
A Novel Cre/lox71-Based System for Inducible Expression of Recombinant Proteins and Genome Editing

 ,
,  , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
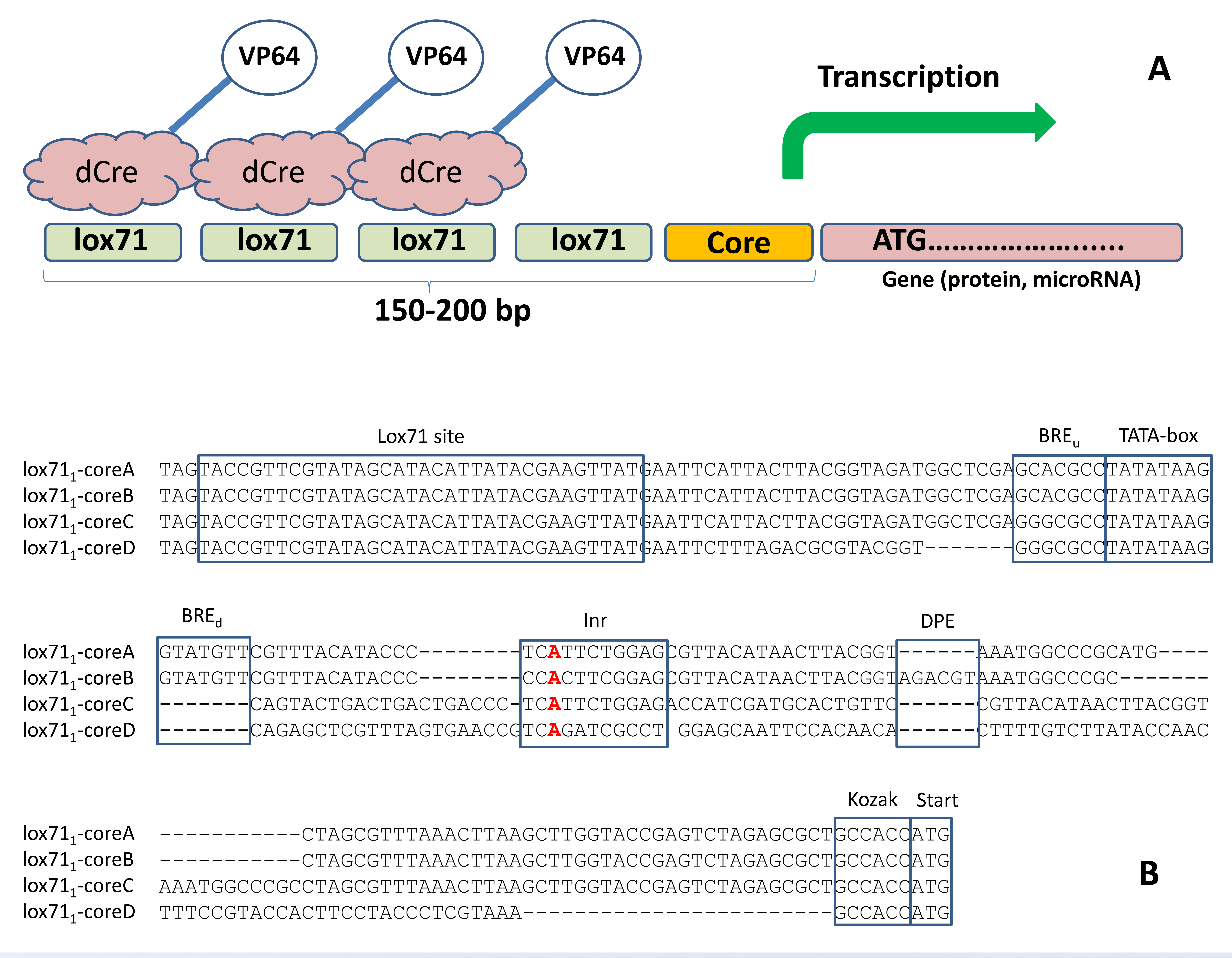
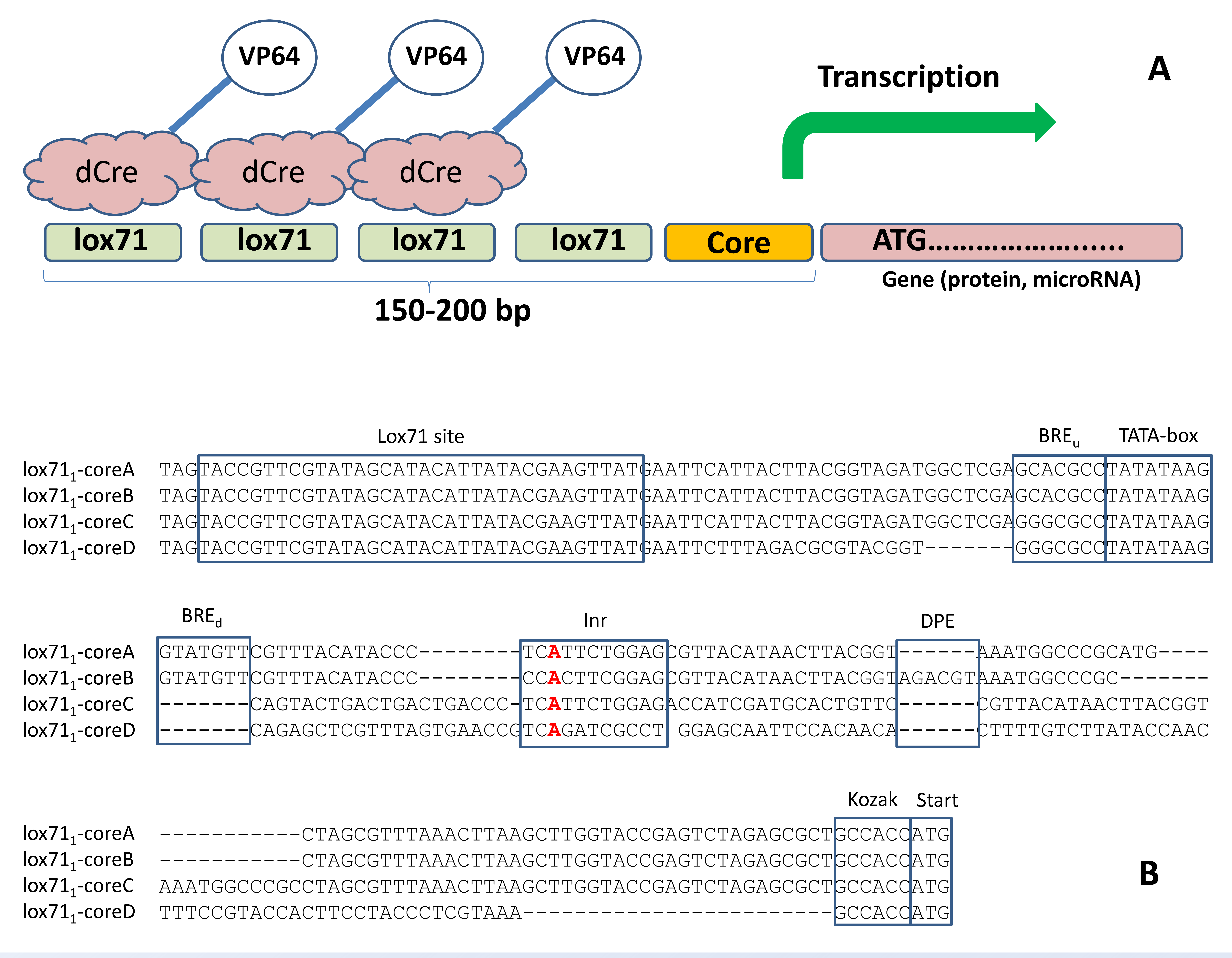
2.1. Genetic Constructs Encoding Cre-VP64 Transactivator and lox71-Based Promote
2.2. Cell Culture and Transfection
2.3. Inducible Transgene Expression Using Cre-VP64/lox71 System
2.4. Inducible Genome Editing Using Cre-VP64/lox71 System
2.5. Statistical Analysis
3. Results
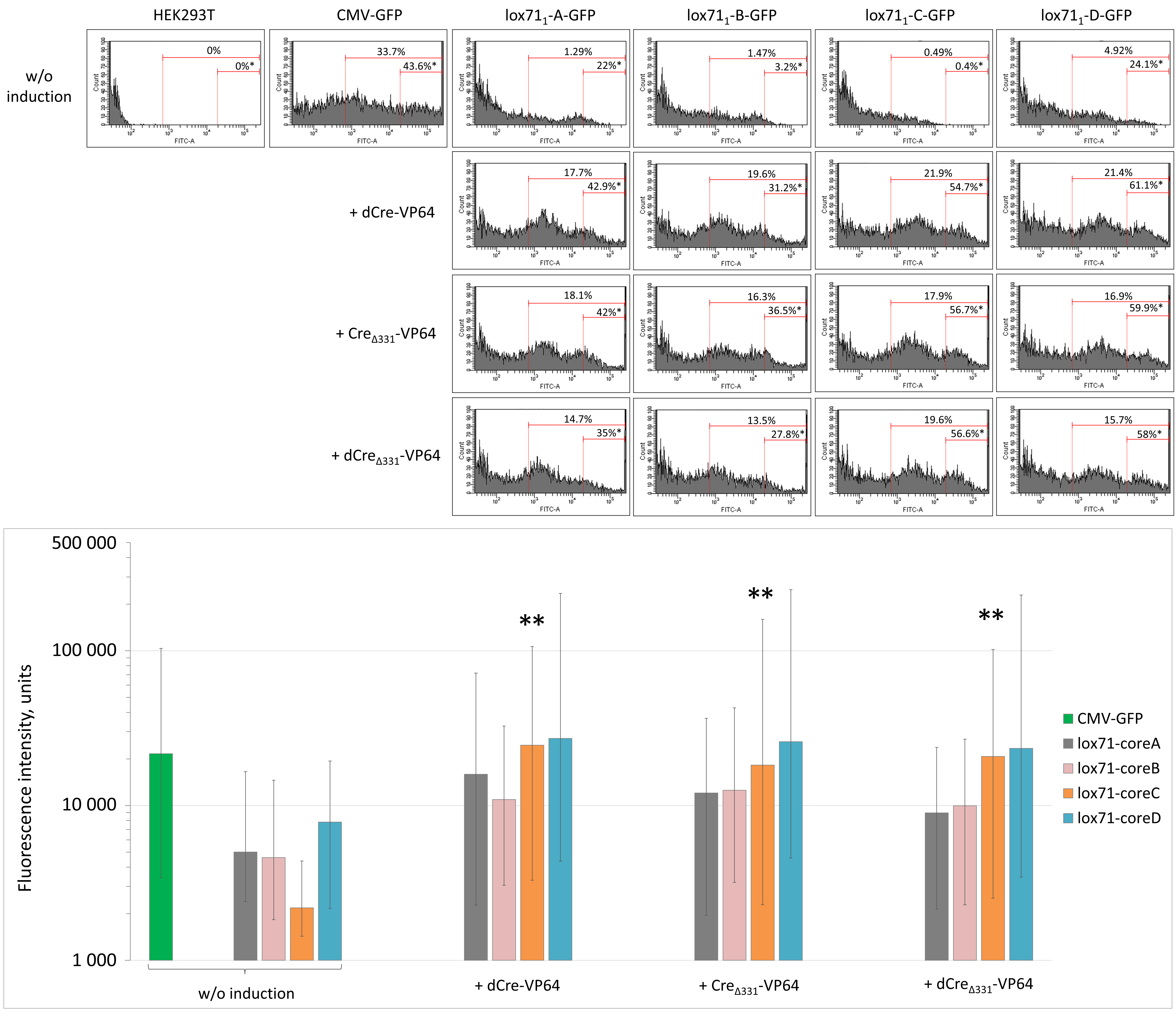
3.1. Characterization of Cre-VP64/lox71 System
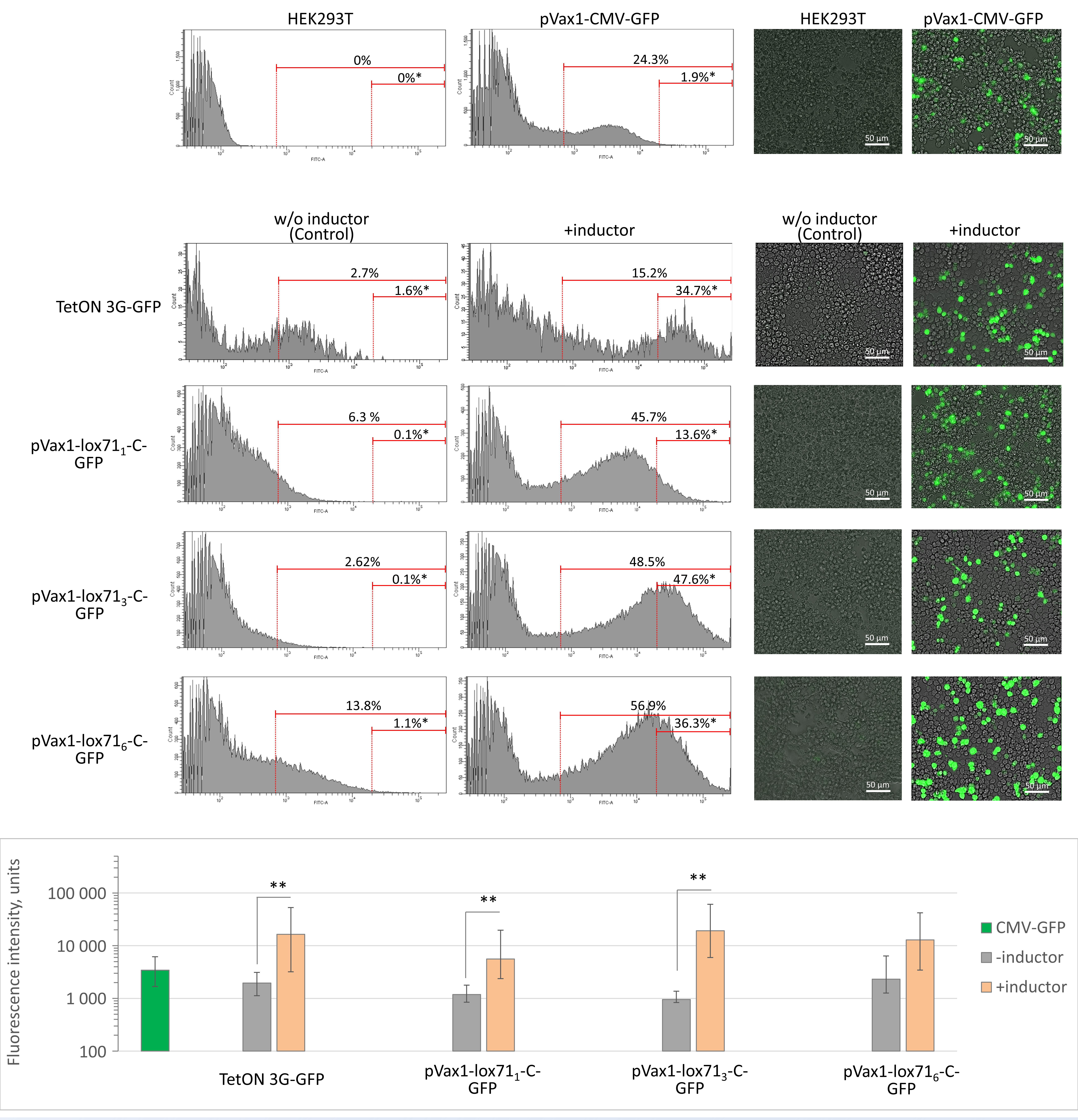
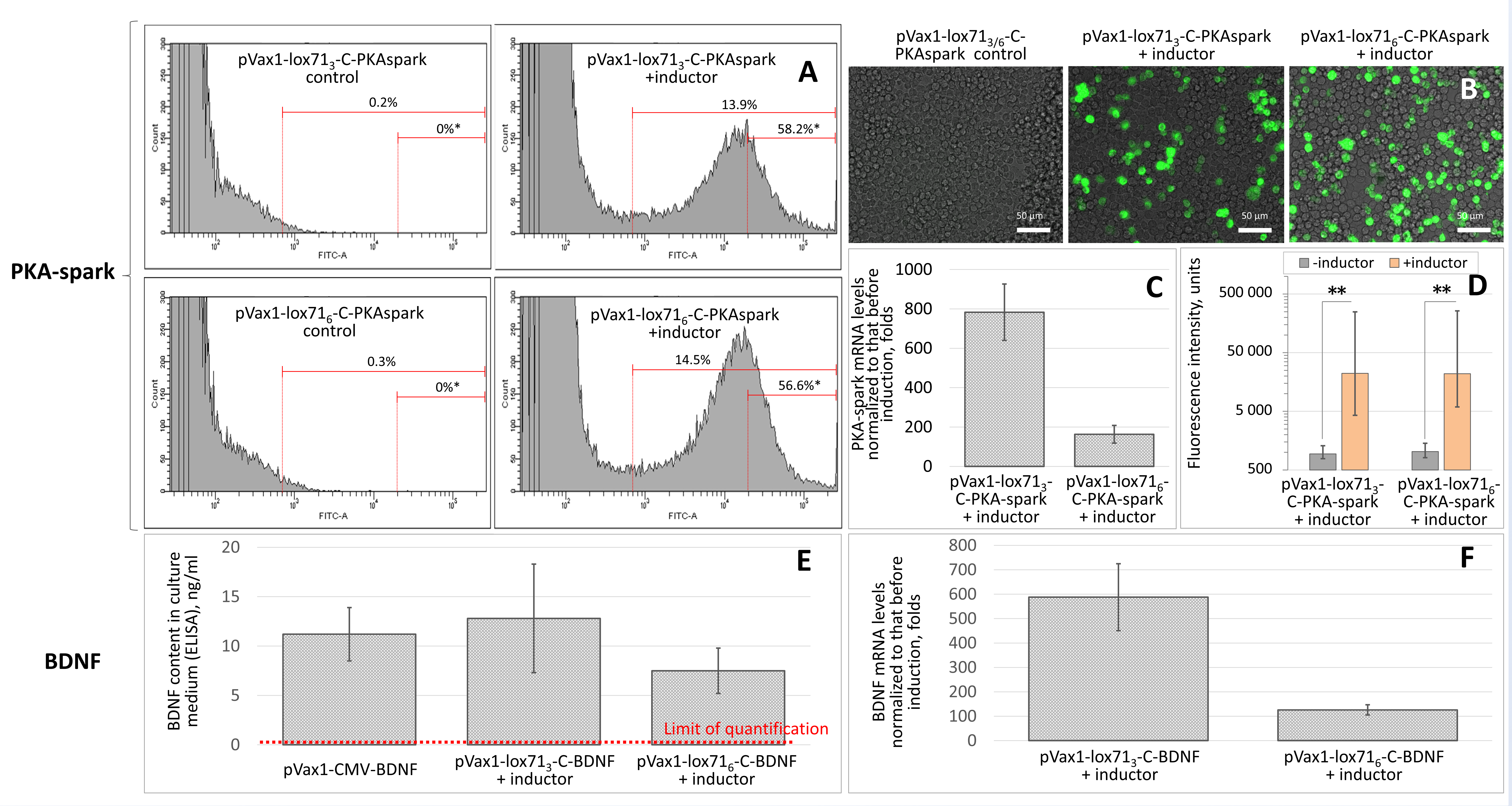
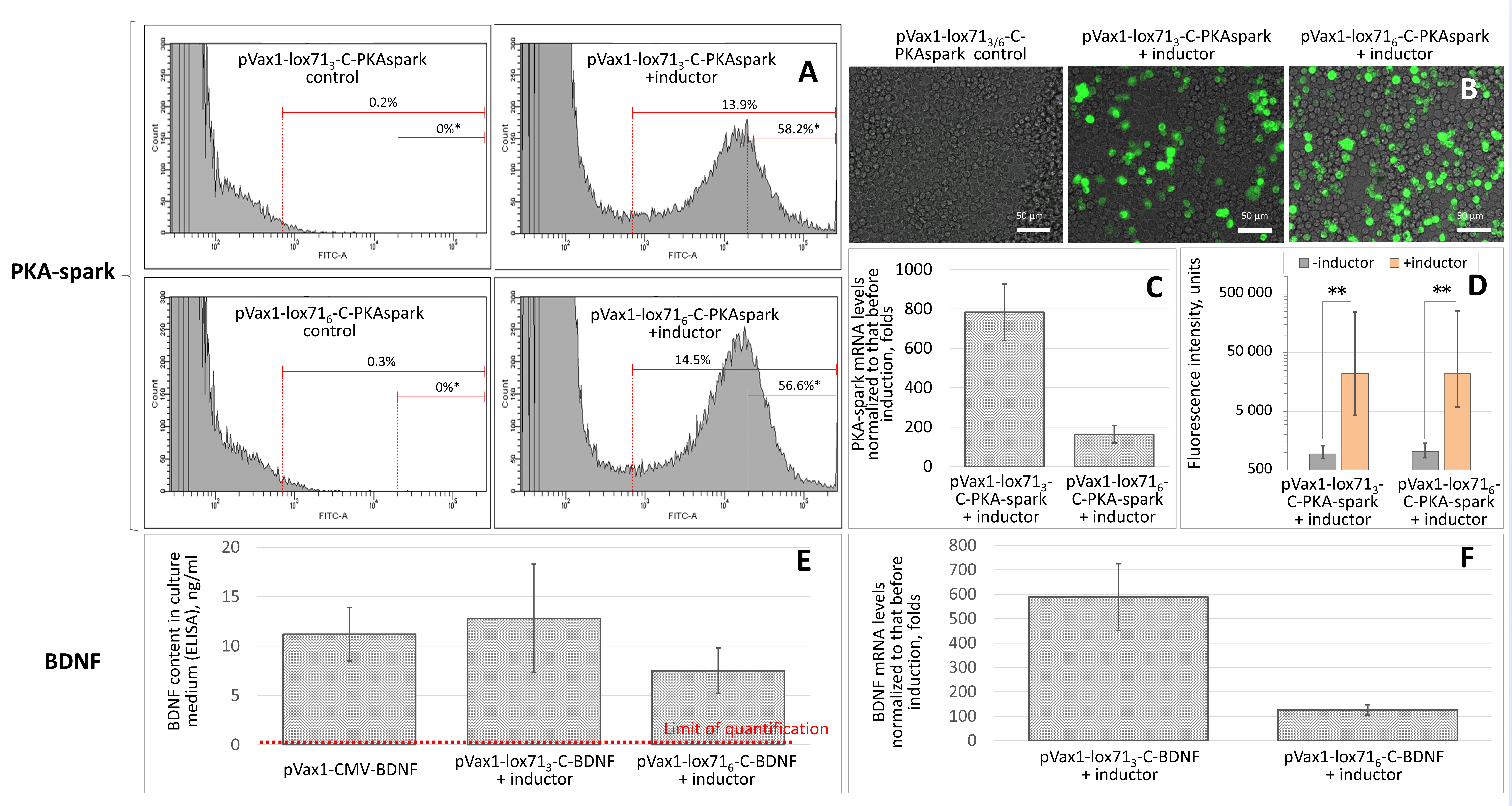
3.2. Cre-VP64/lox71 System Provides a High Level of Transgene Expression
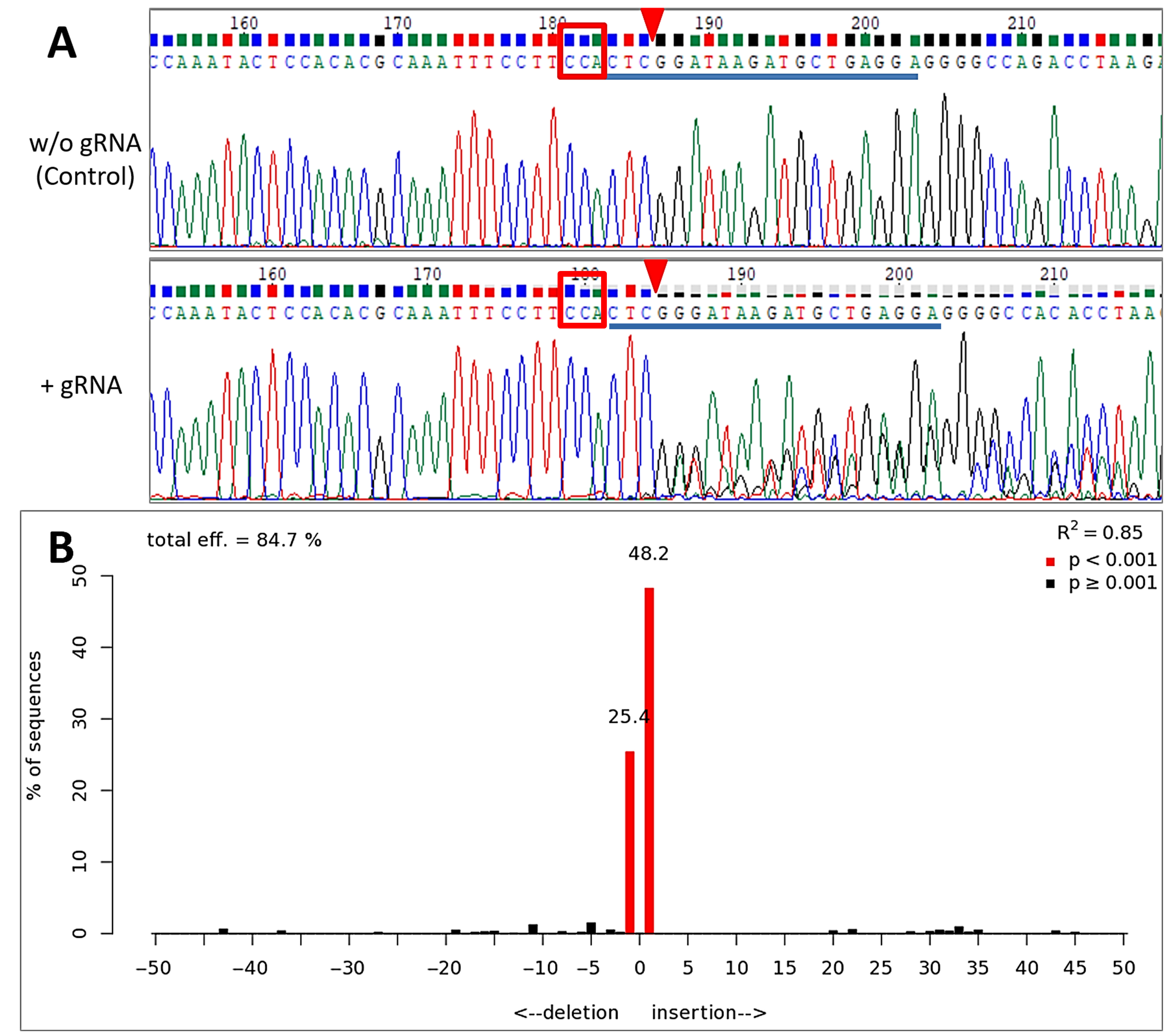
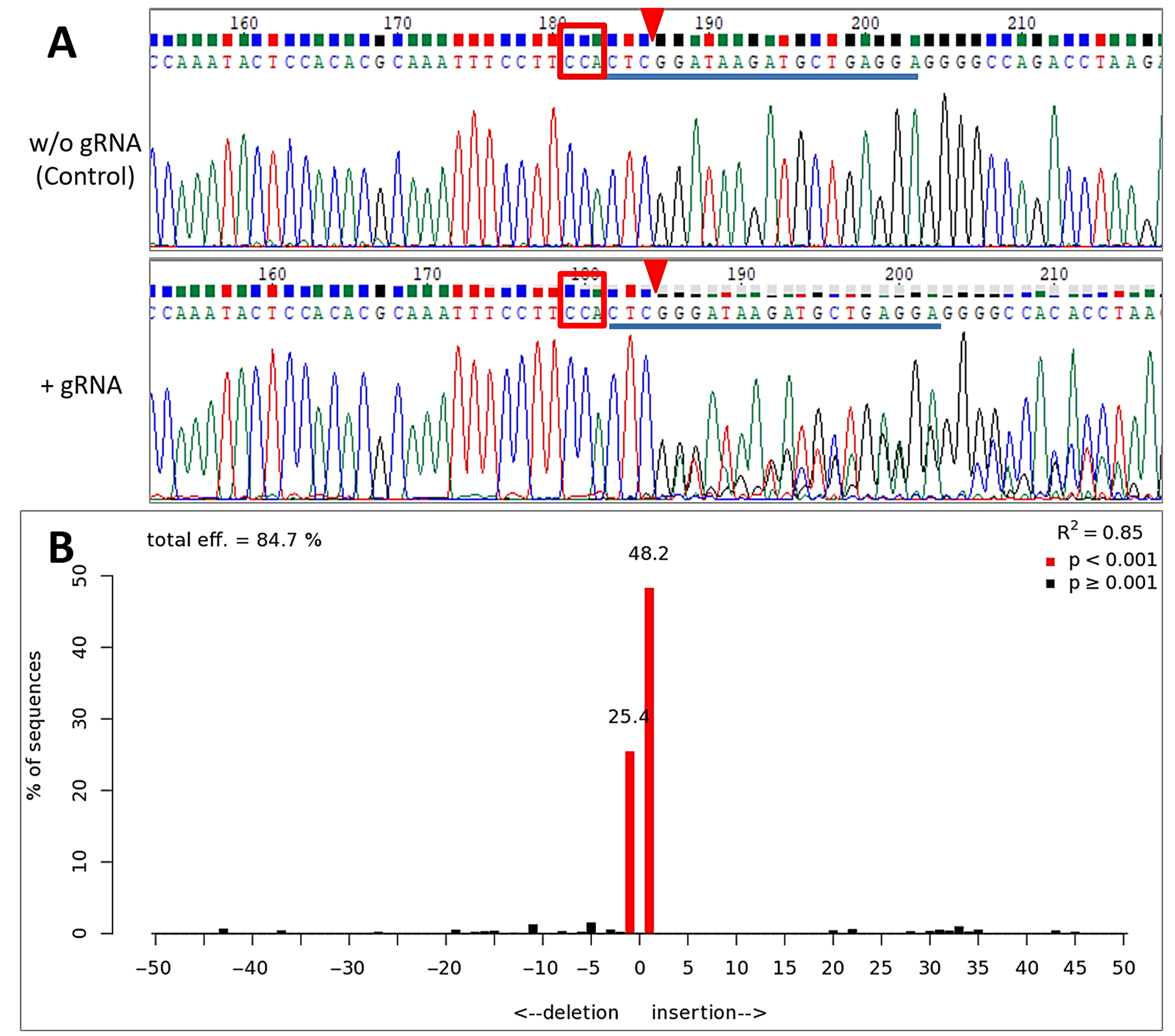
3.3. Inducible Genome Editing Using Cre-VP64/lox71 System
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Das, A.T.; Tenenbaum, L.; Berkhout, B. Tet-On systems for doxycycline-inducible gene expression. Curr. Gene Ther. 2016, 16, 156–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Riesterer, C.; Ayrall, A.M.; Sablitzky, F.; Littlewood, T.D.; Reth, M. Inducible site-directed recombination in mouse embryonic stem cells. Nucleic Acids Res. 1996, 24, 543–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- No, D.; Yao, T.P.; Evans, R.M. Ecdysone-inducible gene expression in mammalian cells and transgenic mice. Proc. Natl. Acad. Sci. USA 1996, 93, 3346–3351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frasor, J.; Chang, E.C.; Komm, B.; Lin, C.Y.; Vega, V.B.; Liu, E.T.; Miller, L.D.; Smeds, J.; Bergh, J.; Katzenellenbogen, B.S. Gene expression preferentially regulated by tamoxifen in breast cancer cells and correlations with clinical outcome. Cancer Res. 2006, 66, 7334–7340. [Google Scholar] [CrossRef] [Green Version]
- Danielian, P.S.; Muccino, D.; Rowitch, D.H.; Michael, S.K.; McMahon, A.P. Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr. Biol. 1998, 8, 1323–1326. [Google Scholar] [CrossRef] [Green Version]
- Oehme, I.; Bösser, S.; Zörnig, M. Agonists of an ecdysone-inducible mammalian expression system inhibit Fas Ligand- and TRAIL-induced apoptosis in the human colon carcinoma cell line RKO. Cell Death Differ. 2006, 13, 189–201. [Google Scholar] [CrossRef] [Green Version]
- Constantino, S.; Santos, R.; Gisselbrecht, S.; Gouilleux, F. The ecdysone inducible gene expression system: Unexpected effects of muristerone A and ponasterone A on cytokine signaling in mammalian cells. Eur. Cytokine Netw. 2001, 12, 365–367. [Google Scholar]
- Ahler, E.; Sullivan, W.J.; Cass, A.; Braas, D.; York, A.G.; Bensinger, S.J.; Graeber, T.G.; Christofk, H.R. Doxycycline alters metabolism and proliferation of human cell lines. PLoS ONE 2013, 8, e64561. [Google Scholar] [CrossRef]
- Dijk, S.N.; Protasoni, M.; Elpidorou, M.; Kroon, A.M.; Taanman, J.W. Mitochondria as target to inhibit proliferation and induce apoptosis of cancer cells: The effects of doxycycline and gemcitabine. Sci. Rep. 2020, 10, 4363. [Google Scholar] [CrossRef]
- Mali, P.; Aach, J.; Stranges, P.B.; Esvelt, K.M.; Moosburner, M.; Kosuri, S.; Yang, L.; Church, G.M. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 2013, 31, 833–838. [Google Scholar] [CrossRef]
- García-Otín, A.L.; Guillou, F. Mammalian genome targeting using site-specific recombinases. Front. Biosci. 2006, 11, 1108–1136. [Google Scholar] [CrossRef] [Green Version]
- Tian, X.; Zhou, B. Strategies for site-specific recombination with high efficiency and precise spatiotemporal resolution. J. Biol. Chem. 2021, 296, 100509. [Google Scholar] [CrossRef]
- Kang, Q.; Sun, Z.; Zou, Z.; Wang, M.; Li, Q.; Hu, X.; Li, N. Cell-penetrating peptide-driven Cre recombination in porcine primary cells and generation of marker-free pigs. PLoS ONE 2018, 13, e0190690. [Google Scholar] [CrossRef] [Green Version]
- Kadonaga, J.T. Perspectives on the RNA polymerase II core promoter. Wiley Interdiscip. Rev. Dev. Biol. 2012, 1, 40–51. [Google Scholar] [CrossRef] [Green Version]
- Sloutskin, A.; Danino, Y.M.; Orenstein, Y.; Zehavi, Y.; Doniger, T.; Shamir, R.; Juven-Gershon, T. ElemeNT: A computational tool for detecting core promoter elements. Transcription 2015, 6, 41–50. [Google Scholar] [CrossRef] [Green Version]
- FlyBase. Available online: http://flybase.org/reports/FBto0000364.html (accessed on 16 May 2022).
- Zhang, Q.; Huang, H.; Zhang, L.; Wu, R.; Chung, C.I.; Zhang, S.Q.; Torra, J.; Schepis, A.; Coughlin, S.R.; Kornberg, T.B.; et al. Visualizing Dynamics of Cell Signaling In Vivo with a Phase Separation-Based Kinase Reporter. Mol. Cell 2018, 69, 334–346.e4. [Google Scholar] [CrossRef] [Green Version]
- Karagyaur, M.; Rostovtseva, A.; Semina, E.; Klimovich, P.; Balabanyan, V.; Makarevich, P.; Popov, V.; Stambolsky, D.; Tkachuk, V. A Bicistronic plasmid encoding brain-derived neurotrophic factor and urokinase plasminogen activator stimulates peripheral nerve regeneration after injury. J. Pharmacol. Exp. Ther. 2020, 372, 248–255. [Google Scholar] [CrossRef]
- Longo, P.A.; Kavran, J.M.; Kim, M.S.; Leahy, D.J. Transient mammalian cell transfection with polyethylenimine (PEI). Methods Enzymol. 2013, 529, 227–240. [Google Scholar] [CrossRef] [Green Version]
- Rusanov, A.L.; Romashin, D.D.; Kozhin, P.M.; Karagyaur, M.N.; Loginov, D.S.; Tikhonova, O.V.; Zgoda, V.G.; Luzgina, N.G. Impact of p53 knockout on protein data set of HaCaT cells in confluent and subconfluent conditions. Data 2022, 7, 27. [Google Scholar] [CrossRef]
- Karagyaur, M.N.; Rubtsov, Y.P.; Vasiliev, P.A.; Tkachuk, V.A. Practical recommendations for improving efficiency and accuracy of the CRISPR/Cas9 genome editing system. Biochemistry 2018, 83, 629–642. [Google Scholar] [CrossRef]
- Brinkman, E.K.; Chen, T.; Amendola, M.; van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014, 42, e168. [Google Scholar] [CrossRef]
- Ghosh, K.; Guo, F.; Van Duyne, G.D. Synapsis of loxP sites by Cre recombinase. J. Biol. Chem. 2007, 282, 24004–24016. [Google Scholar] [CrossRef] [Green Version]
- Lee, L.; Sadowski, P.D. Identification of Cre residues involved in synapsis, isomerization, and catalysis. J. Biol. Chem. 2003, 278, 36905–36915. [Google Scholar] [CrossRef] [Green Version]
- Karagyaur, M.; Dyikanov, D.; Makarevich, P.; Semina, E.; Stambolsky, D.; Plekhanova, O.; Kalinina, N.; Tkachuk, V. Non-viral transfer of BDNF and uPA stimulates peripheral nerve regeneration. Biomed. Pharmacother. 2015, 74, 63–70. [Google Scholar] [CrossRef]
- Cheng, A.W.; Wang, H.; Yang, H.; Shi, L.; Katz, Y.; Theunissen, T.W.; Rangarajan, S.; Shivalila, C.S.; Dadon, D.B.; Jaenisch, R. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res. 2013, 23, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Goverdhana, S.; Puntel, M.; Xiong, W.; Zirger, J.M.; Barcia, C.; Curtin, J.F.; Soffer, E.B.; Mondkar, S.; King, G.D.; Hu, J.; et al. Regulatable gene expression systems for gene therapy applications: Progress and future challenges. Mol. Ther. 2005, 12, 189–211. [Google Scholar] [CrossRef]
- Bock, C.; Datlinger, P.; Chardon, F.; Coelho, M.A.; Dong, M.B.; Lawson, K.A.; Lu, T.; Maroc, L.; Norman, T.M.; Song, B.; et al. High-content CRISPR screening. Nat. Rev. Methods Primers 2022, 2, 8. [Google Scholar] [CrossRef]
- Dong, M.B.; Tang, K.; Zhou, X.; Zhou, J.J.; Chen, S. Tumor immunology CRISPR screening: Present, past, and future. Trends Cancer 2022, 8, 210–225. [Google Scholar] [CrossRef] [PubMed]
- Ibraheim, R.; Tai, P.W.L.; Mir, A.; Javeed, N.; Wang, J.; Rodríguez, T.C.; Namkung, S.; Nelson, S.; Khokhar, E.S.; Mintzer, E.; et al. Self-inactivating, all-in-one AAV vectors for precision Cas9 genome editing via homology-directed repair in vivo. Nat. Commun. 2021, 12, 6267. [Google Scholar] [CrossRef] [PubMed]
- Ortinski, P.I.; O’Donovan, B.; Dong, X.; Kantor, B. Integrase-deficient lentiviral vector as an all-in-one platform for highly efficient CRISPR/Cas9-mediated gene editing. Mol. Ther. Methods Clin. Dev. 2017, 5, 153–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, S.G.; Sayers, E.J.; He, L.; Narayan, R.; Williams, T.L.; Mills, E.M.; Allemann, R.K.; Luk, L.Y.P.; Jones, A.T.; Tsai, Y.H. Cell-penetrating peptide sequence and modification dependent uptake and subcellular distribution of green florescent protein in different cell lines. Sci. Rep. 2019, 9, 6298. [Google Scholar] [CrossRef]
- Hoersten, J.; Ruiz-Gómez, G.; Lansing, F.; Rojo-Romanos, T.; Schmitt, L.T.; Sonntag, J.; Pisabarro, M.T.; Buchholz, F. Pairing of single mutations yields obligate Cre-type site-specific recombinases. Nucleic Acids Res. 2022, 50, 1174–1186. [Google Scholar] [CrossRef]
- Soni, A.; Augsburg, M.; Buchholz, F.; Pisabarro, M.T. Nearest-neighbor amino acids of specificity-determining residues influence the activity of engineered Cre-type recombinases. Sci. Rep. 2020, 10, 13985. [Google Scholar] [CrossRef]
- Nagy, A. Cre recombinase: The universal reagent for genome tailoring. Genesis 2000, 26, 99–109. [Google Scholar] [CrossRef]
- Hayashi, S.; McMahon, A.P. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: A tool for temporally regulated gene activation/inactivation in the mouse. Dev. Biol. 2002, 244, 305–318. [Google Scholar] [CrossRef] [Green Version]
- Bessen, J.L.; Afeyan, L.K.; Dančík, V.; Koblan, L.W.; Thompson, D.B.; Leichner, C.; Clemons, P.A.; Liu, D.R. High-resolution specificity profiling and off-target prediction for site-specific DNA recombinases. Nat. Commun. 2019, 10, 1937. [Google Scholar] [CrossRef]
- Zhang, Z.; Lutz, B. Cre recombinase-mediated inversion using lox66 and lox71: Method to introduce conditional point mutations into the CREB-binding protein. Nucleic Acids Res. 2002, 30, e90. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karagyaur, M.; Dyikanov, D.; Tyurin-Kuzmin, P.; Dzhauari, S.; Skryabina, M.; Vigovskiy, M.; Primak, A.; Kalinina, N.; Tkachuk, V. A Novel Cre/lox71-Based System for Inducible Expression of Recombinant Proteins and Genome Editing. Cells 2022, 11, 2141. https://doi.org/10.3390/cells11142141
Karagyaur M, Dyikanov D, Tyurin-Kuzmin P, Dzhauari S, Skryabina M, Vigovskiy M, Primak A, Kalinina N, Tkachuk V. A Novel Cre/lox71-Based System for Inducible Expression of Recombinant Proteins and Genome Editing. Cells. 2022; 11(14):2141. https://doi.org/10.3390/cells11142141
Chicago/Turabian StyleKaragyaur, Maxim, Daniyar Dyikanov, Pyotr Tyurin-Kuzmin, Stalik Dzhauari, Mariya Skryabina, Maksim Vigovskiy, Alexandra Primak, Natalia Kalinina, and Vsevolod Tkachuk. 2022. "A Novel Cre/lox71-Based System for Inducible Expression of Recombinant Proteins and Genome Editing" Cells 11, no. 14: 2141. https://doi.org/10.3390/cells11142141
APA StyleKaragyaur, M., Dyikanov, D., Tyurin-Kuzmin, P., Dzhauari, S., Skryabina, M., Vigovskiy, M., Primak, A., Kalinina, N., & Tkachuk, V. (2022). A Novel Cre/lox71-Based System for Inducible Expression of Recombinant Proteins and Genome Editing. Cells, 11(14), 2141. https://doi.org/10.3390/cells11142141