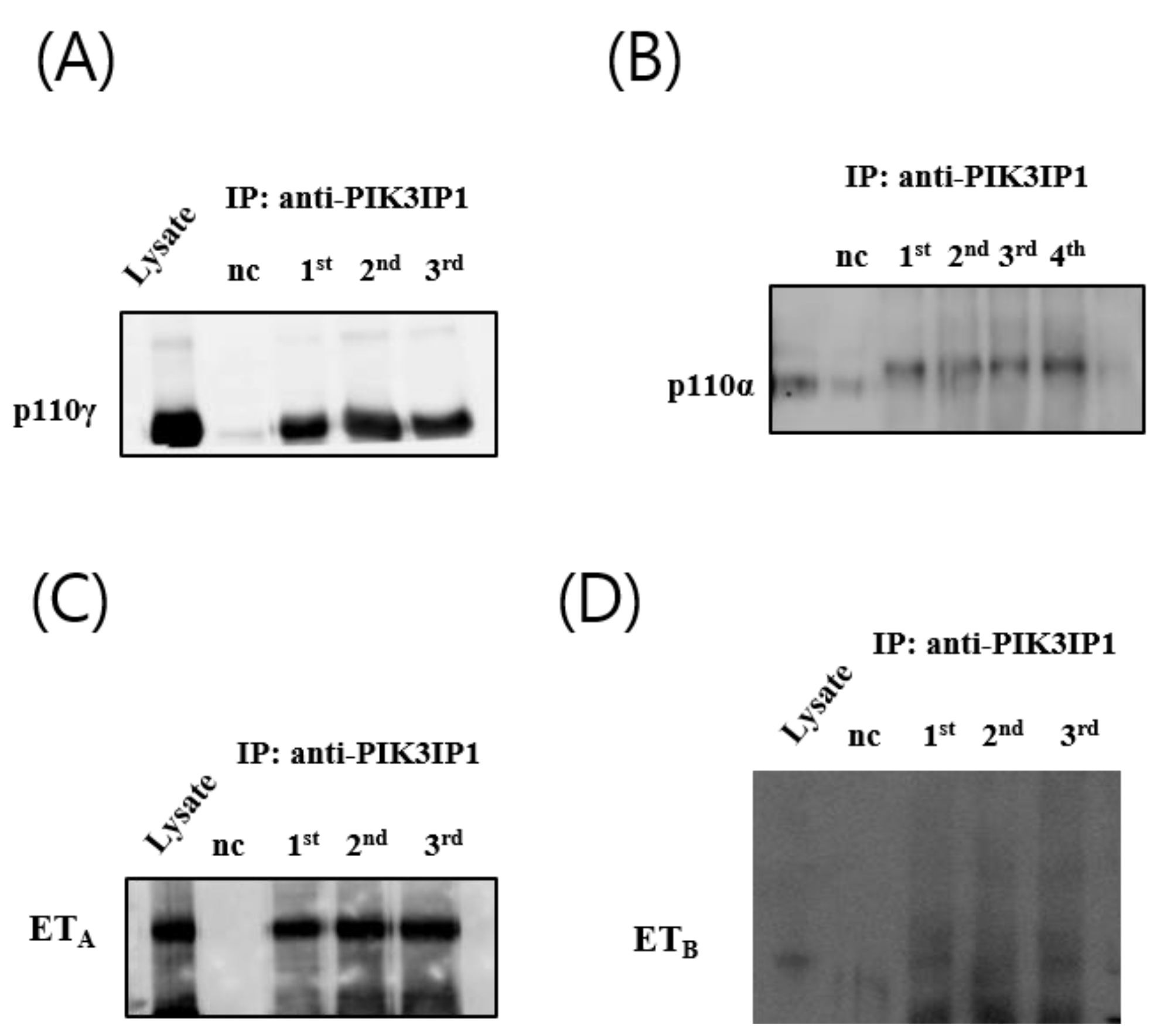
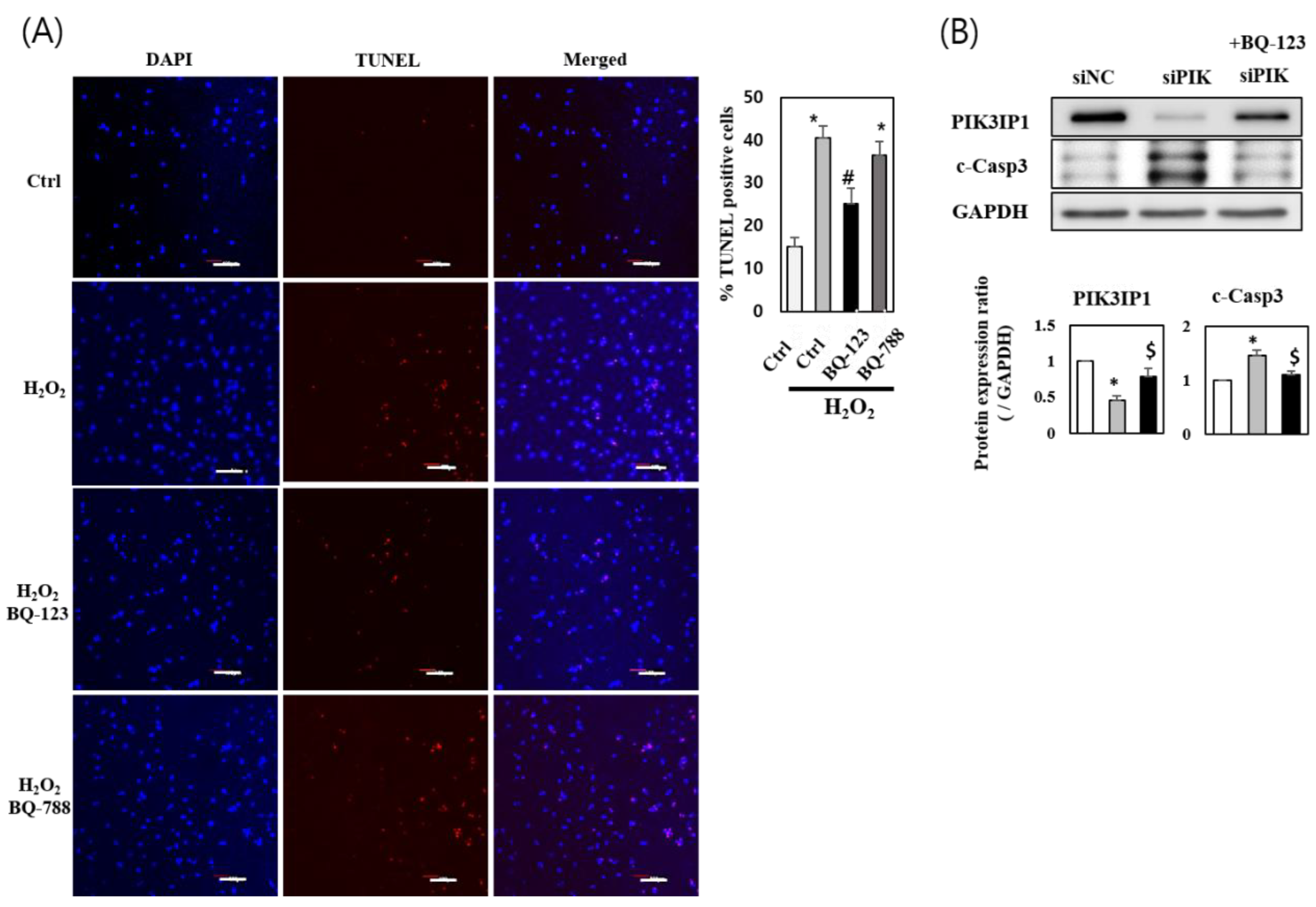
3.4. H2O2 Induced Cardiomyocyte Cell Death via ETA-PIK3IP1 Binding
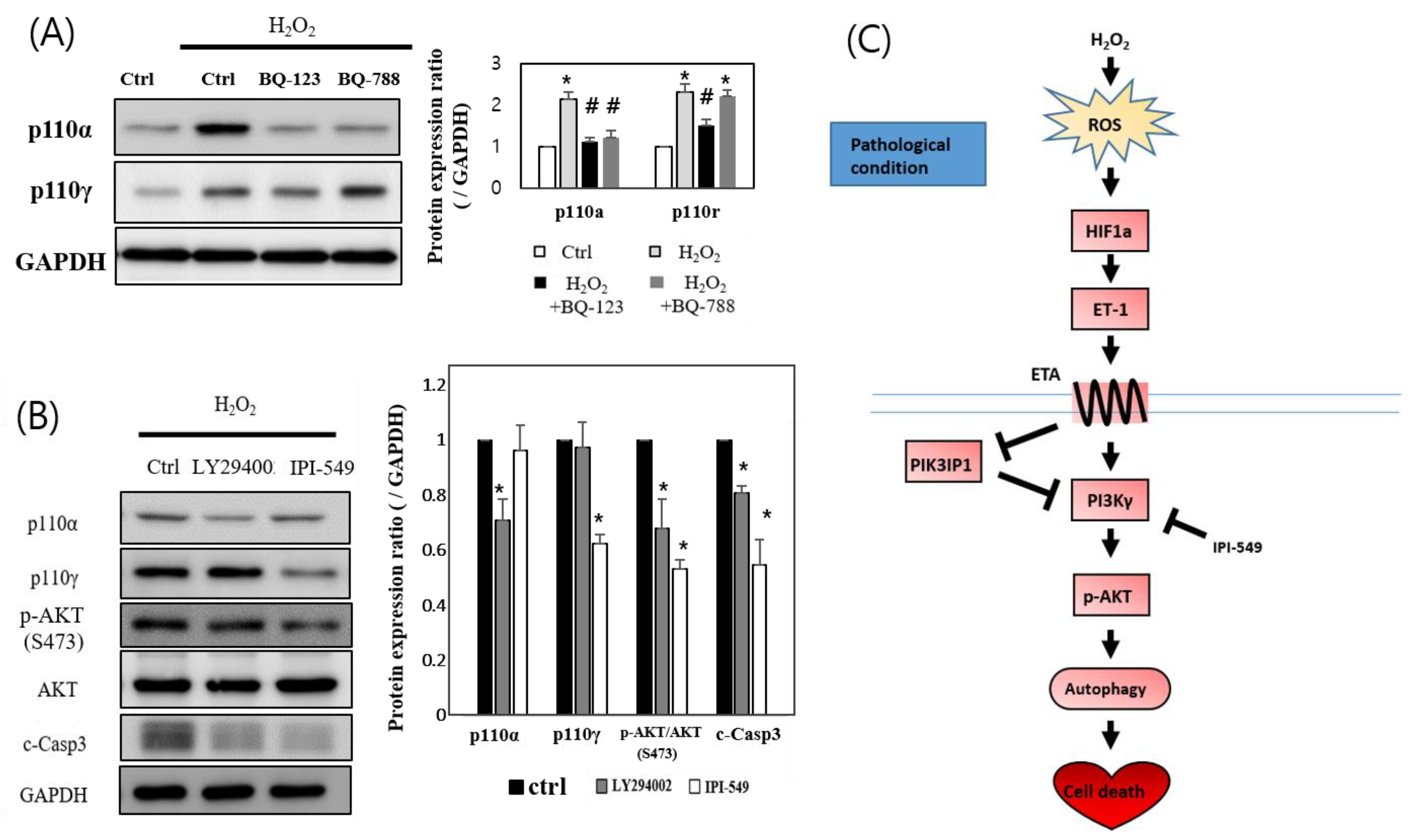
3.5. H2O2 Induces H9c2 Cell Death through the ETA-PIK3IP1-PI3Kγ Axis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef]
- Aoyagi, T.; Matsui, T. Phosphoinositide-3 kinase signaling in cardiac hypertrophy and heart failure. Curr. Pharm. Des. 2011, 17, 1818–1824. [Google Scholar] [CrossRef]
- Alessi, D.R.; James, S.R.; Downes, C.P.; Holmes, A.B.; Gaffney, P.R.; Reese, C.B.; Cohen, P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr. Biol. 1997, 7, 261–269. [Google Scholar] [CrossRef]
- Damilano, F.; Perino, A.; Hirsch, E. PI3K kinase and scaffold functions in heart. Ann. N. Y. Acad. Sci. 2010, 1188, 39–45. [Google Scholar] [CrossRef]
- Dorn, G.W., 2nd; Force, T. Protein kinase cascades in the regulation of cardiac hypertrophy. J. Clin. Investig. 2005, 115, 527–537. [Google Scholar] [CrossRef]
- Patrucco, E.; Notte, A.; Barberis, L.; Selvetella, G.; Maffei, A.; Brancaccio, M.; Marengo, S.; Russo, G.; Azzolino, O.; Rybalkin, S.D.; et al. PI3Kgamma modulates the cardiac response to chronic pressure overload by distinct kinase-dependent and -independent effects. Cell 2004, 118, 375–387. [Google Scholar] [CrossRef]
- Zhabyeyev, P.; McLean, B.; Patel, V.B.; Wang, W.; Ramprasath, T.; Oudit, G.Y. Dual loss of PI3Kalpha and PI3Kgamma signaling leads to an age-dependent cardiomyopathy. J. Mol. Cell Cardiol. 2014, 77, 155–159. [Google Scholar] [CrossRef]
- He, X.; Zhu, Z.; Johnson, C.; Stoops, J.; Eaker, A.E.; Bowen, W.; DeFrances, M.C. PIK3IP1, a negative regulator of PI3K, suppresses the development of hepatocellular carcinoma. Cancer Res. 2008, 68, 5591–5598. [Google Scholar] [CrossRef]
- Song, H.K.; Kim, J.; Lee, J.S.; Nho, K.J.; Jeong, H.C.; Kim, J.; Ahn, Y.; Park, W.J.; Kim, D.H. Pik3ip1 modulates cardiac hypertrophy by inhibiting PI3K pathway. PLoS ONE 2015, 10, e0122251. [Google Scholar] [CrossRef]
- Beghetti, M.; Black, S.M.; Fineman, J.R. Endothelin-1 in congenital heart disease. Pediatr Res. 2005, 57, 16R–20R. [Google Scholar] [CrossRef]
- Klainguti, M.; Aigner, S.; Kilo, J.; Eppenberger, H.M.; Mandinova, A.; Aebi, U.; Schaub, M.C.; Shaw, S.G.; Luscher, T.F.; Atar, D. Lack of nuclear apoptosis in cardiomyocytes and increased endothelin-1 levels in a rat heart model of myocardial stunning. Basic Res. Cardiol. 2000, 95, 308–315. [Google Scholar] [CrossRef]
- Ebersole, J.L.; Novak, M.J.; Orraca, L.; Martinez-Gonzalez, J.; Kirakodu, S.; Chen, K.C.; Stromberg, A.; Gonzalez, O.A. Hypoxia-inducible transcription factors, HIF1A and HIF2A, increase in aging mucosal tissues. Immunology 2018, 154, 452–464. [Google Scholar] [CrossRef]
- Pollock, D.M. Endothelin, angiotensin, and oxidative stress in hypertension. Hypertension 2005, 45, 477–480. [Google Scholar] [CrossRef]
- Weidemann, A.; Johnson, R.S. Biology of HIF-1alpha. Cell Death Differ. 2008, 15, 621–627. [Google Scholar] [CrossRef]
- Stewart, D.J.; Kubac, G.; Costello, K.B.; Cernacek, P. Increased plasma endothelin-1 in the early hours of acute myocardial infarction. J. Am. Coll. Cardiol. 1991, 18, 38–43. [Google Scholar] [CrossRef]
- Lechleitner, P.; Genser, N.; Mair, J.; Maier, J.; Artner-Dworzak, E.; Dienstl, F.; Puschendorf, B. Plasma immunoreactive endothelin in the acute and subacute phases of myocardial infarction in patients undergoing fibrinolysis. Clin. Chem. 1993, 39, 955–959. [Google Scholar] [CrossRef]
- Pawlus, M.R.; Wang, L.; Hu, C.J. STAT3 and HIF1alpha cooperatively activate HIF1 target genes in MDA-MB-231 and RCC4 cells. Oncogene 2014, 33, 1670–1679. [Google Scholar] [CrossRef]
- Hori, M.; Nishida, K. Oxidative stress and left ventricular remodelling after myocardial infarction. Cardiovasc. Res. 2009, 81, 457–464. [Google Scholar] [CrossRef]
- Watkins, S.J.; Borthwick, G.M.; Arthur, H.M. The H9C2 cell line and primary neonatal cardiomyocyte cells show similar hypertrophic responses in vitro. In Vitro Cell Dev. Biol. Anim. 2011, 47, 125–131. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Javadov, S.; Sickinger, S.; Frotschnig, S.; Grimm, M. H9c2 and HL-1 cells demonstrate distinct features of energy metabolism, mitochondrial function and sensitivity to hypoxia-reoxygenation. Biochim. Biophys. Acta 2015, 1853, 276–284. [Google Scholar] [CrossRef]
- Matsui, Y.; Takagi, H.; Qu, X.; Abdellatif, M.; Sakoda, H.; Asano, T.; Levine, B.; Sadoshima, J. Distinct roles of autophagy in the heart during ischemia and reperfusion: Roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ. Res. 2007, 100, 914–922. [Google Scholar] [CrossRef]
- Wang, E.Y.; Gang, H.; Aviv, Y.; Dhingra, R.; Margulets, V.; Kirshenbaum, L.A. p53 mediates autophagy and cell death by a mechanism contingent on Bnip3. Hypertension 2013, 62, 70–77. [Google Scholar] [CrossRef]
- Siragusa, M.; Katare, R.; Meloni, M.; Damilano, F.; Hirsch, E.; Emanueli, C.; Madeddu, P. Involvement of phosphoinositide 3-kinase gamma in angiogenesis and healing of experimental myocardial infarction in mice. Circ. Res. 2010, 106, 757–768. [Google Scholar] [CrossRef]
- Doukas, J.; Wrasidlo, W.; Noronha, G.; Dneprovskaia, E.; Fine, R.; Weis, S.; Hood, J.; Demaria, A.; Soll, R.; Cheresh, D. Phosphoinositide 3-kinase gamma/delta inhibition limits infarct size after myocardial ischemia/reperfusion injury. Proc. Natl. Acad. Sci. USA 2006, 103, 19866–19871. [Google Scholar] [CrossRef]
- McMullen, J.R.; Amirahmadi, F.; Woodcock, E.A.; Schinke-Braun, M.; Bouwman, R.D.; Hewitt, K.A.; Mollica, J.P.; Zhang, L.; Zhang, Y.; Shioi, T.; et al. Protective effects of exercise and phosphoinositide 3-kinase(p110alpha) signaling in dilated and hypertrophic cardiomyopathy. Proc. Natl. Acad. Sci. USA 2007, 104, 612–617. [Google Scholar] [CrossRef]
- Ghigo, A.; Morello, F.; Perino, A.; Hirsch, E. Therapeutic applications of PI3K inhibitors in cardiovascular diseases. Future Med. Chem. 2013, 5, 479–492. [Google Scholar] [CrossRef]
- Perino, A.; Ghigo, A.; Ferrero, E.; Morello, F.; Santulli, G.; Baillie, G.S.; Damilano, F.; Dunlop, A.J.; Pawson, C.; Walser, R.; et al. Integrating cardiac PIP3 and cAMP signaling through a PKA anchoring function of p110gamma. Mol. Cell 2011, 42, 84–95. [Google Scholar] [CrossRef]
- Haubner, B.J.; Neely, G.G.; Voelkl, J.G.; Damilano, F.; Kuba, K.; Imai, Y.; Komnenovic, V.; Mayr, A.; Pachinger, O.; Hirsch, E.; et al. PI3Kgamma protects from myocardial ischemia and reperfusion injury through a kinase-independent pathway. PLoS ONE 2010, 5, e9350. [Google Scholar] [CrossRef]
- Kaushal, G.P.; Liu, L.; Kaushal, V.; Hong, X.; Melnyk, O.; Seth, R.; Safirstein, R.; Shah, S.V. Regulation of caspase-3 and -9 activation in oxidant stress to RTE by forkhead transcription factors, Bcl-2 proteins, and MAP kinases. Am. J. Physiol. Renal. Physiol. 2004, 287, F1258–F1268. [Google Scholar] [CrossRef]
- Li, D.; Ni, S.; Miao, K.S.; Zhuang, C. PI3K/Akt and caspase pathways mediate oxidative stress-induced chondrocyte apoptosis. Cell Stress Chaperones 2019, 24, 195–202. [Google Scholar] [CrossRef]
- Hunter, J.C.; Zeidan, A.; Javadov, S.; Kilic, A.; Rajapurohitam, V.; Karmazyn, M. Nitric oxide inhibits endothelin-1-induced neonatal cardiomyocyte hypertrophy via a RhoA-ROCK-dependent pathway. J. Mol. Cell Cardiol. 2009, 47, 810–818. [Google Scholar] [CrossRef]
- Liu, V.W.; Huang, P.L. Cardiovascular roles of nitric oxide: A review of insights from nitric oxide synthase gene disrupted mice. Cardiovasc. Res. 2008, 77, 19–29. [Google Scholar] [CrossRef]
- Leung, E.L.; Wong, J.C.; Johlfs, M.G.; Tsang, B.K.; Fiscus, R.R. Protein kinase G type Ialpha activity in human ovarian cancer cells significantly contributes to enhanced Src activation and DNA synthesis/cell proliferation. Mol. Cancer Res. 2010, 8, 578–591. [Google Scholar] [CrossRef]
- Wong, J.C.; Fiscus, R.R. Protein kinase G activity prevents pathological-level nitric oxide-induced apoptosis and promotes DNA synthesis/cell proliferation in vascular smooth muscle cells. Cardiovasc. Pathol. 2010, 19, e221–e231. [Google Scholar] [CrossRef]
- Chen, L.; Daum, G.; Chitaley, K.; Coats, S.A.; Bowen-Pope, D.F.; Eigenthaler, M.; Thumati, N.R.; Walter, U.; Clowes, A.W. Vasodilator-stimulated phosphoprotein regulates proliferation and growth inhibition by nitric oxide in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1403–1408. [Google Scholar] [CrossRef]
- Zhu, Z.; He, X.; Johnson, C.; Stoops, J.; Eaker, A.E.; Stoffer, D.S.; Bell, A.; Zarnegar, R.; DeFrances, M.C. PI3K is negatively regulated by PIK3IP1, a novel p110 interacting protein. Biochem. Biophys. Res. Commun. 2007, 358, 66–72. [Google Scholar] [CrossRef]
- Uche, U.U.; Piccirillo, A.R.; Kataoka, S.; Grebinoski, S.J.; D'Cruz, L.M.; Kane, L.P. PIK3IP1/TrIP restricts activation of T cells through inhibition of PI3K/Akt. J. Exp. Med. 2018, 215, 3165–3179. [Google Scholar] [CrossRef]
- DeFrances, M.C.; Debelius, D.R.; Cheng, J.; Kane, L.P. Inhibition of T-cell activation by PIK3IP1. Eur. J. Immunol. 2012, 42, 2754–2759. [Google Scholar] [CrossRef]
- Shin, S.Y.; Kim, T.; Lee, H.S.; Kang, J.H.; Lee, J.Y.; Cho, K.H.; Kim, D.H. The switching role of beta-adrenergic receptor signalling in cell survival or death decision of cardiomyocytes. Nat. Commun. 2014, 5, 5777. [Google Scholar] [CrossRef]
- Jeong, D.; Cha, H.; Kim, E.; Kang, M.; Yang, D.K.; Kim, J.M.; Yoon, P.O.; Oh, J.G.; Bernecker, O.Y.; Sakata, S.; et al. PICOT inhibits cardiac hypertrophy and enhances ventricular function and cardiomyocyte contractility. Circ. Res. 2006, 99, 307–314. [Google Scholar] [CrossRef]
- Rehsia, N.S.; Dhalla, N.S. Potential of endothelin-1 and vasopressin antagonists for the treatment of congestive heart failure. Heart Fail. Rev. 2010, 15, 85–101. [Google Scholar] [CrossRef] [PubMed]
- Baltogiannis, G.G.; Tsalikakis, D.G.; Mitsi, A.C.; Hatzistergos, K.E.; Elaiopoulos, D.; Fotiadis, D.I.; Kyriakides, Z.S.; Kolettis, T.M. Endothelin receptor--a blockade decreases ventricular arrhythmias after myocardial infarction in rats. Cardiovasc. Res. 2005, 67, 647–654. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ozdemir, R.; Parlakpinar, H.; Polat, A.; Colak, C.; Ermis, N.; Acet, A. Selective endothelin a (ETA) receptor antagonist (BQ-123) reduces both myocardial infarct size and oxidant injury. Toxicology 2006, 219, 142–149. [Google Scholar] [CrossRef]
- Zhao, T.X.; Kostapanos, M.; Griffiths, C.; Arbon, E.L.; Hubsch, A.; Kaloyirou, F.; Helmy, J.; Hoole, S.P.; Rudd, J.H.F.; Wood, G.; et al. Low-dose interleukin-2 in patients with stable ischaemic heart disease and acute coronary syndromes (LILACS): Protocol and study rationale for a randomised, double-blind, placebo-controlled, phase I/II clinical trial. BMJ Open 2018, 8, e022452. [Google Scholar] [CrossRef] [PubMed]
- Nicolazzi, M.A.; Carnicelli, A.; Fuorlo, M.; Scaldaferri, A.; Masetti, R.; Landolfi, R.; Favuzzi, A.M.R. Anthracycline and trastuzumab-induced cardiotoxicity in breast cancer. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 2175–2185. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Wang, S.; Kang, W.; Chu, Q.; Liu, Z.; Sun, L.; Ji, Y.; Esteban, C.R.; Yao, Y.; Belmonte, J.C.I.; et al. FOXO3-engineered human mesenchymal progenitor cells efficiently promote cardiac repair after myocardial infarction. Protein Cell 2021, 12, 145–151. [Google Scholar] [CrossRef]
- Yan, P.; Li, Q.; Wang, L.; Lu, P.; Suzuki, K.; Liu, Z.; Lei, J.; Li, W.; He, X.; Wang, S.; et al. FOXO3-Engineered Human ESC-Derived Vascular Cells Promote Vascular Protection and Regeneration. Cell Stem Cell 2019, 24, 447–461.e8. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, J.H.; Nho, K.J.; Lee, J.Y.; Yoo, Y.J.; Park, W.J.; Cho, C.; Kim, D.H. Anti-Ischemic Effects of PIK3IP1 Are Mediated through Its Interactions with the ETA-PI3Kγ-AKT Axis. Cells 2022, 11, 2162. https://doi.org/10.3390/cells11142162
Park JH, Nho KJ, Lee JY, Yoo YJ, Park WJ, Cho C, Kim DH. Anti-Ischemic Effects of PIK3IP1 Are Mediated through Its Interactions with the ETA-PI3Kγ-AKT Axis. Cells. 2022; 11(14):2162. https://doi.org/10.3390/cells11142162
Chicago/Turabian StylePark, Jei Hyoung, Kyoung Jin Nho, Ji Young Lee, Yung Joon Yoo, Woo Jin Park, Chunghee Cho, and Do Han Kim. 2022. "Anti-Ischemic Effects of PIK3IP1 Are Mediated through Its Interactions with the ETA-PI3Kγ-AKT Axis" Cells 11, no. 14: 2162. https://doi.org/10.3390/cells11142162
APA StylePark, J. H., Nho, K. J., Lee, J. Y., Yoo, Y. J., Park, W. J., Cho, C., & Kim, D. H. (2022). Anti-Ischemic Effects of PIK3IP1 Are Mediated through Its Interactions with the ETA-PI3Kγ-AKT Axis. Cells, 11(14), 2162. https://doi.org/10.3390/cells11142162